

Abstract
A role for mitochondria and oxidative stress is emerging in acquired epilepsies such as temporal lobe epilepsy (TLE). TLE is characterized by chronic unprovoked seizures arising from an inciting insult with a variable seizure-free “latent period”. The mechanisms by which inciting injury induces chronic epilepsy, known as epileptogenesis involves multiple cellular, molecular and physiological changes resulting in altered hyperexcitable circuitry. Whether mitochondrial and redox mechanisms contribute to epileptogenesis remains to be fully clarified. Mitochondrial impairment is revealed in studies from human imaging and tissue analysis from TLE patients. The collective data from animal models suggest that steady-state mitochondrial reactive oxygen species and resultant oxidative damage to cellular macromolecules occurs during different phases of epileptogenesis. This review discusses evidence for the role of mitochondria and redox changes occurring in human and experimental TLE. Potential mechanisms by which mitochondrial energetic and redox mechanisms contribute to increased neuronal excitability and therapeutic approaches to target TLE are delineated.
Epilepsy is a common neurological disorder that affects approximately 0.6% of the entire population. Recurrent spontaneous convulsive or non-convulsive seizures are the hallmark of epilepsy. A seizure is characterized by synchronized abnormal electrical discharges from a locus in the brain. Epilepsy is defined by a condition in which recurrent unprovoked seizures occur as a result of genetic disposition or acquired factors such as brain injury. Epilepsies occur throughout the lifespan with the highest incidence in children younger than 5 and precipitously rising in the elderly after 65 years of age [1]. Temporal lobe epilepsy (TLE) is the most prominent of the acquired epilepsies and is commonly preceded by an initial brain injury such as an episode of prolonged seizures or status epilepticus (SE), complicated childhood febrile seizures, hypoxia or trauma which leads to chronic epilepsy or spontaneous recurrent seizures. The process whereby physiological neuronal characteristics and circuitry are altered by a precipitating event is known as epileptogenesis. Animal models of acquired epilepsy attempt to recapitulate several of the features of human TLE and usually involve an initial insult which is followed by a variable “latent period” that results in recurrent, spontaneous seizure activity. The majority of epilepsy research is focused on ion channels and receptors with attempts to understand and control altered network excitability. A key shift in current epilepsy research emphasis is the prevention of chronic epilepsy development and disease progression rather than the traditional focus on controlling seizures per se with antiepileptic drugs. Many different approaches have been taken in this renewed focus of research with a primary purpose of identifying anti-epileptogenic or disease-modifying therapies. Towards this goal, understanding mechanisms by which injury mediates the epileptogenic process and comorbid states such as depression and memory loss that coexist with TLE is important. This review will cover the major strategies employed to implicate the role of mitochondria and oxidative stress in human and experimental TLE and potential mechanisms by which altered metabolism can increase neuronal excitability.
Mitochondrial Function and Neuronal Excitability
Mitochondria serve several key cellular functions that may have a direct and/or indirect impact on neuronal hyperexcitability such as the generation of ATP, metabolite/neurotransmitter biosynthesis, calcium homeostasis, control of cell death and they are the primary site of reactive oxygen species (ROS) production. Given the bioenergetics of seizures themselves and injury processes that trigger epileptogenesis, the role of mitochondria and oxidative stress is gaining increased recognition in the progression of epileptogenesis [2,3]. In fact, several key events initiated by the injury process such as hippocampal cell loss, inflammation and cell signaling suggest a role for mitochondria and redox processes in epileptogenesis. The brain's unique susceptibility to oxidative stress and bioenergetic insults likely drives or at least exacerbates neuronal excitability during epileptogenesis because of a high metabolic demand in hypersynchronous circuits. In addition, mitochondria are a critical interface between environmental factors such as diet, disease and proper cell function. Metabolic control of neuronal excitability is evident from the broad antiepileptic efficacy of the ketogenic diet (KD), a high fat, low carbohydrate dietary therapy in children and adolescents [4] which is based on providing alternative mitochondrial fuels i.e. ketones and fatty acids vs glycolytic substrates to control intractable seizures. Metabolic control of seizures and epileptogenesis is also suggested by their regulation by epigenetic mechanisms through histone modifications as this requires high energy intermediates such as ATP, acetyl-CoA and S-adenosyl-methionine [5]. Altering mitochondrial functions therefore becomes a potentially important area of interest in contemporary epilepsy research.
Metabolic Alterations Implicating Mitochondrial Involvement in Human TLE
The most compelling evidence for the role of mitochondria in epilepsy arises from rare inherited mitochondrial disorders associated with epileptic seizures e.g. myoclonic epilepsy with ragged red fibers (MERRF), and mitochondrial encephalopathy with lactic acidosis (MELAS). Mitochondrial DNA mutations and spontaneous seizure activity are evident in both disorders [6]. The role of mitochondria in acquired epilepsy is largely indirect and based on observed changes in known mitochondrial functions in human tissue and experimental models.
A major suggestion of mitochondrial involvement in human TLE comes from observations of metabolic and bioenergetic changes following acute seizures and during various phases of chronic epilepsy. Glycolytic rates, activity-matched cerebral blood flow, and lactate/pyruvate ratios are acutely increased during seizure activity [7]. This ictal hypermetabolism in the human epileptic foci is followed by inter-ictal hypometabolism. This hypometabolism may reflect a “metabolically stressed circuit” in which mitochondrial bioenergetic capacity may be depleted. Mitochondrial involvement in epilepsy has been suggested based on the loss of mitochondrial N-acetyl aspartate in human epileptic tissue [8,9]. Finally, severe metabolic dysfunction characterized by biphasic abnormal NAD(P)H fluorescence transients and changes in mitochondrial membrane potential have been observed in ex vivo preparations from both chronically epileptic rats and human subjects [10].
Neuronal loss in the hippocampal formation or hippocampal sclerosis is a distinct pathological finding in human TLE and thought to be important because inciting injury can result in the development and progression of TLE. Understanding the mechanisms by which seizures result in neuronal loss and contribute to TLE is important and underscores the relevance of evaluating the role of oxidative damage and mitochondrial involvement in these processes. The vulnerability of the hippocampal principal neurons in TLE is largely due to the involvement of the structure in the seizure circuit and excessive glutamatergic neurotransmission resulting in excitotoxic cell death. Neuropathological findings in human TLE provide evidence for morphological changes in mitochondria of epileptic patients. For instance, neuronal damage incurred by ischemia and seizure-related events cause mitochondrial swelling and disruption [11]. Apoptotic machinery proteins as well as excitotoxic mechanisms are also known to be activated by seizures. For example, there are increases in the anti-apoptotic molecule Bcl-2 in the serum of children with TLE [12], and changes in proapoptotic molecules in epileptic brain tissue [13], highlighting the importance of mitochondrial involvement in the cell death process [14]. Additionally, ROS have been suggested in seizure-induced apoptotic cell death in the hippocampus [15]. Calcium transients associated with seizure activity represent a potential role by which mitochondria might contribute to neuronal injury in TLE [16] based on a crucial role of mitochondria in buffering cytosolic calcium.
Literature attempting to link mitochondrial (dys)function in acquired epilepsies, such as TLE, is based on measurement of mitochondrial enzyme activities, indices of mitochondrial oxidative damage, bioenergetic alterations, secondary processes such as cell death involving mitochondria, alteration of redox status [17] and inhibition of oxidative phosphorylation (OXPHOS) enzyme complexes of mitochondria in animal models and human tissue [18]. Redox status measured by reduced and oxidized forms of glutathione (GSH/GSSG) shifts to a more oxidized state in brain regions and plasma of epileptic patients [19]. Another study shows decreased copy numbers for mitochondrial DNA and decreased aconitase activity in the CA3 [20]. The hypometabolism seen in the epileptic focus in inter-ictal phases of seizure activity may therefore be attributed more to mitochondrial dysfunction than to neuronal cell loss [21]. The impact that mitochondrial dysfunction can have in human TLE is becoming more generally accepted while work in experimental models of TLE will continue to help elucidate mechanisms of how mitochondria are involved. In sum, evidence for alterations in metabolic functions directly and indirectly suggesting mitochondrial involvement is mounting in human TLE. However, evidence for oxidative damage in human TLE is yet to be clearly established.
Oxidative Stress and Mitochondrial Involvement in Experimental TLE
The majority of data supporting the role of mitochondria and oxidative stress in the epilepsies including TLE comes from experimental models. The difficulties associated with assessment of ROS and reactive nitrogen species (RNS) in tissues from human samples and animals arises due to their unstable, evanescent and interchangeable nature as well as the need for respiring mitochondria for functional analysis. Nevertheless, animal models of TLE have provided sufficient data to date of profound alterations in mitochondrial function and ROS. Key features of experimental models for the study of human TLE include a precipitating injury such as traumatic brain injury, infection or status epilepticus (SE), followed by a “latent” period after acute changes due to injury in which seizure probability is low akin to the human condition, and finally, the appearance of chronic spontaneous electrographic and behavioral seizures that are progressive (see figure 1). Chemoconvulsant or SE models initiated by the glutamatergic agonist kainic acid or the cholinergic agonist pilocarpine replicate these features and are therefore widely used as experimental models of TLE. The kindling model refers to an electrical stimulus applied repeatedly to a limbic brain structure sufficient to evoke a short after-discharge duration, which, over time, evokes an electrographic after-discharge that initiates a generalized convulsive seizure. While kindling results in permanent alteration in limbic circuitry, it lacks a true latent period and the impact of a precipitating injury. Therefore, it is not usually associated with spontaneous seizures but rather a lowered seizure threshold. Both chemoconvulsant models as well as the kindling model in adult animals show evidence of increased steady-state ROS levels [2,22–25]. In addition, hippocampal slices and slice cultures are also used to assess changes associated with TLE ex vivo and in vitro. In the following sections, evidence of oxidative damage to cellular macromolecules in animal and ex vivo models of TLE is discussed.
Figure 1.

Mitochondrial oxidative stress during the acute, latent and chronic phases of epileptogenesis. During the acute phase, shortly following an inciting injury increased production of reactive oxygen species (ROS) such as superoxide, hydrogen peroxide and hydroxyl radicals via Fenton chemistry and oxidative damage to mitochondrial targets and altered redox status occur. ROS production returns to control values during the latent period during which seizure probability is low; however mitochondrial and cellular redox environment remains persistently altered. ROS production and oxidative damage returns during the chronic phase accompanied by spontaneous recurrent seizures. Mitochondrial repair enzyme expression is activated shortly after injury during the acute phase and remains elevated during the latent phase. Failure of repair enzymes may further exacerbate oxidative damage during the chronic phase of epileptogenesis. GSH=glutathione; GSSG=glutathione disulfide; α-KGDH= α-ketoglutarate dehydrogenase; CI= complex I; ATP= adenosine triphosphate; mtDNA= mitochondrial DNA; [Ca++]m= mitochondrial calcium concentration; ROS=reactive oxygen species.
Protein oxidation
Proteins can undergo altered primary, secondary, and tertiary structures, spontaneous fragmentation and increased proteolytic susceptibility following exposure to ROS. The amino acid side chains of proteins are particularly susceptible to various forms of reversible or irreversible oxidation that can lead to functional impairment. One way to estimate protein oxidation is to quantify protein carbonyls, or the addition of ketone and aldehyde groups to protein side chains. Elevated carbonyl levels are observed during aging and neurodegenerative disorders [26,27]. Increase in carbonyl content has also been observed following kainate injection in the rat piriform cortex and hippocampus as early as 8 hour post-treatment [28].
An important mechanism by which ROS can alter cellular and mitochondrial function is via post-translational modification of critical proteins leading to changes in function. Mitochondrial aconitase, alpha ketoglutarate dehydrogenase (α-KGDH) and complex I (C1) are among the proteins that are known to be specifically posttranslationally modified or inhibited by ROS during epileptogenesis. Mitochondrial aconitase is a TCA cycle enzyme that contains a labile iron motif in its iron-sulfer (Fe-S) center that is susceptible to oxidative damage by superoxide (O2−) and related species (see Figure 2). The measurement of endogenous aconitase activity can serve both as an index of steady state O2− levels and evidence of oxidative damage to proteins [29,30]. Inactivation of aconitase normalized to fumarase or citrate synthetase has been used to determine oxidative damage to mitochondrial proteins in animal models of TLE and human TLE. Kainate-induced seizures inactivate mitochondrial aconitase but not fumarase, with maximal inactivation occurring 16 hours post-treatment, several hours after SE and preceding neuronal death of susceptible hippocampal neurons [2]. Inactivation of mitochondrial aconitase at times following the onset of behavioral seizures suggests that mitochondrial O2− production occurs as a consequence of prolonged seizure activity. Mice that are partially deficient in the primary mitochondrial antixodant, MnSOD (SOD2−/+), show exacerbation of KA-induced mitochondrial aconitase inactivation and hippocampal neuronal loss [31], while both indices are attenuated in mice overexpressing SOD2. Manganese (III) tetrakis (4-benzoic acid) porphyrin (MnTBAP), a broad spectrum antioxidant, protected rats against seizure-induced mitochondrial aconitase inactivation and hippocampal damage without decreasing behavioral seizure intensity [31]. Aconitase and α-KGDH activities in the rat hippocampus are decreased 16–44 hours post-SE induced by pilocarpine, and return to normal values by 8 days [32]. Finally, aconitase activity is decreased in the CA3 region of human cases of TLE [8]. One consequence of oxidative inactivation of aconitase is the release of iron and generation of H2O2 which can form ˙OH and thereby result in further oxidative damage. Increases of hippocampal mitochondrial iron coinciding with inactivation of mitochondrial aconitase and neuroprotection by an iron chelator have been demonstrated in the kainate model [17]. This suggests that O2−-mediated posttranslational inactivation of mitochondrial aconitase may be one source of iron accumulation and H2O2 following seizures leading to a vicious cycle of ROS-induced ROS formation.
Figure 2.

Normal mitochondrial functions (depicted above) can be impaired following acute injury and during the chronic phase of epileptogenesis (depicted below). In normal brain mitochondria, superoxide (O2−.) produced via electron transport chain (ETC) Complexes I (CI) and III is detoxified by manganese superoxide dismutase (SOD2) while the majority of oxygen consumed is reduced to water; H2O2 resulting from enzymatic and spontaneous dismutation is detoxified via the glutathione and thioredoxin/peroxiredoxin (Trx/Prx) pathways such that low levels emitted from mitochondria play a redox signaling role. Following an inciting injury such as status epilepticus or trauma, steady-state levels of mitochondrial (O2− and H2O2) are elevated under conditions of low ATP production and inhibited CI[2, 25]. Inhibition of CI and aconitase are among two mechanisms that provide a feed-forward mechanism of reactive oxygen species (ROS)-induced ROS production by increasing O2− and release of redox-active iron and H2O2, respectively [34,86]. Oxidative damage to vicinal targets such as mtDNA, lipids and proteins ensues[53]. Higher levels of H2O2 emitted from mitochondria may damage plasmalemmal synaptic targets such as glutamate transporters and/or the ion channels potentially affecting neuronal excitability and cell death. mPTP=mitochondrial permeability transition pore; GSH=glutathione; GSSG=glutathione disulfide; GR=glutathione reductase; GPx=glutathione peroxidase.
CI of the mitochondria receives a lot of attention in neurodegenerative disease [33] due to the importance of this enzyme in the initiation of electron flow in the mitochondrial electron transport chain (ETC). In TLE, CI inhibition is of particular significance since its activity has been demonstrated to be inhibited in hippocampal CA3 area from human TLE tissue [18]. Recently Ryan et al. [34] demonstrated for the first time an oxidative post-translational modification via carbonylation of the 75kDa subunit of CI in experimental TLE. The pattern of CI carbonylation occurred in a biphasic manner during epileptogenesis and coincided with periods following/accompanied by high seizure activity and inhibition of CI activity. Mass spectrometric analysis of the oxidation of the 75kDa subunit in rat hippocampi during acute, latent and chronic phases of epileptogenesis revealed a specific type of carbonylation classified as metal catalyzed oxidation (MCO), which is typically generated in the presence of oxidative stress and increased chelatable metals such as iron (Figure 3). Although MCO may not be the most common type of carbonyl addition in vivo, its occurrence in experimental TLE is supported by previous reports that mitochondrial Fenton reactants, H2O2 and Fe2+, are increased in the kainate model [17,35] and HBED, an iron chelator is neuroprotective. These data suggest that MCO products may exist in other neurological conditions with increased mitochondrial oxidative stress and elevated redox active chelatable metals such as Parkinson's disease [35–37].
Figure 3.
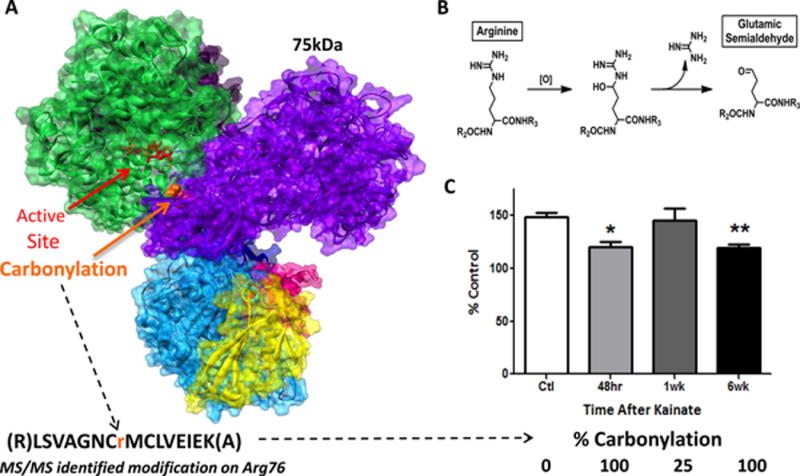
Oxidative post-translational modification (carbonylation) of the 75kDa subunit in complex I correlates with loss of activity during epileptogenesis. [A] Homology model of the rat hydrophilic domain of mitochondrial complex I highlighting the enzyme active site (NADH binding in red) and location of carbonylation on Arginine 76 (Arg 76 - orange) at the n-terminus of the 75kDa subunit (purple). Mass spectrometry results indicate that carbonylation occurs in a single peptide 100% of the time at the acute and chronic stages of epileptogenesis and 25% during the latent period of kainate-treated adult male Sprague Dawley rats. [B] Schematic of the chemical oxidation of Arg76 to glutamic semialdehyde. [C] Mitochondrial complex I activity was significantly decreased at acute (48 hour) and chronic (6 week) stages of epileptogenesis in accordance with carbonylation at Arg 76 as shown in Ryan et al. 2012, J. Neuroscience.
The nature of post-translational modification suggests that MCO primarily occurs on lysine (K) and arginine (R) protein AA, converting them to aminoadipic semialdehyde and glutamic semialdehyde chemical modification products [38]. Specific conversion of arginine 76 (Arg76) to glutamic semialdehyde 76 (GSA76) occurred within the 75kDa subunit 100% of the time at acute and chronic stages of epileptogenesis. Further, molecular modeling studies predicted that carbonylation at this site within the 75kDa subunit results in altered surface structure and charge at the interaction interface within the CI active site, its interaction with the 51kDa subunit of CI, and the position of nearby Fe-S centers proximal to the CI active site which can impair electron transfer from NADH to the proximal Fe-S center, and thus CI function. Together, these data identify a post-translational oxidative lesion on Arg76 in an important subunit of mitochondrial CI during epileptogenesis. This demonstrates a specific ROS-mediated oxidative post-translational modification in the 75kDa subunit of CI during the acute and chronic phases of epilepsy development which can impair mitochondrial function during epileptogenesis. Carbonylation and inhibition of CI during epileptogenesis suggests that CI is not only a target of oxidative stress via post-translational modifications, but may also be a source of O2− production when inhibited, generating excess reactive species and further propagating mitochondrial and cellular damage [39–42]. Thus, CI may be both a source and target of ROS during epileptogenesis leading to a vicious cycle of seizures and ROS production. Additionally, this suggests that oxidative damage may underlie specific CI deficiency observed in tissue resected from TLE patients [18].
Membrane proteins susceptible to oxidative damage and/or alterations in ATP which are also known to be coupled to increased neuronal excitability include glutamate transporters, Na+/K+-ATPase and glutamine synthetase (GS). Of particular interest are neuronal and glial glutamate transporters which limit glutamate from reaching neurotoxic levels in the synapse [43]. Improper handling of glutamate can lead to hyperexcitability and neuronal death. GLT-1 is regulated by extracellular levels of ATP in the hippocampus [44] and GLT-1 and GLAST are highly sensitive to oxidative insults resulting in lowered transport activity [45]. This makes GLT-1 and GLAST attractive targets for ongoing oxidative stress during epileptogenesis as well as potential links to increased neuronal excitability. In Sod2−/+ mice, decreased GLT-1 and GLAST expression coincides with increased susceptibility to seizures and mitochondrial oxidative stress [31]. Epileptic human patients have been shown to have less glutamate transporters with an overall deficiency in glutamate transport [46].
Oxidative Mitochondrial DNA Damage
Mitochondrial DNA (mtDNA) damage from oxidative stress and resultant impairment are implicated in the pathogenesis of many human mitochondrial disorders [47]. MtDNA damage has been implicated in the progression of epileptogenesis. mtDNA is especially susceptible to oxidative damage for many reasons. Primarily, mtDNA lack protective histones and is located near the inner mitochondrial membrane in close proximity to where ROS can be generated. Increased production of ROS from seizures can cause mtDNA damage that potentially leads to decreased activities of electron transport complexes [48]. Therefore, the mtDNA is specifically vulnerable to damage induced by ROS [49] under normal circumstances and is exacerbated after seizures. One month after pilocarpine treatment, one study shows a decrease of mtDNA copy number in the CA3 and CA1 areas of the hippocampus [50]. Another method of determining mtDNA damage is using the markers thymine glycol (TG) and 8-hydroxy 2-deoxyguanine (8-OHdG), an oxidized guanine adduct most commonly formed from oxidation of DNA [51]. Reports show that a time dependent increase in mitochondrial 8-OHdG to total 2-deoxyguanine (2-dG) occurs 16-48 hours following kainate administration with a corresponding increase in mtDNA lesion frequency [17]. Additionally, basic excision DNA repair systems known to be activated by 8-OHdG are also activated during epileptogenesis initiated by kainate administration. The activation of these repair pathways after an epileptogenic insult suggests ongoing adaptive repair and response to oxidative stress.
Lipid Peroxidation
Lipids are another major target of oxidative damage that can occur during epileptogenesis as a consequence of seizures. Polyunsaturated fatty acids present in phospholipids of biological membranes are highly susceptible to oxidation by ROS. Lipid peroxidation occurs when hydrogen is abstracted from polyunsaturated fatty acids by ˙OH [52,53]. As with damage to proteins and mtDNA, the brain is particularly susceptible to lipid peroxidation after seizures and increased ROS production. Following seizure activity, Ca2+-dependent activation of phospholipase A2 releases arachidonic acid [54,55] yielding ROS via enzymatic and non-enzymatic mechanisms [53]. F2isoprostanes (F2-IsoPs) and isofurans (IsoFs) are produced by a non-cyclooxygenase and free radical catalyzed mechanism involving the peroxidation of arachidonic acid. F2-IsoPs are used as an index of lipid peroxidation in vivo because of their formation in disease states associated with oxidative stress, stable nature which allows detection in bodily tissues and fluids, they are modulated by antioxidants and unaffected by dietary lipids [56]. Seizure-induced increases in F2-IsoPs and IsoFs have been reported in micro-dissected dentate gyrus, CA1, and CA3 regions at distinct time points post-kainate administration [3,57]. The mechanism(s) by which seizure activity results in F2-IsoP and IsoF likely results from calcium-dependent activation of phospholipase A2 (PLA2), release of arachidonic acid (AA), formation of prostaglandins including prostaglandin- F2α, precursor of αF2-IsoP, and subsequent free-radical catalyzed formation that coincide with seizure activity. Therefore, the dentate gyrus, which serves as a “gate” for seizure activity, shows the highest increases followed by CA3 and CA1 areas. Further investigation regarding the factors that control F2-IsoPs and IsoFs and any role of mitochondria was revealed by correlation of their formation with tissue pO2 levels and indices of mitochondria-specific oxidative stress. Since IsoFs but not F2-IsoP formation is favored by high oxygen tensions, this enabled examination of any correlation of seizure-induced IsoFs and its relationship with hippocampal pO2 measured by in vivo electron paramagnetic resonance oximetry. Interestingly, this revealed that seizure-induced F2-IsoP formation coincided with the peak hypoxia phase, whereas IsoF formation coincided with the “re-oxygenation” phase. Furthermore, IsoF formation correlated closely with indices of mitochondrial oxidative stress e.g. aconitase inactivation [3]. Other indices of lipid peroxidation include the formation of reactive lipid mediators such as 4-hydroxy-2-(E)-nonenal (4-HNE) and malondialdehyde (MDA) [58] and confirm ongoing lipid peroxidation in the cortex, hippocampus and other brain regions as early as 4 hours into SE and lasting until 24 hours after SE in a variety of seizure models including chemoconvulsant models [23,24,28,59]. These results suggest that oxidative damage to lipids occurs as a result of seizures and implicate an important role in the progression of epilepsy. Interestingly, the precise role of seizure-induced lipid peroxidation remains unclear particularly due to the high extent in the dentate gyrus region in which cell death-resistant granule cells reside. This suggests that reactive lipids may have roles other than mediating neuronal injury in the epileptic brain such as signal transduction.
Changes in Redox Status
Redox couples such as GSH and its disulfide (GSSG) serve as biomarkers of oxidative stress [60,61]. Coenzyme A (CoASH) and its disulfide with GSH (CoASSG), primary compartmentalized within mitochondria, have been measured and utilized as a marker of mitochondrial specific redox status [61,62]. GSSG levels have been found to increase as early as 4 hours post-SE in the cortex [24]. A greater time-dependent decrease in the GSH/GSSG ratio and depletion of total glutathione following KA-induced SE is observed in mitochondrial fractions than whole hippocampal homogenates [61]. This altered redox status was accompanied by a moderate increase in glutathione peroxidase activity and a decrease in glutathione reductase (GR) activity in hippocampal homogenates and mitochondria, respectively. Ratios of hippocampal GSH/GSSG and CoASH/CoASSG following lithium-pilocarpine-induced SE have been recently demonstrated to decrease by 24 hours and remain permanently impaired throughout epileptogenesis and chronic epilepsy even during the latent phase when measurements of H2O2 production and mtDNA damage returned to control levels [63]. Both GSH levels and GR activity are lowered in brain regions and plasma of epileptic patients [19,64]. A profound and persistent oxidation of GSH to GSSG and depletion of total GSH during epileptogenesis, including the latent period, would favor protein post-translational modifications such as S-glutathionylation and/or S-nitrosylation of sensitive targets, e.g. ion channels and energy-dependent transporters that could ultimately alter neuronal excitability. Therefore, altered cellular and mitochondrial redox status may initiate neuronal injury and redox signaling events. Whether they provide an important mechanistic link between acute and chronic stages of epilepsy remains to be determined.
Mitochondria Disproportionately Contribute to Seizure-Induced ROS
The above studies clearly demonstrate that target oxidation occurs during various phases of epileptogenesis and suggest a disproportionate contribution of mitochondria to the increased ROS. First, inactivation of oxidant-sensitive proteins i.e. mitochondrial vs cytosolic aconitase during the acute phase of epileptogenesis clearly demonstrates sensitivity towards the mitochondrial enzyme and not the cytosolic [2]. Similarly, increases in mitochondrial 8OHdG/2dg and not nuclear 8OHdG/2DG levels in the acute phase of epileptogenesis are suggestive of mitochondria but not the nucleus as both a source and target of ROS. Finally, studies in SOD mutant mice provide further evidence of the importance of subcellular O2− in seizure-induced neuronal death as well as seizure susceptibility. The relative importance of mitochondrial vs cytosolic and extracellular O2− is evident from knowledge that genetic inactivation of SOD1 or SOD3 results in mild phenotypes [65,66] while mice lacking SOD2 are neonatal lethal by three weeks regardless of their background strain [67,68]. In earlier studies it has been demonstrated that mice deficient in SOD3 (SOD3−/− mice) or SOD2−/+ mice are more sensitive to kainate-induced hippocampal damage [61,69] and overexpression of SOD3 and SOD2 is protective. A small but significant source of seizure-induced O2− revealed by the kainate model was Nox-2 derived and SOD3-sensitive [70]. This pool of ROS most likely contributes to seizure-induced inflammation and may also have links or “cross-talk” with the mitochondrial pool of ROS [71]. Moreover, SOD2−/− mice in a mixed background have recently shown to exhibit spontaneous seizures and profound oxidative stress which can be rescued by administration of a catalytic antioxidant [72]. This suggests that steady-state increase in mitochondrial O2− and resultant mitochondrial impairment is sufficient to result in epilepsy. Furthermore, SOD2−/+ in either a C57B6 or CD-1 background have age-related seizure and show greater susceptibility to chemoconvulsant and handling-induced seizures. Hence, mitochondrial O2− appears to be a critical mediator of both seizure-induced neuronal damage as well as seizure susceptibility.
Mechanism(s) of Seizure-Induced ROS Generation
Steady-state levels of ROS increase and oxidative damage ensues when antioxidant systems and repair processes are overwhelmed [73]. Prolonged seizure activity increases steady-state O2− as evidenced by oxidative inactivation of oxidant-sensitive aconitase but not fumarase and measurement by dihyrdoethidium in situ [2,17,25]. The extent and mechanism of seizure-induced O2− formation may vary during the acute, latent and chronic phases of epileptogenesis as well as during ictal and interictal periods. During the acute phase of epileptogenesis shortly after an inciting event such as SE or trauma, and in the chronic phase of epileptogenesis when spontaneous seizures occur, increased mitochondrial O2− and H2O2 occur as evidenced by inactivation of O2− sensitive aconitase but not fumarase and resorufin fluorescence [17]. The mechanism of ROS production during these phases may result from increased OXPHOS from over activation of glycolysis during the ictal period leading to accumulation of electrons at the ETC and thereby reduction of O2 to O2− and H2O2. Inhibition of ETC enzymes such as CI and TCA cycle enzymes α-KGDH and aconitase further exacerbates ROS production during the acute and chronic phases of epileptogenesis [32,34]. Additionally, transient hypoxia-reperfusion associated with the inciting injury may contribute to increased production of mitochondrial O2− and H2O2 [2]. Measurement of pO2 in rat brain using oxygen-sensitive probes [57] indicated a transient lowering of pO2 shortly following kainate-induced SE (6 hours) which returned to normal values by 16 hours and coinciding with increased ROS production [2,61]. The decrease in SE induced change in pO2 was similar in magnitude to that observed in the ischemic penumbra suggesting that ischemia-reperfusion may be one mechanism by which seizure activity increases O2− production. After prolonged seizures, glucose levels are decreased around 50% in the hippocampus [74] and brain imaging shows increases in glycolytic activity, especially in the temporal lobe, during ictal phases [75–77]. A similar phenomenon occurs in hyperglycemia, where increased glycolytic rates increase ROS production due to large flux of electron donors from the TCA cycle, generating a high mitochondrial membrane potential that can inhibit electron transport and therefore increase the production of O2− [78]. Therefore, increased glycolytic activity stimulated by ictal events and partially inhibited ETC complexes can increase steady-state ROS levels due to electron accumulation on OXPHOS complexes that are transferred to O2 and ultimately overwhelm endogenous antioxidant defenses, potentially deplete reserve capacity and lead to oxidative damage of vulnerable targets mentioned previously [2,3,17,57]. Adaptive mechanisms may be important among the reasons why mitochondrial O2− and H2O2 production occurs during acute and chronic phases of epileptogenesis but not during the latent period. During the latent phase when seizure probability is low, antioxidant defenses and repair processes are activated as demonstrated by elevated mtDNA repair enzymes, Ogg1 and Polγ mRNA and protein levels shortly after the inciting injury and lasting ∼ 3 wk thereafter, followed by improvement in mtDNA repair [17]. Interestingly, spontaneous seizures associated with the chronic phase of epileptogenesis coincided with accumulation of mtDNA damage, increased mitochondrial H2O2, decreased Ogg1 and Polγ, and impaired mtDNA repair. This suggests that ongoing seizure activity and/or failure to adapt/repair may contribute to oxidative stress and mitochondrial injury as epileptogenesis progresses. A vicious cycle is also created by the occurrence of chronic seizures which produce more ROS rising above threshold levels once chronic epilepsy is established.
Mechanisms by Which Mitochondria and Oxidative Damage can Impact Epileptogenesis
There are many potential ways that mitochondrial function and ROS production during epilepsy can contribute to increased hyperexcitability. Two potential links between mitochondrial oxidative stress and increased neuronal excitability are: 1) bioenergetic failure and 2) fuel utilization. There are many areas of research that could fit into these two links that may encompass all or many of the deficits discussed previously with a larger emphasis on the effects of energy failure and ATP depletion.
Bioenergetic Failure
There is a large demand for neuronal mitochondria to produce cellular energy (ATP) for maintenance of proper neuronal membrane potential and an imbalance in ATP levels is associated with seizure activity. One of the primary consumers of neuronal mitochondria-derived ATP is the Na+/K+ ATPase, which typically requires 40-50% of total cellular ATP [79]. Decreased Na+/K+ ATPase activity has been detected in post-mortem epileptic human brain tissue [80]. Furthermore, neuronal excitability is controlled by glutamate and GABA which depend on mitochondria for their biosynthesis, as well as apoptosis, and Ca2+ homeostasis [10]. Mitochondria are therefore involved in a diverse array of cellular functions that if unregulated, can lead to increased disease progression in TLE. Mitochondrial ROS have the potential to impact epileptogenesis both via redox signaling as well as oxidative damage to macromolecules. Oxidative inactivation of TCA cycle enzymes (aconitase, α-KGDH) and carbonylation and inhibition of CI during epileptogenesis suggests that these are not only a targets of oxidative stress, but may also be a source of ROS production when inhibited, generating excess reactive species and further propagating mitochondrial and cellular damage [39–42]. Thus, several key proteins may be both a source and target of ROS during epileptogenesis leading to a vicious cycle of seizures and ROS production. A consequence of this ongoing cycle of oxidative damage to mitochondria in epilepsy is energy failure and ATP depletion. Decreased levels of ATP could potentially increase neuronal excitability by lowering neuronal resting membrane potential. This would occur through inhibition of the Na+/K+ ATPase that requires ATP to restore a normal resting potential. An alternative explanation would be the importance of presynaptic ATP levels for normal synaptic transmission. Many types of inhibitory interneurons contain high amounts of mitochondria [81] where ATP levels are important for mobilization of the synaptic vesicle reserve pool [82]. Therefore, as mentioned previously over time there would be increased mitochondrial oxidative stress due to a potential lack of mitochondrial reserve capacity leading to further ATP depletion and lack of inhibitory signaling. Over the course of epileptogenesis this could increase overall neuronal excitability. However, at this time these ideas remain unproven necessitating further work to confirm these hypotheses.
Mitochondrial reserve capacity or spare respiratory capacity has been shown to be important in maintaining ATP levels during a metabolic insult or challenge [83]. Interestingly, one study showed the importance of reserve capacity during Complex I (CI) inhibition [84]. Proof of mitochondrial reserve capacity changes in epilepsy have yet to be shown but it should be considered due to the highly metabolic activity of a seizure and the CI inhibition that occurs in both human and experimental TLE [18,34]. Therefore, between changes in enzyme complexes due to energy failure and ATP depletion we can see the many ways that oxidative stress can affect macromolecules and overall mitochondrial function.
Fuel Utilization
Bioenergetic functions of mitochondria are linked to neuronal excitability at the level of substrate utilization via TCA cycle, fatty acid metabolism and OXPHOS activity leading to ATP production to maintain plasmalemmal Na+/K+ATPase function. The TCA cycle recently has received more attention as a potential therapeutic target in epilepsy based on anapleurotic substrates being effective in seizure control [85]. Additionally, oxidative inactivation of the TCA cycle aconitase and α-KGDH occurs in TLE models [2,17,32]. Oxidative inactivation of TCA cycle enzymes such as aconitase is known to generate ROS providing an additional mechanism of already ongoing damage [32]. In the case of aconitase, oxidative inactivation results in both redox active iron and H2O2 which via Fenton chemistry generate ˙OH radicals and propagate damage [86]. Surprisingly, decreasing aconitase expression decreased OXPHOS activity but also prevented damage from exogenous oxidative insults suggesting a key role for posttranslational oxidative modifications [87].
The importance of metabolic fuels in controlling seizures is underscored by both clinical and experimental observations attesting to the broad spectrum efficacy of the KD. The KD remains the main-stay method of altering fuels to control epilepsies. The KD is a high-fat/low-carbohydrate diet used to treat children and adolescents who are refractory to normal anticonvulsant drugs. The KD appears to have broader efficacy than any currently available anti-epileptic drug, suggestive of a broad mechanism of action. A group of studies conducted from 1925-1998 revealed that out of 720 patients placed on the KD, 37% experienced greater than 90% reduction in their seizures, and an additional 30% achieved 50-90% seizure control [88]. This type of general efficacy is highly suggestive of a central mechanism that could affect oxidative stress pathways previously discussed. Studies on the mechanism of action for the KD are varied and there are many different theories. However, one of these theories is the effect that the KD has on mitochondria and ROS production. Included in this theory is the similar mechanism of glycolysis inhibition that is accomplished pharmacologically with 2-Deoxy-D-Glucose (2-DG) and fructose-1,6-bisphosphate (FBP) [89]. The KD has been shown to produce mitochondrial biogenesis [90] and decrease mitochondrial ROS [91]. Additionally, rats fed the diet for a period of 3 weeks improved mitochondrial redox status including GSH levels [4] that could help the brain to resist metabolic insults that occur during seizures. The KD is a very important treatment in pediatric epilepsy and knowledge from the application of this diet has led to increased research on mitochondrial bioenergetics and epilepsy. However, all of the contributing mechanisms of the KD remain unknown and further research is needed to apply current knowledge toward an effective treatment strategy.
There is renewed resurgence of interest in the research community to explore ways to modify neuronal excitability by altering metabolic flux. Therefore, targeted investigation of mechanisms by which the KD exerts an antiepileptic effect and attempting to substitute its beneficial effects with single molecules are ongoing. One such approach is using 2-DG, a non-metabolizable analog of glucose which shows remarkable benefits in models of acquired epilepsy [92]. The effects of 2-DG seem to mimic caloric restriction. Simply reducing daily caloric intake by 40% can extend life span by 30-50% over ad libitum fed animals [93]. The precise mechanisms of how 2-DG exerts its effect remain to be further clarified since a narrow margin of therapeutic effect has been observed in its ability to be anti-convulsive vs pro-convulsive [94]. Consistent with this line of investigation, FBP, which shunts the flux of glucose into the pentose phosphate pathway [95], has been shown to lower time to seizure onset, seizure duration, and seizure severity in the kainate, pilocarpine, and pentylenetetrazole (PTZ) models [96]. Further research is needed with these compounds to understand the mechanism by which they attenuate seizures, and whether or not they are anti-convulsive only or anti-epileptogenic.
Giménez-Cassina and colleagues (2012) recently made an intriguing mechanistic link between alternative mitochondrial fuel utilization, apoptotic machinery and neuronal excitability. They demonstrated that the Bcl-2-associated agonist of cell death (BAD), controls mitochondrial fuel utilization and neuronal excitability via plasmalemmal KATP channels. BAD therefore is an optimal target for interfering with epilepsy progression due to the effects it has on cellular function, classically in promoting apoptosis but also in regulating glucose metabolism. BAD exists in either phosphorylated or dephosphorylated states, resulting in opposing effects on cell death. By manipulating the phosphorylation state of BAD they were able to induce a metabolic switch comparable to what occurs with the KD and 2-DG administration and confer resistance to seizures [97].
Potential Therapeutic Approaches to Targeting Metabolic Pathways and ROS in Epilepsy
Even with a myriad of anti-epileptic drugs approximately one-third of patients are refractory to these medications. Therefore, much of the focus in drug development has switched from treating or alleviating seizures to prevention or progression of the disease. The focus of current research is to develop treatments that are anti-epileptogenic as opposed to merely anti-seizure. Although it is evident that ROS and oxidative stress play a role in epileptogenesis, the exact pathogenesis and progression of free radicals and their effects on seizures remains unclear. Importantly, whether targeting mitochondrial processes has any effects on epileptogenesis remains to be determined. Two major issues associated with the therapeutic targeting of metabolic pathways is the complex interaction between 1) mitochondrial functions and cell signaling and 2) redundancies and adaptive control of metabolic perturbation. Despite the difficulties in specifically targeting metabolic pathways, it is clear that increased mitochondrial ROS occurs throughout epileptogenesis and contributes to seizure-induced neuronal injury. Additionally, global changes in metabolic function by switching fuels achieved with the KD can be antiepileptic. Collectively, this suggests that targeting mitochondria and ROS may be beneficial in controlling seizures and epileptogenesis by exerting neuroprotection and via alternate fuel utilization, both of which are operative during epileptogenesis. With respect to neuroprotection, preventing mitochondria-mediated damage may be directly beneficial or aid in the beneficial effects of neuroprotective drugs that also have anticonvulsant effects e.g. topiramate and leviteracitam. Preventing ROS formation and mitochondrial dysfunction may be an important avenue to prevent or delay the development of epilepsy and/or its comorbidities such as cognitive impairment. Levetiracetam, a novel antiepileptic drug, is unique to other drugs because of the effect it has on mitochondria. Leveritacetam has rescued mitochondrial deficits seen in GSH, aconitase, citrate synthase, CI, and α-KGDH in a model of experimental or electrically induced SE [98]. These effects however did not attenuate seizure severity or decrease the frequency of seizures as seen from EEG parameters.
With respect to antioxidant therapies, three types of therapies may be considered 1) bulk or stoichiometric scavengers such as vitamin E or C or nitroxide spin traps, 2) catalytic antioxidants such as salen, macrocyclic or metalloporphyrins or 3) indirect antioxidants such as iron chelators or KD which activates antioxidant pathways such as Nrf2, increases mitochondrial GSH and decreases ROS production [4]. Vitamin E has also been shown to have antioxidant and neuroprotective effects in the pilocarpine model. In these studies, vitamin E increased catalase levels along with brain free fatty acid levels [99]. Vitamin E given as a co-medication to children with refractory epilepsy had an effect of decreasing seizure frequency over 3 months [100]. However, subsequent clinical trials of vitamin E have been controversial with many failed attempts to lessen the occurrence of seizures in pediatric patients and in animal models of epilepsy. A variety of other antioxidant compounds have been used in many models with varying degrees of success. The variability in results shows that although certain antioxidants have been effective in certain models, these effects need increased research to understand why these effects often don't translate to humans and/or other rodent models [101,102]. The use of antioxidants that directly affect or improve mitochondrial function or oxidative stress pathways may provide new approaches for management of epilepsies.
The use of synthetic catalytic antioxidants capable of detoxifying ROS such as such as O2−, H2O2, ONOO−, and lipid peroxide radicals [103], have been shown to protect mitochondrial targets and decrease neuronal death in animal models of epilepsy [2]. The metalloporpyhrin MnTBAP and salen compounds (e.g. EUK-134) have been shown to lessen oxidative stress and neuronal damage induced by SE [104, 105]. Acute administration of MnTBAP did not attenuate the severity of behavioral seizures but did protect from mitochondrial aconitase inactivation, 8-OHdG formation, and hippocampal neuronal loss in the kainate model [2]. More recently it was shown that SOD2−/− mice from a mixed genetic background (B6D2F2) that lived approximately 3 weeks, exhibited frequent spontaneous seizures by the second to third postnatal week which were attenuated by a blood-brain barrier permeable metalloporphyrin catalytic antioxidant [72]. Additionally, these mice had increased lifespan and were protected from neuronal death while indices of mitochondrial oxidative stress were all partially rescued. Whether antioxidant therapies or alternative fuel substrates alter progression of epileptogenesis and/or comorbidities associated with TLE remain to be determined. Regardless, mitochondria remain an attractive target for epilepsy therapeutics.
Conclusion
In summary, there is ample evidence for increased ROS production and oxidative damage to cellular targets in experimental models of TLE. In human TLE, the evidence for oxidative damage is limited and more studies are needed using stable and informative biomarkers and methodologies. With respect to mitochondrial dysfunction, studies to date in human and experimental models have demonstrated alterations in various functions of mitochondria including apoptotic factors, ETC enzyme activities, ROS production/detoxification etc. Although alterations in these individual mitochondrial functions may herald “dysfunctional” mitochondria, it has been argued that direct assessment of the physiological function of mitochondria in generating ATP via OXPHOS is necessary to truly demonstrate mitochondrial “dysfunction” in a system [106]. Based on lack of bioenergetic assessment of mitochondria in experimental models of TLE, it is difficult to conclude that mitochondrial “dysfunction” occurs following seizures and/or epileptogenesis. Work from several laboratories including ours has shown oxidative damage to individual mitochondrial enzyme complexes as well as increased production of ROS during epileptogenesis. However, whether these individual events are adaptable via repair and/or antioxidant defenses and result in loss of ATP generation via OXPHOS remains to be established. Therefore, assigning a role for mitochondrial dysfunction in TLE based on currently available data should be cautiously interpreted.
Highlights.
We examine mitochondrial function and neuronal excitability in human and experimental models of temporal lobe epilepsy.
Mitochondria contribute to seizure-induced reactive oxygen species formation.
More work is needed to determine the mechanisms of how reactive oxygen species contributes to epileptogenesis.
We examine potential metabolic therapies and targets for epileptogenesis.
Acknowledgments
The authors acknowledge grants from CURE and NIH (NS039578) and help from Drs. Kristen Ryan and Donald Backos and the molecular modeling core at the University of Colorado Anschutz Medical Campus in preparing Figure 3.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Hauser WA, Annegers JF. Risk factors for epilepsy. Epilepsy research Supplement. 1991 Jan;4:45–52. [PubMed] [Google Scholar]
- 2.Liang LP, Ho YS, Patel M. Mitochondrial superoxide production in kainate-induced hippocampal damage. Neuroscience. 2000 Jan;101(3):563–70. doi: 10.1016/s0306-4522(00)00397-3. [DOI] [PubMed] [Google Scholar]
- 3.Patel M, Liang LP, Roberts LJ. Enhanced hippocampal F2-isoprostane formation following kainate-induced seizures. Journal of neurochemistry. 2001 Dec;79(5):1065–9. doi: 10.1046/j.1471-4159.2001.00659.x. [DOI] [PubMed] [Google Scholar]
- 4.Jarrett SG, Milder JB, Liang LP, Patel M. The ketogenic diet increases mitochondrial glutathione levels. Journal of neurochemistry. 2008 Aug;106(3):1044–51. doi: 10.1111/j.1471-4159.2008.05460.x. [DOI] [PubMed] [Google Scholar]
- 5.Garriga-Canut M, Schoenike B, Qazi R, Bergendahl K, Daley TJ, Pfender RM, Morrison JF, Ockuly J, Stafstrom C, Sutula T, Roopra A. 2-Deoxy-D-glucose reduces epilepsy progression by NRSF-CtBP-dependent metabolic regulation of chromatin structure. Nature neuroscience. 2006 Nov;9(11):1382–7. doi: 10.1038/nn1791. [DOI] [PubMed] [Google Scholar]
- 6.Rahaman S. Mitochondrial disease and epilepsy. Developmental Medicine & Child Neurology. 2012 May;54(5):397–406. doi: 10.1111/j.1469-8749.2011.04214.x. [DOI] [PubMed] [Google Scholar]
- 7.Kawai N, Miyake K, Kuroda Y, Yamashita S, Nishiyama Y, Monden T, Sasakawa Y, Nagao S. Magnetic resonance imaging and positron emission tomography findings in status epilepticus following severe hypoglycemia. Annals of nuclear medicine. 2006 Jun;20(5):371–6. doi: 10.1007/BF02987250. [DOI] [PubMed] [Google Scholar]
- 8.Vielhaber S, Niessen HG, Debska-Vielhaber G, Kudin AP, Wellmer J, Kaufmann J, Schönfeld MA, Fendrich R, Willker W, Leibfritz D, Schramm J, Elger CE, Heinze HJ, Kunz WS. Subfield-specific loss of hippocampal N-acetyl aspartate in temporal lobe epilepsy. Epilepsia. 2008 Jan;49(1):40–50. doi: 10.1111/j.1528-1167.2007.01280.x. [DOI] [PubMed] [Google Scholar]
- 9.Savic I, Thomas AM, Ke Y, Curran J, Fried I, Engel J. In vivo measurements of glutamine + glutamate (Glx) and N-acetyl aspartate (NAA) levels in human partial epilepsy. Acta neurologica Scandinavica. 2000 Sep;102(3):179–88. doi: 10.1034/j.1600-0404.2000.102003179.x. [DOI] [PubMed] [Google Scholar]
- 10.Kann O, Kovács R, Njunting M, Behrens CJ, Otáhal J, Lehmann TN, Gabriel S, Heinemann U. Metabolic dysfunction during neuronal activation in the ex vivo hippocampus from chronic epileptic rats and humans. Brain : a journal of neurology. 2005 Oct;128(Pt 10):2396–407. doi: 10.1093/brain/awh568. [DOI] [PubMed] [Google Scholar]
- 11.Meldrum BS. Excitotoxicity and selective neuronal loss in epilepsy. Brain pathology (Zurich, Switzerland) 1993 Oct;3(4):405–12. doi: 10.1111/j.1750-3639.1993.tb00768.x. [DOI] [PubMed] [Google Scholar]
- 12.Kilany A, Raouf ERA, Gaber AA, Aloush TK, Aref HA, Anwar M, Henshall DC, Abdulghani MO. Elevated serum Bcl-2 in children with temporal lobe epilepsy. Seizure : the journal of the British Epilepsy Association. 2012 May;21(4):250–3. doi: 10.1016/j.seizure.2012.01.004. [DOI] [PubMed] [Google Scholar]
- 13.Henshall DC, Clark RS, Adelson PD, Chen M, Watkins SC, Simon RP. Alterations in bcl-2 and caspase gene family protein expression in human temporal lobe epilepsy. Neurology. 2000 Jul;55(2):250–7. doi: 10.1212/wnl.55.2.250. [DOI] [PubMed] [Google Scholar]
- 14.Duchen MR. Mitochondria and calcium: from cell signalling to cell death. The Journal of physiology. 2000 Nov;529(Pt 1):57–68. doi: 10.1111/j.1469-7793.2000.00057.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Chuang YC, Chen SD, Liou CW, Lin TK, Chang WN, Chan SHH, Chang AYW. Contribution of nitric oxide, superoxide anion, and peroxynitrite to activation of mitochondrial apoptotic signaling in hippocampal CA3 subfield following experimental temporal lobe status epilepticus. Epilepsia. 2009 Apr;50(4):731–46. doi: 10.1111/j.1528-1167.2008.01778.x. [DOI] [PubMed] [Google Scholar]
- 16.Kovács R, Kardos J, Heinemann U, Kann O. Mitochondrial calcium ion and membrane potential transients follow the pattern of epileptiform discharges in hippocampal slice cultures. The Journal of neuroscience : the official journal of the Society for Neuroscience. 2005 Apr;25(17):4260–9. doi: 10.1523/JNEUROSCI.4000-04.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Jarrett SG, Liang LP, Hellier JL, Staley KJ, Patel M. Mitochondrial DNA damage and impaired base excision repair during epileptogenesis. Neurobiology of disease. 2008 Apr;30(1):130–8. doi: 10.1016/j.nbd.2007.12.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Kunz WS, Kudin AP, Vielhaber S, Blümcke I, Zuschratter W, Schramm J, Beck H, Elger CE. Mitochondrial complex I deficiency in the epileptic focus of patients with temporal lobe epilepsy. Annals of neurology. 2000 Nov;48(5):766–73. [PubMed] [Google Scholar]
- 19.Mueller SG, Trabesinger AH, Boesiger P, Wieser HG. Brain glutathione levels in patients with epilepsy measured by in vivo (1)H-MRS. Neurology. 2001 Oct;57(8):1422–7. doi: 10.1212/wnl.57.8.1422. [DOI] [PubMed] [Google Scholar]
- 20.Baron M, Kudin AP, Kunz WS. Mitochondrial dysfunction in neurodegenerative disorders. Biochemical Society transactions. 2007 Dec;35(Pt 5):1228–31. doi: 10.1042/BST0351228. [DOI] [PubMed] [Google Scholar]
- 21.O'Brien TJ, Newton MR, Cook MJ, Berlangieri SU, Kilpatrick C, Morris K, Berkovic SF. Hippocampal atrophy is not a major determinant of regional hypometabolism in temporal lobe epilepsy. Epilepsia. 1997 Jan;38(1):74–80. doi: 10.1111/j.1528-1157.1997.tb01080.x. [DOI] [PubMed] [Google Scholar]
- 22.Sashindranath M, McLean KJ, Trounce IA, Cotton RGH, Cook MJ. Early hippocampal oxidative stress is a direct consequence of seizures in the rapid electrical amygdala kindling model. Epilepsy research. 2010 Aug;90(3):285–94. doi: 10.1016/j.eplepsyres.2010.06.005. [DOI] [PubMed] [Google Scholar]
- 23.Frantseva MV, Perez Velazquez JL, Tsoraklidis G, Mendonca AJ, Adamchik Y, Mills LR, Carlen PL, Burnham MW. Oxidative stress is involved in seizure-induced neurodegeneration in the kindling model of epilepsy. Neuroscience. 2000 Jan;97(3):431–5. doi: 10.1016/s0306-4522(00)00041-5. [DOI] [PubMed] [Google Scholar]
- 24.Gluck MR, Jayatilleke E, Shaw S, Rowan AJ, Haroutunian V. CNS oxidative stress associated with the kainic acid rodent model of experimental epilepsy. Epilepsy research. 2000 Mar;39(1):63–71. doi: 10.1016/s0920-1211(99)00111-4. [DOI] [PubMed] [Google Scholar]
- 25.Peterson SL, Morrow D, Liu S, Liu KJ. Hydroethidine detection of superoxide production during the lithium-pilocarpine model of status epilepticus. Epilepsy research. 2002 May;49(3):226–38. doi: 10.1016/s0920-1211(02)00047-5. [DOI] [PubMed] [Google Scholar]
- 26.Forster MJ, Dubey A, Dawson KM, Stutts WA, Lal H, Sohal RS. Age-related losses of cognitive function and motor skills in mice are associated with oxidative protein damage in the brain. Proceedings of the National Academy of Sciences of the United States of America. 1996 May;93(10):4765–9. doi: 10.1073/pnas.93.10.4765. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Floor E, Wetzel MG. Increased protein oxidation in human substantia nigra pars compacta in comparison with basal ganglia and prefrontal cortex measured with an improved dinitrophenylhydrazine assay. Journal of neurochemistry. 1998 Jan;70(1):268–75. doi: 10.1046/j.1471-4159.1998.70010268.x. [DOI] [PubMed] [Google Scholar]
- 28.Bruce AJ, Baudry M. Oxygen free radicals in rat limbic structures after kainate-induced seizures. Free radical biology & medicine. 1995 Jun;18(6):993–1002. doi: 10.1016/0891-5849(94)00218-9. [DOI] [PubMed] [Google Scholar]
- 29.Gardner PR, Fridovich I. Inactivation-reactivation of aconitase in Escherichia coli. A sensitive measure of superoxide radical. The Journal of biological chemistry. 1992 May;267(13):8757–63. [PubMed] [Google Scholar]
- 30.Gardner PR, Costantino G, Szabó C, Salzman AL. Nitric oxide sensitivity of the aconitases. The Journal of biological chemistry. 1997 Oct;272(40):25071–6. doi: 10.1074/jbc.272.40.25071. [DOI] [PubMed] [Google Scholar]
- 31.Liang LP, Patel M. Mitochondrial oxidative stress and increased seizure susceptibility in Sod2(−/+) mice. Free radical biology & medicine. 2004 Mar;36(5):542–54. doi: 10.1016/j.freeradbiomed.2003.11.029. [DOI] [PubMed] [Google Scholar]
- 32.Cock HR, Tong X, Hargreaves IP, Heales SJR, Clark JB, Patsalos PN, Thom M, Groves M, Schapira AHV, Shorvon SD, Walker MC. Mitochondrial dysfunction associated with neuronal death following status epilepticus in rat. Epilepsy research. 2002 Feb;48(3):157–68. doi: 10.1016/s0920-1211(01)00334-5. [DOI] [PubMed] [Google Scholar]
- 33.Wallace DC. A mitochondrial paradigm of metabolic and degenerative diseases, aging, and cancer: a dawn for evolutionary medicine. Annual review of genetics. 2005 Jan;39:359–407. doi: 10.1146/annurev.genet.39.110304.095751. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Ryan K, Backos DS, Reigan P, Patel M. Post-Translational Oxidative Modification and Inactivation of Mitochondrial Complex I in Epileptogenesis. Journal of Neuroscience. 2012 Aug;32(33):11250–11258. doi: 10.1523/JNEUROSCI.0907-12.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Liang LP, Jarrett SG, Patel M. Chelation of mitochondrial iron prevents seizure-induced mitochondrial dysfunction and neuronal injury. The Journal of neuroscience : the official journal of the Society for Neuroscience. 2008 Nov;28(45):11550–6. doi: 10.1523/JNEUROSCI.3016-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Gerlach M, Ben-Shachar D, Riederer P, Youdim MB. Altered brain metabolism of iron as a cause of neurodegenerative diseases? Journal of neurochemistry. 1994 Sep;63(3):793–807. doi: 10.1046/j.1471-4159.1994.63030793.x. [DOI] [PubMed] [Google Scholar]
- 37.Lee DW, Andersen JK. Iron elevations in the aging Parkinsonian brain: a consequence of impaired iron homeostasis? Journal of neurochemistry. 2010 Jan;112(2):332–9. doi: 10.1111/j.1471-4159.2009.06470.x. [DOI] [PubMed] [Google Scholar]
- 38.Amici A, Levine RL, Tsai L, Stadtman ER. Conversion of amino acid residues in proteins and amino acid homopolymers to carbonyl derivatives by metal-catalyzed oxidation reactions. The Journal of biological chemistry. 1989 Feb;264(6):3341–6. [PubMed] [Google Scholar]
- 39.Kudin AP, Bimpong-Buta NYB, Vielhaber S, Elger CE, Kunz WS. Characterization of superoxide-producing sites in isolated brain mitochondria. The Journal of biological chemistry. 2004 Feb;279(6):4127–35. doi: 10.1074/jbc.M310341200. [DOI] [PubMed] [Google Scholar]
- 40.Fato R, Bergamini C, Bortolus M, Maniero AL, Leoni S, Ohnishi T, Lenaz G. Differential effects of mitochondrial Complex I inhibitors on production of reactive oxygen species. Biochimica et biophysica acta. 2009 May;1787(5):384–92. doi: 10.1016/j.bbabio.2008.11.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Hirst J. Towards the molecular mechanism of respiratory complex I. The Biochemical journal. 2010 Jan;425(2):327–39. doi: 10.1042/BJ20091382. [DOI] [PubMed] [Google Scholar]
- 42.Murphy MP. How mitochondria produce reactive oxygen species. The Biochemical journal. 2009 Jan;417(1):1–13. doi: 10.1042/BJ20081386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Rothstein JD, Martin L, Levey AI, Dykes-Hoberg M, Jin L, Wu D, Nash N, Kuncl RW. Localization of neuronal and glial glutamate transporters. Neuron. 1994 Sep;13(3):713–25. doi: 10.1016/0896-6273(94)90038-8. [DOI] [PubMed] [Google Scholar]
- 44.Frizzo ME, Frizzo JK, Amadio S, Rodrigues JM, Perry ML, Bernardi G, Volonté C. Extracellular adenosine triphosphate induces glutamate transporter-1 expression in hippocampus. Hippocampus. 2007 Jan;17(4):305–15. doi: 10.1002/hipo.20269. [DOI] [PubMed] [Google Scholar]
- 45.Trotti D, Danbolt NC, Volterra A. Glutamate transporters are oxidant-vulnerable: a molecular link between oxidative and excitotoxic neurodegeneration? Trends in pharmacological sciences. 1998 Aug;19(8):328–34. doi: 10.1016/s0165-6147(98)01230-9. [DOI] [PubMed] [Google Scholar]
- 46.Meldrum BS, Akbar MT, Chapman AG. Glutamate receptors and transporters in genetic and acquired models of epilepsy. Epilepsy research. 1999 Sep;36(2–3):189–204. doi: 10.1016/s0920-1211(99)00051-0. [DOI] [PubMed] [Google Scholar]
- 47.Tritschler HJ, Medori R. Mitochondrial DNA alterations as a source of human disorders. Neurology. 1993 Feb;43(2):280–8. doi: 10.1212/wnl.43.2.280. [DOI] [PubMed] [Google Scholar]
- 48.Esposito LA, Melov S, Panov A, Cottrell BA, Wallace DC. Mitochondrial disease in mouse results in increased oxidative stress. Proceedings of the National Academy of Sciences of the United States of America. 1999 Apr;96(9):4820–5. doi: 10.1073/pnas.96.9.4820. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Bohr VA, Stevnsner T, de Souza-Pinto NC. Mitochondrial DNA repair of oxidative damage in mammalian cells. Gene. 2002 Mar;286(1):127–34. doi: 10.1016/s0378-1119(01)00813-7. [DOI] [PubMed] [Google Scholar]
- 50.Kudin AP, Kudina TA, Seyfried J, Vielhaber S, Beck H, Elger CE, Kunz WS. Seizure-dependent modulation of mitochondrial oxidative phosphorylation in rat hippocampus. The European journal of neuroscience. 2002 Apr;15(7):1105–14. doi: 10.1046/j.1460-9568.2002.01947.x. [DOI] [PubMed] [Google Scholar]
- 51.Dizdaroglu M. Chemical determination of free radical-induced damage to DNA. Free radical biology & medicine. 1991 Jan;10(3–4):225–42. doi: 10.1016/0891-5849(91)90080-m. [DOI] [PubMed] [Google Scholar]
- 52.Halliwell B, Gutteridge JM. Oxygen free radicals and iron in relation to biology and medicine: some problems and concepts. Archives of biochemistry and biophysics. 1986 May;246(2):501–14. doi: 10.1016/j.abb.2022.109246. [DOI] [PubMed] [Google Scholar]
- 53.Coyle JT, Puttfarcken P. Oxidative stress, glutamate, and neurodegenerative disorders. Science (New York, NY) 1993 Oct;262(5134):689–95. doi: 10.1126/science.7901908. [DOI] [PubMed] [Google Scholar]
- 54.Eraković V, Zupan G, Mrsić J, Simonić A, Varljen J. The influence of nicardipine and ifenprodil on the brain free arachidonic acid level and behavior in hypoxia-exposed rats. Progress in neuro-psychopharmacology & biological psychiatry. 1997 May;21(4):633–47. doi: 10.1016/s0278-5846(97)00037-7. [DOI] [PubMed] [Google Scholar]
- 55.Mrsić J, Zupan G, Eraković V, Simonić A, Varljen J. The influence of nimodipine and MK-801 on the brain free arachidonic acid level and the learning ability in hypoxia-exposed rats. Progress in neuro-psychopharmacology & biological psychiatry. 1997 Feb;21(2):345–58. doi: 10.1016/s0278-5846(97)00005-5. [DOI] [PubMed] [Google Scholar]
- 56.Roberts LJ, Morrow JD. Measurement of F(2)-isoprostanes as an index of oxidative stress in vivo. Free radical biology & medicine. 2000 Feb;28(4):505–13. doi: 10.1016/s0891-5849(99)00264-6. [DOI] [PubMed] [Google Scholar]
- 57.Patel M, Liang LP, Hou H, Williams BB, Kmiec M, Swartz HM, Fessel JP, Roberts LJ. Seizure-induced formation of isofurans: novel products of lipid peroxidation whose formation is positively modulated by oxygen tension. Journal of neurochemistry. 2008 Jan;104(1):264–70. doi: 10.1111/j.1471-4159.2007.04974.x. [DOI] [PubMed] [Google Scholar]
- 58.Cini M, Moretti A. Studies on lipid peroxidation and protein oxidation in the aging brain. Neurobiology of aging. 16(1):53–7. doi: 10.1016/0197-4580(95)80007-e. [DOI] [PubMed] [Google Scholar]
- 59.Tejada S, Sureda A, Roca C, Gamundí A, Esteban S. Antioxidant response and oxidative damage in brain cortex after high dose of pilocarpine. Brain research bulletin. 2007 Jan;71(4):372–5. doi: 10.1016/j.brainresbull.2006.10.005. [DOI] [PubMed] [Google Scholar]
- 60.Reed DJ, Savage MK. Influence of metabolic inhibitors on mitochondrial permeability transition and glutathione status. Biochimica et biophysica acta. 1995 May;1271(1):43–50. doi: 10.1016/0925-4439(95)00008-r. [DOI] [PubMed] [Google Scholar]
- 61.Liang LP, Patel M. Seizure-induced changes in mitochondrial redox status. Free radical biology & medicine. 2006 Jan;40(2):316–22. doi: 10.1016/j.freeradbiomed.2005.08.026. [DOI] [PubMed] [Google Scholar]
- 62.O'Donovan DJ, Rogers LK, Kelley DK, Welty SE, Ramsay PL, Smith CV. CoASH and CoASSG levels in lungs of hyperoxic rats as potential biomarkers of intramitochondrial oxidant stresses. Pediatric research. 2002 Mar;51(3):346–53. doi: 10.1203/00006450-200203000-00014. [DOI] [PubMed] [Google Scholar]
- 63.Waldbaum S, Liang LP, Patel M. Persistent impairment of mitochondrial and tissue redox status during lithium-pilocarpine-induced epileptogenesis. Journal of neurochemistry. 2010 Dec;115(5):1172–82. doi: 10.1111/j.1471-4159.2010.07013.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Sudha K, Rao AV, Rao A. Oxidative stress and antioxidants in epilepsy. Clinica chimica acta; international journal of clinical chemistry. 2001 Jan;303(1–2):19–24. doi: 10.1016/s0009-8981(00)00337-5. [DOI] [PubMed] [Google Scholar]
- 65.Carlsson LM, Jonsson J, Edlund T, Marklund SL. Mice lacking extracellular superoxide dismutase are more sensitive to hyperoxia. Proceedings of the National Academy of Sciences of the United States of America. 1995 Jul;92(14):6264–8. doi: 10.1073/pnas.92.14.6264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Reaume AG, Elliott JL, Hoffman EK, Kowall NW, Ferrante RJ, Siwek DF, Wilcox HM, Flood DG, Beal MF, Brown RH, Scott RW, Snider WD. Motor neurons in Cu/Zn superoxide dismutase-deficient mice develop normally but exhibit enhanced cell death after axonal injury. Nature genetics. 1996 May;13(1):43–7. doi: 10.1038/ng0596-43. [DOI] [PubMed] [Google Scholar]
- 67.Li Y, Huang TT, Carlson EJ, Melov S, Ursell PC, Olson JL, Noble LJ, Yoshimura MP, Berger C, Chan PH, Wallace DC, Epstein CJ. Dilated cardiomyopathy and neonatal lethality in mutant mice lacking manganese superoxide dismutase. Nature genetics. 1995 Dec;11(4):376–81. doi: 10.1038/ng1295-376. [DOI] [PubMed] [Google Scholar]
- 68.Huang TT, Carlson EJ, Kozy HM, Mantha S, Goodman SI, Ursell PC, Epstein CJ. Genetic modification of prenatal lethality and dilated cardiomyopathy in Mn superoxide dismutase mutant mice. Free radical biology & medicine. 2001 Nov;31(9):1101–10. doi: 10.1016/s0891-5849(01)00694-3. [DOI] [PubMed] [Google Scholar]
- 69.Patel MN. Metalloporphyrins improve the survival of Sod2-deficient neurons. Aging cell. 2003 Aug;2(4):219–22. doi: 10.1046/j.1474-9728.2003.00055.x. [DOI] [PubMed] [Google Scholar]
- 70.Patel M, Li QY, Chang LY, Crapo J, Liang LP. Activation of NADPH oxidase and extracellular superoxide production in seizure-induced hippocampal damage. Journal of neurochemistry. 2005 Jan;92(1):123–31. doi: 10.1111/j.1471-4159.2004.02838.x. [DOI] [PubMed] [Google Scholar]
- 71.Hwang J, Saha A, Boo YC, Sorescu GP, McNally JS, Holland SM, Dikalov S, Giddens DP, Griendling KK, Harrison DG, Jo H. Oscillatory shear stress stimulates endothelial production of O2- from p47phox-dependent NAD(P)H oxidases, leading to monocyte adhesion. The Journal of biological chemistry. 2003 Nov;278(47):47291–8. doi: 10.1074/jbc.M305150200. [DOI] [PubMed] [Google Scholar]
- 72.Liang LP, Waldbaum S, Rowley S, Huang TT, Day BJ, Patel M. Mitochondrial oxidative stress and epilepsy in SOD2 deficient mice: attenuation by a lipophilic metalloporphyrin. Neurobiology of disease. 2012 Mar;45(3):1068–76. doi: 10.1016/j.nbd.2011.12.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.James AM, Murphy MP. How mitochondrial damage affects cell function. Journal of biomedical science. 2002 Jan;9(6 Pt 1):475–87. doi: 10.1159/000064721. [DOI] [PubMed] [Google Scholar]
- 74.Galella G, Turano C, Girvin J. Metabolic changes in the hippocampus after prolonged epileptic discharge. Journal of neurosurgical sciences. 27(2):69–71. [PubMed] [Google Scholar]
- 75.Handforth A, Treiman DM. Functional mapping of the late stages of status epilepticus in the lithium-pilocarpine model in rat: a 14C-2-deoxyglucose study. Neuroscience. 1995 Feb;64(4):1075–89. doi: 10.1016/0306-4522(94)00377-h. [DOI] [PubMed] [Google Scholar]
- 76.Folbergrová J, Ingvar M, Nevander G, Siesjö BK. Cerebral metabolic changes during and following fluorothyl-induced seizures in ventilated rats. Journal of neurochemistry. 1985 May;44(5):1419–26. doi: 10.1111/j.1471-4159.1985.tb08778.x. [DOI] [PubMed] [Google Scholar]
- 77.Nakamoto Y, Nakayama S, Suzuki J. Cerebral uptake of [14C]deoxyglucose during the entire seizure and the recovery period in an El mouse. Epilepsy research. 5(1):43–8. doi: 10.1016/0920-1211(90)90064-3. [DOI] [PubMed] [Google Scholar]
- 78.Nishikawa T, Edelstein D, Du XL, Yamagishi S, Matsumura T, Kaneda Y, Yorek MA, Beebe D, Oates PJ, Hammes HP, Giardino I, Brownlee M. Normalizing mitochondrial superoxide production blocks three pathways of hyperglycaemic damage. Nature. 2000 Apr;404(6779):787–90. doi: 10.1038/35008121. [DOI] [PubMed] [Google Scholar]
- 79.Erecińska M, Silver IA. Ions and energy in mammalian brain. Progress in neurobiology. 1994 May;43(1):37–71. doi: 10.1016/0301-0082(94)90015-9. [DOI] [PubMed] [Google Scholar]
- 80.Grisar T. Glial and neuronal Na+-K+ pump in epilepsy. Annals of neurology. 1984 Jan;16(Suppl):S128–34. doi: 10.1002/ana.410160719. [DOI] [PubMed] [Google Scholar]
- 81.Gulyás AI, Buzsáki G, Freund TF, Hirase H. Populations of hippocampal inhibitory neurons express different levels of cytochrome c. The European journal of neuroscience. 2006 May;23(10):2581–94. doi: 10.1111/j.1460-9568.2006.04814.x. [DOI] [PubMed] [Google Scholar]
- 82.Verstreken P, Ly CV, Venken KJT, Koh TW, Zhou Y, Bellen HJ. Synaptic mitochondria are critical for mobilization of reserve pool vesicles at Drosophila neuromuscular junctions. Neuron. 2005 Aug;47(3):365–78. doi: 10.1016/j.neuron.2005.06.018. [DOI] [PubMed] [Google Scholar]
- 83.Vesce S, Jekabsons MB, Johnson-Cadwell LI, Nicholls DG. Acute glutathione depletion restricts mitochondrial ATP export in cerebellar granule neurons. The Journal of biological chemistry. 2005 Nov;280(46):38720–8. doi: 10.1074/jbc.M506575200. [DOI] [PubMed] [Google Scholar]
- 84.Yadava N, Nicholls DG. Spare respiratory capacity rather than oxidative stress regulates glutamate excitotoxicity after partial respiratory inhibition of mitochondrial complex I with rotenone. The Journal of neuroscience : the official journal of the Society for Neuroscience. 2007 Jul;27(27):7310–7. doi: 10.1523/JNEUROSCI.0212-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Kovac S, Abramov AY, Walker MC. Energy depletion in seizures: Anaplerosis as a strategy for future therapies. Neuropharmacology. 2012 May; doi: 10.1016/j.neuropharm.2012.05.012. [DOI] [PubMed] [Google Scholar]
- 86.Cantu D, Schaack J, Patel M. Oxidative inactivation of mitochondrial aconitase results in iron and H2O2-mediated neurotoxicity in rat primary mesencephalic cultures. PloS one. 2009 Jan;4(9):e7095. doi: 10.1371/journal.pone.0007095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Cantu D, Fulton RE, Drechsel DA, Patel M. Mitochondrial aconitase knockdown attenuates paraquat-induced dopaminergic cell death via decreased cellular metabolism and release of iron and H2O2. Journal of neurochemistry. 2011 Jul;118(1):79–92. doi: 10.1111/j.1471-4159.2011.07290.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Thiele EA. Assessing the efficacy of antiepileptic treatments: the ketogenic diet. Epilepsia. 2003 Jan;44(Suppl 7):26–9. doi: 10.1046/j.1528-1157.44.s7.4.x. [DOI] [PubMed] [Google Scholar]
- 89.Schwechter EM, Velísková J, Velísek L. Correlation between extracellular glucose and seizure susceptibility in adult rats. Annals of neurology. 2003 Jan;53(1):91–101. doi: 10.1002/ana.10415. [DOI] [PubMed] [Google Scholar]
- 90.Bough KJ, Wetherington J, Hassel B, Pare JF, Gawryluk JW, Greene JG, Shaw R, Smith Y, Geiger JD, Dingledine RJ. Mitochondrial biogenesis in the anticonvulsant mechanism of the ketogenic diet. Annals of neurology. 2006 Aug;60(2):223–35. doi: 10.1002/ana.20899. [DOI] [PubMed] [Google Scholar]
- 91.Maalouf M, Sullivan PG, Davis L, Kim DY, Rho JM. Ketones inhibit mitochondrial production of reactive oxygen species production following glutamate excitotoxicity by increasing NADH oxidation. Neuroscience. 2007 Mar;145(1):256–64. doi: 10.1016/j.neuroscience.2006.11.065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Stafstrom CE, Ockuly JC, Murphree L, Valley MT, Roopra A, Sutula TP. Anticonvulsant and antiepileptic actions of 2-deoxy-D-glucose in epilepsy models. Annals of neurology. 2009 Apr;65(4):435–47. doi: 10.1002/ana.21603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Weindruch R, Naylor PH, Goldstein AL, Walford RL. Influences of aging and dietary restriction on serum thymosin alpha 1 levels in mice. Journal of gerontology. 1988 Mar;43(2):B40–2. doi: 10.1093/geronj/43.2.b40. [DOI] [PubMed] [Google Scholar]
- 94.Gasior M, Yankura J, Hartman AL, French A, Rogawski MA. Anticonvulsant and proconvulsant actions of 2-deoxy-D-glucose. Epilepsia. 2010 Aug;51(8):1385–94. doi: 10.1111/j.1528-1167.2010.02593.x. [DOI] [PubMed] [Google Scholar]
- 95.Vexler ZS, Wong A, Francisco C, Manabat C, Christen S, Täuber M, Ferriero DM, Gregory G. Fructose-1,6-bisphosphate preserves intracellular glutathione and protects cortical neurons against oxidative stress. Brain research. 2003 Jan;960(1–2):90–8. doi: 10.1016/s0006-8993(02)03777-0. [DOI] [PubMed] [Google Scholar]
- 96.Lian XY, Khan FA, Stringer JL. Fructose-1,6-bisphosphate has anticonvulsant activity in models of acute seizures in adult rats. The Journal of neuroscience : the official journal of the Society for Neuroscience. 2007 Oct;27(44):12007–11. doi: 10.1523/JNEUROSCI.3163-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Giménez-Cassina A, Martínez-François JR, Fisher JK, Szlyk B, Polak K, Wiwczar J, Tanner GR, Lutas A, Yellen G, Danial NN. BAD-dependent regulation of fuel metabolism and K(ATP) channel activity confers resistance to epileptic seizures. Neuron. 2012 May;74(4):719–30. doi: 10.1016/j.neuron.2012.03.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Gibbs JE, Walker MC, Cock HR. Levetiracetam: antiepileptic properties and protective effects on mitochondrial dysfunction in experimental status epilepticus. Epilepsia. 2006 Mar;47(3):469–78. doi: 10.1111/j.1528-1167.2006.00454.x. [DOI] [PubMed] [Google Scholar]
- 99.Barros DO, Xavier SML, Barbosa CO, Silva RF, Freitas RLM, Maia FD, Oliveira AA, Freitas RM, Takahashi RN. Effects of the vitamin E in catalase activities in hippocampus after status epilepticus induced by pilocarpine in Wistar rats. Neuroscience letters. 2007 Apr;416(3):227–30. doi: 10.1016/j.neulet.2007.01.057. [DOI] [PubMed] [Google Scholar]
- 100.Ogunmekan AO, Hwang PA. A randomized, double-blind, placebo-controlled, clinical trial of D-alpha-tocopheryl acetate (vitamin E), as add-on therapy, for epilepsy in children. Epilepsia. 30(1):84–9. doi: 10.1111/j.1528-1157.1989.tb05287.x. [DOI] [PubMed] [Google Scholar]
- 101.Levy SL, Burnham WM, Bishai A, Hwang PA. The anticonvulsant effects of vitamin E: a further evaluation. The Canadian journal of neurological sciences. Le journal canadien des sciences neurologiques. 1992 May;19(2):201–3. [PubMed] [Google Scholar]
- 102.Xu K, Stringer JL. Antioxidants and free radical scavengers do not consistently delay seizure onset in animal models of acute seizures. Epilepsy & behavior : E&B. 2008 Jul;13(1):77–82. doi: 10.1016/j.yebeh.2008.03.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Patel M, Day BJ. Metalloporphyrin class of therapeutic catalytic antioxidants. Trends in pharmacological sciences. 1999 Sep;20(9):359–64. doi: 10.1016/s0165-6147(99)01336-x. [DOI] [PubMed] [Google Scholar]
- 104.Melov S, Doctrow SR, Schneider JA, Haberson J, Patel M, Coskun PE, Huffman K, Wallace DC, Malfroy B. Lifespan extension and rescue of spongiform encephalopathy in superoxide dismutase 2 nullizygous mice treated with superoxide dismutase-catalase mimetics. The Journal of neuroscience : the official journal of the Society for Neuroscience. 2001 Nov;21(21):8348–53. doi: 10.1523/JNEUROSCI.21-21-08348.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Rong Y, Doctrow SR, Tocco G, Baudry M. EUK-134, a synthetic superoxide dismutase and catalase mimetic, prevents oxidative stress and attenuates kainate-induced neuropathology. Proceedings of the National Academy of Sciences of the United States of America. 1999 Aug;96(17):9897–902. doi: 10.1073/pnas.96.17.9897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Brand MD, Nicholls DG. Assessing mitochondrial dysfunction in cells. The Biochemical journal. 2011 Apr;435(2):297–312. doi: 10.1042/BJ20110162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Freitas RM, Vasconcelos SMM, Souza FCF, Viana GSB, Fonteles MMF. Oxidative stress in the hippocampus after pilocarpine-induced status epilepticus in Wistar rats. The FEBS journal. 2005 Mar;272(6):1307–12. doi: 10.1111/j.1742-4658.2004.04537.x. [DOI] [PubMed] [Google Scholar]