

Abstract
We have determined the solution NMR structure of the intermembrane space domain (IMSD) of the human mitochondrial ATPase associated with various activities (AAA) protease known as AFG3-like protein 2 (AFG3L2). Our structural analysis and molecular dynamics results indicate that the IMSD is peripherally bound to the membrane surface. This is a modification to the location of the six IMSDs in a model of the full length yeast hexaoligomeric homolog of AFG3L2 determined at low resolution by electron cryomicroscopy 1. The predicted protein-protein interaction surface, located on the side furthest from the membrane, may mediate binding to substrates as well as prohibitins.
Keywords: m-AAA protease, Molecular dynamics, NMR structure
INTRODUCTION
Here we report the solution NMR structure of the intermembrane space domain (IMSD, residues) of human mitochondrial ATPase associated with various activities (AAA) protease AFG3-like protein 2 (AFG3L2). In humans, AFG3L2 forms homo- or hetero-oligomeric complexes with paraplegin (49% identical) in the mitochondrial inner membrane (IM). They are named m-AAA proteases, meaning that their catalytic domains are located in the mitochondrial matrix. The catalytic domains of both AFG3L2 and paraplegin are made up of two subunits, the ATPase associated AAA domain and the metallopeptidase domain (PD) responsible for substrate proteolysis. A single chain of AFG3L2 or paraplegin has two transmembrane (TM) helices, so that the topology in the IM results in the short N-terminus and large catalytic domain on the matrix side and a small ~70-residue IMSD on the lumen side. Established and putative functions for m-AAA proteases have been extensively characterized and several recent reviews are available 2–4. Although the crucial functions of m-AAA proteases are proteolytic processing and degradation of both non-membrane and membrane-embedded substrates, the role of IMSDs remains to be determined.
AFG3L2 and paraplegin have mammalian, plant, and fungal homologs, including yeast Yta10 and Yta12 (59% identical to AFG3L2) in addition to the well-studied bacterial homolog, FtsH (Filamentous temperature sensitive H, 45% sequence identity), which forms only homo-oligomers, and is essential for survival in Escherichia coli. The IMSD of AFG3L2 belongs to a conserved protein family (Pfam), Pfam PF06480.
Prior to the NMR structure reported here, no atomic resolution information was available for domains in this diverse Pfam. However, the overall architecture of a m-AAA protease was recently determined in a 12 Å-resolution cryo-electron tomography (CET) map of yeast Yta10/Yta12 hetero-oligomer solubilized in a detergent micelle 1. The authors observed symmetrical hexamers that rely on residues in PD for oligomerization. In their model, the six IMSDs splayed-out in a hexagon around 12 clustered TM helices and have no contact with IMSDs on other subunits 1. Prediction of the IM position based on the location of the TM helices resulted in the IMSDs being half buried in the membrane. Our results, however, are inconsistent with this aspect of the model, rather, our data suggests that this domain has only peripheral contacts with the IM. We propose a modification to the previous model that is consistent with our structural data and molecular dynamics (MD) simulations.
MATERIALS AND METHODS
Cloning, Expression and Purification
The selected IMSD fragment of the afg3l2 gene was cloned into a pET15 expression vector (NESG Clone ID HR6741A-15.1) as described elsewhere 5,6. The IMSD, residues 164–251 of AFG3L2, included 11 non-native N-terminal residues (MGHHHHHHSHM). Expression and purification were conducted following standard protocols of the Northeast Structural Genomics Consortium (NESG) to prepare [U-13C, 15N]- and U-15N, 5% biosynthetically-directed 13C (NC5) samples 5,6 and details can be found in Supplemental Procedures. The final NMR samples were 0.7–1.0 mM in the NMR buffer: 20 mM MES, 100 mM NaCl, 5 mM CaCl2, 10 mM dithiothreitol, and 0.02% NaN3 at pH 6.5. The protein was monomeric under the conditions used in the NMR experiments based on analytical static light scattering in-line with gel filtration chromatography and correlation time estimates 15N relaxation data (τc 7.1 ± 0.5 ns, Fig. S1).
NMR and structure determination
NMR data were collected at 298 K on NC and NC5 samples of ~300 µL in 5 mm Shigemi NMR tubes on 600 MHz Varian Inova spectrometer with a 5-mm HCN cold probe and 850 MHz Bruker Avance III spectrometer equipped with a conventional 5-mm HCN probe. A description of NMR experiments and methods for structure determination and refinement can be found in Supplemental Procedures. The assigned 1H-15N HSQC spectrum is provided as Fig. S2. Chemical shifts, NOESY peak lists, and raw FIDS were deposited in the BioMagResDB ID 18156). The final ensemble of 20 models and NMR resonance assignments were deposited to the Protein Data Bank ID 2LNA.
MD simulations
MD studies of the lowest energy structure from the NMR ensemble (residues 16 – 95) with a 1-Palmitoyl-2-oleoylphosphatidylcholine (POPC) membrane were performed using NAMD version 2.9 7 with the CHARMM27 8 force field. The Visual Molecular Dynamics software, VMD version 1.8.7 9, was used for simulation setup and MD trajectory analysis. A pre-equilibrated POPC bilayer with an 80 Å 2 surface was generated with the VMD Membrane Builder plugin. The IMSD was positioned > 8 Å above the membrane and the system was solvated into a water box and neutralize with NaCl. Equilibration and simulations were performed similar to those reported in the KcsA tutorial for VMD and NAMD 10 and is described in detail in the Supplemental Procedures.
RESULTS
Solution NMR structure
The human AFG3L2 IMSD features an uncommon mixed α+β fold comprised of two α-helices (α1, 20–30; α2, 67–81) and a five-stranded β-sheet (β1, 16–17; β2, 33–39; β3, 43–48; β4, 60–63; β5, 91–93;) arranged in a βαβββαβ topology (Fig. 1A–C). The first helix (α1) has a break at Tyr27 resulting in a bend in the helix. The β-sheet has an up-up-down-up-up topology with the order β1-β4-β3-β2-β5 and packs against the two helices to form a compact structure, whereas a poorly-defined loop is located between β3 and β4 (loop3–4; Thr49–Gln58). Structural statistics for the NMR ensemble are presented in Supplemental Table S1.
Figure 1. Structure and Topology of AFG3L2 IMSD.
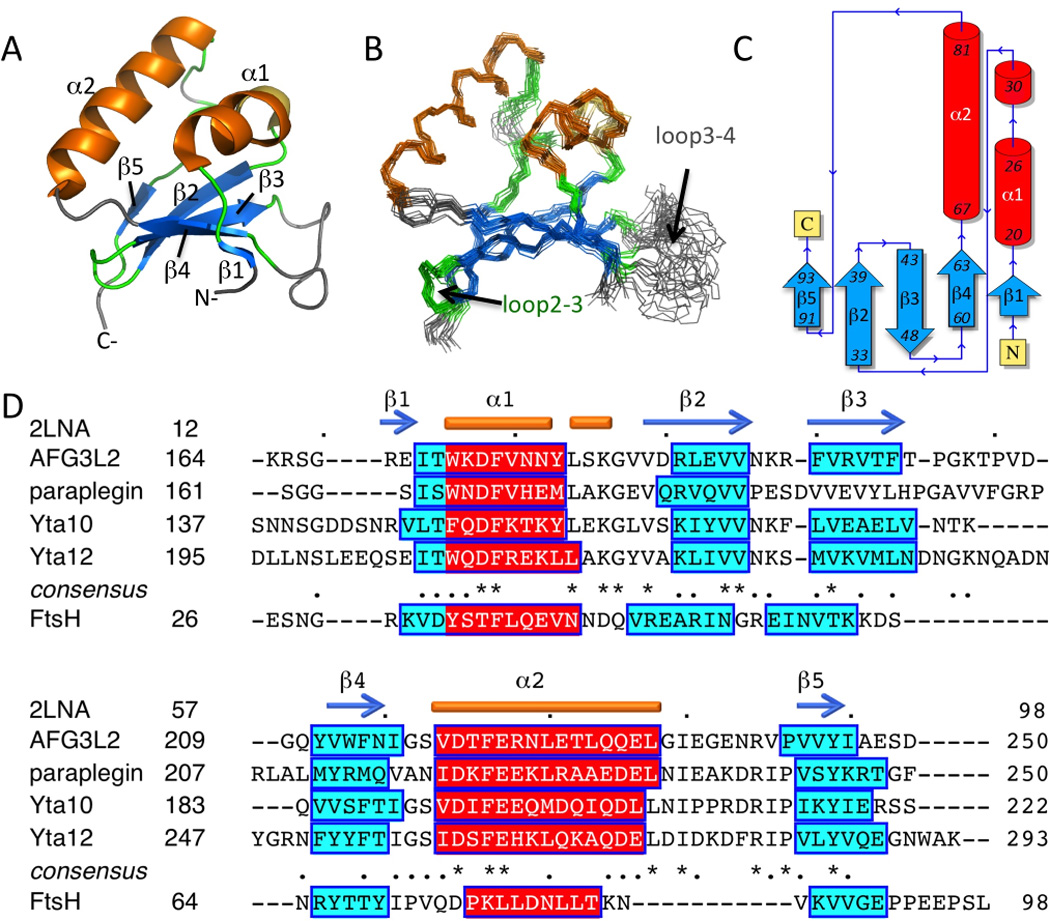
(A) Cartoon representation of the lowest energy NMR structure, α-helices and β-strands are shown in blue and orange, and loops are colored green or grey, if they were not ordered residues. (B) Superposition of the final ensemble of 20 models (residues 14–96). (C) Domain topology obtained using PDBSum 35. (D) IMSDs from human AFG3L2 and paraplegin, yeast Yta10 and Yta12, and E. coli FtsH. Alignment of human and yeast sequences was performed with Clustal W 2.0 36 and the consensus is reported. FtsH alignment is based on secondary structure prediction by PSIPRED 3.0 11. Secondary structure for the NMR structure of human AFG3L2 IMSD, PDB ID 2LNA, is shown above the sequences.
Homologs have the same predicted secondary structure
IMSDs of human paraplegin, yeast Yta10 and Yta12, share 36%, 40%, and 43% sequence identity with that of human AFG3L2. However, these ~85-residue domains were all predicted to have the same order of secondary structural elements by PSIPRED 3.0 11 (Fig. 1D). The prediction for the human AFG3L2 IMSD was in good agreement with the NMR structure with the exception that the first short β-strand was slightly shifted from the PSIPRED prediction (Fig. 1D). Similarly, the ~70-residue E. coli FtsH periplasmic domain had an equivalent secondary structure prediction although it is ~14 residues shorter (Fig. 1D). This result suggests that the IMSD structure will be conserved within Pfam PF06480, including bacterial homologs.
Structural alignment comparison and protein-protein interaction prediction
Structural alignment analysis of AFG3L2 IMSD by Dali 12 found no protein structures with RMSD < 2.5 Å or sequences with > 20% identity. Dali identified the 60S eukaryotic ribosome subunit protein L38 (PDB ID 3U5E:k and 4A18:p, Z-scores 4–5) as a similar topology protein. L38 is part of a large complex of ribosomal proteins. According to predictions of protein-protein interactions by PredUS 13, residues near α1 (Asn 26) and part of the α2–loop-β5 (Gly82–Glu86, with the highest score for Gly85) are likely to interact with other proteins (Fig.3C and 3D). The prediction is primarily due to structural alignment with the N-terminal domain of the molecular chaperone heat shock protein Hsp90 (Fig. S4). This domain contains an ATP binding site and binds co-chaperones (reviewed in 14). In human cells, Hsp90 binds to more than 10 known co-chaperones that together with Hsp90 regulate the stability and activation of other proteins, many of which are involved in cellular signaling pathways. For example, the co-chaperone Cdc37 regulates Hsp90 interactions with kinases and the stability of the respective kinases determined the extent of association with Hsp90, with unstable kinases having increased affinity 15. Taken together, the structural alignment results indicate a role in the assembly of protein-protein complexes that could regulate substrate recognition.
Figure 3. (A) MD simulations of IMSD and POPC membrane.
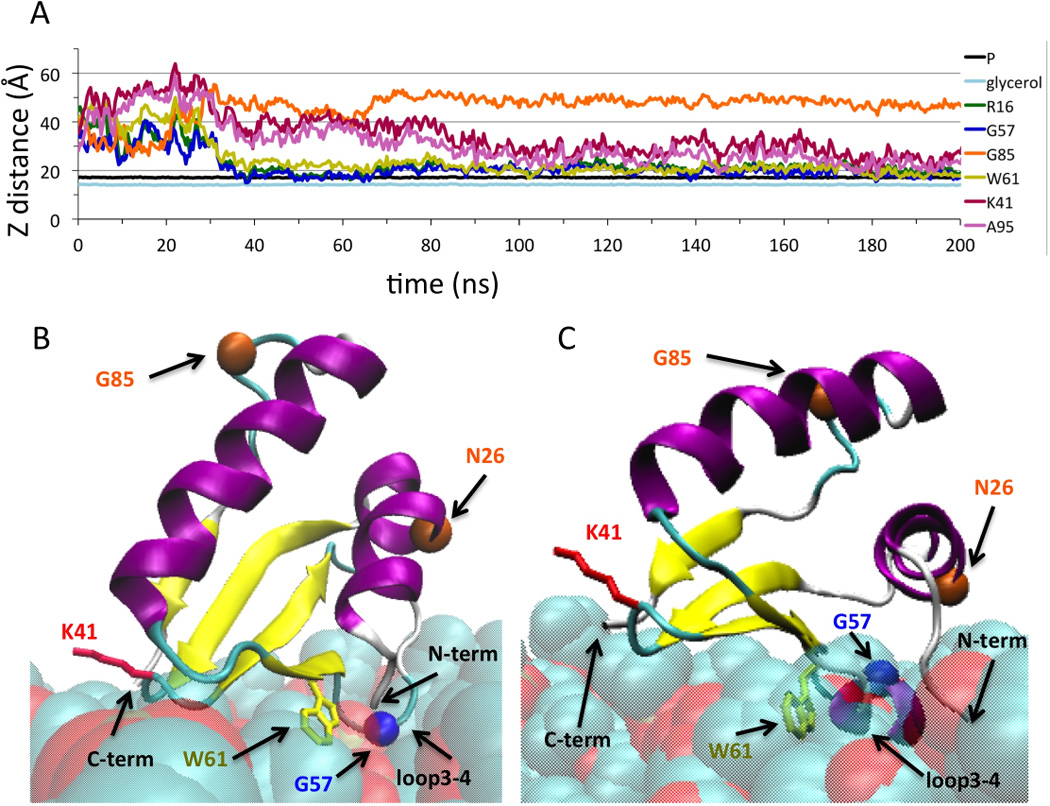
To characterize the extent of membrane insertion, the distance in the Z-dimension from the center of mass of the lipid atoms to selected lipid atoms or residues atoms was calculated as a function of simulation time. Select protein atoms used for the analysis were the Arg16 (atom N) and A95 (atom C) to represent the N- and C-termini, respectively, Lys41 side chain (atom NZ) to represent loop2–3, Gly57 (atom CA) to represent loop3–4, Trp61 side chain (atom CZ2), and Gly85 (atom CA). Lipid atoms used were glycerol backbone (atoms C1–C3) and phosphates (atom P1). (B) and (C) Cartoon representation at 200 ns from the first and second MD simulation trajectories. The residues and atoms depicted in (A) are indicated as well as Asn26 and Gly85, shown with orange circles, that are predicted by PredUS 13 to have a highly propensity for protein-protein interactions. Figures were generated using VMD software and scripts 9.
Membrane interaction surface
In AFG3L2, the interaction of the IMSD with the membrane is governed, in part, by the tethering to the TM helices at its N- and C-termini. Analysis of the IMSD surface electrostatic surface potential 16 revealed a patch of basic and hydrophobic residues at one end of the protein that constitutes a putative membrane interaction surface (Fig. 2A–D). It is primarily made up of loops between secondary structural elements, including loop2–3 and loop3–4 (Fig. 2A–D). Solvent exposed hydrophobic residues, as well as basic residues from this surface are shown in Fig.2A and 2B. It is well known that Trp and Tyr residues have a preferred localization at membrane interfaces and that Arg and Lys residues can interact with phospholipid headgroups.
Figure 2. Structure and membrane interaction surface of IMSD.
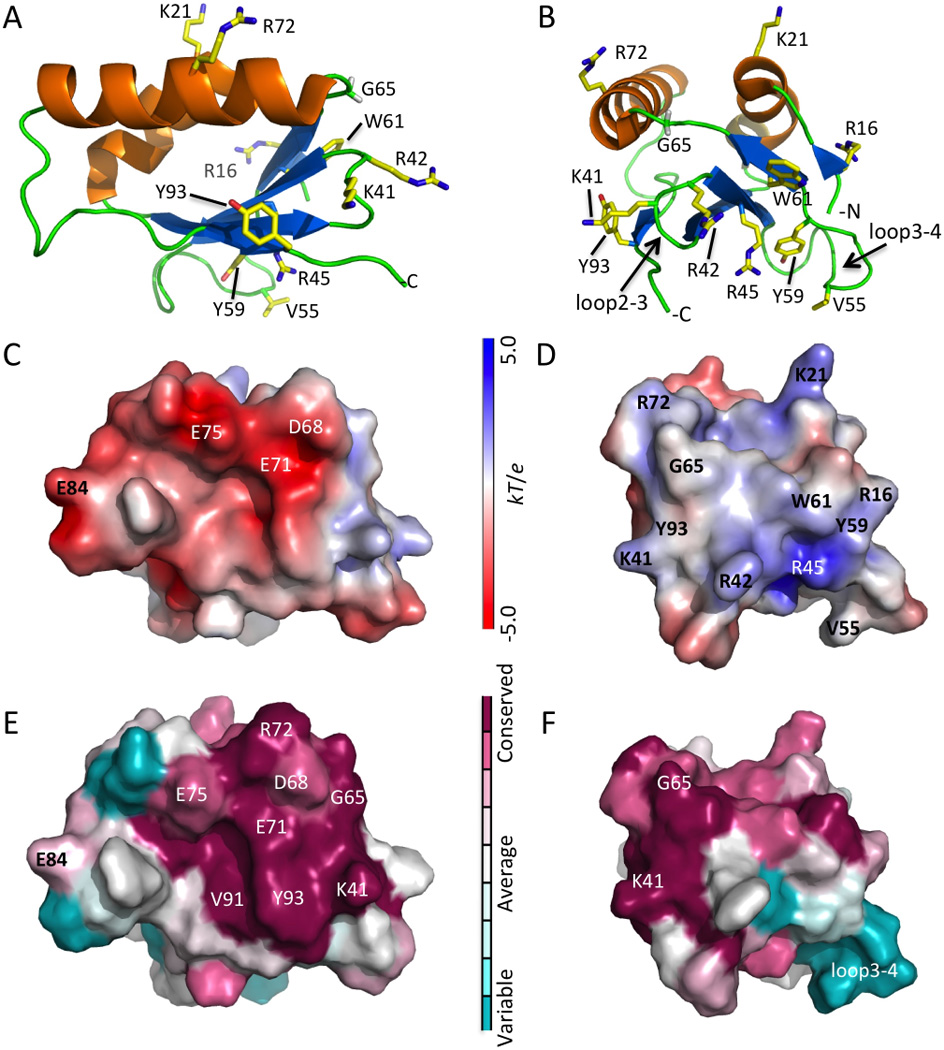
(A) Solution NMR structure of the human IMSD from AFG3L2 (residues 14–96), selected side chains are shown. (B) Cartoon figure rotated from A to show the proposed membrane interaction residues. (C and D) APBS 16 solvent accessible electrostatic surface potential with same orientation as in A and B showing negative (red), neutral (white), and positive (blue) charges. (E and F) ConSurf 17 image showing the conserved surface residues. Residue coloring, reflecting the degree of residue conservation for selected eukaryotic homologs from Pfam06480 (42 sequences). All figures were rendered using PyMOL (DeLano Scientific).
ConSurf 17 analysis revealed that conserved residues across the eukaryotic homologs are located on one side and that the putative membrane binding surface has few conserved residues. The conserved surface patch has an acidic surface resulting primarily from residues Glu71 and Glu75 (Fig.2C and 2E), but the function of this patch is unknown. On the putative membrane-binding surface, only a few residues are conserved (Lys41 and G65), and loop3–4 is variable among the sequence homologs (Fig. 2F).
MD simulations of IMSD and POPC membrane
In order to elucidate the specific membrane interactions and orientation of the IMSD relative to the mitochondrial IM, extensive all-atom MD simulations were carried out. A lipid bilayer membrane of POPC was chosen since phosphatidylcholines are the most abundant phospholipids in the mitochondrial IM (~38% overall)18. All-atom simulations of peripheral protein-membrane interactions have been used to determine the extent of the interactions are typically performed for tens to thousands of ns (several are reviewed in 19,20). During the MD simulation, the IMSD spontaneously approached and interacted with the membrane within 40 ns. We ran the MD trajectory for 200 ns, in three independent computations, until the protein orientation with respect to the membrane appeared to converge.
At the end of the three MD trajectories, IMSD orientations were in agreement with our proposed membrane interaction surface based on the surface charge distribution (Fig. 2D). The C-terminal end of α2 was positioned furthest from the POPC membrane patch (Gly85 immediately follows this helix) (Fig. 3A). The Trp61 sidechain was near, or embedded in, the membrane at the end of each simulation (Fig. 3A). Lys41 in loop2–3 was close enough to interact with the phosphate moity of the lipids at certain times in the MD simulations. Near the end of the MD trajectories, loop3–4 (Gly57), and both N- and C-termini were within 10 Å of the membrane surface, demonstrating that these regions of the IMSD can simultaneously interact with the membrane (Fig.3B and 3C). In the second MD run only, loop3–4 formed a helix at 50 ns and remained helical for the rest of the simulation (Fig 3C), indicating that this loop has helical propensity when in proximity to the membrane. The results from all three MD runs are provided in Fig. S3.
Model for full-length AFG3L2
Since experimental characterization of the membrane interaction has not been successful, our membrane interaction model of the IMSD is based on the solution structure of the domain and MD simulation results. Titration of the NMR sample with dodecyl phosphocholine micelles resulted in complete unfolding of the protein and addition of POPC liposomes resulted in no change in the 1H-15N HSQC spectrum (data not shown), although disappearance of NH peak intensities would be expected of the IMSD was in slow exchange with the liposomes. These results further suggest that this water-soluble domain is not likely to be half-buried in the mitochondrial membrane.
The IMSD structure is consistent with the shape of the domain obtained from the CET map of the Yta10/Yta12 hexaoligomer solubilized in n-Octyl-β-D-Glucopyranoside (β-OG) detergent micelles (Fig.4A and 4B). Modeling of the IM based on the location of the 12 TM helices resulted in the IMSDs being approximately half-buried in the membrane (Fig. 4B). As noted by the authors, the cryo-EM structure determined in a micelle environment may differ from the native structure in the mitochondrial IM 1. Indeed, since the β-OG micelles have a radius of gyration of ~30 Å21, the micelle surface surrounding the TM helices has sharp curvature compared to an membrane bilayer, and this could influence the position of IMSDs relative to the TM helices. In our new model, the IMSDs are reorganized so that they are positioned at the surface of the membrane (Fig. 4C). According to UniProt predictions 22 of the TM helix sequences (UniProt ID Q9Y4W6), there is a 4–5 residue “spacer” on either side of the domain (Arg16–Tyr93) to connect to the respective TM helices. Although the structure of this linker is unknown, this length appears sufficient to allow the required movement of the IMSDs. Also, the IMSDs may be situated closer together as well as above the membrane in the structure of the m-AAA protease in its native membrane (not shown in model).
Figure 4. Model of AFG3L2.
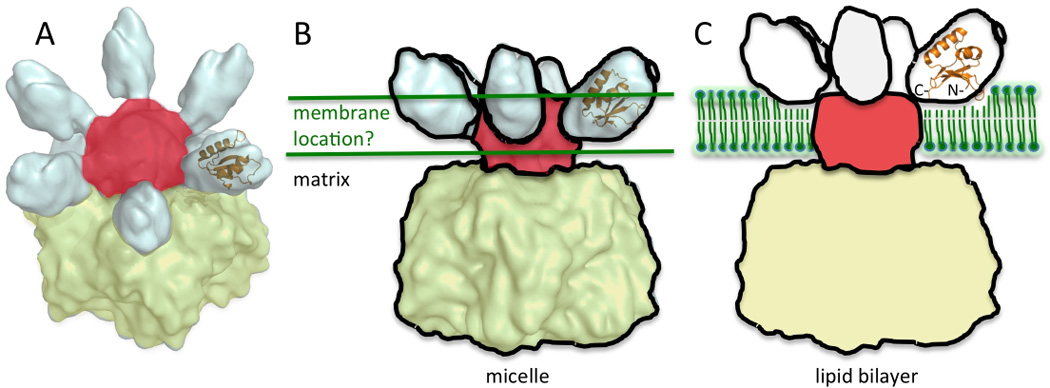
(A) Cryo-EM structure of the yeast m-AAA protease with the human AFG3L2 shown as an orange cartoon manually fit into the density. The cryo-EM data was obtained from EMDataBank (ID 1712) 1, and PyMOL was used to generate the image. The matrix located AAA and PD domains are colored yellow, TM helices are colored red, and IMSDs are in white. (B) The membrane, depicted as green lines, is drawn based on the location of the TM helices in the detergent micelle. (C) Model of m-AAA protease in a lipid bilayer. The six IMSDs are positioned above the membrane depicted as green phospholipids. The IMSD cartoon is shown as predicted by MD simulations.
DISCUSSION
Prohibitin interactions with IMSDs
The m-AAA proteases have many experimentally determined protein substrates (reviewed in 2–4). However, the only confirmed interaction partner for an IMSD is between the E. coli FtsH periplasmic domain and the HflK and HflC membrane protein complex (HflKC) 23, where HflK and HflC are related to eukaryotic prohibitin 1 (Phb1) and prohibitin 2 (Phb2). Prohibitins have a short hydrophobic stretch at their N-termini that anchors them to the membrane and the remaining ~30 kDa resides in the intermembrane space. This region of prohibitin consists of a so-called prohibitin (PHB) and a predicted coiled-coil (CC) segment at the C-terminal end 24. Purified prohibitin complexes from yeast assumed large ~1 MDa ring-shaped structures with a diameter of ~20–27 nm containing equal amounts of Phb1 and Phb2 subunits by single particle electron microscopy 25. The CC segment was required for assembly of the prohibitin complex 25. However, it is not known if the CC segment and/or the PHB domain interact with IMSDs.
In human mitochondria, both homo-oligomers of AFG3L2 and hetero-oligomers of AFG3L2 and paraplegin were found in a ~900 kDa protein complex by gel filtration and Blue Native PAGE 26,27, significantly larger than the ~400–500 kDa expected for m-AAA hexaoligomers. These complexes contain prohibitins as was demonstrated in mouse mitochondria 27, and in yeast, where prohibitins were associated with Yta10 and Yta12 in high molecular weight complexes 28. In E. coli a molecular composition of (FtsH)6(HflKC)6 was proposed 29, a result that supports a 1:1 interaction with the IMSD and each pair of prohibitin proteins. The human (AFG3L2)6(Phb1–Phb2)6 complex would be 866 kDa, which is consistent with the experimentally determined molecular weight of the complex. This supports a complex with 12 copies of Phb1–Phb2 in a ring around the m-AAA protease; a structure that seems likely based on the spread-out six-fold positioning of IMSDs in the m-AAA hexaoligomer with a 13 nm diameter 1. This would fit nicely inside the prohibitin central cavity of 9–16 nm 25. It is also consistent with our model with the IMSD protein-protein interaction interface on the top, outside surface of the hexaoligomer and the membrane-interaction surface at the bottom (Fig. 4). However, it remains to be experimentally determined whether the protease will be inside or outside the prohibitin ring structure 24. Estimations of the number of copies of Phb1–Phb2 in the yeast complex vary from 12–16 or 16–20 25,30; it should be noted that the ring complexes had variability in their shape and size 25.
The role of prohibitins and their interactions m-AAA proteases is not well understood. In yeast mitochondria, prohibitins may act as negative regulators of m-AAA proteolysis of nonassembled IM proteins or newly synthesized proteins because deletion of Phb1 or Phb2 results in accelerated proteolysis by the m-AAA protease 28,30. Prohibitins could prevent accessibility of targets to m-AAAs by binding to the substrate or by changing the protease activity. A direct interaction between prohibitins and newly synthesized mitochondria proteins was demonstrated in yeast by co-immunoprecipitation indicating a role of prohibitins as chaperones that stabilize these proteins 30. Furthermore, human Phb1 shares sequence similarity with the bacterial chaperone GroEL, called Hsp60 in eukaryotes 30. It has also been suggested that prohibitins may act as a scaffold to maintain the correct organization of proteins in a lipid raft environment in order to physically separate the substrate from the protease 24,31. Taken together, m-AAA proteases and prohibitins may play a role in stabilization of membrane protein complexes and other chaperone-like activities 2–4.
Possible role for IMSD in substrate recognition
In general, recognition involves substrate binding to the AAA domain, followed by ATP-dependent unfolding and translocation, and progressive proteolysis by the PD domain. However, substrate recognition by m-AAA proteases is not entirely understood. In E. coli, FtsH recognized membrane proteins with 10–20 residue unstructured N- or C-terminal ends 32. Recognition may not have sequence specificity but rather seems to target a broad range of unstructured polypeptides 32. These would include misfolded, damaged, or non-assembled subunits. In yeast mitochondria, deletion of the IMSD along with the two TM segments impaired proteolysis of integral membrane proteins, indicating a possible role for IMSDs in recognition of membrane proteins 33. For proteins that have residues in the intermembrane space this role could be direct or indirect through interactions with adaptor/chaperone proteins.
Another complexity is that substrate recognition may be different for different targets. For example, deletion of the periplasmic domain in E. coli resulted in FtsH that could still degrade the membrane protein SecY but not the cytosolic protein CII 23. This FtsH mutant could no longer bind to HflKC, suggesting that prohibitins can modulate interactions of some protein targets with the m-AAA protease. In addition to prohibitins, it is likely that other specific adaptors/chaperones interact with diverse m-AAA target proteins as is the case for other AAA proteases such as the prokaryotic Clp proteases 34. Based on our data and current understanding of m-AAA function, we suggest that protein-protein interactions with IMSDs will modulate degradation of a subset of protein targets.
Supplementary Material
HIGHLIGHTS.
The first atomic-resolution structure of the IMSD of a m-AAA protease is presented
MD simulations reveal the peripheral membrane interaction of the IMSD
We model the location of the six IMSDs in the full-length AFG3L2 hexaoligomer
ACKNOWLEDGEMENT
Grant sponsor: National Institute of General Medical Sciences Protein Structure Initiative - Biology (PSI-Biology); Grant numbers: U54-GM094597 in support of the NESG (G. T. M. and M.K.) and U01-GM094622 in support of the Mitochondrial Protein Partnership (MPP) (John L. Markley, PI, and David J. Pagliarini, Co-PI). The target was nominated by the MPP, University of Wisconsin-Madison. The NAMD work was supported by computing time allocation from the Ohio Supercomputer Center (OSC), start-up project PMIU0117 to T.A.R.
We thank J. Mueller for assistance with paralellization of NAMD on the Miami University Redhawk Cluster and OSC. We thank Y.J. Huang and J.K. Everett for construct design, and H. Janua, E. Kohan, R. Shastry, and T. Acton at the Rutgers’ protein production facility for technical support. Most NMR data collection was conducted at the Ohio Biomedicine Center of Excellence in Structural Biology and Metabonomics at Miami University.
Footnotes
SUPPLEMENTAL INFORMATION
Four supplemental figures and methods associated with this article are in the Supplementary data.
References
- 1.Lee S, et al. Electron cryomicroscopy structure of a membrane-anchored mitochondrial AAA protease. J. Biol. Chem. 2011;286:4404–4411. doi: 10.1074/jbc.M110.158741. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Janska H, Kwasniak M, Szczepanowska J. Protein quality control in organelles - AAA/FtsH story. Biochim. Biophys. Acta. 2013;1833:381–387. doi: 10.1016/j.bbamcr.2012.03.016. [DOI] [PubMed] [Google Scholar]
- 3.Gerdes F, Tatsuta T, Langer T. Mitochondrial AAA proteases--towards a molecular understanding of membrane-bound proteolytic machines. Biochim. Biophys. Acta. 2012;1823:49–55. doi: 10.1016/j.bbamcr.2011.09.015. [DOI] [PubMed] [Google Scholar]
- 4.Langklotz S, Baumann U, Narberhaus F. Structure and function of the bacterial AAA protease FtsH. Biochim. Biophys. Acta. 2012;1823:40–48. doi: 10.1016/j.bbamcr.2011.08.015. [DOI] [PubMed] [Google Scholar]
- 5.Acton TB, et al. Preparation of protein samples for NMR structure, function, and small-molecule screening studies. Method. Enzymol. 2011;493:21–60. doi: 10.1016/B978-0-12-381274-2.00002-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Xiao R, et al. The high-throughput protein sample production platform of the Northeast Structural Genomics Consortium. J. Struct. Biol. 2010;172:21–33. doi: 10.1016/j.jsb.2010.07.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Phillips JC, et al. Scalable molecular dynamics with NAMD. J. Comput. Chem. 2005;26:1781–1802. doi: 10.1002/jcc.20289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.MacKerell AD, Jr, et al. All-atom empirical potential for molecular modeling and dynamics studies of proteins. J. Phys. Chem. B. 1998;102:3586–3616. doi: 10.1021/jp973084f. [DOI] [PubMed] [Google Scholar]
- 9.Humphrey W, Dalke A, Schulten K. VMD: Visual Molecular Dynamics. J. Mol. Graph. 1996;14:33–38. doi: 10.1016/0263-7855(96)00018-5. [DOI] [PubMed] [Google Scholar]
- 10.Aksimentiev A, Sotomayor M, Wells D. 2012 http://www.ks.uiuc.edu/Training/Tutorials/science/membrane/mem-tutorial.pdf. [Google Scholar]
- 11.Buchan DWA, et al. Protein annotation and modelling servers at University College London. Nucleic Acids Res. 2010;38:W563–W568. doi: 10.1093/nar/gkq427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Holm L, Rosenstrom P. Dali server: conservation mapping in 3D. Nucleic Acids Res. 2010;38(Suppl):W545–W549. doi: 10.1093/nar/gkq366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Zhang QC, et al. PredUs: a web server for predicting protein interfaces using structural neighbors. Nucleic Acids Res. 2011;39:W283–W287. doi: 10.1093/nar/gkr311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Taipale M, Jarosz DF, Lindquist S. HSP90 at the hub of protein homeostasis: emerging mechanistic insights. Nat. Rev. Mol. Cell. Bio. 2010;11:515–528. doi: 10.1038/nrm2918. [DOI] [PubMed] [Google Scholar]
- 15.Taipale M, et al. Quantitative analysis of Hsp90-client interactions reveals principles of substrate recognition. Cell. 2012;150:987–1001. doi: 10.1016/j.cell.2012.06.047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Baker NA, Sept D, Joseph S, Holst MJ, McCammon JA. Electrostatics of nanosystems: application to microtubules and the ribosome. Proc. Natl. Acad. Sci. U S A. 2001;98:10037–10041. doi: 10.1073/pnas.181342398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Ashkenazy H, Erez E, Martz E, Pupko T, Ben-Tal N. ConSurf 2010: calculating evolutionary conservation in sequence and structure of proteins and nucleic acids. Nucleic Acids Res. 2010;38:W529–W533. doi: 10.1093/nar/gkq399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Zinser E, et al. Phospholipid synthesis and lipid composition of subcellular membranes in the unicellular eukaryote Saccharomyces cerevisiae. J. Bacteriol. 1991;173:2026–2034. doi: 10.1128/jb.173.6.2026-2034.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Lindahl E, Sansom MS. Membrane proteins: molecular dynamics simulations. Curr. Opin. Struc. Biol. 2008;18:425–431. doi: 10.1016/j.sbi.2008.02.003. [DOI] [PubMed] [Google Scholar]
- 20.Stansfeld PJ, Sansom MSP. Molecular simulation approaches to membrane proteins. Structure. 2011;19:1562–1572. doi: 10.1016/j.str.2011.10.002. [DOI] [PubMed] [Google Scholar]
- 21.Lipfert J, Columbus L, Chu VB, Lesley SA, Doniach S. Size and shape of detergent micelles determined by small-angle X-ray scattering. J. Phys. Chem. B. 2007;111:12427–12438. doi: 10.1021/jp073016l. [DOI] [PubMed] [Google Scholar]
- 22.Apweiler R, et al. Reorganizing the protein space at the Universal Protein Resource (UniProt) Nucleic Acids Res. 2012;40:D71–D75. doi: 10.1093/nar/gkr981. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Akiyama Y, Kihara A, Mori H, Ogura T, Ito K. Roles of the periplasmic domain of Escherichia coli FtsH (HflB) in protein interactions and activity modulation. J. Biol. Chem. 1998;273:22326–22333. doi: 10.1074/jbc.273.35.22326. [DOI] [PubMed] [Google Scholar]
- 24.Merkwirth C, Langer T. Prohibitin function within mitochondria: Essential roles for cell proliferation and cristae morphogenesis. Biochim. Biophys. Acta. 2009;1793:27–32. doi: 10.1016/j.bbamcr.2008.05.013. [DOI] [PubMed] [Google Scholar]
- 25.Tatsuta T, Model K, Langer T. Formation of membrane-bound ring complexes by prohibitins in mitochondria. Mol. Biol. Cell. 2005;16:248–259. doi: 10.1091/mbc.E04-09-0807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Atorino L, et al. Loss of m-AAA protease in mitochondria causes complex I deficiency and increased sensitivity to oxidative stress in hereditary spastic paraplegia. J. Cell Biol. 2003;163:777–787. doi: 10.1083/jcb.200304112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Koppen M, Metodiev MD, Casari G, Rugarli EI, Langer T. Variable and tissue-specific subunit composition of mitochondrial m-AAA protease complexes linked to hereditary spastic paraplegia. Mol. Cell. Biol. 2007;27:758–767. doi: 10.1128/MCB.01470-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Steglich G, Neupert W, Langer T. Prohibitins regulate membrane protein degradation by the m-AAA protease in mitochondria. Mol. Cell. Biol. 1999;19:3435–3442. doi: 10.1128/mcb.19.5.3435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Saikawa N, Akiyama Y, Ito K. FtsH exists as an exceptionally large complex containing HflKC in the plasma membrane of Escherichia coli. J. Struct. Biol. 2004;146:123–129. doi: 10.1016/j.jsb.2003.09.020. [DOI] [PubMed] [Google Scholar]
- 30.Nijtmans LGJ, et al. Prohibitins act as a membrane-bound chaperone for the stabilization of mitochondrial proteins. EMBO J. 2000;19:2444–2451. doi: 10.1093/emboj/19.11.2444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Merkwirth C, et al. Prohibitins control cell proliferation and apoptosis by regulating OPA1-dependent cristae morphogenesis in mitochondria. Genes Dev. 2008;22:476–488. doi: 10.1101/gad.460708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Chiba S, Akiyama Y, Ito K. Membrane protein degradation by FtsH can be initiated from either end. J. Bacteriol. 2002;184:4775–4782. doi: 10.1128/JB.184.17.4775-4782.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Korbel D, Wurth S, Käser M, Langer T. Membrane protein turnover by the m-AAA protease in mitochondria depends on the transmembrane domains of its subunits. EMBO Rep. 2004;5:698–703. doi: 10.1038/sj.embor.7400186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Kirstein J, Molière N, Dougan DA, Turgay K. Adapting the machine: adaptor proteins for Hsp100/Clp and AAA+ proteases. Nature Rev. Microbiol. 2009;7:589–599. doi: 10.1038/nrmicro2185. [DOI] [PubMed] [Google Scholar]
- 35.Laskowski RA. PDBsum new things. Nucleic Acids Res. 2009;37:D355–D359. doi: 10.1093/nar/gkn860. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Larkin MA, et al. Clustal W and Clustal X version 2.0. Bioinformatics. 2007;23:2947–2948. doi: 10.1093/bioinformatics/btm404. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.