

Abstract
Breast cancer resistance protein (BCRP) is an efflux transporter expressed in tissues that act as barriers to drug entry. Given that single nucleotide polymorphisms (SNPs) in the ABCG2 gene encoding BCRP are common, the possibility exists that these genetic variants may be a determinant of interindividual variability in drug response. The objective of this study is to confirm the human BCRP-mediated transport of sulfasalazine in vitro, evaluate interindividual variation in BCRP expression in human intestine and to determine the role of ABCG2 SNPs to drug disposition in healthy patients using sulfasalazine as a novel in vivo probe. To evaluate these objectives, pinch biopsies were obtained from 18 patients undergoing esophagogastro–duodenoscopy or colonoscopy for determination of BCRP expression in relation to genotype. Wild-type and variant BCRP were expressed in a heterologous expression system to evaluate the effect of SNPs on cell-surface trafficking. A total of 17 healthy individuals participated in a clinical investigation to determine the effect of BCRP SNPs on sulfasalazine pharmacokinetics. In vitro, the cell surface protein expression of the common BCRP 421 C>A variant was reduced in comparison with the wild-type control. Intestinal biopsy samples revealed that BCRP protein and mRNA expression did not significantly differ between patients with 34GG/421CC versus patients with 34GG/421CA genotypes. Remarkably, in subjects with 34GG/421CA genotype, sulfasalazine area under the concentration-time curve was 2.4-fold greater compared with 34GG/421CC subjects (P<0.05). This study links commonly occurring SNPs in BCRP with significantly increased oral sulfasalazine plasma exposure in humans. Accordingly, sulfasalazine may prove to have utility as in vivo probe for assessing the clinical impact of BCRP for the disposition and efficacy of drugs.
Keywords: breast cancer resistance protein/ATP binding cassette, drug transport, member 2, pharmacogenetics, pharmacokinetics, subfamily G (WHITE), sulfasalazine
Introduction
Breast cancer resistance protein (BCRP) is a member of the ATP binding cassette family of transporters and was first identified in a drug resistant breast cancer cell line [1]. BCRP mediates the efflux transport of a wide variety of xenobiotic compounds and endogenous molecules. It is widely expressed in tissues including heart [2], brain [1,3], placenta [1], kidney [4], mammary gland [5], stem cells [6], liver [1] and intestine [1]. BCRP is increasingly recognized for its role in regulating drug disposition and environmental carcinogen exposure owing to its broad substrate specificity and expression in tissues that act as barriers to drug entry and drug distribution. Unlike P-glycoprotein (multiple drug resistance 1/ P-gp/ ABCB1) and members of the multidrug resistance-associated protein (ABCC) subfamily that contain 12–18 transmem-brane domains and two nucleotide binding domains, BCRP is considered a ‘half transporter’, which appears to function as a homo-oligomer to mediate ATP-dependent drug efflux [7].
Studies in abcg2-deficient mice have delineated an important role for intestinal BCRP in determining the oral bioavailability of drugs, such as, topotecan [8,9], nitrofurantoin [10], fluoroquinolones [11] and imatinib [12]. These results, in addition to studies that have localized the transporter on the apical membrane of enterocytes [13], indicate that BCRP acts to efflux solutes back into the intestinal lumen to limit absorption. Therefore, in humans it is likely that the interindividual differences in intestinal expression and function of BCRP contribute to variability in drug bioavailability, exposure and pharmacological response. Although BCRP is expressed in the duodenum, ileum, jejunum and colon [14], the extent of interindividual difference in expression of BCRP in the gastrointestinal tract of humans has not been systematically assessed.
In addition, several nonsynonymous single nucleotide polymorphisms (SNPs) of the ABCG2 gene have been described including ABCG2 34G > A (V12M) located in exon 2 and 421C > A (Q141K) in exon 5. The allele frequency of the 34 G>A SNP varies from as low as 2% in European–American’s to between 15–20% in Asian–Americans, whereas the 421 C > A SNP is more common, being found at a frequency of 14% in European–Americans and up to 35% in Asian–Americans [15]. A number of in vitro studies have shown that the 421C > A variant has reduced efflux transport function owing to low cellular expression, poor membrane sorting, or altered activity [16–18]. These findings are consistent with studies that demonstrate a substantial effect of the 421 C > A genotype on the pharmacokinetics of substrate drugs, such as diflomotecan [19] and rosuvastatin [20], or predict risk for drug toxicity, such as gefitinib-associated diarrhea [21]. Hence, ABCG2 genotype appears to have phenotypic consequences for a number of drugs.
Probe drugs are often administered to obtain an in vivo phenotypic measure of specific drug metabolism or transport pathways in individuals. Ideally, a transporter probe drug substrate would display little or predictable hepatic metabolism. In the case of BCRP, relatively safe and validated probe drugs have not yet been described despite that such tools would be useful in understanding mechanisms of variation in drug response. Most drugs studied to date in relation to BCRP activity in vivo have been anticancer agents that are not innocuous and others, such as rosuvastatin, are subject to multiple transporter pathways whose activities are genetically determined [22]. A recent study demonstrated that long-term treatment of a human T lymphoblast cell line with the anti-inflammatory agent, sulfasalazine, causes the development of cellular drug resistance mediated by the induction of BCRP, suggesting that sulfasalazine is a substrate of human BCRP [23]. When sulfasalazine pharmacokinetics were assessed in a Bcrp knockout mouse model, oral administration of 20 mg/kg sulfasalazine resulted in a striking 111-fold increase in the area under the plasma concentration-time curve (AUC) in Bcrp knockout mice compared with the wild-type control [24]. Importantly, sulfasalazine AUC in mdr1a knockout mice was not significantly different than wild-type mice following 20 mg/kg sulfasalazine, suggesting sulfasalazine as a potential sensitive and selective in vivo probe of BCRP activity in humans. Interestingly, oral sulfasalazine exposure was highly variable in early human studies and this variability was not associated with its metabolite profile [25].
In this report, we show significant variability in intestinal BCRP expression in human patients. Further, we demonstrate in vitro sulfasalazine transport by human BCRP. Finally, in a study of healthy human volunteers, we establish that the wide variability in sulfasalazine pharmacokinetics is at least partially attributed to common SNPs in BCRP. Taken together, we demonstrate that BCRP polymorphisms are associated with significant alterations in attained sulfasalazine plasma levels, suggesting that sulfasalazine may be a useful in vivo probe of BCRP activity. Furthermore, these findings raise the possibility that sulfasalazine phenotyping may aid in predicting response to other BCRP-substrate drugs, including many used for cancer chemotherapy.
Methods
Materials
[3H] sulfasalazine (specific activity 115.33 μCi/mg, > 99% purity) was obtained from Pfizer Global Research and Development (Kalamazoo, Michigan, USA). All other reagents and chemicals were obtained from Sigma-Aldrich (St Louis, Missouri, USA) unless otherwise stated.
Determination of intestinal breast cancer resistance protein expression
A total of 18 patients (eight men and 10 women) undergoing diagnostic esophagogastro–duodenoscopy or colonoscopy participated in this study. During the patient’s scheduled routine endoscopic procedure, two pinch biopsies were collected in addition to those obtained for diagnostic purposes. The first sample was immediately flash frozen in liquid nitrogen and stored at − 80°C for protein analysis. The second sample for mRNA expression was stored in RNAlater according to the manufacturer’s instructions (Qiagen, Valencia, California, USA). The study protocol was approved by the Health Sciences Research Ethics Board at the University of Western Ontario and all the patients provided written informed consent before the procedure.
Samples for protein analysis were homogenized in 1 mmol/l EDTA, 10 mmol/l Tris, pH=7.4 with protease inhibitor. Homogenates were prepared in loading buffer and heat denatured at 70°C for 10 min. Samples were separated on 4–12% bis-tris gradient gels (Invitrogen, Carlsbad, California, USA) and subsequently transferred onto nitrocellulose membranes. Following blocking with 5% milk powder, the membranes were probed with the primary mouse monoclonal antibody BXP-21 (Alexis, Lausen, Switzerland) followed by a goat antimouse HRP conjugated secondary antibody (Bio-Rad, Hercules, California, USA). Proteins were visualized by chemiluminescence detection using the ECL Plus system (Amersham, Piscataway, New Jersey, USA) and images digitally captured using the Kodak 4000 mm Image Station (Mandel, Guelph, Ontario, Canada). For detection of villin, blots were incubated for 15 min in stripping buffer (Pierce, Rockford, Illinois, USA), washed and the above steps repeated using a primary antibody raised against human villin (Ab-1, Thermo Scientific, Waltham, Massachusetts). Mean optical density of BCRP and villin was determined using Kodak Molecular Imaging Software Version 4.0 and expressed as a normalized ratio of BCRP to villin.
RNAlater stored tissue was homogenized and RNA extracted in Trizol (Invitrogen) following standard methods. The cDNA synthesis was performed with a total of 500 ng of RNA. Quantitative reverse transcriptase-PCR for BCRP was performed using a SYBR green assay (Applied Biosystems, Foster City, California, USA) with the following primers: 5′-TGGCTGTCATGGCTTCAGTA-3′ (forward) and 5′-GCCACGTGATTCTTCCACAA-3′ (reverse). All samples were compared with a standard curve of the BCRP amplicon cloned into pCR2.1 TOPO (Invitrogen) for quantitative determination of copy number. BCRP expression was normalized to expression of the enterocyte-specific marker, villin, which was amplified using the following primers: 5′-CTGGCAACCTTAGGGACTGG-3′ (forward) and 5′-GTTAGCATTGAACACGTCCACTTT-3′ (reverse), which was compared with a standard curve of the villin amplicon cloned into pCR2.1 TOPO.
Wild-type and variant breast cancer resistance protein plasmid construction
The full open reading frame of human BCRP cDNA was obtained by PCR, using a PCR Optimizer Kit (Invitrogen) with AmpliTaq DNA polymerase (PerkinElmer Life Sciences, Waltham, Massachusetts, USA) from a cDNA library synthesized from human liver mRNA using oligonucleotide primers 5′-CTGTGGAGGAACTGGGTAGGATTTAGGAACG-3′ (forward) and 5′-GTGATGGCAAGGGAACAGAAAACAACAAAAA-3′ (reverse). The PCR product was ligated into the pEF6/V5-His-TOPO vector (Invitrogen) and transformed in E.coli. A pEF6/V5-His-TOPO/BCRP was fully sequenced using an ABI 3700 DNA Analyzer (Applied Biosystems Inc.), and found to match the published reference sequence (GenBank accession number NM_004827). This clone was termed wild-type. Site-directed mutagenesis was utilized to create the nonsynonymous allelic variants, 34G > A (V12M) and 421C > A (Q141K). The appropriate point mutations were introduced individually into wild-type BCRP using the QuikChange site-directed mutagenesis kit (Stratagene, La Jolla, California, USA).
Epitope-tagged wild-type and variant breast cancer resistance protein construction
We utilized a 14 amino acid C-terminus epitope (V5) in the pEF6/V5-His-TOPO vector (Invitrogen) to generate V5-tagged wild-type and variant BCRP proteins. The QuikChange site-directed mutagenesis kit (Stratagene) was used to introduce a point mutation, which converted the stop codon of BCRP to a tyrosine residue (TAA→TAT) utilizing the following oligonucleotide sense and antisense primers: 5′-CTTAAAAAATATTCTTATAATTTCCCCTTAATTCAG-3′ and 5′-CTGAATTAAGGGGAAATTATAAGAATATTTTTTAAG-3′. The presence of the mutation was verified by full sequencing. The C-terminal V5 tag was generated for wild-type BCRP and each of the nonsynonymous BCRP variants, and these were used for characterization of total protein and cell surface expression.
Breast cancer resistance protein expression in HeLa Cells
HeLa cells were grown in six-well plates (approximately 0.8 × 106 cells/well) and infected with vaccinia (vtf-7) at a multiplicity of infection of 10 PFU/cell in serum-free Opti-MEM I medium (Life Technologies, Inc., Gaithersburg, Maryland, USA) and allowed to adsorb for 30 min at 37°C. Cells in each well were then transfected with 1 μg of wild-type or variant BCRP cDNA along with Lipofectin (Invitrogen) and incubated at 37°C for 16 h. The parental plasmid lacking any insert was used as control. HeLa cells transfected with BCRP cDNA were scraped off plates, and the resulting suspension was centrifuged at 21000g for 3 min. The cell pellet was reconstituted with HED buffer (25 mmol/l HEPES, 1.5 mmol/l EDTA, 1 mmol/l dithiothreitol, pH 7.4) containing protease inhibitors (Complete, Roche Molecular Biochemicals, Indianapolis, Indiana, USA) and lysed by sonication. Samples were diluted with 2X Laemmli buffer, and 20 μl of total cell protein were separated by SDS-PAGE on 10% gels. Following transfer onto nitrocellulose membranes, blots were probed with a monoclonal antiV5 antibody (1: 5000 dilution) (Invitrogen) and appropriate secondary antibody. To normalize sample loading, blots were stripped and reprobed with anti-calnexin antibody (StressGen, Victoria, British Colombia, Canada). Bands were visualized using enhanced chemiluminescence (Amersham Pharmacia Biotech, Piscataway, New Jersey, USA).
Breast cancer resistance protein cell surface expression
HeLa cells were grown on six-well plates and transfected with BCRP cDNAs, tagged with the V5 epitope. Sixteen hours post-transfection, cells were washed with ice-cold, phosphate-buffered saline Ca/Mg and then treated with a membrane-impermeable biotinylating agent (sulfo-NHS-SS-biotin, 1.5 mg/ml, Pierce, Rockford, Illinois, USA) at 4°C for 1 h. Subsequently, the cells were washed three times with ice-cold, phosphate-buffered saline Ca/Mg containing 100 mmol/l glycine and then incubated for 20 min at 4°C with the same buffer to remove the remaining labeling agent. After washing with phosphate-buffered saline Ca/Mg, cells were disrupted with 700 μl of lysis buffer (10 mmol/l Tris-base, 150 mmol/l NaCl, 1 mmol/l EDTA, 0.1% SDS, 1% Triton X-100, pH 7.4) containing protease inhibitors (Complete, Roche Molecular Biochemicals) at 4°C for 1 h with constant agitation. Following centrifugation, 140 μl of streptavidin agarose beads (Pierce) were added to 600 μl of cell lysate and incubated for 1 h at room temperature. Beads were washed four times with ice-cold lysis buffer, and the biotinylated proteins were released by incubation of the beads with 2X Laemmli buffer for 30 min at room temperature. Similar to total cell lysates, samples of the biotinylated fractions (20 μl) were subjected to Western blot analysis for BCRP detection.
Transport of sulfasalazine across Caco-2 monolayers
Caco-2 cells were, seeded at a density of 2.2 × 105 cells per well, onto 0.4 μm Transwell inserts (Corning Life Sciences, Acton, Massachusetts, USA) and maintained in DMEM (Lonza, Walkersville, Maryland, USA) containing 10% FBS (Invitrogen), penicillin (50 U/ml) (Invitrogen), streptomycin (50 μg/ml) (Invitrogen) and L-glutamine (2 mmol/l) (Invitrogen). Twenty-one days after seeding, the transepithelial electrical resistance between chambers was measured. Cells were deemed ready for a transport experiment if the transepithelial electrical resistance was ≥ 300 Ωcm2. To ensure integrity of the monolayer, [14C] inulin and [3H] digoxin transport experiments were performed preceding experiments with [3H] sulfasalazine. Before experimentation, the apical and basal chambers were washed with prewarmed serum reduced Opti-MEM (Invitrogen) and then incubated at 371C for 1 h in fresh Opti-MEM. The transport experiment was initiated by removing the Opti-MEM and replacing with Opti-MEM with or without [3H] sulfasalazine. In a select number of wells, the cells were also treated with 5 μmol/l fumitremorgin C, a selective BCRP inhibitor, in both apical and basolateral reservoirs. At selected time intervals, 25 μl aliquots were removed from the appropriate chamber and added to scintillation vials containing 5 ml of scintillation cocktail (Ultima Gold, PerkinElmer Life Sciences) and total radioactivity was measured by liquid scintillation spectrophotometry.
Apparent permeability (Papp; 10−6cm/s) from transwell experiments was calculated using equation (1) where dX/dt is the slope of the line fit through the [3H] sulfasalazine transport versus time profile, SA is the surface area of the transwell insert and C0 is the initial concentration.
| (1) |
Evaluation of sulfasalazine as an in vivo probe of breast cancer resistance protein
Seventeen healthy patients (10 men, 7 women) were recruited to participate in this study. All patients were deemed healthy following a physical examination (including routine serum chemistry and hematology) and assessment of their medical history by a physician. No patient had significant illness in the two weeks leading up to the study day and no history of gastrointestinal, cardiovascular, renal disease or sulfa drug hypersensitivity was recorded. Patients were instructed to abstain from grapefruit and citrus products as well as herbal or natural products for one week leading up to the study. No patients were taking any concurrent medications before or during the study. The mean age and weight range of the patients were 18–50 years and 54.5–95.0 kg, respectively. All patients provided written informed consent and the protocol was approved by the Health Sciences Research Ethics Board at the University of Western Ontario.
To determine the effect of drug formulation on sulfasalazine pharmacokinetics, three patients participated in an initial pilot investigation to determine the influence of drug formulation on sulfasalazine pharmacokinetics. On two days, separated by one month, patients received 1000 mg sulfasalazine, first as two 500 mg tablets (PMS-Sulfasalazine, Pharmascience, Montreal, Canada) and then two tablets crushed and suspended in 100 ml of suspension vehicle consisting of 30% simple syrup and 70% methylcellulose with sodium benzoate (methylcellulose CPS 1500 powder; 5 g, sodium benzoate; 1 g, sterile water; QS 500 ml). After comparison of the AUC0 → ∞ of suspension and tablet formulation by paired t-test, it was determined that the suspension was the optimal dose formulation (see Results) and the full study proceeded using a suspension of sulfasalazine.
Patients arrived at the clinic after an overnight fast and an indwelling catheter was inserted into an appropriate arm vein for blood sampling. Patients were then instructed to drink 1000 mg sulfasalazine prepared as a suspension and blood samples were drawn at selected intervals over the next 24 h. Patients were fed a standardized lunch at t = 4.0 h, snack at t = 7.0 h and dinner at t = 10.0 h. All blood samples were centrifuged at 2000 g and immediately frozen at −80°C until analysis.
Genomic DNA was extracted from the buffy coat of blood samples using the DNA blood mini kit (Qiagen) and quantified by Pico Green assay (Invitrogen). The presence of SNPs in BCRP (34 G > A and 421 C > A) were determined using TaqMan SNP genotyping assays (Applied Biosystems Inc.). The 34 G > A SNP assay was custom designed and used the following reporters, reporter 1: allele G, VIC AAGTTTTTATCCCAGTGTCACA and reporter 2: allele A, FAM AAGTTTTTATCCCAATGTCACA. The 421 C > A SNP assay was predesigned by Applied Biosystems and is commercially available (assay ID C__15854163_70).
Sample analysis
LC/MS/MS analysis was carried out using a high-performance liquid chromatography system consisting of a Shimadzu binary pump (Shimadzu Scientific Instruments, Columbia, Maryland, USA) with CTC PAL autosampler (Leap Technologies, Carrboro, North Carolina, USA) interfaced to an API 4000 LC/MS/MS triple quadrupole tandem mass spectrometer (Applied Biosystems, MDS Sciex Inc., Ontario, Canada). Methanolic stock solutions were prepared for sulfasalazine at 100 μg/ml. From this stock solution a 5000 ng/ml high calibration standard was prepared in human plasma. Calibration standards were then prepared from the high calibration standard by serial dilution at the following levels: 2000, 1000, 500, 200, 100, 50, 20, 10 and 5 ng/ml in human plasma.
Sample preparation for sulfasalazine in human plasma was as follows: (i) 50 μl plasma aliquot was precipitated with 150 μl of acetonitrile containing internal standard (diclofenac) using a Strata Protein Precipitation Plate; (ii) samples were vortexed for 30 s, then a vacuum of 15 mmHg was applied for an additional 30 s and the supernatant was collected in a clean 96-well collection plate; (iii) the samples were then dried under N2 and reconstituted with mobile phase and (iv) the plate was then vortexed for 15 s and 5 μl was injected onto the HPLC for LC/MS/MS analysis.
Sulfasalazine and the internal standard (diclofenac) were separated on a Hypersil Gold column (2.1 × 50 mm, Thermo Electron, Waltham, Massachusetts, USA). The mobile phase consisted of solvent A (5 mmol/l ammonium formate in water) and solvent B (acetonitrile). The gradient was as follows: solvent B was held at 10% for 0.3 min, then linearly ramped from 10 to 75% in 1.7 min, ramped from 75 to 95% in 0.7 min and then immediately brought back down to 10% for re-equilibration. Total run time was 3 min with a flow rate of 0.30 ml/min. The mass spectrometer was operated in positive ion ESI for the detection of sulfasalazine and diclofenac. Multiple reaction monitoring analysis was performed with the transitions, m/z 399.0 → 381.0 for sulfasalazine and m/z 296.0 → 215.0 for diclofenac. All raw data were processed using Analyst Software, version 1.4.1 (Applied Biosystems/MDS Sciex Inc.).
Area under the plasma concentration time curve (AUC) was calculated by the linear trapezoidal rule. The elimination rate constant (kel) was calculated by determining the slope of the terminal elimination phase of the semilog transformed plasma concentration time data and half-life was calculated using equation (2). The AUC24 → ∞ was calculated by dividing the concentration at 24 h by the kel.
| (2) |
Pharmacokinetic parameters were compared using unpaired t-test. A P < 0.05 was considered significant.
Results
Intestinal breast cancer resistance protein expression
The first objective of this report was to determine the interindividual variability of intestinal BCRP expression. Intestinal biopsies were obtained from 13 patients undergoing routine esophagogastro–duodenoscopy and five patients undergoing colonoscopy. Of the 13 patients who had duodenal biopsies, nine were determined to have wild-type BCRP (34GG/421CC) and four were heterozygous for the 421 C > A genotype (34GG/421CA). Western blotting was performed to compare duodenal BCRP protein expression between wild-type patients to those heterozygous for 421 C > A (Fig. 1a). Normalized BCRP protein and mRNA expression differed 1.8-fold (Fig. 1b) and 2.7-fold (Fig. 1d), respectively, between patients and this variability was not related to ABCG2 genotype (Fig. 1b and d). Biopsy samples were also taken from terminal ileum (n = 4) and colon (n = 5) of patients undergoing colonoscopy and compared with duodenal expression. Regional protein (Fig. 1c) and mRNA (Fig. 1e) BCRP expression did not significantly differ between the various regions of the intestinal tract.
Fig. 1.
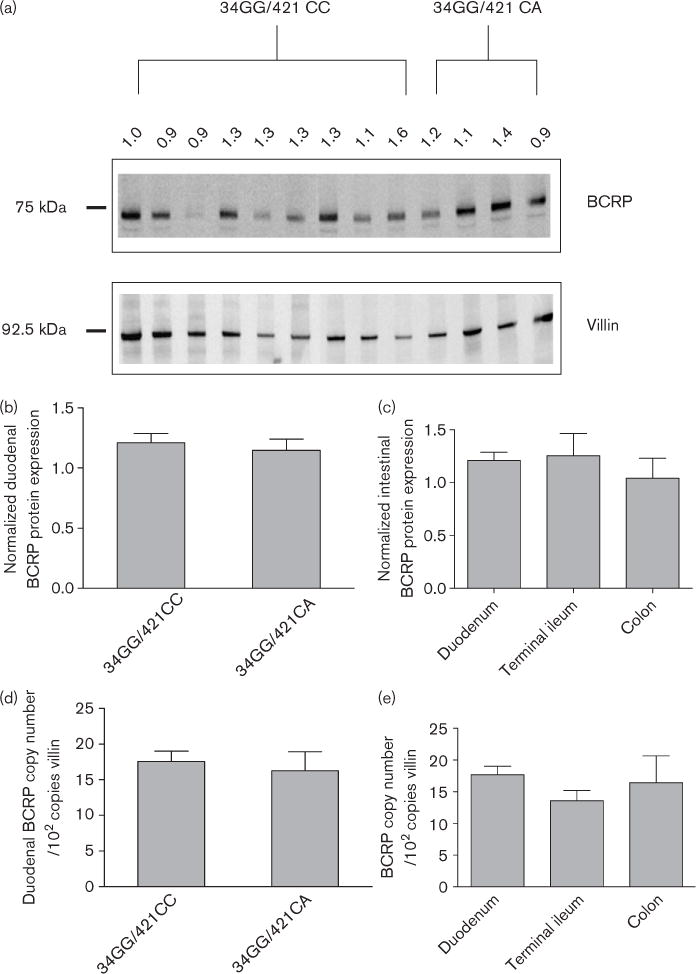
Intestinal BCRP expression. (a) BCRP expression was determined in 13 duodenal biopsy samples by Western blot and compared with the intestinal protein villin. Blots were digitally captured using the Kodak 4000 mm Image Station. Band intensity was determined and the normalized BCRP: villin ratio is shown above the blot. A total of 5 μg of protein lysate was loaded in each lane. (b) Summary of the normalized BCRP: villin ratio segregated by BCRP genotype. Results are mean ± SEM, ABCG2 34GG/421CC, n = 9; ABCG2 34GG/421CA, n = 4. (c) Regional intestinal BCRP protein expression. Results are mean ± SEM, duodenum (n = 9); terminal ileum (n = 4); colon (n = 5). All samples are from patients with 34GG/421CC genotype. (d) BCRP mRNA was determined in 13 duodenal biopsy samples by quantitative reverse transcriptase-PCR and normalized to the intestinal protein villin. Results are mean ± SEM, ABCG2 34GG/421CC, n = 9; ABCG2 34GG/421CA, n = 4. (e) Regional intestinal BCRP mRNA expression. Results are mean ± SEM, duodenum (n = 9); terminal ileum (n =4); colon (n = 5). All the samples are from patients with ABCG2 34GG/421CC genotype. BCRP, breast cancer resistance protein; SEM, standard error of the mean.
Cellular localization of breast cancer resistance protein variants
In order to evaluate the effect of BCRP polymorphisms on cellular BCRP expression, we heterologously expressed wild-type and variant BCRP (34G > A and 421 C > A) in human cervical carcinoma cells (HeLa). With this expression system, total cellular expression of BCRP variants was similar to wild-type (Fig. 2). Cell surface expression of 421 C > A, however, was markedly lower than that of the wild-type (Fig. 2). These results indicate that total 34G > A expression and subcellular 34G > A expression is similar to wild-type, whereas 421 C > A appears to have a sorting defect that limits cell surface expression in vitro.
Fig. 2.
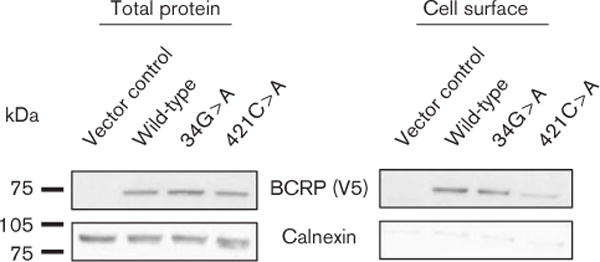
Total and cell surface BCRP protein expression. Wild-type and variant BCRP were transiently transfected in HeLa cells and total BCRP protein expression and cell surface BCRP protein expression determined by Western blot. Blots were stripped and reprobed with the intracellular marker protein, calnexin, to confirm specificity of cell surface protein enrichment. BCRP, breast cancer resistance protein.
Polarized transport of sulfasalazine in human intestinal Caco-2 cells
Vectorial transport of [3H] sulfasalazine was assessed in the intestinal Caco-2 model. Consistent with the apical expression of BCRP in Caco-2 cells [26], there was negligible apical to basal movement of [3H] sulfasalazine, whereas significant and time-dependent basal to apical transport was readily detected (Fig. 3). In the presence of the specific inhibitor of BCRP, fumitremorgin C [27], [3H] sulfasalazine transport in the basal to apical direction was attenuated, whereas apical to basal flux was enhanced. The ratio of apparent permeability (Papp) (Papp B → A/Papp A → B) differed 28-fold, with a value of 36.4 and 1.3 in the absence and presence of fumitremorgin C, respectively. These results indicate that sulfasalazine is a substrate of human BCRP. The Papp for [3H] sulfasalazine along with [3H] inulin and [14C] digoxin controls are summarized in Table 1.
Fig. 3.
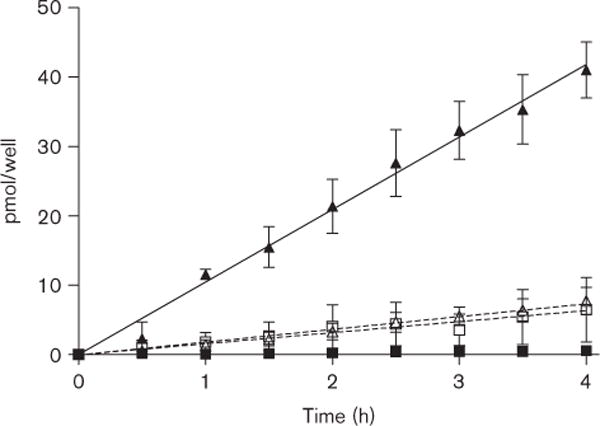
In vitro transport of [3H] sulfasalazine across Caco-2 monolayers. Results are mean ± SEM, n = 3, ■ = [3H] sulfasalazine apical to basal, ▲ = [3H] sulfasalazine basal to apical □ = [3H] sulfasalazine +5 μmol/l FTC, apical to basal, △ [3H] sulfasalazine +5 μmol/l FTC, basal to apical. FTC, fumitremorgin C.
Table 1.
Apparent permeability of [3H] sulfasalazine across Caco-2 monolayers in the presence and absence of fumitremorgin C (FTC)
| Papp (A → B) × 10−6cm/s (SD) | Papp (B → A) × 10−6cm/s (SD) | Papp (B → A)/Papp (A → B) | |
|---|---|---|---|
| [3H] Sulfasalazine | 0.28 (0.19) | 10.2 (3.9) | 36.4 |
| [3H] Sulfasalazine + 5 μmol/l FTC | 1.6 (2.0) | 2.1 (1.0) | 1.3 |
| [14C] inulin | 0.24 (0.19) | − | − |
| [3H] digoxin | 1.95 (0.2) | 14.4 (1.3) | 7. 4 |
Papp, apparent permeability; SD, standard deviation.
Sulfasalazine pharmacokinetics in relation to breast cancer resistance protein genotype
The impact of common SNPs in BCRP to the disposition of sulfasalazine in healthy subjects was assessed. We first evaluated whether sulfasalazine disposition was affected by drug formulation in three patients. In a crossover design, sulfasalazine disposition after oral administration of two 500 mg film-coated tablets taken with 300 ml of water was compared with sulfasalazine disposition after oral administration of 1000 mg of sulfasalazine as a suspension (100 ml) followed by 300 ml of water. Maximum plasma concentration (Cmax) and time to maximum plasma concentration (tmax) were significantly greater when sulfasalazine was administered as a suspension than as tablets. The average AUC0 → ∞ for sulfasalazine as a suspension was 1.6-fold higher when given as a suspension in comparison with tablets, although this was not statistically significant (P = 0.07). These results indicate that dosage form influenced the plasma concentration time-course of sulfasalazine, thus the remainder of the study was carried out using sulfasalazine suspension in 17 healthy subjects.
Oral sulfasalazine exposure in healthy subjects (Table 2) was highly variable with AUCs differing 20.5-fold (16.5–338.5 μg/ml × h), and Cmax varying 17.2-fold (2.3–39.5 μg/ml). Intersubject variability in tmax was observed and ranged between three to six hours. Mean sulfasalazine half-life (t1/2) was, however, consistent between patients (4.4 ± 1.1 h). Of the 17 study patients, nine patients were determined to be wild-type BCRP (34GG/421CC), two of them had the 34 G > A polymorphism (one homozygous, 34AA and one heterozygous 34GA), five patients were heterozygous for 421 C > A (34GG/421CA) and one carrying both the 34A and 421A allele (34GA/421CA). Sulfasalazine pharmacokinetic parameters were associated with BCRP genotype (Fig. 4). In subjects with 34GG/421CA genotype, sulfasalazine AUC0 → ∞ and Cmax values (166.7 ± 54.3 μg/ml × h and 15.2 ± 4.5 μg/ml, respectively) were significantly greater than those obtained from wild-type 34GG/421CC subjects (70.2 ± 37.8 μg/ml × h and 8.9 ± 4.8 μg/ml, respectively, P < 0.01). Interestingly, the single subject carrying both variant alleles, 34A and 421A (subject with symbol Δ in Fig. 4), exhibited a 4.8-fold increase in the AUC and a 4.4-fold increase in Cmax compared with wild-type controls. Pharmacokinetic data categorized by genotype are summarized in Table 2. A linear relationship was observed when plasma sulfasalazine concentration at 6 h was compared with AUC0 → ∞ (R2 =0.9271, P < 0.0001) suggesting that a sparse sampling approach could be used in future studies to obtain estimates of drug exposure (Fig. 5).
Table 2.
Summary of sulfasalazine pharmacokinetics stratified by ABCG2 genotype
|
ABCG2 34GG/421CC (n = 9) |
ABCG2 34GA/421CC (n = 1) |
ABCG2 34AA/421CC (n = 1) |
ABCG2 34GG/421CA (n = 5) |
ABCG2 34GA/421CA (n = 1) |
|
|---|---|---|---|---|---|
| AUC0 → ∞ (μg/ml × h) | 70.2 (37.8) | 54.1 | 113.6 | 166.7 (54.3)* | 338.5 |
| t1/2 (h) | 4.2 (1.0) | 3.5 | 3.6 | 4.8 (1.0) | 4.1 |
| Cmax (μg/ml) | 8.9 (4.8) | 13.8 | 10.0 | 15.2 (4.5)* | 39.5 |
| tmax (h) | 5.0 (1.2) | 3.5 | 4.0 | 5.5 (1.1) | 6.0 |
The genotype ABCG2 34GA/421CA represents a patient with one A allele at position 34 and one A allele at position 421. Results are mean (± SD), * denotes P < 0.05 compared with ABCG2 34GG/421CC. AUC, area under the plasma concentration time curve; Cmax, maximum plasma concentration; SD, standard deviation; tmax, time to maximum plasma concentration; t1/2, half-life.
Fig. 4.
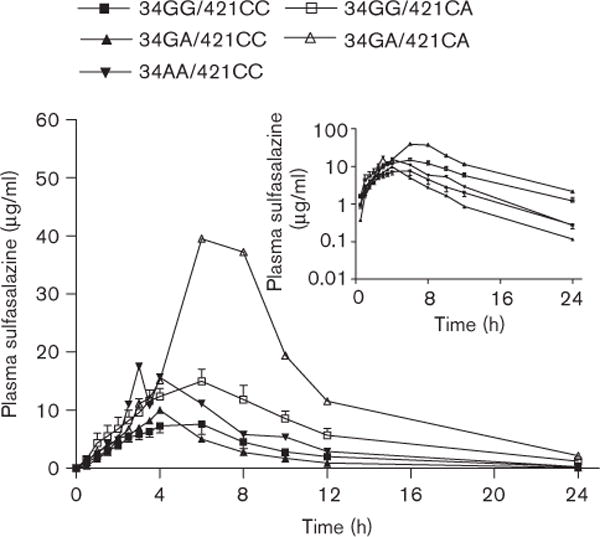
Plasma concentration versus time profile of 1g sulfasalazine (as a suspension) segregated by BCRP genotype. Results are mean ± SEM. ■ = ABCG2 34GG/421CC, n = 9; ▲ = ABCG2 34GA/421CC, n = 1; ▼ = ABCG2 34AA/421CC, n = 1; □ = ABCG2 34GG/421CA, n = 5; △ = ABCG2 34GA/421CA, n = 1. The genotype ABCG2 34GA/421CA represents a patient with one A allele at position 34 and one A allele at position 421. Inset: Plasma concentration versus time profile of 1 g sulfasalazine on log-linear scale. BCRP, breast cancer resistance protein; SEM, standard error of the mean.
Fig. 5.
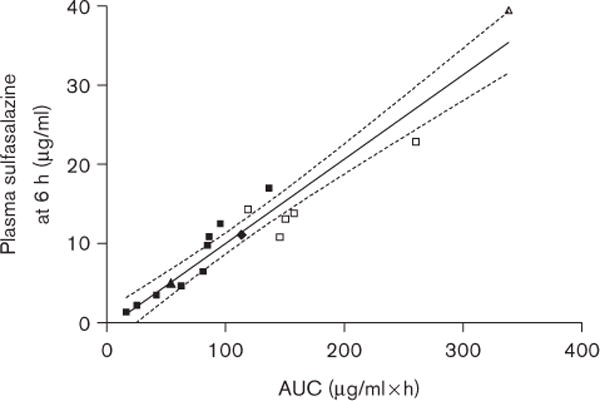
Correlation of plasma sulfasalazine at 6 h with area under the plasma concentration time curve (AUC0 → ∞). R2 = 0.9271, P < 0.0001. 95% confidence interval is shown as dashed lines. ■ = ABCG2 34GG/421CC, ▲ = ABCG2 34GA/421CC, ▼ = ABCG2 34AA/421CC, □ = ABCG2 34GG/421CA, △ = ABCG2 34GA/421CA.
Discussion
The efflux transporter BCRP is increasingly recognized for its role in oral drug absorption and distribution of substrates to target sites of action. Although there is recognition that variation in BCRP activity may alter drug levels or efficacy, there have been few studies in humans, to date, that demonstrate a pharmacokinetic impact of differences in BCRP tissue expression, modulation of BCRP activity by drug interactions or through the effects of functional BCRP genetic variations. In this report, we systematically assessed the intestinal expression of BCRP with respect to genotype and region in an attempt to better understand the interindividual variability in response to drug substrates. Our results indicate an overall low variability in intestinal BCRP expression among patients. In addition, we found no significant difference in duodenal BCRP mRNA or protein expression in patients with the common 421C > A polymorphism when compared with those with genotype 34GG/ 421CC, a finding that is in agreement with earlier reports [15]. Similarly, longitudinal intestinal BCRP expression did not differ between proximal and distal sections of the intestine suggesting that BCRP is an important mediator of drug efflux throughout the intestine.
We now show, using Caco-2 cells, that sulfasalazine is a substrate of human BCRP through the use of a selective BCRP inhibitor. These findings are consistent with earlier results of a profound increase in plasma level of sulfasalazine in Bcrp knockout mice [24], indicating that sulfasalazine disposition in humans would be sensitive to BCRP activity. Therefore, we tested the hypothesis that sulfasalazine pharmacokinetics in humans would be influenced by common BCRP polymorphisms that were previously shown to be transport defective in vitro. Indeed the large increase in AUC and Cmax in patients with the 421C > A polymorphism in comparison with patients with 34GG/421CC genotype supports this hypothesis. In vitro expression data suggest reduced cell surface expression of the 421C > A variant as a key determinant of the reduced BCRP mediated transport of sulfasalazine in patients carrying an A allele at position 421.
Sulfasalazine is used in the treatment of ulcerative colitis and is a disease modifying antirheumatic drug for rheumatoid arthritis. After oral intake and disintegration of the tablet, sulfasalazine is converted to its metabolites, sulfapyridine and 5-aminosalicylate by bacterial azoreductases in the distal GI tract, which represents the ratelimiting step of metabolic clearance of this compound. The presumed mechanism of action relates to the antiinflammatory properties of 5-aminosalicylate; thus, in ulcerative colitis therapeutic activity depends on minimal sulfasalazine absorption in the upper GI tract and subsequent cleavage in the distal intestine [28]. Decreased BCRP activity, associated with the 421C > A polymorphism, appears to enhance sulfasalazine absorption and therefore, reduce the amount available for bioactivation in the lower GI tract. In contrast, although the mechanism of sulfasalazine action in the treatment of rheumatoid arthritis remains controversial, it is generally agreed that sulfasalazine must be absorbed into the systemic circulation to mediate the anti-inflammatory effect [29]. In such a clinical setting, patients with the 421C > A polymorphism would be likely to absorb more sulfasalazine, and may obtain greater therapeutic benefit. This observation may help explain the high variability of response to sulfasalazine in rheumatoid arthritis patients.
In addition, BCRP is expressed in several tissues that may represent therapeutic targets of substrate drugs or potential sites of unwanted toxicity. Such tissues include the liver, heart, breast, various cancers, bone marrow, placenta, brain and kidney [1–6]. Thus, it is likely BCRP polymorphisms may alter tissue distribution and efficacy of many substrate drugs.
Indeed, a large fraction of BCRP substrates are chemotherapeutic agents, and the role of BCRP in multidrug resistance is widely appreciated, although the clinical significance is highly debated. Cells expressing or overexpressing BCRP are known to be resistant to chemotherapeutic agents [30–33] and therefore, decreased BCRP mediated transport in tumors of patients, who harbor the 421C > A polymorphism, would be expected to result in greater tumor sensitivity to chemotherapy. Our results would suggest that patients with the 421C > A polymorphism would likely be more prone to side effects during chemotherapy with BCRP substrate chemotherapeutic agents owing to higher drug exposure and Cmax. Indeed this is the case for the BCRP substrate, gefitinib, where patients with the 421C > A polymorphism were noted to have higher steady state plasma concentrations associated with a higher incidence of diarrhea, a concentration dependent side effect of this therapy [21,34].
BCRP is expressed on the apical domain of epithelial cells of the mammary gland during pregnancy and lactation. At the level of the mammary gland, BCRP has been implicated in enhancing the concentration of drugs, toxins, carcinogens and vitamins into breast milk [5,35]. Our results suggest that lactating women, who have the 421C > A polymorphism, would have decreased ability to concentrate BCRP substrate drugs and xenobiotics into milk. Although this is clearly beneficial when considering newborn exposure to toxins and carcinogens such as pheophorbide and 2-amino-1-methyl-6-phenylimidazo (4,5-b) pyridine, the potential for decreased vitamin secretion into breast milk may contribute to unwanted deficiencies [36].
In conclusion, we demonstrate a prominent role of BCRP polymorphisms in the intestinal absorption of sulfasalazine. Hence, sulfasalazine may be a useful and relatively safe in vivo probe of BCRP activity in humans. Importantly, our findings suggest that the clinical utility of sulfasalazine in future studies assessing the relevance of BCRP in drug–drug interactions and response to drugs. The results of this study further support the notion that polymorphisms of BCRP significantly impact drug disposition.
Acknowledgments
The authors are grateful to nurse Linda Asher and thank Robert Polzer and Steven Michael of PGRD, Michigan for their support in this project. This work was supported in part by a United States Public Health Service Grant GM31304 (RBK), a Vanderbilt University Physician Scientist Development Award (RHH) and a fellowship from the Canadian Society for Clinical Pharmacology (BLU).
References
- 1.Doyle LA, Yang W, Abruzzo LV, Krogmann T, Gao Y, Rishi AK, et al. A multidrug resistance transporter from human MCF-7 breast cancer cells. Proc Natl Acad Sci U S A. 1998;95:15665–15670. doi: 10.1073/pnas.95.26.15665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Meissner K, Heydrich B, Jedlitschky G, Meyer Zu SH, Mosyagin I, Dazert P, et al. The ATP-binding cassette transporter ABCG2 (BCRP), a marker for side population stem cells, is expressed in human heart. J Histochem Cytochem. 2006;54:215–221. doi: 10.1369/jhc.5A6750.2005. [DOI] [PubMed] [Google Scholar]
- 3.Cooray HC, Blackmore CG, Maskell L, Barrand MA. Localisation of breast cancer resistance protein in microvessel endothelium of human brain. NeuroReport. 2002;13:2059–2063. doi: 10.1097/00001756-200211150-00014. [DOI] [PubMed] [Google Scholar]
- 4.Staud F, Pavek P. Breast cancer resistance protein (BCRP/ABCG2) Int J Biochem Cell Biol. 2005;37:720–725. doi: 10.1016/j.biocel.2004.11.004. [DOI] [PubMed] [Google Scholar]
- 5.Jonker JW, Merino G, Musters S, van Herwaarden AE, Bolscher E, Wagenaar E, et al. The breast cancer resistance protein BCRP (ABCG2) concentrates drugs and carcinogenic xenotoxins into milk. Nat Med. 2005;11:127–129. doi: 10.1038/nm1186. [DOI] [PubMed] [Google Scholar]
- 6.Zhou S, Schuetz JD, Bunting KD, Colapietro AM, Sampath J, Morris JJ, et al. The ABC transporter Bcrp1/ABCG2 is expressed in a wide variety of stem cells and is a molecular determinant of the side-population phenotype. Nat Med. 2001;7:1028–1034. doi: 10.1038/nm0901-1028. [DOI] [PubMed] [Google Scholar]
- 7.Xu J, Peng H, Chen Q, Liu Y, Dong Z, Zhang JT. Oligomerization domain of the multidrug resistance-associated transporter ABCG2 and its dominant inhibitory activity. Cancer Res. 2007;67:4373–4381. doi: 10.1158/0008-5472.CAN-06-3169. [DOI] [PubMed] [Google Scholar]
- 8.Merino G, van Herwaarden AE, Wagenaar E, Jonker JW, Schinkel AH. Sexdependent expression and activity of the ATP-binding cassette transporter breast cancer resistance protein (BCRP/ABCG2) in liver. Mol Pharmacol. 2005;67:1765–1771. doi: 10.1124/mol.105.011080. [DOI] [PubMed] [Google Scholar]
- 9.Jonker JW, Buitelaar M, Wagenaar E, van DV, Scheffer GL, Scheper RJ, et al. The breast cancer resistance protein protects against a major chlorophyll-derived dietary phototoxin and protoporphyria. Proc Natl Acad Sci U S A. 2002;99:15649–15654. doi: 10.1073/pnas.202607599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Merino G, Jonker JW, Wagenaar E, van Herwaarden AE, Schinkel AH. The breast cancer resistance protein (BCRP/ABCG2) affects pharmacokinetics, hepatobiliary excretion, and milk secretion of the antibiotic nitrofurantoin. Mol Pharmacol. 2005;67:1758–1764. doi: 10.1124/mol.104.010439. [DOI] [PubMed] [Google Scholar]
- 11.Merino G, Alvarez AI, Pulido MM, Molina AJ, Schinkel AH, Prieto JG. Breast cancer resistance protein (BCRP/ABCG2) transports fluoroquinolone antibiotics and affects their oral availability, pharmacokinetics, and milk secretion. Drug Metab Dispos. 2006;34:690–695. doi: 10.1124/dmd.105.008219. [DOI] [PubMed] [Google Scholar]
- 12.Breedveld P, Pluim D, Cipriani G, Wielinga P, van TO, Schinkel AH, et al. The effect of Bcrp1 (Abcg2) on the in vivo pharmacokinetics and brain penetration of imatinib mesylate (Gleevec): implications for the use of breast cancer resistance protein and P-glycoprotein inhibitors to enable the brain penetration of imatinib in patients. Cancer Res. 2005;65:2577–2582. doi: 10.1158/0008-5472.CAN-04-2416. [DOI] [PubMed] [Google Scholar]
- 13.Maliepaard M, Scheffer GL, Faneyte IF, van Gastelen MA, Pijnenborg AC, Schinkel AH, et al. Subcellular localization and distribution of the breast cancer resistance protein transporter in normal human tissues. Cancer Res. 2001;61:3458–3464. [PubMed] [Google Scholar]
- 14.Englund G, Rorsman F, Ronnblom A, Karlbom U, Lazorova L, Grasjo J, et al. Regional levels of drug transporters along the human intestinal tract co-expression of ABC and SLC transporters and comparison with Caco-2 cells. Eur J Pharm Sci. 2006;29:269–277. doi: 10.1016/j.ejps.2006.04.010. [DOI] [PubMed] [Google Scholar]
- 15.Zamber CP, Lamba JK, Yasuda K, Farnum J, Thummel K, Schuetz JD, et al. Natural allelic variants of breast cancer resistance protein (BCRP) and their relationship to BCRP expression in human intestine. Pharmacogenetics. 2003;13:19–28. doi: 10.1097/00008571-200301000-00004. [DOI] [PubMed] [Google Scholar]
- 16.Imai Y, Nakane M, Kage K, Tsukahara S, Ishikawa E, Tsuruo T, et al. C421A polymorphism in the human breast cancer resistance protein gene is associated with low expression of Q141 K protein and low-level drug resistance. Mol Cancer Ther. 2002;1:611–616. [PubMed] [Google Scholar]
- 17.Mizuarai S, Aozasa N, Kotani H. Single nucleotide polymorphisms result in impaired membrane localization and reduced atpase activity in multidrug transporter ABCG2. Int J Cancer. 2004;109:238–246. doi: 10.1002/ijc.11669. [DOI] [PubMed] [Google Scholar]
- 18.Yanase K, Tsukahara S, Mitsuhashi J, Sugimoto Y. Functional SNPs of the breast cancer resistance protein-therapeutic effects and inhibitor development. Cancer Lett. 2006;234:73–80. doi: 10.1016/j.canlet.2005.04.039. [DOI] [PubMed] [Google Scholar]
- 19.Sparreboom A, Gelderblom H, Marsh S, Ahluwalia R, Obach R, Principe P, et al. Diflomotecan pharmacokinetics in relation to ABCG2 421 C > A genotype. Clin Pharmacol Ther. 2004;76:38–44. doi: 10.1016/j.clpt.2004.03.003. [DOI] [PubMed] [Google Scholar]
- 20.Zhang W, Yu BN, He YJ, Fan L, Li Q, Liu ZQ, et al. Role of BCRP 421 C > A polymorphism on rosuvastatin pharmacokinetics in healthy Chinese males. Clin Chim Acta. 2006;373:99–103. doi: 10.1016/j.cca.2006.05.010. [DOI] [PubMed] [Google Scholar]
- 21.Cusatis G, Gregorc V, Li J, Spreafico A, Ingersoll RG, Verweij J, et al. Pharmacogenetics of ABCG2 and adverse reactions to gefitinib. J Natl Cancer Inst. 2006;98:1739–1742. doi: 10.1093/jnci/djj469. [DOI] [PubMed] [Google Scholar]
- 22.Ho RH, Tirana RG, Leake BF, Glaeser H, Lee W, Lemke CJ, et al. Drug and bile Acid transporters in rosuvastatin hepatic uptake function, expression, and pharmacogenetics. Gastroenterology. 2006;130:1793–1806. doi: 10.1053/j.gastro.2006.02.034. [DOI] [PubMed] [Google Scholar]
- 23.van der Heijden J, de Jong MC, Dijkmans BA, Lems WF, Oerlemans R, Kathmann I, et al. Development of sulfasalazine resistance in human T cells induces expression of the multidrug resistance transporter ABCG2 (BCRP) and augmented production of TNFalpha. Ann Rheum Dis. 2004;63:138–143. doi: 10.1136/ard.2002.005249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Zaher H, Khan AA, Palandra J, Brayman TG, Yu L, Ware JA. Breast cancer resistance protein (Bcrp/abcg2) is a major determinant of sulfasalazine absorption and elimination in the mouse. Mol Pharm. 2006;3:55–61. doi: 10.1021/mp050113v. [DOI] [PubMed] [Google Scholar]
- 25.Schroder H, Campbell DE. Absorption, metabolism, and excretion of salicylazosulfapyridine in man. Clin Pharmacol Ther. 1972;13:539–551. doi: 10.1002/cpt1972134539. [DOI] [PubMed] [Google Scholar]
- 26.Xia CQ, Liu N, Yang D, Miwa G, Gan LS. Expression, localization, and functional characteristics of breast cancer resistance protein in Caco-2 cells. Drug Metab Dispos. 2005;33:637–643. doi: 10.1124/dmd.104.003442. [DOI] [PubMed] [Google Scholar]
- 27.Rabindran SK, Ross DD, Doyle LA, Yang W, Greenberger LM. Fumitremorgin C reverses multidrug resistance in cells transfected with the breast cancer resistance protein. Cancer Res. 2000;60:47–50. [PubMed] [Google Scholar]
- 28.Peppercorn MA. Sulfasalazine. Pharmacology, clinical use, toxicity, and related new drug development. Ann Intern Med. 1984;101:377–386. doi: 10.7326/0003-4819-101-3-377. [DOI] [PubMed] [Google Scholar]
- 29.Plosker GL, Croom KF. Sulfasalazine: a review of its use in the management of rheumatoid arthritis. Drugs. 2005;65:1825–1849. doi: 10.2165/00003495-200565130-00008. [DOI] [PubMed] [Google Scholar]
- 30.Bates SE, Robey R, Miyake K, Rao K, Ross DD, Litman T. The role of half-transporters in multidrug resistance. J Bioenerg Biomembr. 2001;33:503–511. doi: 10.1023/a:1012879205914. [DOI] [PubMed] [Google Scholar]
- 31.Kamiyama N, Takagi S, Yamamoto C, Kudo T, Nakagawa T, Takahashi M, et al. Expression of ABC transporters in human hepatocyte carcinoma cells with cross-resistance to epirubicin and mitoxantrone. Anticancer Res. 2006;26:885–888. [PubMed] [Google Scholar]
- 32.Litman T, Brangi M, Hudson E, Fetsch P, Abati A, Ross DD, et al. The multidrug-resistant phenotype associated with overexpression of the new ABC half-transporter, MXR (ABCG2) J Cell Sci. 2000;113(Pt 11):2011–2021. doi: 10.1242/jcs.113.11.2011. [DOI] [PubMed] [Google Scholar]
- 33.Priebsch A, Rompe F, Tonnies H, Kowalski P, Surowiak P, Stege A, et al. Complete reversal of ABCG2-depending atypical multidrug resistance by RNA interference in human carcinoma cells. Oligonucleotides. 2006;16:263–274. doi: 10.1089/oli.2006.16.263. [DOI] [PubMed] [Google Scholar]
- 34.Li J, Cusatis G, Brahmer J, Sparreboom A, Robey RW, Bates SE, et al. Association of variant ABCG2 and the pharmacokinetics of epidermal growth factor receptor tyrosine kinase inhibitors in cancer patients. Cancer Biol Ther. 2007;6:432–438. doi: 10.4161/cbt.6.3.3763. [DOI] [PubMed] [Google Scholar]
- 35.van Herwaarden AE, Schinkel AH. The function of breast cancer resistance protein in epithelial barriers, stem cells and milk secretion of drugs and xenotoxins. Trends Pharmacol Sci. 2006;27:10–16. doi: 10.1016/j.tips.2005.11.007. [DOI] [PubMed] [Google Scholar]
- 36.van Herwaarden AE, Wagenaar E, Merino G, Jonker JW, Rosing H, Beijnen JH, et al. Multidrug transporter ABCG2/breast cancer resistance protein secretes riboflavin (vitamin B2) into milk. Mol Cell Biol. 2007;27:1247–1253. doi: 10.1128/MCB.01621-06. [DOI] [PMC free article] [PubMed] [Google Scholar]