

Abstract
The long time scale of adaptive evolution makes it difficult to directly observe the spread of most beneficial mutations through natural populations. Therefore, inferring attributes of beneficial mutations by studying the genomic signals left by directional selection is an important component of population genetics research. One kind of signal is a trough in nearby neutral genetic variation due to selective fixation of initially rare alleles, a phenomenon known as “genetic hitchhiking.” Accumulated evidence suggests that a considerable fraction of substitutions in the Drosophila genome results from positive selection, most of which are expected to have small selection coefficients and influence the population genetics of sites in the immediate vicinity. Using Drosophila melanogaster population genomic data, we found that the heterogeneity in synonymous polymorphism surrounding different categories of coding fixations is readily observable even within 25 bp of focal substitutions, which we interpret as the result of small-scale hitchhiking effects. The strength of natural selection on different sites appears to be quite heterogeneous. Particularly, neighboring fixations that changed amino acid polarities in a way that maintained the overall polarities of a protein were under stronger selection than other categories of fixations. Interestingly, we found that substitutions in slow-evolving genes are associated with stronger hitchhiking effects. This is consistent with the idea that adaptive evolution may involve few substitutions with large effects or many substitutions with small effects. Because our approach only weakly depends on the numbers of recent nonsynonymous substitutions, it can provide a complimentary view to the adaptive evolution inferred by other divergence-based evolutionary genetic methods.
Keywords: genetic hitchhiking, natural selection, natural variation, adaptive evolution, Drosophila melanogaster
Introduction
Despite the central importance of natural selection in evolution, important properties of the selection-driven dynamics of beneficial mutations through populations remain poorly understood. For example, the relative roles of beneficial mutations of small and large effect are still debated, and the extent to which adaptive evolution may be mutation limited is unclear (Orr and Coyne 1992; Orr 2005, 2009; Barrett and Schluter 2008; Radwan and Babik 2012; Messer and Petrov 2013). The intersection of these issues with the extent of variation in adaptive landscapes (e.g., the number of fitness optima and whether such optima are constant or moving over time due to changing environment) and the organization of particular biological functions are also important. Identifying adaptive substitutions is a natural step toward empirically addressing all of these questions. A population genetic approach is important because those beneficial mutations that can be directly genetically analyzed are of unusually large effect (Eyre-Walker and Keightley 2007) and constitute a relatively biased sample of all adaptive variants.
The fixation of an initially rare, beneficial mutation leads to reduced polymorphism at linked neutral sites through a process known as “genetic hitchhiking” (Maynard Smith and Haigh 1974). The width of the region being influenced depends positively on the strength of selection acting on the beneficial mutation and negatively on the recombination rate between the beneficial mutation and linked neutral mutations (Maynard Smith and Haigh 1974; Kaplan et al. 1989; Stephan et al. 1992). A region affected by the hitchhiking effect will gradually recover neutral variation through the spread of newly arising mutations. With the same strength of selection and recombination rate, the reduction in nearby neutral variation will be greater for beneficial substitutions that were fixed recently than those that were fixed in the distant past. Approaches using this “footprint” of directional selection to detect adaptive evolution or to measure parameters of the process, such as the strength of selection, have been successfully applied to natural variation data (Thornton et al. 2007). However, other biological processes, such as selection against deleterious mutations (background selection [Charlesworth et al. 1993, 1995; Hudson and Kaplan 1995; Charlesworth 2012]) and biased gene conversion (Gutz and Leslie 1976; Nagylaki 1983a, 1983b; Marais 2003), can also be associated with locally reduced level of polymorphism. Distinguishing between these competing hypotheses for observed heterogeneity in polymorphism has been difficult (Andolfatto 2001; Stephan 2010; Cutter and Payseur 2013). In addition, it has been theoretically shown that fixations of slightly deleterious mutations have a comparably reduced distribution of sojourn time (Maruyama 1974), from which one can surmise that they will have a hitchhiking effect similar to beneficial mutations with the same magnitude of selection coefficient. Observed reduction in neutral variation around substitutions could be attributed to fixation of either beneficial or deleterious mutations, even though the latter is expected less likely to happen in species with very large population size (such as Drosophila melanogaster).
There is considerable evidence that directional selection plays an important role in the evolution of the Drosophila genome. This evidence comes not only from the existence of regions with reduced polymorphism as described earlier but also from other types of analyses. For example, Drosophila protein divergence appears to be strongly influenced by directional selection as inferred from contrasts of polymorphic and fixed synonymous and nonsynonymous variation (McDonald and Kreitman 1991; Begun et al. 2007; Langley et al. 2012). Indeed, an estimated 35–87% of the amino acid substitutions have been fixed by positive selection in Drosophila (Fay et al. 2002; Smith and Eyre-Walker 2002; Bierne and Eyre-Walker 2003; Welch 2006; Andolfatto 2007; Begun et al. 2007; Shapiro et al. 2007; Haddrill et al. 2010; Langley et al. 2012; Kousathanas and Keightley 2013). Further supporting this view, genes with greater amino acid divergence or genomic regions with larger number of amino acid substitutions have lower levels of nearby/genic variation in Drosophila (Ometto et al. 2005; Andolfatto 2007; Begun et al. 2007; Macpherson et al. 2007; Langley et al. 2012). These analyses suggest that the influence of positive selection on the level of genetic variation due to hitchhiking effects must be widespread in the Drosophila genome because, for example, there is roughly one amino acid substitution every 300 nucleotides in the coding region in the D. melanogaster lineage (Langley et al. 2012).
If a considerable fraction of genomic divergence resulted from positive selection, the aggregate effects of even weakly selected but numerous beneficial fixations could be studied through investigating the particular patterns of polymorphism at sites very close to sites that have experienced a fixation. This approach was first applied in the pregenomic era to a study of polymorphism near sites that had fixed in the D. simulans lineage (Kern et al. 2002). However, the available data were so limited that no strong conclusions could be drawn. A recent study by Sattath et al. (2011) extended this approach to whole-genome data from D. simulans and found a significantly stronger local reduction of neutral variation around nonsynonymous than synonymous substitutions. An especially useful attribute of this conceptual framework is that it allows for the comparisons of population genetic variation across different categories of fixation events as defined by genome annotation, thereby potentially revealing heterogeneity in substitution processes across classes of mutations that are functionally diverse. This approach has advantages over several widely used evolutionary genetic tests (e.g., dN/dS ratio [Yang 2007] and McDonald-Kreitman test [McDonald and Kreitman 1991]), which heavily depends on the number of recent substitutions to have strong enough statistical power.
Here, we used recently generated whole-genome sequences from D. melanogaster population (Langley et al. 2012) to investigate small-scale hitchhiking effects, with an emphasis on substitutions in coding sequences. The physical scale of hitchhiking is expected to have a wide distribution. The influence from larger scale processes that affect polymorphism should be taken into account when attempting to understand how the population genetics of sites near fixations differ from random sites in such regions. For example, both theoretical predictions and empirical observations suggest that genetic variation correlates with both local recombination rate (Maynard Smith and Haigh 1974; Kaplan et al. 1989; Begun et al. 2007; Langley et al. 2012) and the rate of possibly beneficial substitutions (i.e., amino acid fixations) in a given region (Kaplan et al. 1989; Ometto et al. 2005; Andolfatto 2007; Begun et al. 2007; Macpherson et al. 2007; Langley et al. 2012). Also, selection on other coding substitutions and functional elements that have different selective constraint and/or probability of under positive selection from coding regions (such as untranslated regions [UTRs] and introns) is expected to influence the observed signals of hitchhiking effects (selective interference [Hill and Robertson 1966; Felsenstein 1974]). The genetic linkage between the focal and other selected variants/functional elements is a critical parameter in determining the extent of this effect. Indeed, selective interference may be weaker on the exon–intron boundaries than in the center of exons (Comeron and Kreitman 2002; Comeron and Guthrie 2005; Loewe and Charlesworth 2007). To deal with these issues, we used a model fitting approach by performing generalized linear regression analysis to correct for the effects of possibly confounding factors on local genetic variation near fixations and used the remaining variation (residuals) as our target for further analysis.
Results
Regression Analysis to Correct for the Possible Effects of Other Factors on Polymorphism
We estimated the level of synonymous polymorphism (π) using 4-fold degenerate sites within a fixed window size surrounding coding substitutions with six African D. melanogaster genomes. Under the infinite site model, the level of variation depends on the total branch length of a genealogy and the mutation rate of a particular genomic region (Tajima 1983). The former can be significantly influenced by the impact of selection (Kaplan et al. 1989; Charlesworth et al. 1993, 1995), leading to deviation from the expected level of variation under neutral model. To reflect the variation in the total branch length of genealogies associated with neutral sites surrounding different categories of coding fixations, the estimated π was divided by the D. simulans–D. yakuba synonymous divergence (estimated for 4-fold degenerate sites) in the corresponding window, which is taken as a proxy of mutation rate. This estimate is referred to as “normalized π” or πnor. Because several factors were significantly correlated with πnor (table 1 and supplementary table S1, Supplementary Material online), we performed regression analysis to correct for their effects and used the residuals of the regression model (επnor) as the major subject of our subsequent analyses. επnor can be interpreted as the distance of the observed πnor to the predicted πnor of the fitted regression model. A lower επnor suggests that πnor around a substitution deviates negatively from the predicted πnor with a greater extent, which could be attributable to the effect of hitchhiking. It is expected that selection on other linked sites can also influence the population genetics of synonymous sites near the focal coding substitution, and the extent of this effect depends on the genetic linkage among them. We therefore included distance from the focal substitution to other substitution or functional elements in the regression analysis. Selection on codon usage bias (Akashi 1995, 1996) and/or GC-biased gene conversion (Galtier et al. 2006; Haddrill and Charlesworth 2008) influences the evolutionary dynamics of synonymous sites and local GC content. Therefore, we also included base composition in our regression model.
Table 1.
List of Factors Correlated with πnor.
| Correlated Factor | Sign of Correlation | |
|---|---|---|
| recomb | Recombination rate around the focal substitution | + |
| nns | Number of nonsynonymous substitutions other than the focal substitution in a window | − |
| ns | Number of synonymous substitutions other than the focal substitution in a window | − |
| dns | Distance from the focal substitution to the nearest nonsynonymous substitution | + |
| ds | Distance from the focal substitution to the nearest synonymous substitution | + |
| dintron | Distance from the focal substitution to intron–exon boundary | +/− |
| d5UTR | Distance from the focal substitution to the edge of 5'-UTR | + |
| d3UTR | Distance from the focal substitution to the edge of 3'-UTR | + |
| GC | GC content of the 4-fold degenerate sites of a window | + |
The distribution of πnor is far from normal (an overall exponential distribution with a gap between zero and nonzero πnor; supplementary fig. S1, Supplementary Material online), which would violate the assumptions of a generalized linear regression model and lead to difficulties in interpreting the residuals. We therefore used two separate regression analyses: one considering only nonzero πnor (linear regression) and the other considering all πnor that are coded as “0” and “1” for being zero or nonzero values, respectively (logistic regression; see Materials and Methods). For both regression analyses, a smaller επnor would correspond to lower than the predicted πnor near the focused fixation based on our fitted model.
It is worth noting that a significant result is sometimes observed only for larger window sizes because of the greater variance when using smaller windows (fig. 1A and B). The number of substitutions with nonzero πnor also decreases with smaller window size, leading to decreased statistical power (fig. 1C). The επnor of the two regression analyses would capture very different signals of the hitchhiking effects, with επnor from linear regression detecting differences in levels of variation, whereas επnor from logistic regression detecting presence or absence of polymorphism. Therefore, patterns may sometimes be apparent for only one of the two regression analyses. Also, because we could not normalize the variation with respect to divergence in windows without polymorphism (πnor = 0), we expect greater noise associated with επnor from logistic regression.
Fig. 1.

The properties of πnor that change over window sizes. Standard deviations of πnor of all substitutions (A) and of substitutions with nonzero windows (B) both decrease with window size. Proportion of windows with zero πnor , which is shown as solid column in (C), decreases with window size.
Nonsynonymous Substitutions Have Lower Nearby Polymorphism than Synonymous Substitutions
At the time of fixation for a neutral mutation, there is around 40% of reduction in nearby neutral variation (in the absence of recombination) (Tajima 1990). Even though at the time of substitution, the reduction in nearby neutral variation associated with a beneficial mutation would be much stronger (100% in the absence of recombination), it is necessary to control for the possible effects of recently fixed neutral mutations. Accordingly, we first compared the level of variation in windows centering on nonsynonymous substitutions to those centering on synonymous substitutions to investigate whether the expected heterogeneity in hitchhiking effects is detectable on a small physical scale. The former had significantly lower nonzero πnor (Mann–Whitney U test, P = 0.001 for 50-bp window and P < 10−6 for all other window sizes, table 2) and a larger proportion of windows having zero πnor (Fisher’s exact test, P = 0.004 for 50-bp window and P < 0.001 for all other window sizes, table 2) than windows centered on synonymous substitutions. Comparisons between nonsynonymous and synonymous substitutions using επnor from linear regression gave the same result (fig. 2A) and were statistically significant for all except for the smallest window size (Mann–Whitney U test, P < 0.003 for 100–1,600 bp windows; see figure 3A for statistical significant levels associated with individual window size). επnor from logistic regression showed consistent patterns although it was only statistically significant for larger window sizes (fig. 3A; Mann–Whitney U test, P = 0.03 for 200-bp window and P < 10−5 for 800 bp and 1,600 bp windows). Analyses using επnor of regression models can avoid the confounding effect of factors that are not of interest to us and are expected to detect heterogeneity in local polymorphism more accurately. Accordingly, we used επnor of regression models in the following analyses.
Table 2.
Comparisons between πnor of Nonsynonymous and Synonymous Substitutions without Performing Regression Analysis.
| Window size | Median of Nonzero πnor |
Proportion of Windows with Zero πnor |
|||||
|---|---|---|---|---|---|---|---|
| Nonsyn. | Syn. | % Reductiona | P valueb | Nonsyn. | Syn. | P valuec | |
| 50 bp | 0.327 | 0.333 | 1.56 | 1.25E-03 | 0.822 | 0.811 | 4.27E-03 |
| 100 bp | 0.192 | 0.215 | 10.84 | 2.49E-07 | 0.705 | 0.688 | 5.53E-04 |
| 200 bp | 0.125 | 0.137 | 8.78 | <2.20E-16 | 0.548 | 0.523 | 1.95E-10 |
| 400 bp | 0.087 | 0.097 | 9.91 | <2.20E-16 | 0.379 | 0.358 | 8.91E-10 |
| 800 bp | 0.067 | 0.074 | 9.84 | <2.20E-16 | 0.240 | 0.219 | 1.84E-11 |
| 1,600 bp | 0.052 | 0.061 | 13.62 | <2.20E-16 | 0.137 | 0.129 | 2.61E-06 |
a% of reduction is calculated as the πnor reduction in windows centered around nonsynonymous substitutions as compared with windows centered around synonymous substitutions.
bMann–Whitney U test.
cFisher’s exact test.
Fig. 2.

Medians of επnor from linear regression for different categories of substitutions. Medians of επnor from linear regression are shown for (A) nonsynonymous and synonymous substitutions in all genes, (B) nonsynonymous and synonymous substitutions that are in single-substitution genes and other genes, (C) nonsynonymous substitutions that changed amino acid chemical properties in different ways, (D) nonsynonymous substitutions increasing basicity or acidity of the amino acids, and (E) nonsynonymous substitutions that compensate or reinforce the chemical property changes of the nearest amino acid substitutions. Data of 400-bp window are shown, for which all comparisons are statistically significant with Mann–Whitney U test except for the comparisons between “compensatory–change charge” and “reinforcing–change charge”. Numbers of substitutions of each category are in parenthesis.
Fig. 3.
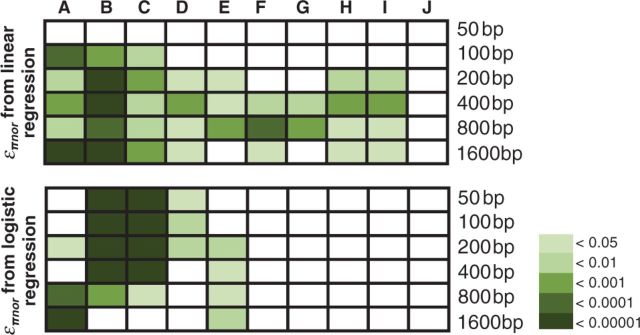
Statistical significance for comparisons of επnor of fixations associated with different biological categories. Upper and lower graphs are for comparisons using επnor from linear regression and logistic regression, respectively. All P values were calculated using Mann–Whitney U test unless otherwise specified. Increased statistical significance is represented with darker color. Comparisons shown are (A) nonsynonymous substitutions (lower επnor) versus synonymous substitutions, (B) nonsynonymous substitutions of single-substitution genes (lower επnor) versus synonymous substitutions of single-substitution genes, (C) nonsynonymous substitutions of single-substitution genes (lower επnor) versus nonsynonymous substitutions of other genes, (D) synonymous substitutions of single-substitution gene versus synonymous substitutions of other genes (lower επnor), (E) nonsynonymous substitutions that changed amino acid chemical properties in different ways (Kruskal–Wallis test), (F) nonsynonymous substitutions that did not change amino acid charges versus nonsynonymous substitutions that changed amino acid charges (lower επnor), (G) nonsynonymous substitutions that increased amino acid acidity versus nonsynonymous substitutions that increased amino acid basicity (lower επnor), (H) nonsynonymous substitutions that reinforced the chemical property changes of the nearest amino acid substitutions on the same linage (both on D. melanogaster) versus those compensated for these changes (lower επnor), (I) nonsynonymous substitutions that reinforced the polarities changes of the nearest amino acid substitutions on the same linage (both on D. melanogaster) versus those compensated for these changes (lower επnor), and (J) nonsynonymous substitutions that reinforced the charge changes of the nearest amino acid substitutions on the same linage (both on D. melanogaster) versus those compensated for these changes.
Nonsynonymous Substitutions in Slowly Evolving Genes Exhibit Lower Nearby Polymorphism than Those in Rapidly Evolving Genes
Genes with only one amino acid fixation (single-substitution genes) are usually excluded from evolutionary genetic analyses seeking to detect adaptive amino acid divergence because of lack of statistical power. They are generally considered highly constrained and it is unknown whether the fixation process of nonsynonymous mutations in these genes is different from other amino acid substitutions. In our data set, there are 1,952 genes with only one amino acid substitution and, as expected, these genes have significantly lower dN/dS ratio than other genes (median of dN/dS: 0.041 [single-substitution] versus 0.0848 [other], Mann–Whitney U test, P < 10−16). For these single-substitution genes, windows centering on nonsynonymous substitutions have significantly lower επnor than those centering on synonymous substitutions for most window sizes (figs. 2B and 3B; Mann–Whitney U test, P = 0.0003 for 100-bp window and P < 10−4 for 200–1,600 bp windows [επnor from linear regression]; P < 10−6 for 50–400 bp windows and P = 0.0001 for 800-bp window [επnor from logistic regression]).
To investigate possible heterogeneity of hitchhiking effects of nonsynonymous fixations in slower versus faster evolving proteins, we compared the heterozygosity associated with amino acid fixations in both types of genes. We found that windows centered on amino acid fixations in single-substitution genes have significantly lower επnor than those in other genes (figs. 2B and 3C; Mann–Whitney U test, P < 0.006 for 100–1,600 bp windows [επnor from linear regression]; P < 10−6 for 50–400 bp windows; and P = 0.02 for 800-bp window [επnor from logistic regression]). Selection against deleterious mutations can also lead to reduction in nearby linked variation (background selection [Charlesworth et al. 1993, 1995; Hudson and Kaplan 1995; Charlesworth 2012]). The lower επnor around amino acid substitutions in single-substitution genes may simply reflect their greater functional constraint and thus a stronger influence of background selection. Under this hypothesis, windows centered on synonymous fixations in these single-substitution genes should have lower επnor than those in other genes. However, we observed the opposite pattern: windows centered on synonymous fixations in single-substitution genes had higher επnor than those of other genes (figs. 2B and 3D; Mann–Whitney U test, P < 0.05 for 200–1,600 bp windows [επnor from linear regression]; P < 0.05 for 50–200 bp windows [επnor from logistic regression]). This result suggests that our observations cannot be explained by background selection.
One potential caveat associated with our observation is that substitutions in single-substitution genes tend to occur in smaller exons compared to substitutions in other genes (median of the size of exons substitutions located in: 615 bp [single-substitution genes] versus 746 bp [other genes], Mann–Whitney U test, P < 10−16). Previous studies suggested that because of the weaker effects of linked selection at the edge of exons, shorter exons have higher polymorphism than longer exons (Comeron and Kreitman 2002; Loewe and Charlesworth 2007). We used two strategies to address this issue. First, we controlled the effect of exon size on επnor by comparing substitutions that are located in exons with similar sizes (supplementary fig. S2, Supplementary Material online, and explanations therein). Second, we used linear regression to jointly test the effect of being in single-substitution gene and the size of exons (supplementary table S2, Supplementary Material online, and explanations therein). Both analyses supported the conclusion that amino acid fixations in single-substitution genes have lower nearby neutral variation, whereas synonymous substitutions showed the opposite pattern.
Local Reduction in Polymorphism Yields Distinct Gene Ontology Enrichment from Other Signatures of Positive Selection
Most previous functional enrichment analyses for genes putatively experiencing positive selection were based on genes with excess of amino acid fixations under the McDonald-Kreitman test framework (Begun et al. 2007; Langley et al. 2012). Under this framework, mainly genes experienced recurrent directional selection while retaining sufficient polymorphism will have enough statistical power to detect positive selection. Several categories of genes, such as single-substitution genes, will be excluded from this approach. Investigation of possible hitchhiking effects of nonsynonymous fixations in genes with low rates of amino acid substitution may thus contain additional important information about adaptive protein evolution. Assuming classes of amino acid substitutions associated with lower nearby neutral polymorphism experienced stronger positive selection, functional enrichment analysis based on population genetics of these regions may provide a complementary view of biological functions influenced by adaptive protein evolution. To control the across-genome variation of other factors that are known to influence the level of neutral variation, we used επnor to perform our GO enrichment analysis.
We used our window-based GO enrichment analysis and permutations to determine whether the επnor associated with nonsynonymous fixations of each GO term is lower than expected (see Materials and Methods). We reported GO terms that are significant (P < 0.05) for at least three window sizes after multiple-test correction. For επnor from linear regression, biological functions related to mitosis (mitotic cell cycle, regulation of the rate of mitosis, mitotic spindle organization, and cytokinesis), DNA metabolic process, movement within cell (including transportation of mRNA out of nucleus), gene silencing (including silencing at chromatin, transcriptional, and translational level), and double-strand break repair, cellular locations of chromatin, centromeric region of condensed chromosome, nucleus, and nuclear pore, and molecular processes related to nucleic acid-binding (DNA binding and RNA binding), transcription by core RNA polymerase, and GTPase bindings showed reduced average επnor. Enrichment analysis based on επnor from logistic regression identified several similar categories (movement within cell, nucleic acid metabolism, and GTPase bindings) and additional categories (regulation of transcription and translation, pole cell fate determination, meiotic chromosome organization [biological function], pole plasm differentiation [cellular location], and zinc ion binding, sulfite bond formation, and protein dimerization [molecular process]). Interestingly, several of these categories are involved in the basic cellular processes and have previously been considered highly constrained. In addition, the majority of these categories were not identified by the GO enrichment analysis of genes with significant McDonald–Kreitman tests using the same data set (Langley et al. 2012), which reported functions related to male and female reproduction, stem cell maintenance, and neural and neuromuscular junction development.
Substitutions Changing Amino Acid Chemical Properties Have Greater Locally Reduced Polymorphism
There are four major amino acid R groups that differ in their chemical properties: nonpolar, uncharged polar, acidic, and basic. According to whether the substituted amino acid has an R group with different chemical properties from that of the ancestral amino acid, we classified amino acid substitutions into four categories: preserved amino acid charge/polarity (“no change”), changed from nonpolar to polar amino acid or vice versa (“change polarity”), changed from charged (acidic or basic) to polar amino acid or vice versa (“change polarity and charge”), and changed from nonpolar to charged amino acid, from charged to nonpolar amino acid, or from basic to acidic amino acid or vice versa (“change charge”; see Materials and Methods for details). The observed numbers of each type of substitution in our data set decreases with the above order (6,940 [44.50%, “no change”], 4,456 [28.59%, “change polarity”], 2,635 [10.91%, “change polarity and charge”] and 1,556 [9.98%, “change charge”]), which is consistent with the general findings that radical substitutions are less common.
Although επnor in windows surrounding amino acid substitutions with different R groups (nonpolar, polar, basic, and acidic) are not significantly different (Kruskal–Wallis test, P > 0.05 for all window sizes and both regression methods), επnor around amino acid substitutions that led to different changes in R group chemical properties is significantly different (figs. 2C and 3E; Kruskal–Wallis test, P < 0.02 for 200–800 bp windows [επnor from linear regression]; P < 0.05 for 200–1,600 bp windows [επnor from logistic regression]). The median of επnor is lowest for “change charge” amino acid fixations, followed by “change charge and polarities,” “change polarity,” and, lastly, “no change” amino acids. Pairwise comparisons between four categories of substitutions found that the heterogeneity in nearby neutral variation mainly comes from the differences between substitutions that resulted in changes in R group charges (“change charge” and “change charge and polarities”) versus those that did not (“no change” and “change polarity”; pairwise Mann–Whitney U tests [with Holm–Bonferroni multiple test correction] are significant for επnor from linear regression, fig. 4). Indeed, the difference between amino acid substitutions that led to changes in R group charges and those that did not is statistically significant (fig. 3F; Mann–Whitney U test, P < 0.002 for 400–800 bp windows and P < 0.05 for 1,600-bp window [επnor from linear regression]; no significant results for επnor from logistic regression). A confounding factor for our observation is that nonsynonymous substitutions that led to changes in R group charges tend to be in genes with greater dN (median of dN/dS: 0.13 [no R group charge changes] and 0.20 [with R group charge changes]; Mann–Whitney U test, P < 10−16), which are known to have lower overall level of synonymous polymorphism (Ometto et al. 2005; Andolfatto 2007; Begun et al. 2007; Langley et al. 2012). Interestingly, among substitutions that changed amino acid charges, fixations leading to increased basicity (from acidic to uncharged [nonpolar and polar], acidic to basic, or uncharged to basic amino acids) have lower nearby επnor than those increasing acidity (figs. 2D and 3G; Mann–Whitney U test, P < 0.03 for 400–800-bp window [επnor from linear regression]; no significant results for επnor from logistic regression). This pattern is unlikely to be driven by nonrandom distribution of these two classes of substitutions among genes with different rates of evolution (median of dN/dS: 0.200 [increase basicity] and 0.190 [decrease basicity]; Mann–Whitney U test, P > 0.05).
Fig. 4.
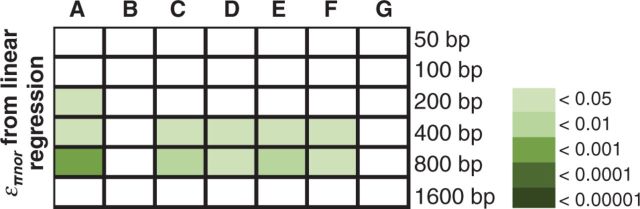
Statistical significance for comparisons of επnor (from linear regression) of amino acid fixations that changed R group chemical properties differently. (A) P values of Kruskal–Wallis test for comparing nonsynonymous substitutions that changed amino acid chemical properties in different ways (the same as fig. 3E). (B–G) Pairwise Mann–Whitney U test P values with Holm–Bonferroni multiple-test correction for comparisons between (B) “no change” versus “change polarity,” (C) “no change” versus “change charge” (lower επnor), (D) “no change” versus “change charge and polarity” (lower επnor), (E) “change polarity” versus “change charge” (lower επnor), (F) “change polarity” versus “change charge and change polarity” (lower επnor), and (G) “change charge” versus “change charge and polarity.”
Amino Acid Substitutions Compensating for Nearby Chemical Changes Have Lower Nearby Polymorphism than Those Reinforcing Such Changes
The fixation of individual amino acid substitutions might not only lead to changes in important functional sites of a protein but also lead to changes in the overall protein chemical properties (charges and polarities). It is interesting to investigate whether, conditioning on the presence of an adjacent fixation changing amino acid polarity or charge, the substitution process of a nonsynonymous mutation varies according to whether it maintains the ancestral polarity or charge of the protein (compensatory) or reinforces the change in polarity or charge (reinforcing). Amino acid substitutions with nearest neighbor compensating the polarity or charge change have lower επnor than substitutions with reinforcing neighbors (figs. 2E and 3H; Mann–Whitney U test, P < 0.004 for 200–400 bp windows and P < 0.05 for 800–1,600 bp windows [επnor from linear regression]; result not significant for επnor from logistic regression). This observation did not result from nonrandom distribution of these types of substitutions among genes with different rates of protein evolution (median of dN/dS: 0.176 [compensatory] and 0.172 [reinforcing]; Mann–Whitney U test, P > 0.05). We only observed these differences when the two nearest substitutions were both fixed on the D. melanogaster lineage but not so when one substitution occurred in D. melanogaster and the nearest substitution occurred in D. simulans (Mann–Whitney U test, P > 0.05). This suggested that stronger reduction in variation only occurs when the two compensatory substitutions happened in the same protein.
These compensatory and reinforcing substitutions were further classified according to whether they changed amino acid polarity or charge. In both cases, the compensatory substitutions still had lower επnor than reinforcing substitutions (fig. 2E). However, these differences were only significant for substitutions that changed polarities (fig. 3I; Mann–Whitney U test, P < 0.005 for 200–400 bp windows and P < 0.02 for 800–1,600 bp windows [επnor from linear regression]; result not significant for επnor from logistic regression) but not for substitutions that changed charges (fig. 3J; Mann–Whitney U test, P > 0.05). This may be attributable to the much smaller number of observations of amino acids changing charges (total number of observations [including substitutions with zero and nonzero πnor] change polarities: 360 [compensatory] and 494 [reinforcing]; change charges: 40 [compensatory] and 41 [reinforcing]).
Discussion
Conditioning on coding substitutions on the D. melanogaster branch, we found significant heterogeneity in the level of nearby synonymous polymorphism that we attribute to variation in strength of positive selection. This interpretation is based on the assumption that the time since fixation and the variability of strength and directionality of selection through time due to environmental fluctuations (Takahata et al. 1975; Gillespie 1994; Mustonen and Lässig 2007) are similar for different categories of substitutions. We also assumed that the proportion of substitutions in the D. melanogaster genome that were fixed from newly arising mutations instead of pre-existing segregating alleles (Orr and Betancourt 2001; Hermisson and Pennings 2005; Prezeworski et al. 2005) is homogeneous across classes of substitutions. In addition, it is worth noting that our approach mainly detects reduction in polymorphisms from beneficial substitutions that were fixed in the recent past (fewer than Ne [the effective population size] generations ago [Kaplan et al. 1989; Przeworski 2002]).
Local reductions in heterozygosity are significantly greater for nonsynonymous fixations than for synonymous fixations even on a very small physical scale, suggesting that our approach can be an effective alternative to other evolutionary genetic analyses in detecting variation in the relative effect of positive selection. The spatial scale at which hitchhiking effects are analyzed has a significant effect on the range of strengths of positive selection that can be inferred (Andolfatto 2007; Macpherson et al. 2007; Sella et al. 2009). We used windows that are as small as possible because our main focus is to understand the impact of positive selection on the population genetics of sites very near to the focal substitution. Our result suggests that hitchhiking effects may occur on the scale of a few hundred base pairs or less, which is consistent with other work suggesting that many selected substitutions may have relatively small selection coefficients (Andolfatto 2007; Sattath et al. 2011; Schneider et al. 2011; but see Macpherson et al. 2007). Even though we mainly concentrated on the heterogeneity of hitchhiking effects associated with coding substitutions, this approach can be readily extended to fixation in noncoding sequences.
Several scenarios other than genetic hitchhiking could also lead to the observed heterogeneity in linked synonymous variation around different categories of substitutions. A nonrandom distribution of biased gene conversion (Nagylaki 1983a, 1983b; Marais 2003) could potentially generate these correlated patterns of reduced polymorphism. Note that the length of gene conversion tracks in Drosophila (several hundred base pairs [Hilliker et al. 1994; Comeron et al. 2012; Miller et al. 2012]) is roughly the physical scale of our study. However, the definitive evidence for the significant role of biased gene conversion in Drosophila is still lacking. In addition, the distribution of the biases of gene conversion would need to be highly correlated with our a priori categorization of substitutions to lead to our observations, which seems highly unlikely. Purifying selection against deleterious mutations, known as background selection (Charlesworth et al. 1993, 1995; Hudson and Kaplan 1995; Charlesworth 2012), is a more likely alternative that could contribute to the observed variation in the level of polymorphism around different substitutions. In humans, studies comparing the diversity around nonsynonymous and synonymous fixations have suggested that most of the troughs in polymorphism around amino acid substitutions could be attributable to background selection instead of positive selection (Hernandez et al. 2011), even though similar analysis (Sattath et al. 2011) and other genome-wide studies in Drosophila concluded differently. Empirically distinguishing the influence of genetic hitchhiking from that of background selection is not always straightforward because the underlying key parameters (such as rate of deleterious mutations and beneficial mutations) are not known (Andolfatto 2001; Stephan 2010; Cutter and Payseur 2013). If the effects of background selection vary among our categories of substitutions, it might explain some of our observed patterns (but see later).
Perhaps our strongest finding is that amino acid substitutions in slowly evolving genes, on average, showed stronger hitchhiking effects than those in fast evolving genes. This would be expected to occur if the fitness benefits of nonsynonymous substitutions in highly constrained genes are on average larger than those in genes under recurrent directional selection, suggesting that some proteins adapt by few mutations with large effects, whereas others adapt through multiple mutations with weaker effects. Background selection is unlikely a primary cause for this observation because the same analyses of synonymous substitutions in the same sets of genes found the opposite pattern, which contradicts the prediction of background selection. Another alternative that might explain our observations is that the influence of selective interference (Hill and Robertson 1966; Felsenstein 1974), which is known to weaken the effect of genetic hitchhiking on linked neutral variation (Barton 1995; Kim and Stephan 2000, 2003; Hartfield and Otto 2011), might vary for these two categories of substitutions. Although the substitution processes of beneficial mutations in fast-evolving genes are most prone to selective interference from other positively selected mutations, those in slow-evolving genes are subjected to strong selective interference from purifying selection. Further modeling would be required to evaluate how variation in the relative contribution of selective interference from positive and negative selections may lead to systematic biases in our observation.
In addition to the influence of single amino acid changes, the biochemical interactions among amino acid residues within a protein are crucial in maintaining the higher order structures, stabilities and chemical properties of proteins (DePristo et al. 2005). We found that substitutions compensating for the polarity or charge change of the nearest amino acid substitution showed stronger hitchhiking effects than substitutions reinforcing the polarity or charge change. The hitchhiking effects associated with compensatory amino acid substitutions could result from selection on the second substitution that compensates the pleiotropic deleterious effect (on overall protein polarities/charges in our case) of the first positively fixed amino acid change (Kulathinal et al. 2004). On the other hand, weakly deleterious mutations can drift to fixation, and the subsequent substitutions that restore the fitness impact caused by the polarity/charge changes of the first substitutions will be positively selected for (Gillespie 1984; Hartl and Taubes 1996; Osada and Akashi 2012). Note, however, previous theoretical analysis indicates that this scenario may be rare and that simultaneous substitution of compensatory substitutions may be common (Innan and Stephan 2001). Nevertheless, with either scenarios of sequential fixation of mutations, we could not distinguish which substitution happened first and had to include both of them in the analyses. This is expected to reduce the effect we could observe and the actual strength of positive selection on compensatory substitutions could be stronger. After further categorizing changes into those affecting charge and those affecting polarity, the signals were still statistically significant only for compensatory substitutions that maintained polarity, whereas previous studies based on between-species divergence only found evidence supporting selection for compensatory evolution of amino acid substitutions that maintained charges (Neher 1994; Fukami-Kobayashi et al. 2002; Callahan et al. 2011). Our observed statistical insignificance may be attributable to the small number of observed substitutions that confer compensatory charge changes in one species. On the other hand, the new observation of hitchhiking effects for amino acid substitutions compensatory for polarity changes suggests that our approach may be able to detect subtler differences in the relative impact of selection than methods based on fixed differences between species or simply reflects the heterogeneity of substitution processes across the phylogeny. The importance of maintaining protein polarities is suggested by the heavy dependence of protein stability on the retention of the hydrogen bonds formed between polar side chains of amino acids (Takano et al. 1999; Pace 2001) and on the overall amino acid volumes (Altschuh et al. 1987; Atchley et al. 2000; Fares and Travers 2006; Yeang and Haussler 2007) in the core of proteins. However, unlike compensatory charge changes, whose biochemical models have been well proposed and studied (Kumar and Nussinov 2002), the biochemical basis for compensatory polarity change has not been well formulated.
At a given rate of crossing over, the reduction in neutral variation around a substitution depends on the sojourn time of the mutation in the population. Theoretical predictions for how the genealogy of neutral linked sites is perturbed by the fixation process of beneficial mutations are widely known and discussed (Kaplan et al. 1989; Braverman et al. 1995; Barton 1998; Fay and Wu 2000; Kim and Stephan 2002). However, it must be noted that, the sojourn time for the substitutions of mildly deleterious mutations will be very similar to that of beneficial mutations having the same magnitude of selection coefficient (Maruyama 1974). Therefore, the trough in polymorphism associated with the fixation of a deleterious mutation would be indistinguishable from that of a beneficial mutation with equal size of selection coefficient. Although the probability of fixing adaptive mutations is much greater than that for deleterious mutations, especially in species with large effective population size, it is also true that such favored mutations are less frequent. Furthermore, it is unclear how the proportion of fixations that came from beneficial or deleterious mutations varies according categories of substitutions. Our observed stronger reduction in nearby neutral variation of nonsynonymous substitutions that are in single-substitution genes or that changed the charges of amino acids could reflect the fact that these amino acid mutations are expected to have stronger deleterious effects. Whether the reduction of local variation is the result of fixing beneficial or deleterious mutations, our observations would indicate that the fitness distribution of newly arising mutations of functionally more constrained sites/genes is coarser than that of less constrained sites/genes.
Based on our analyses of patterns of polymorphism surrounding coding substitutions on the D. melanogaster lineage, it is clear that the dynamics of nonsynonymous substitutions lead to local reductions. Hitchhiking on this small scale is the simplest and best-supported interpretation. The differences in local hitchhiking associated with chemically distinct classes of nonsynonymous substitutions (especially compensatory polarity changes) extend earlier conclusions based solely on divergence. Most importantly, the unique aspect of this approach to detect the impact of selection is its weak dependence on the numbers of recent nonsynonymous substitutions, thus affording an opportunity to make inferences about highly conserved (slowly evolving) genes. However, alternative mechanisms that we currently are not included in our analyses may also contribute to the observed heterogeneity in variation. This situation points to the importance of further development of theoretical models that jointly analyze mechanisms that are often considered separately on their influence on the level of variation.
Materials and Methods
Estimation of Polymorphism around Substitutions on the D. melanogaster Branch
We used 43 D. melanogaster genomes (Langley et al. 2012) and six D. simulans genomes (Begun et al. 2007) to call fixed nonsynonymous and synonymous coding differences between the two species. Bases with quality lower than 30 were treated as missing data. Only sites with allelic coverage (number of individuals with data at a particular site) above 29 in D. melanogaster samples and above three in D. simulans samples are included in the analysis. The D. yakuba allele was used in a parsimony framework to infer fixations on the D. melanogaster lineage. The multispecies alignment we used is from Langley et al. (2012) and included D. melanogaster, D. simulans, D. yakuba, and D. erecta.
The 43 D. melanogaster genomes from Langley et al. (2012) consist of six African (Malawi) and 37 North American strains (NC). Previous studies have identified sub-Saharan Africa as the potential ancestral range of D. melanogaster (Lachaise et al. 1988; Veuille et al. 2004; Pool and Aquadro 2006). This suggests that African D. melanogaster populations are less likely to be disturbed by the recent out-of-Africa demographic history of the species, which is known to generate population genetic signals similar to that of selective sweeps (Barton 1998; Wall et al. 2002; Jensen et al. 2005; Thornton et al. 2007). Therefore, estimates of heterozygosity around each coding substitution on the D. melanogaster branch were made using the six African D. melanogaster genomes. Nucleotide heterozygosity (π) was estimated as average pairwise divergence (Nei 1987) across all 4-fold degenerate sites that were within 25, 50, 100, 200, 400, and 800 bp of the focal substitution (referred to as window size 50, 100, 200, 400, and 1,600 bp, respectively). Sites with allelic coverage lower than four were removed from the estimation of π. Note this is a different criterion from that of calling the fixed differences between D. melanogaster and D. simulans, which used all D. melanogaster alleles and required at least 30 out of 43 individuals with data at a site. To correct for variation in the mutation rates, we divided π in each window by the average divergence of 4-fold degenerate sites between D. simulans (the mosaic assembly [Begun et al. 2007]) and D. yakuba for the same window, a ratio we referred to as “normalized π” and denoted as πnor. We used D. simulans rather than D. melanogaster to avoid the contribution of D. melanogaster within species polymorphism to divergence. Because D. simulans and D. yakuba are closely related to D. melanogaster, we assumed that the variation in mutation rate across the genome is similar among these species. The reported evidence for selection on codon usage bias in these species (Akashi 1995, 1996; McVean and Vieira 2001; Akashi et al. 2006; Nielsen et al. 2007) might invalidate this assumption. However, unless there is a major lineage x gene interaction, selection on codon usage bias is unlikely to compromise our analysis, especially given that most selection on codon usage bias is likely to be weak (Ns ∼ 1) and thus unlikely to significantly influence signals of positive selection on amino acids. Indeed, we found that Fop (frequency of optimum codon) of D. melanogaster, D. simulans, and D. yakuba lineages are highly correlated (Spearman’s rank ρ = 0.96 [D. melanogaster vs. D. simulans], 0.91 [D. simulans vs. D. yakuba], 0.91 [D. melanogaster vs. D. yakuba], P value < 10−16 for all).
Windows that had fewer than five sites included in the estimation of either heterozygosity or divergence, windows with zero divergence, or windows located in genomic region with zero recombination rate (see later) were removed from the analysis. For windows centering on coding substitutions that are near the edge of exons or for windows that are larger than the length of exons, the distal part of the window will be noncoding sequences that are not included in the estimation of variation or divergence. The average distance between focal substitutions and 4-fold degenerate sites will, accordingly, be nonhomogeneous across windows. To investigate the influences of this on our observation, we performed all analyses excluding substitutions whose windows contain noncoding sites and found consistent results (supplementary fig. S7, Supplementary Material online), suggesting that the variability of average distance between focal substitution and 4-fold degenerate sites does not substantially bias our observations.
Accounting for Factors Correlated with πnor
Variables that are known or expected to be correlated with the level of polymorphism but are not our main interest include (table 1): recombination rate in the region near a focal substitution (recomb, cM/Mbp), the number of nonfocal nonsynonymous substitutions (nns) and synonymous substitutions (ns) that are in the same window as a focal substitution, the physical distance from a focal substitution to the nearest nonsynonymous (dns, bp) or synonymous (ds, bp) substitution, the physical distance from a focal substitution to the nearest exon–intron boundary (dintron, bp), and the physical distance from a focal substitution to the edge of UTRs (d5UTR and d3UTR, bp), and GC content of the 4-fold generate sites in a window (GC). We considered variables related not only to nonsynonymous but also to synonymous substitutions on the D. melanogaster branch because of the expected reduction in nearby polymorphism at the time of substitution even when the mutation is selectively neutral (Tajima 1990).
We used recombination rate (cM/Mbp) estimated by Comeron et al. (2012), which estimated recombination rate at 100 kb scale. Because recombination rate estimates at corresponding scale to our analyses are still not available, we used the physical distance (bp) as a proxy for the genetic distance of variables that we included (dns, ds, dintron, d5UTR, and d3UTR). The positions of introns and UTRs were defined according to D. melanogaster reference genome annotation 5.16, see supplementary figures S3–S5, Supplementary Material online, for the distribution of these factors and supplementary table S1, Supplementary Material online, for the Spearman’s rank ρ of each factor with πnor.
We performed regression analysis on πnor with the above variables as predictors and used the deviation from the regression model (residuals of πnor, denoted as επnor) instead of πnor for subsequent analyses. The distribution of πnor is far from normal: an overall exponential distribution (supplementary fig. S1A, Supplementary Material online) and a gap between zero and nonzero πnor (supplementary fig. S1B, Supplementary Material online). Therefore, we used two regression analyses to acquire επnor. 1) Using only nonzero πnor: The nonzero πnor is highly nonnormal (supplementary fig. S6A, Supplementary Material online), which will lead to problems in interpreting the residuals of the least-squares linear regression model. Accordingly, we replaced each πnor with the corresponding quantile value of a normal distribution (denoted as πquan) and performed linear regression on πquan. 2) Using all πnor: We performed logistic regression on zero and nonzero πnor coded as “0” and “1,” respectively (denoted as π0,1). Even though the effects of several of these factors on polymorphism have been theoretically investigated before (Kaplan et al. 1989; Barton 1998; Comeron and Kreitman 2002), there have not been models considering all their effects jointly. We took an empirical approach and first did regression analysis that included only one predictor at a time to determine the regression model (linear, quadratic, or logarithmic; see supplementary figs. S8 and S9 [and explanations therein] and table S3, Supplementary Material online). We chose the model that has the largest R2 (linear regression) or smallest AIC (Akaike information criterion; logistic regression). We then additively combined the individually determined regression model of all variables and used backward model selection based on AIC (implemented in R) to select for the most appropriate model. One exception is, for 50-bp window size linear regression, recomb was not chosen to be included in the regression model based on backward model selection. However, we still include recomb in the regression model because of our prior knowledge of the relationship between recombination rate and heterozygosity. For factors that perform similarly well in several regression models (supplementary figs. S8 and S9, and table S3, Supplementary Material online), we performed regression using additional models. We found that our results, particularly those based on επnor from the linear regression, are generally insensitive to the regression model chosen (see supplementary figs. S10 and S11, Supplementary Material online, for the P values of comparisons using επnor from other regression models).
The regression models we used (before performing model selection) are as follows:
substitutions with nonzero πnor (linear regression):
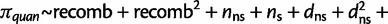
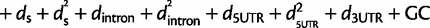
all substitutions (logistic regression):
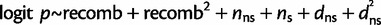
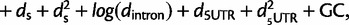
where logit p is the log odds of having π0,1 = 1. See supplementary tables S4 and S5, Supplementary Material online, for the chosen regression model based on backward selection for each window size, statistical significance of the regression coefficients, and proportion of variation explained of the linear regression model (R2).
Categorization of Substitutions
Substitutions on the D. melanogaster branch were categorized to functional classes (e.g., nonsynonymous vs. synonymous) according to D. melanogaster reference genome annotation version 5.16. We only analyzed πnor of coding substitutions that are included in the conservative gene set of Langley et al. (2012), which are genes whose D. melanogaster, D. simulans, and the outgroup alleles all have canonical (i.e., the same as the reference annotation) initiation codons, splice junctions, and stop codons and at least 100 bp of high-quality data (bases with Q30 or above with no gaps). Genes that do not pass these filtering criteria either have low sequencing quality or have potentially experienced relaxed selection (Langley et al. 2012; Lee and Reinhardt 2012). Lineage-specific divergences for individual genes were estimated by maximum likelihood using PAML version 4 (Yang 2007) on the branch leading to D. melanogaster, using D. yakuba as the outgroup. Genes with fewer than 100 sites included in the PAML analysis or with a dS value smaller than 0.0001 were excluded from the analysis. On the basis of the chemical properties of the amino acid R groups, we categorized amino acid substitutions into following four groups: no change (from nonpolar to nonpolar, polar to polar, basic to basic, or acidic to acidic R group), change polarity (from nonpolar to polar or polar to nonpolar R group), change polarity and charge (from polar to basic, polar to acidic, basic to polar and acidic to polar R group), and change charge (from nonpolar to basic, nonpolar to acidic, basic to nonpolar, acidic to nonpolar, basic to acidic, and acidic to basic R group).
GO Enrichment Analysis
We combined the full GO list and the GO slim list (from http://www.geneontology.org/, last accessed December 31, 2013) for gene ontology annotation. For each GO term, we calculated the average of επnor for nonsynonymous substitutions in genes associated with that GO term. We only considered GO terms associated with at least 10 nonsynonymous substitutions. The P value of each GO term was determined by sampling uniformly without replacement an equivalent number of nonsynonymous substitutions, calculating the average of επnor of windows surrounding them and repeating this process 10,000 times to obtain the empirical distribution of P values for any random subset of nonsynonymous substitutions of equal size. The analysis was done separately for επnor from linear regression and logistic regression. To correct for multiple testing, we used the qvalue package for R (Dabney and Storey 2010) and false-discovery rate of 5%. We reported GO categories that are significant for at least three window sizes.
Supplementary Material
Supplementary tables S1–S5 and figures S1–S11 are available at Molecular Biology and Evolution online (http://www.mbe.oxfordjournals.org/).
Acknowledgments
The authors greatly appreciate Peter Ralph and Graham Coop for extensive and helpful discussions and Hsiao-Han Chang for statistical assistance. They thank Peter Ralph, Richard Hudson, Wynn Meyer, and Nicholas VanKuren for critically reading the manuscript. They are grateful to Daniel Falush and three anonymous reviewers whose helpful and constructive comments greatly improved this work. This work is supported by Center for Population Biology Research Award of UC Davis to Y.C.G.L. and NIH HG02942 grant to C.H.L.
References
- Akashi H. Inferring weak selection from patterns of polymorphism and divergence at “silent” sites in Drosophila DNA. Genetics. 1995;139:1067–1076. doi: 10.1093/genetics/139.2.1067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Akashi H. Molecular evolution between Drosophila melanogaster and D. simulans: reduced codon bias, faster rates of amino acid substitution, and larger proteins in D. melanogaster. Genetics. 1996;144:1297–1307. doi: 10.1093/genetics/144.3.1297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Akashi H, Ko W-Y, Piao S, John A, Goel P, Lin C-F, Vitins AP. Molecular evolution in the Drosophila melanogaster species subgroup: frequent parameter fluctuations on the timescale of molecular divergence. Genetics. 2006;172:1711–1726. doi: 10.1534/genetics.105.049676. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Altschuh D, Lesk AM, Bloomer AC, Klug A. Correlation of co-ordinated amino acid substitutions with function in viruses related to tobacco mosaic virus. J Mol Biol. 1987;193:693–707. doi: 10.1016/0022-2836(87)90352-4. [DOI] [PubMed] [Google Scholar]
- Andolfatto P. Adaptive hitchhiking effects on genome variability. Curr Opin Genet Dev. 2001;11:635–641. doi: 10.1016/s0959-437x(00)00246-x. [DOI] [PubMed] [Google Scholar]
- Andolfatto P. Hitchhiking effects of recurrent beneficial amino acid substitutions in the Drosophila melanogaster genome. Genome Res. 2007;17:1755–1762. doi: 10.1101/gr.6691007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Atchley WR, Wollenberg KR, Fitch WM, Terhalle W, Dress AW. Correlations among amino acid sites in bHLH protein domains: an information theoretic analysis. Mol Biol Evol. 2000;17:164–178. doi: 10.1093/oxfordjournals.molbev.a026229. [DOI] [PubMed] [Google Scholar]
- Barrett RDH, Schluter D. Adaptation from standing genetic variation. Trends Ecol Evol. 2008;23:38–44. doi: 10.1016/j.tree.2007.09.008. [DOI] [PubMed] [Google Scholar]
- Barton NH. Linkage and the limits to natural selection. Genetics. 1995;140:821–841. doi: 10.1093/genetics/140.2.821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barton NH. The effect of hitch-hiking on neutral genealogies. Genet Res. 1998;72:123–133. [Google Scholar]
- Begun DJ, Holloway AK, Stevens K, Hillier LW, Poh Y, Hahn MW, Nista PM, Jones CD, Kern AD, Dewey CN, et al. Population genomics: whole-genome analysis of polymorphism and divergence in Drosophila simulans. PLoS Biol. 2007;5:e310. doi: 10.1371/journal.pbio.0050310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bierne N, Eyre-Walker A. The problem of counting sites in the estimation of the synonymous and nonsynonymous substitution rates: implications for the correlation between the synonymous substitution rate and codon usage bias. Genetics. 2003;165:1587–1597. doi: 10.1093/genetics/165.3.1587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Braverman JM, Hudson RR, Kaplan NL, Langley CH, Stephan W. The hitchhiking effect on the site frequency spectrum of DNA polymorphisms. Genetics. 1995;140:783–796. doi: 10.1093/genetics/140.2.783. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Callahan B, Neher RA, Bachtrog D, Andolfatto P, Shraiman BI. Correlated evolution of nearby residues in drosophilid proteins. PLoS Genet. 2011;7:e1001315. doi: 10.1371/journal.pgen.1001315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Charlesworth B. The effects of deleterious mutations on evolution at linked sites. Genetics. 2012;190:5–22. doi: 10.1534/genetics.111.134288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Charlesworth B, Morgan MT, Charlesworth D. The effect of deleterious mutations on neutral molecular variation. Genetics. 1993;134:1289–1303. doi: 10.1093/genetics/134.4.1289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Charlesworth D, Charlesworth B, Morgan MT. The pattern of neutral molecular variation under the background selection model. Genetics. 1995;141:1619–1632. doi: 10.1093/genetics/141.4.1619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Comeron JM, Guthrie TB. Intragenic Hill-Robertson interference influences selection intensity on synonymous mutations in Drosophila. Mol Biol Evol. 2005;22:2519–2530. doi: 10.1093/molbev/msi246. [DOI] [PubMed] [Google Scholar]
- Comeron JM, Kreitman M. Population, evolutionary and genomic consequences of interference selection. Genetics. 2002;161:389–410. doi: 10.1093/genetics/161.1.389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Comeron JM, Ratnappan R, Bailin S. The many landscapes of recombination in Drosophila melanogaster. PLoS Genet. 2012;8:e1002905. doi: 10.1371/journal.pgen.1002905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cutter AD, Payseur BA. Genomic signatures of selection at linked sites: unifying the disparity among species. Nat Rev Genet. 2013;14:262–274. doi: 10.1038/nrg3425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dabney A, Storey JD. 2010. qvalue: Q-value estimation for false discovery rate control. R package version 1.22.0. [Google Scholar]
- DePristo MA, Weinreich DM, Hartl DL. Missense meanderings in sequence space: a biophysical view of protein evolution. Nat Rev Genet. 2005;6:678–687. doi: 10.1038/nrg1672. [DOI] [PubMed] [Google Scholar]
- Eyre-Walker A, Keightley PD. The distribution of fitness effects of new mutations. Nat Rev Genet. 2007;8:610–618. doi: 10.1038/nrg2146. [DOI] [PubMed] [Google Scholar]
- Fares MA, Travers SAA. A novel method for detecting intramolecular coevolution: adding a further dimension to selective constraints analyses. Genetics. 2006;173:9–23. doi: 10.1534/genetics.105.053249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fay JC, Wu C-I. Hitchhiking under positive Darwinian selection. Genetics. 2000;155:1405–1413. doi: 10.1093/genetics/155.3.1405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fay JC, Wyckoff GJ, Wu C-I. Testing the neutral theory of molecular evolution with genomic data from Drosophila. Nature. 2002;415:1024–1026. doi: 10.1038/4151024a. [DOI] [PubMed] [Google Scholar]
- Felsenstein J. The evolutionary advantage of recombination. Genetics. 1974;78:737–756. doi: 10.1093/genetics/78.2.737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fukami-Kobayashi K, Schreiber DR, Benner SA. Detecting compensatory covariation signals in protein evolution using reconstructed ancestral sequences. J Mol Biol. 2002;319:729–743. doi: 10.1016/S0022-2836(02)00239-5. [DOI] [PubMed] [Google Scholar]
- Galtier N, Bazin E, Bierne N. GC-biased segregation of noncoding polymorphisms in Drosophila. Genetics. 2006;172:221–228. doi: 10.1534/genetics.105.046524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gillespie JH. Molecular evolution over the mutational landscape. Evolution. 1984;38:1116–1129. doi: 10.1111/j.1558-5646.1984.tb00380.x. [DOI] [PubMed] [Google Scholar]
- Gillespie JH. The causes of molecular evolution. New York: Oxford University Press; 1994. [Google Scholar]
- Gutz H, Leslie JF. Gene conversion: a hitherto overlooked parameter in population genetics. Genetics. 1976;83:861–866. doi: 10.1093/genetics/83.4.861. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haddrill PR, Charlesworth B. Non-neutral processes drive the nucleotide composition of non-coding sequences in Drosophila. Biol Lett. 2008;4:438–441. doi: 10.1098/rsbl.2008.0174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haddrill PR, Loewe L, Charlesworth B. Estimating the parameters of selection on nonsynonymous mutations in Drosophila pseudoobscura and D. miranda. Genetics. 2010;185:1381–1396. doi: 10.1534/genetics.110.117614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hartfield M, Otto SP. Recombination and hitchhiking of deleterious alleles. Evolution. 2011;65:2421–2434. doi: 10.1111/j.1558-5646.2011.01311.x. [DOI] [PubMed] [Google Scholar]
- Hartl DL, Taubes CH. Compensatory nearly neutral mutations: selection without adaptation. J Theor Biol. 1996;182:303–309. doi: 10.1006/jtbi.1996.0168. [DOI] [PubMed] [Google Scholar]
- Hermisson J, Pennings PS. Soft sweeps. Genetics. 2005;169:2335–2352. doi: 10.1534/genetics.104.036947. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hernandez RD, Kelley JL, Elyashiv E, Melton SC, Auton A, McVean G, Sella G, Przeworski M. Classic selective sweeps were rare in recent human evolution. Science. 2011;331:920–924. doi: 10.1126/science.1198878. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hill WG, Robertson A. The effect of linkage on limits to artificial selection. Genet Res. 1966;8:269–294. [PubMed] [Google Scholar]
- Hilliker AJ, Harauz G, Reaume AG, Gray M, Clark SH, Chovnick A. Meiotic gene conversion tract length distribution within the rosy locus of Drosophila melanogaster. Genetics. 1994;137:1019–1026. doi: 10.1093/genetics/137.4.1019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hudson RR, Kaplan NL. Deleterious background selection with recombination. Genetics. 1995;141:1605–1617. doi: 10.1093/genetics/141.4.1605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Innan H, Stephan W. Selection intensity against deleterious mutations in RNA secondary structures and rate of compensatory nucleotide substitutions. Genetics. 2001;159:389–399. doi: 10.1093/genetics/159.1.389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jensen JD, Kim Y, DuMont VB, Aquadro CF, Bustamante CD. Distinguishing between selective sweeps and demography using DNA polymorphism data. Genetics. 2005;170:1401–1410. doi: 10.1534/genetics.104.038224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaplan NL, Hudson RR, Langley CH. The “hitchhiking effect” revisited. Genetics. 1989;123:887–899. doi: 10.1093/genetics/123.4.887. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kern AD, Jones CD, Begun DJ. Genomic effects of nucleotide substitutions in Drosophila simulans. Genetics. 2002;162:1753–1761. doi: 10.1093/genetics/162.4.1753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim Y, Stephan W. Joint effects of genetic hitchhiking and background selection on neutral variation. Genetics. 2000;155:1415–1427. doi: 10.1093/genetics/155.3.1415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim Y, Stephan W. Detecting a local signature of genetic hitchhiking along a recombining chromosome. Genetics. 2002;160:765–777. doi: 10.1093/genetics/160.2.765. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim Y, Stephan W. Selective sweeps in the presence of interference among partially linked loci. Genetics. 2003;164:389–398. doi: 10.1093/genetics/164.1.389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kousathanas A, Keightley PD. A comparison of models to infer the distribution of fitness effects of new mutations. Genetics. 2013;193:1197–1208. doi: 10.1534/genetics.112.148023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kulathinal RJ, Bettencourt BR, Hartl DL. Compensated deleterious mutations in insect genomes. Science. 2004;306:1553–1554. doi: 10.1126/science.1100522. [DOI] [PubMed] [Google Scholar]
- Kumar S, Nussinov R. Close-range electrostatic interactions in proteins. Chembiochem. 2002;3:604–617. doi: 10.1002/1439-7633(20020703)3:7<604::AID-CBIC604>3.0.CO;2-X. [DOI] [PubMed] [Google Scholar]
- Lachaise D, Carious ML, David JR, Lemeunier F, Tsacas L, Ashburner M. Historical biogeography of the Drosophila melanogaster species subgroup. Evol Biol. 1988;22:159–225. [Google Scholar]
- Langley CH, Stevens K, Cardeno C, Lee YCG, Schrider DR, Pool JE, Langley SA, Suarez C, Corbett-Detig RB, Kolaczkowski B, et al. Genomic variation in natural populations of Drosophila melanogaster. Genetics. 2012;192:533–598. doi: 10.1534/genetics.112.142018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee YCG, Reinhardt J. Widespread polymorphism in the positions of stop codons in Drosophila melanogaster. Genome Biol Evol. 2012;4:533–549. doi: 10.1093/gbe/evr113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Loewe L, Charlesworth B. Background selection in single genes may explain patterns of codon bias. Genetics. 2007;175:1381–1393. doi: 10.1534/genetics.106.065557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Macpherson JM, Sella G, Davis JC, Petrov DA. Genomewide spatial correspondence between nonsynonymous divergence and neutral polymorphism reveals extensive adaptation in Drosophila. Genetics. 2007;177:2083–2099. doi: 10.1534/genetics.107.080226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marais G. Biased gene conversion: implications for genome and sex evolution. Trends Genet. 2003;19:330–338. doi: 10.1016/S0168-9525(03)00116-1. [DOI] [PubMed] [Google Scholar]
- Maruyama T. The age of an allele in a finite population. Genet Res. 1974;23:137–143. doi: 10.1017/s0016672300014750. [DOI] [PubMed] [Google Scholar]
- Maynard Smith J, Haigh J. The hitch-hiking effect of a favourable gene. Genet Res. 1974;23:23–35. [PubMed] [Google Scholar]
- McDonald JH, Kreitman M. Adaptive protein evolution at the Adh locus in Drosophila. Nature. 1991;351:652–654. doi: 10.1038/351652a0. [DOI] [PubMed] [Google Scholar]
- McVean GAT, Vieira J. Inferring parameters of mutation, selection and demography from patterns of synonymous site evolution in Drosophila. Genetics. 2001;157:245–257. doi: 10.1093/genetics/157.1.245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Messer PW, Petrov DA. Population genomics of rapid adaptation by soft selective sweeps. Trends Ecol Evol. 2013;28:659–669. doi: 10.1016/j.tree.2013.08.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miller DE, Takeo S, Nandanan K, Paulson A, Gogol MM, Noll AC, Perera AG, Walton KN, Gilliland WD, Li H, et al. A whole-chromosome analysis of meiotic recombination in Drosophila melanogaster. G3. 2012;2:249–260. doi: 10.1534/g3.111.001396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mustonen V, Lässig M. Adaptations to fluctuating selection in Drosophila. Proc Natl Acad Sci U S A. 2007;104:2277–2282. doi: 10.1073/pnas.0607105104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nagylaki T. Evolution of a finite population under gene conversion. Proc Natl Acad Sci U S A. 1983a;80:6278–6281. doi: 10.1073/pnas.80.20.6278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nagylaki T. Evolution of a large population under gene conversion. Proc Natl Acad Sci U S A. 1983b;80:5941–5945. doi: 10.1073/pnas.80.19.5941. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neher E. How frequent are correlated changes in families of protein sequences? Proc Natl Acad Sci U S A. 1994;91:98–102. doi: 10.1073/pnas.91.1.98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nei M. Molecular evolutionary genetics. New York: Columbia University Press; 1987. [Google Scholar]
- Nielsen R, Bauer DuMont VL, Hubisz MJ, Aquadro CF. Maximum likelihood estimation of ancestral codon usage bias parameters in Drosophila. Mol Biol Evol. 2007;24:228–235. doi: 10.1093/molbev/msl146. [DOI] [PubMed] [Google Scholar]
- Ometto L, Glinka S, De Lorenzo D, Stephan W. Inferring the effects of demography and selection on Drosophila melanogaster populations from a chromosome-wide scan of DNA variation. Mol Biol Evol. 2005;22:2119–2130. doi: 10.1093/molbev/msi207. [DOI] [PubMed] [Google Scholar]
- Orr HA. The genetic theory of adaptation: a brief history. Nat Rev Genet. 2005;6:119–127. doi: 10.1038/nrg1523. [DOI] [PubMed] [Google Scholar]
- Orr HA. Fitness and its role in evolutionary genetics. Nat Rev Genet. 2009;10:531–539. doi: 10.1038/nrg2603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Orr HA, Betancourt AJ. Haldane’s Sieve and adaptation from the standing genetic variation. Genetics. 2001;157:875–884. doi: 10.1093/genetics/157.2.875. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Orr HA, Coyne JA. The genetics of adaptation: a reassessment. Am Nat. 1992;140:725–742. doi: 10.1086/285437. [DOI] [PubMed] [Google Scholar]
- Osada N, Akashi H. Mitochondrial–nuclear interactions and accelerated compensatory evolution: evidence from the primate cytochrome C oxidase complex. Mol Biol Evol. 2012;29:337–346. doi: 10.1093/molbev/msr211. [DOI] [PubMed] [Google Scholar]
- Pace CN. Polar group burial contributes more to protein stability than nonpolar group burial. Biochemistry. 2001;40:310–313. doi: 10.1021/bi001574j. [DOI] [PubMed] [Google Scholar]
- Pool JE, Aquadro CF. History and structure of sub-Saharan populations of Drosophila melanogaster. Genetics. 2006;174:915–929. doi: 10.1534/genetics.106.058693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prezeworski M, Coop G, Wall JD. The signature of positive selection on standing genetic variation. Evolution. 2005;59:2312–2323. [PubMed] [Google Scholar]
- Przeworski M. The signature of positive selection at randomly chosen loci. Genetics. 2002;160:1179–1189. doi: 10.1093/genetics/160.3.1179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Radwan J, Babik W. The genomics of adaptation. Proc Biol Sci. 2012;279:5024–5028. doi: 10.1098/rspb.2012.2322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sattath S, Elyashiv E, Kolodny O, Rinott Y, Sella G. Pervasive adaptive protein evolution apparent in diversity patterns around amino acid substitutions in Drosophila simulans. PLoS Genet. 2011;7:e1001302. doi: 10.1371/journal.pgen.1001302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schneider A, Charlesworth B, Eyre-Walker A, Keightley PD. A method for inferring the rate of occurrence and fitness effects of advantageous mutations. Genetics. 2011;189:1427–1437. doi: 10.1534/genetics.111.131730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sella G, Petrov DA, Przeworski M, Andolfatto P. Pervasive natural selection in the Drosophila genome? PLoS Genet. 2009;5:e1000495. doi: 10.1371/journal.pgen.1000495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shapiro JA, Huang W, Zhang C, Hubisz MJ, Lu J, Turissini DA, Fang S, Wang H, Hudson RR, Nielsen R, et al. Adaptive genic evolution in the Drosophila genomes. Proc Natl Acad Sci U S A. 2007;104:2271–2276. doi: 10.1073/pnas.0610385104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith NGC, Eyre-Walker A. Adaptive protein evolution in Drosophila. Nature. 2002;415:1022–1024. doi: 10.1038/4151022a. [DOI] [PubMed] [Google Scholar]
- Stephan W. Genetic hitchhiking versus background selection: the controversy and its implications. Philos Trans R Soc Lond B Biol Sci. 2010;365:1245–1253. doi: 10.1098/rstb.2009.0278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stephan W, Wiehe THE, Lenz MW. The effect of strongly selected substitutions on neutral polymorphism: analytical results based on diffusion theory. Theor Popul Biol. 1992;41:237–254. [Google Scholar]
- Tajima F. Evolutionary relationship of DNA sequences in finite populations. Genetics. 1983;105:437–460. doi: 10.1093/genetics/105.2.437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tajima F. Relationship between DNA polymorphism and fixation time. Genetics. 1990;125:447–454. doi: 10.1093/genetics/125.2.447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takahata N, Ishii K, Matsuda H. Effect of temporal fluctuation of selection coefficient on gene frequency in a population. Proc Natl Acad Sci U S A. 1975;72:4541–4545. doi: 10.1073/pnas.72.11.4541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takano K, Yamagata Y, Funahashi J, Hioki Y, Kuramitsu S, Yutani K. Contribution of intra- and intermolecular hydrogen bonds to the conformational stability of human lysozyme. Biochemistry. 1999;38:12698–12708. doi: 10.1021/bi9910169. [DOI] [PubMed] [Google Scholar]
- Thornton KR, Jensen JD, Becquet C, Andolfatto P. Progress and prospects in mapping recent selection in the genome. Heredity. 2007;98:340–348. doi: 10.1038/sj.hdy.6800967. [DOI] [PubMed] [Google Scholar]
- Veuille M, Baudry E, Cobb M, Derome N, Gravot E. Historicity and the population genetics of Drosophila Melanogaster and D. Simulans. Genetica. 2004;120:61–70. doi: 10.1023/b:gene.0000017630.69020.32. [DOI] [PubMed] [Google Scholar]
- Wall JD, Andolfatto P, Przeworski M. Testing models of selection and demography in Drosophila simulans. Genetics. 2002;162:203–216. doi: 10.1093/genetics/162.1.203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Welch JJ. Estimating the genomewide rate of adaptive protein evolution in Drosophila. Genetics. 2006;173:821–837. doi: 10.1534/genetics.106.056911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang Z. PAML 4: phylogenetic analysis by maximum likelihood. Mol Biol Evol. 2007;24:1586–1591. doi: 10.1093/molbev/msm088. [DOI] [PubMed] [Google Scholar]
- Yeang C-H, Haussler D. Detecting coevolution in and among protein domains. PLoS Comput Biol. 2007;3:e211. doi: 10.1371/journal.pcbi.0030211. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.