

Abstract
Bioactive compounds are considered safe and have been shown to alter genetic and epigenetic profiles of tumor cells. However, many of these changes have been reported at molecular concentrations higher than physiologically achievable levels. We investigated the role of the combinatorial effects of epigallocatechin gallate (EGCG), a predominant polyphenol in green tea, and sodium butyrate (NaB), a dietary microbial fermentation product of fiber, in the regulation of survivin, which is an overexpressed anti-apoptotic protein in colon cancer cells. For the first time, our study showed that the combination treatment induced apoptosis and cell cycle arrest in RKO, HCT-116 and HT-29 colorectal cancer cells. This was found to be regulated by the decrease in HDAC1, DNMT1, survivin and HDAC activity in all three cell lines. A G2/M arrest was observed for RKO and HCT-116 cells, and G1 arrest for HT-29 colorectal cancer cells for combinatorial treatment. Further experimentation of the molecular mechanisms in RKO colorectal cancer cells revealed a p53-dependent induction of p21 and an increase in nuclear factor kappa B (NF-κB)-p65. An increase in double strand breaks as determined by gamma-H2A histone family member X (γ-H2AX) protein levels and induction of histone H3 hyperacetylation was also observed with combination treatment. Further, we observed a decrease in global CpG methylation. Taken together, these findings suggest that at low and physiologically achievable concentrations, combinatorial EGCG and NaB are effective in promoting apoptosis, inducing cell cycle arrest and DNA-damage in colorectal cancer cells.
Keywords: Epigenetics, Colon cancer, Sodium Butyrate, Epigallocatechine gallate, Methylation, Histone deacetylases
Introduction
Survivin, a member of the inhibitor of apoptosis protein family, plays a bifunctional role, as an anti-apoptotic protein and also as a regulator of cell cycle progression [1, 2]. It associates with the mitotic spindle during the cell cycle and serves as a check point for correct association of the spindle with chromosomes in metaphase [2]. In colorectal cancers (CRC), survivin is overexpressed but its expression in normal adult tissue is undetectable [3]. The expression of survivin has been linked to poor survival, recurrence rate and death due to colorectal cancers [4]. Chemoregulatory expression of this protein is therefore a promising target for cure.
The incidence rate of colon cancer is lower in Asian countries where the diet is predominantly rich in vegetables and fruits [5]. The constituents of these dietary foods provides for a healthy colonic environment. Dietary fiber, an important dietary constituent, ensures that potential carcinogens are removed from the colon and the microbiota within the colon converts the fiber into short chain fatty acids (SCFA) by the process of fermentation [6]. These short chain fatty acids are a major source of energy for the colon cells. Of the SCFAs, butyrate is the predominant energy providing source [7] and is a natural epigenetic regulator functioning as an inhibitor of histone deacetylases (HDACs) [8]. Sodium butyrate (NaB) can induce cell differentiation, apoptosis and histone hyperacetylation [8–10] and these tumor inhibitory properties of butyrate can be exploited as part of a treatment for colorectal cancers.
Another dietary epigenetic molecule, epigallocatechin gallate (EGCG), is a predominant constituent of green tea polyphenols, and regulates epigenetic changes by altering methylation profiles of genes through its DNA methyltransferase 1 (DNMT1) inhibitory activity [11]. Combination therapies incorporating EGCG with other bioactive molecules may be very effective in numerous cancers [12]. However, many of these studies employ high concentrations of the compound that may not be achievable in vivo. Our rational is that when two effective compounds with potent epigenetic properties are used the combined epigenetic effects may be more effective in reducing survivin expression, an upregulated anti-apoptotic molecule in colorectal cancers (CRC), and that this may allow lower concentrations of the compounds for therapy. Studies in various other cancer cell lines have shown that EGCG and NaB can effectively inhibit survivin independently, albeit at higher concentrations [13, 14]. However, the combination effects of these compounds on colon cells, where the availability of the molecules are at the highest physiological levels, are not known.
In our study, we treated RKO, HCT-116 and HT-29 colorectal cancer cells at physiologically achievable concentrations of EGCG and NaB (10 μM and 5 mM, respectively) [15–18] and the combined effects of these epigenetic regulators were observed in terms of survivin down-regulation. RKO and HCT-116 are colorectal carcinoma cell lines and are genetically similar. HT-29 is not genetically similar to RKO or HCT-116 cell lines and is an adenocarcinoma cell line. We sought to determine if the compounds were effective against cell lines that were genetically similar or different, and if p53 would govern the molecular changes observed in the study. We also assessed p21, an important cell cycle regulatory protein that has been reported to regulate survivin expression in other cancer cell types [19, 20]. We asked if the combined therapy of EGCG and NaB could have a greater effect at inducing p21 expression with the concomitant down-regulation of survivin in colon cancer cells, at lower molecular concentrations. NaB alone is potent enough to induce DNA-damage, and when combined with EGCG this damage may be enhanced, stimulating cell cycle arrest in parallel with p21 induction and down-regulation of survivin. We found that the combination of EGCG and NaB arrested cells in the G2/M phase for both the RKO and HCT-116 colon cancer cells and a G1 arrest was observed in HT-29 cells. All cells had a decreased S phase. p21 induction was observed in the RKO colorectal cancer cell which was p53-dependent. Taken together this study provides a novel chemotherapeutic approach in the treatment of colorectal cancers at lower effective doses of natural molecules.
Materials and Methods
Cell culture
RKO (CRL-2577), HCT-116 (CCL-247) and HT-29 (HTB-38) colorectal cells were obtained from American Type Culture Collection (ATCC). RKO colorectal cells were cultured in DMEM 1X medium (Mediatech Inc, Manassas, VA, USA), HCT-116 and HT-29 were cultured in DMEM-F12 (Mediatech Inc, Manassas, VA, USA), and all cell cultures were supplemented with 10% fetal bovine serum (Atlanta Biologicals, Lawrenceville, GA, USA) and 1% penicillin/streptomycin (Mediatech, Herndon, VA, USA). The cells were cultured as per the manufacture’s protocol and were maintained in a humidified 5% CO2 incubator at 37°C. RKO, HCT-116 and HT-29 colorectal cancer cells were treated with 10 μM EGCG (Sigma, St. Louis, MO, USA) or 5 mM sodium butyrate (NaB) (Sigma, St. Louis, MO, USA) for 48 h. EGCG was prepared in DMSO with a stock concentration of 20 mg/ml and NaB was at a stock concentration of 100 mg/ml in sterile water. The concentration of DMSO in medium was less than 0.1% (v/v). Cells treated with DMSO served as a vehicle control. During treatments working solutions were freshly prepared and the medium was changed every 24 h with the freshly prepared compound solutions.
Cell viability assessment
Cell viability was determined by the 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT) assay after treatment with various concentrations of EGCG and NaB and selected concentration of the combined drugs. Approximately 1 × 104 RKO, HCT-116 and HT-29 colorectal cancer cells were seeded in each well in 96-well plates. Cells were treated as indicated after 24 h. At the end of each treatment the cells were washed twice with 100 μL PBS and 100 μL of media containing 10 μL of 1 mg/mL MTT (Sigma, St. Louis, MO, USA) was added to each well before incubation for 1 h at 37°C in a humidified 5% CO2 incubator. At the end of the incubation period, the medium was aspirated and 200 μL DMSO was added to each well to dissolve the formazan crystals. Dye absorbance in each well was measured at 490 nm with a reference wavelength at 620 nm. Cells treated only with media served as negative control and DMSO at a final concentration of 0.01% was used as experimental control.
Analysis of cell cycle progression
Propidium iodide (PI) staining-based flow cytometry cell cycle assay was used to analyze cell cycle distribution. Approximately 5 × 104 RKO, HCT-116 and HT-29 cells were plated in each well of 6-well plates with a 2 mL volume of medium for each well. Medium containing freshly dissolved EGCG or NaB was added 24 h later and changed daily. Harvesting of the cells was carried out at the indicated time points from 6-well plates by trypsinization. After washing with PBS, cells were fixed in 70% ethanol at 4°C overnight and centrifuged and washed with PBS the second day. Before analysis, incubation of cells was carried out in the dark for about 30 min in PBS containing 0.1% Triton X-100, 0.1% RNase, and 50 μg/mL PI. DNA contents in stained nuclei were then analyzed with flow cytometry.
Apoptosis analysis
Annexin V and PI double-staining-based flow cytometry apoptosis assay was used to determine the effect of EGCG and NaB treatment on apoptosis. Cells were harvested with trypsinization followed by washing with PBS and staining with Annexin V and PI (Invitrogen) in the binding buffer for 15 min in the dark. Subsequently, the cells were analyzed through flow cytometry.
Protein expression analysis
Western blotting was employed to assess protein expression. After cells were washed with PBS, protein lysates were obtained in RIPA lysis buffer containing protease inhibitors (Upstate Biotechnology, Charlottesville, VA, USA). Ten micrograms of protein sample was electrophoresed on 4–10% Tris glycine SDS-polyacrylamide gels (Invitrogen) and transferred to nitrocellulose membranes. Nuclear lysates were assessed for p21, p53 and NF-κB-p65. Total protein was used to assess HDAC1, DNMT1, DNMT3A and DNMT3B, survivin, anti-acetylated H3 and γ-H2AX. Total protein analysis instead of acid extraction was performed to determine anti-acetylated H3 and γ-H2AX levels as they have been determined in the similar manner as has been previously reported [21, 22]. Antibodies against p21 and anti-acetylated H3 (Millipore, CA, USA), HDAC1, DNMT1, NF-κB-p65 (Abcam), p53, survivin, DNMT3A, DNMT3B (Santacruz Biotechnology) and γ-H2AX (Cell Signaling) were used to probe the corresponding proteins. Briefly, blots were blocked in 5% Milk powder TBS-T solution for 30 min. Primary antibody was added in 1% Milk powder TBS-T solution for 1 h at room temperature with shaking; for survivin and γ-H2AX, incubation with primary antibody was carried out at 4°C overnight, the rest were performed at room temperature for 1 h. DNMT3A and DNMT3B and survivin were used at 1:100 dilution and the rest were used at 1:1000 dilution. Blots were washed for 30 min at 10 min each wash. Secondary antibody at 1:1000 dilution was added for 1 h at room temperature. Blots were washed with TBS-T for 30 min at 10 min each wash and immunoreactive bands were visualized with the enhanced chemiluminescence detection system (Millipore) using the BIORAD chemidoc XRS image. Proteins were identified based on their molecular weights compared to the standard that was run in parallel and transferred to the blot. The western blots were conducted in triplicates and densitometric analysis of the bands were performed using myImageAnalysis1.1 (Thermo Scientific).
Real-time quantitative PCR
Total cellular RNA was isolated using an RNeasy mini kit (Qiagen, Valencia, CA, USA) according to the manufacturer’s instructions. Total RNA (100 ng) were reverse-transcribed into cDNA using the iScript cDNA synthesis kit (Bio-rad, Hercules, CA, USA). The PCR primer sets that were used are the following: 5′-ACCACAGTCCATGCCATCAC-3′ (F), 5′-TCCACCACCCTGTTGCTGTA-3′ (R) for GAPDH; 5′-CACTCCAAACGCCGGCTGATCTTC-3′ (F), 5′ TGTAGAGCGGGCCTTTGAGGCCCTC-3′ (R) for p21; 5′-ACCGCTTCTACTTCCTCGAGGCCTA-3′ (F) 5′-GTTGCAGTCCTCTGTGAACACTGTGG-3′ (R) for DNMT1; 5′-GACGGGGATGTTGGAAATTA-3′ (F), 5′-CATCTCCTCAGCATTGGCTT-3′ (R) for HDAC1; 5′-ATGGACGATCTGTTTCCCCT-3′ (F), 5′-CGGTTTACTCGGCAGATCTT-3′ (R) for NF-κB-p65; and 5′-GCATGGGTGCCCCGACGTTG-3′ (F), 5′-GCTCCGGCCAGAGGCCTCAA-3′ (R) for survivin. GAPDH was used as an internal loading control. Real-time quantitative PCR was conducted in a CFX96 Real-Time PCR System (Biorad) using SYBR Green detection system (Bio-rad, Hercules, CA, USA). The delta-delta CT method was used to determine the relative level of gene expression. Fold change in gene expression was determined by the formula 2−ΔΔCT where Δ Δ CT = Δ CT(target)normalized − Δ CT(Control)normalized; Δ CT(target) normalized = CT target (gene of interest) −CT target (GADPH) and Δ CT(Control) normalized = CT control (gene of interest) – CT control (GADPH).
Colonogenic assays
The cells were seeded at a density of 100 cells per well in a 24-well plate and allowed to recover for a day prior to the experiment. EGCG and NaB were added to the wells at the appropriate concentrations. After the 48 h time period, the media was removed and fresh media was added and the plates were incubated at 37°C at 5% CO2 for a week. The wells were washed with 1X PBS and the colonies were fixed using glacial acetic acid and methanol at the ratio 1:7 for 5 min. Crystal violet (0.5%) in PBS was added and kept for 30 min. The wells were gently washed and allowed to air dry. Colonogenic assays were performed in two ways: 1) Cells were first treated to the compounds and then colony formation potential was assessed and 2) the plated cells were allowed to form colonies prior to treatment and then exposed to treatment to determine colony reduction potential. Percent values were obtained by comparing to control.
HDAC activity and CpG percent methylation
The epigenetic changes were assessed using the HDAC and Methylflash Epigentek kits as per the manufacture’s protocol. For HDAC assays 20 μg/μl of nuclear protein was used and for percent methylation 100 ng of genomic DNA was used. The absorbance was measured at 450 nm using the Bio rad 680 plate reader. Activities for each were calculated based on manufacturer’s instructions.
Chromatin Immunoprecipitation assay (ChIP)
RKO cells were seeded in a 10 cm dish and were subjected to the respective treatments for 48 h. After treatment the cells were fixed with 37% formaldehyde and then subjected to enzymatic shearing as per the manufacture’s protocol (ChIP-IT express enzymatic kit, Active Motif). p53 antibody (3 μg, mouse monoclonal, ab26 Abcam) was used to pull down the DNA protein complex. After reversing the crosslinks, the p53-enriched DNA and input DNA was purified and then subjected to PCR amplification using primers for p21 promoter sequences that amplify the region near the p53-binding site: 5′-GCACTCTTGTCCCCCAG-3′ and 3′-TCTATGCCAGAGCTCAACAT-5′. The amplified product was electrophoresed on a 3% agarose gel and the density of the bands was measured using the myImageAnalysis software (ThermoScientific). The density of p53-enriched samples was normalized to the input sample.
Statistical analyses
Statistical significance among treatments was evaluated using one-way ANOVA followed by Tukey test. *P < 0.05 and **P < 0.01 was considered significant.
Results
EGCG and NaB suppresses RKO colorectal cancer cell proliferation
As an individual compound, EGCG was less effective than NaB at inhibiting the cell proliferation of RKO colorectal cells (10% at 10 μM) at lower concentrations (Figure 1A). The IC50 for EGCG was about 50 μM. However, the bioavailability of EGCG is relatively poor at 0.1 μM plasma levels although it is delivered at 1–20 μM in the colon based on the dose of green tea consumed [23, 24]. NaB was more effective at inhibiting the cells. At the 5 mM NaB dose, which is considered achievable in the colon [25], about 35% of the cells were inhibited. It is notable that the combined effect of 10 μM EGCG and 5 mM NaB resulted in 50% cellular inhibition suggestive of an additive effect at this IC50 value for the combined treatment. The additive effect of the combined treatment on HCT-116 and HT-29 colorectal cells was significant at higher IC values than at IC50. For consistency and given that at these doses had an additive effect, the same concentrations were used for HCT-116 and HT-29 cells (Figure 1B). All three cell lines chosen are positive for the CpG island methylator phenotype, indicative of a higher degree of genomic methylation in these cells [26]. In addition, RKO and HCT-116 are p53 wild-type (wt) and are colorectal carcinoma cell type. HT-29 cells carry a mutation in the codon 273 in p53 and are adenocarcinoma cell types [26]. We wanted to test the applicability and effectiveness of the combination in cell lines having similar and different genetic characteristics. Morphological changes associated with the respective treatments were observed at 100× using a Leica DM 750 phase contrast microscope (Figure 1C). As compared to non-treated control, the RKO cells in the combination appeared to gain a more spindle, flattened and circular characteristic. However, with EGCG alone a very slight morphological change was observed which was consistent with the efficacy to the cancer cells at the concentration of 10 μM. Prominent morphological changes were visible with 5 mM NaB. The combination of EGCG and NaB resulted in pronounced changes of a larger number of circular shaped cells from the normal epithelial-like morphology, indicative of the enhanced effect of EGCG with NaB (Figure 1C). More pronounced morphological changes were also observed for HCT-116 and HT-29 colon cells with NaB treatment only and the combination treatment as compared to the control and EGCG treatment only.
Figure 1. EGCG and NaB inhibits the proliferation of RKO, HCT-116 and HT-29 colorectal cancer cells.



The effective dose of the combination for EGCG and NaB was determined at IC50 for the combined effect (A). EGCG and NaB independently inhibit the proliferation of RKO colon cancer cells, with NaB having a greater effect at physiological cocnentrations. The combination effect of EGCG and NaB at physiological available doses (10 μM and 5 mM EGCG and NaB, respectively) produced a significant additive effect in terms of inhibiton of cell proliferation in RKO colon cancer cells (A). Significant additive effects was also observed for HCT-116 and HT-29 colon cancer cells at the same doses, albeit at higher IC values for the combination (B). Morphology of RKO, HCT-116 and HT-29 colon cancer cells after various treatments is shown at 100× magnification (C). * < 0.05, ** < 0.01
EGCG and NaB promotes apoptosis
Induction of apoptosis is one of the major pathways through which chemotherapeutic agents inhibit tumor proliferation. We therefore examined the apoptotic potential of the combination treatment of RKO, HCT-116 and HT-29 colorectal cancer cells. We found that the combination of 10 μM EGCG and 5 mM NaB induced significant apoptosis (P < 0.01) in comparison to the individual treatments at 48 h (Figure 2). However, the apoptotic induction was not high enough to be considered the sole mechanism of inhibition of cell proliferation. We therefore believe that a co-mechanism in addition to apoptosis may be involved in the observed inhibition of cell proliferation.
Figure 2. EGCG and NaB combination can induce apoptosis of colon cancer cells.

Cells were treated with 10 μM EGCG and 5 mM NaB and after a 48 h time period the percent apoptotic cells were analyzed by flow cytometry. Apoptosis cells include both early apoptosis and late apoptosis. A significant increase in the percent apoptotic cells was observed for the combination treatment as compared to the individual treatments. (* < 0.05, ** < 0.01). The experiment was repeated three times.
EGCG and NaB arrest RKO, HCT-116 colorectal cells predominantly in the G2/M phase and HT-29 colorectal cells in the G1 phase
Cell cycle progression analysis revealed a shift from S to the G2/M phase (Figure 3). The cells were predominantly in the S phase for EGCG at the 10 μM concentration. However, with NaB treatment at 5 mM a G2/M arrest was observed for RKO cells and in the combination treatment was arrested in the G1 and G2/M phase. In HCT-116 cells a significant G2/M arrest was observed and in HT-29 cells a significant G1 arrest was observed (P < 0.01) as compared to the controls (Figure 3). In comparing all three cell lines a reduction in S-phase was observed indicative of a possible mechanism of preventing cell proliferation that may be associated with DNA damage.
Figure 3. EGCG and NaB in combination can induce cell cycle arrest in colon cancer cells.

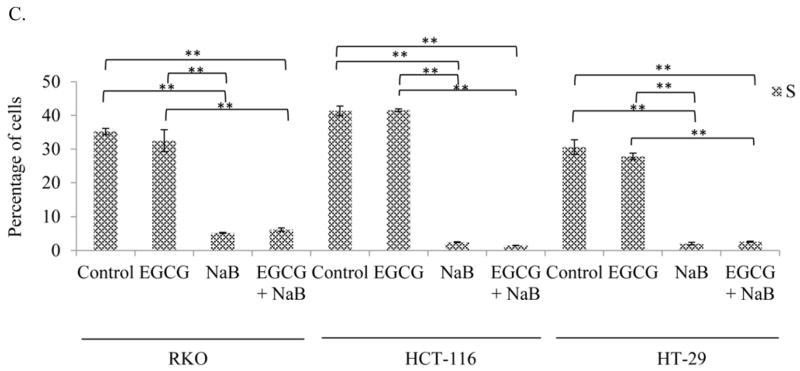
A shift towards the G2/M phase was observed in the combination treatment (10 μM EGCG and 5 mM NaB) as compared to controls for both RKO and HCT-116 colon cancer cells and the change was significant. A significant G1 arrest was observed for HT-29 colon cancer cells. Therefore, depending on the cell line under study the combination treatment was able to arrest cells in G1 and G2/M phases. * < 0.05, ** < 0.01. The experiment was repeated two times and each point indicates the mean ± SEM.
EGCG and NaB effectively inhibit colony formation in RKO colorectal cancer cells
From our apoptotic and cell cycle arrest results we next sought to determine if the combination was effective in inhibiting the colony forming potential of RKO colorectal cancer cells. Our results indicate that the combination was significantly more potent in terms of reducing the number of colonies than individual treatments. Both types of colonogenic assay showed that the combination was successful in inhibiting/reducing RKO cellular tumor forming potential (Figures 4A, 4B and 4C). The combination significantly inhibited colony formation by 80%, while EGCG and NaB administered singly were about 20% and 50% effective, respectively. These changes were found to be significant at P < 0.01. Thus, EGCG significantly enhances the effect of NaB in reducing colony formation. Although slightly different efficacies were observed with respect to Figure 4A, the reverse colonogenic assay showed similar trends in colony reduction potential. EGCG reduced the number of colonies formed by 10%, NaB by 40% and the combination by 70%. These changes were significant with respect to the control (P < 0.01). Also, the combination was significantly different from the individual treatments alone (P < 0.01). These data indicate that the combination is potent in inhibiting as well as reducing colonies formed, activities that are important to effective chemotherapeutic strategies.
Figure 4. Colony formation potential of RKO cells.


Colony forming potential of RKO cells was assessed by treating the cells with 10 μM EGCG and 5 mM NaB for a 48 h time period and colonies formed in the absence of the compounds after a week of incubation were counted. Values are represented as percent control ± SEM of two independent experiments performed in triplicates. Representative photographs are shown from the experiments. (A). Colonies that had greater than 50 cells were counted. A significant decrease in colony formation was observed for the combination treatment as compared to the individual treatments. Morphology and sizes of the colonies formed are shown in (B). Reverse colonogenic assay was also performed to determine compound effectiveness in reducing colonies once formed (C).
EGCG and NaB increases p21, NF-κB-p65, HDAC1; and decreases DNMT1 and survivin in RKO colorectal cancer cells
Real-time PCR analysis of RKO cells revealed that p21 levels increased for the individual treatments and was highly induced with the combination treatment. EGCG was effective in enhancing NaB induction of p21 (Figure 5A). We also determined the effect of the combination treatment on survivin expression as it is transcriptionally regulated by p21. Survivin mRNA levels decreased (Figure 5A) in response to the combination treatment and was significant (P < 0.01) as compared to the control and individual treatment of 10 μM EGCG and 5 mM NaB. HDAC inhibitors are known to transactivate NF-κB and NF-κB is associated with p21 dependent G2/M arrest. We therefore determined the effect of the treatments on NF-κB-p65. Ours is the first study to report that NF-κB-p65 levels were significantly up-regulated for NaB alone and for the combination treatments of EGCG and NaB (Figure 5B). However, the combination was lower than NaB treatment but was significantly higher than EGCG alone (P < 0.01). We examined the effect of the combination on DNMT1 and HDAC1 levels and found that DNMT1 levels decreased for the combination treatment, whereas HDAC1 levels increased (Figure 5C). Findings from other studies point to the existence of a regulatory feedback mechanism that modulates HDAC levels, which was also observed in this study [27].
Figure 5. Effect of EGCG and NaB on the mRNA expression of specific epigenetic, cell cycle and cell proliferation modulators in RKO colon cancer cells.


RKO colorectal cancer cells were treated to 10 μM EGCG and 5 mM NaB and after a 48 h time period RNA was extracted. Real time assessment of mRNA expression of the various genes revealed that DNMT1 and survivin expression decreased significantly for the combination treatment as compared to EGCG and NaB alone (A, B). EGCG is a known DNMT1 inhibitor although its effects are more pronounced at the protein than mRNA level. However, HDAC1, p21, and NF-κB-p65 (A, B, C) increased several fold and the increase was significant when compared to the control and individual treatments. A significant increase in NF-κB-p65 was observed for both the NaB alone and the combination treatment. However, the combination treatment was lower as compared to the NaB alone treatment, and significant and relatively higher when compared to EGCG and the control. Data are in triplicates from three independent experiments. * < 0.05, ** < 0.01
EGCG and NaB inhibits HDAC1, DNMT1 and survivin in all the three colorectal cancer cells tested
EGCG and NaB are epigenetic modulators and therefore we assessed the levels of DNMT1 and HDAC1 for the individual treatments and the combination treatment in all three cell lines. Epigenetic proteins HDAC1 and DNMT1 levels were significantly reduced in all three cell lines for the combination indicative of a combinatorial epigenetic effect of the compounds under question (Figures 6A and 6B, respectively). EGCG directly inhibits the catalytic site of DNMT1 via its gallic acid moiety [11]. Sodium butyrate has been shown to decrease DNMT1 levels in breast and prostate cancer cells via proteasomal degradation and may be a possible mechanism of the inhibition seen in our study [28]. The combination of EGCG and NaB in significantly reducing DNMT1 levels may be due to the combination of inhibiting the catalytic site along with proteasomal degradation.
Figure 6. Effect of the combinatorial compounds on the expression of HDAC1, DNMT1 and survivin in RKO, HCT-116 and HT-29 colorectal cancer cells.


The combination treatment was significantly effective in down-regulating the expression of HDAC1, DNMT1 and survivin in all the three colon cancer cells tested (A, B, C, respectively). The western blot was performed in triplicate and densitometric analysis of the bands was performed. The data represent values normalized to β-actin and then computed as relative to the control. A representative western is shown as an inset in the graph for each protein type tested. C = control; E = EGCG; N = NaB and E + N = EGCG + NaB.
Survivin is highly expressed in colorectal cancers and we therefore determined the effect of the combination in regulating survivin expression. Survivin expression was significantly inhibited with the combination treatment as compared to the control (P < 0.01) in all three cell lines (Figure 6C). In RKO cells the changes in survivin expression was significant in the combination as compared to the individual treatments (Figure 6C).
EGCG and NaB activates nuclear p53, p21, NF-κB-p65; activates γ-H2AX and anti-acetylated H3; inhibits DNMT3A and DNMT3B in total protein assessed in RKO colorectal cells
Densitometric analysis of p53, p21, and NF-κB-p65 from nuclear protein revealed an upregulation of the proteins in the combination treatment and was significant in comparison to the control (P < 0.01) (Figure 7A). NF-κB-p65 induction may be linked to DNA damage and therefore we assessed the levels of γ-H2AX, the expression of which indicates DNA damage. Our results showed an induction of the protein which was significant as compared to the control (P < 0.05) (Figure 7B). Sodium butyrate is a HDAC inhibitor and therefore we assessed at the acetylation status of histone H3 and found a significant increase in the combination treatment as compared to the control. A significant (P < 0.05) decrease in DNMT3A and DNMT3B levels was also observed in the combination treatment as compared to the controls (Figure 7C).
Figure 7. Effect of the combinatorial compounds on the expression of p21, p53 and NF-κB-p65 from nuclear lysates; γ-H2AX, anti-acetylated H3, DNMT3A and DNMT3B from total protein lysates, in RKO colon cancer cells.


Nuclear proteins were analyzed in triplicates for p21, p53, and NF-κB-p65 in RKO colon cancer cells (Figure 7A) and total protein in triplicates was assessed for γ-H2AX and anti-acetylated H3 levels (Figure 7B). At 10 μM EGCG and 5 mM NaB combination, p21 levels increased in RKO colorectal cancer cells. An induction in p53 expression was also observed in the combination treatment. Thus, EGCG may enhance the effect of NaB by inducing p53 expression. NF-κB-p65 levels increased in the combination and NaB treatment when compared to the control and EGCG treatment. This increase may be associated with the double-strand breaks (DSBs) indicated by increased γ-H2AX levels (7B) and G2/M arrest as was found in the study. De novo methylation-associated epigenetic proteins DNMT3A and DNMT 3B levels also decreased for the combination and this was tested only in RKO colon cancer cells (Figure 7C). An increase in anti-acetylated H3 levels was also observed with the combination (Figure 7B), an epigenetic change probably associated by the decrease in HDAC1 levels and HDAC activity. * < 0.05, ** < 0.01.
EGCG and NaB induces p21 through a p53-dependent mechanism in RKO colorectal cancer
Induction of both p21 mRNA and protein were observed in RKO colorectal cancer cells. To determine if the induction of p21 was p53-dependent we performed the ChIP assay using a monoclonal p53 antibody. ChIP-PCR analysis using primers specific to the p21 promoter region showed a significant increase in p21 levels for the combination (Figure 8). These results suggest a p53-dependent mechanism of p21 induction in RKO colorectal cancer cells.
Figure 8. p53-dependent induction of p21 by EGCG and NaB combination treatment in RKO colon cancer cells.

ChIP-PCR analysis of RKO cell p53-enrinched chromatin showed a significant upregulation of p21 in the combination treatment. Primers chosen were specific for the p21 promoter region. The increase in p53-enriched p21 levels in the combination is indicative of a p53-dependent mechanism of p21 induction in RKO colon cancer cells. The result is in agreement with the increase in mRNA and protein levels of p21 observed in the study. PCRs are from a single experiment repeated three times * < 0.05, ** < 0.01.
EGCG and NaB affect global DNA methylation and chromatin structure
Epigenetic mechanisms control gene expression through changes predominantly in DNA methylation and histone acetylation. The EGCG and NaB compounds that we used in the study are potent DNMT1 and HDAC1 inhibitors, respectively, and therefore, the epigenetic effects in the presence of two different types of epigenetic inhibitors were assessed. HDAC activity was determined for all three cell lines and the activity significantly decreased in the combination treatment as compared to the control for all of the three cell lines (P < 0.01) (Figure 8A). No significant changes in HAT activity was observed in the combined drug treatments (data not shown). Percent CpG methylation was assessed only for the RKO cell line as the level of three DNMT proteins were determined through western blots for this cell line only. Percent CpG methylation significantly decreased for the combination treatment of EGCG and NaB for RKO cells (P < 0.01) (Figure 8B). The increase in percent methylation for the NaB treatment may be attributed to an increase in DNMT3B levels as was observed with the protein densitometric analysis. The treatment of the cells that alter dominant epigenetic enzymes may account for the observed changes in percent methylation and HDAC activity.
Discussion
Sodium butyrate (NaB) is non-toxic and is produced naturally within the colon through microbial fermentation. The concentrations of NaB are effective in the millimolar range and 1 mM is considered low, 5 mM intermediate and 10 mM high [29]. We considered the intermediate concentration in terms of treatment as it was effective with 10 μM EGCG in reaching an IC50 value for the combination within 48 h of treatment for RKO cells and these concentrations are achievable in vivo. NaB has shown promise as a suitable chemotherapeutic agent due to its myriad anti-tumorigenic effects, including cell proliferation inhibition, induction of cell cycle arrest and differentiation, promotion of apoptosis, and even more significantly as a potent HDAC inhibitor inducing histone hyperacetylation and altering gene expression [6, 8, 30, 31]. Sodium butyrate has been shown to induce p21 expression that is a prominent cell cycle regulatory protein [32, 33]
In the current study we showed that the potent anti-cancer activities of EGCG and NaB in RKO, HCT-116 and HT-29 colorectal cancer cells. On the contrary, a study showed that EGCG interferes with butyrate-induced differentiation in colon cancer cells by preventing the cellular uptake of NaB [34]. However, in the study conducted, the dose chosen was based on the optimal values of the individual compounds and not on the combination of the compounds. Two given compounds can act differently at different dose combinations. From our MTT assays we showed that at 2 mM NaB and 10 μM EGCG there was no additive or synergistic effect observed in HT-29 cells which was the same cell line chosen for the previous study [34]. Therefore, dose appears to be crucial in determining chemotherapeutic effects. Based on the dose chosen we observed significant changes in morphology, apoptosis, cell cycle arrest and epigenetic proteins for the HT-29 cell line. In addition, survivin, an antiapoptotic protein highly expressed in colon cancers, was down-regulated in the HT-29 cell line with the combination as compared to EGCG alone. RKO and HCT-116 cell lines also showed significant morphological changes after the combination treatment. Epigenetically and genetically RKO and HCT-116 cells are more similar than HT-29. The presence of wt-p53 in RKO and HCT-116 cells may produce an improved response to the combination treatment than HT-29 cells that carry mutated p53. Reinduction of p53 perhaps may be important in encouraging apoptosis or cell-replicative inhibition which may be p53-dependent. The cell lines chosen were also CIMP positive, which is described in detail by the study [26]. Most of our analysis was performed in detail in the RKO wt-p53 colorectal cell line and where comparisons were necessary, we included the other cell lines as well.
From our in vitro colonogenic assays we were able to show that a combination of EGCG and NaB was significantly effective in inhibiting colony formation in RKO colorectal cancer cells by 80%, when many studies show that either EGCG or NaB acting alone require higher concentrations and time periods for the same effective inhibition. Clearly, the combination treatment at lower physiologically relevant doses is able to generate a significant chemotherapeutic effect suggestive of strong activity against colorectal cancer. Our study also showed that the combination treatment of EGCG and NaB was successful in inducing apoptosis in all three cell lines we treated but was in the range of 12–16% and therefore the apoptotic pathway may not be the only mode of tumor-inhibition by the compounds we evaluated.
In addition to the induction of apoptosis, we found that the EGCG and NaB combination induced cell cycle arrest predominantly in the G2/M phase for RKO and HCT-116 cells and G1 phase for HT-29 cells. NaB has been shown to exert cell cycle inhibitory effects in the G1 phase or G2/M phase based on the cell line under study [35]. In addition, down-regulation of survivin via p21 induction and p53 upregulation provides an alternative strategy of cell replicative inhibition through DNA damage. Our study for the first time showed that the combination therapy was successful in down-regulating the expression of survivin both at the mRNA and protein levels with p21 induction and G1 and G2/M arrest. We believe that this novel mechanism involved in the inhibition of colorectal cancer cells could be applicable to tumors that are resistant to apoptosis.
Genetic and epigenetic changes are instrumental in promoting neoplastic transformation in colorectal cancers [36]. The reversibility of these changes presents a suitable treatment strategy for colorectal cancers. Previous studies have shown that EGCC and NaB are potent DNMT1 and HDAC1 inhibitors, respectively, both of which can individually correct epigenetic observations in cancer cells [8, 11]. In our study, NaB was used and is a known HDAC1 inhibitor, and is the most extensively studied HDAC. Contradictory to the norm of gene and protein regulation, our study showed that the HDAC1 mRNA levels were significantly upregulated with the down-regulation of HDAC1 level for the NaB treatment alone and the combination treatment of EGCG and NaB. The upregulation of HDAC1 and the concomitant decrease in protein level may be attributed to the autoregulation of HDAC1 by HDAC inhibitors and could be cell-type specific [37]. HDAC1 has been found to be recruited to its own promoter thereby mediating its own repression by negative feedback loop inhibition [37]. In addition, HDACs are known to undergo several post-translational modifications, of which acetylation, phosphorylation, sumoylation and ubiquitination have been reported to occur with HDAC1 that can affect their activity and stability [38]. Taken together, the binding of NaB to the catalytic site of HDAC1 and possible post-translational effects on the protein may account for the decrease in HDAC1 levels despite the increase in HDAC1.
DNA methyltransferases and histone ennzymes are no longer considered independent epigenetic regulators but are thought to function in tandem or in cohesion to alter the epigenome. Global CpG methylation reponse to NaB treatment has been show to vary on the basis of the cell-type under study [39]. HeLa cells treated with 3 mM NaB for 48 h have shown an increase in global genomic methylation as compared to the control and in glioblastoma multiforme cells a hypomethylated state has been observed at 2 mM NaB for 48 h [40]. The findings from our results showed that percent CpG methylation for NaB increased in comparison to the control. A similar finding has been observed in transformed lung fibroblast cell lines in the presence of 5 mM NaB [39]. At this point we cannot determine the actual reasons for this observation but can infer from the data that the compensatory effects by de novo enzymes may account for this.
A significant decrease in HDAC activity was observed that likely explains the increase in global histone H3 acetylation levels in the combination treatment in comparison to the individual compounds. Histone hyperacetylation has been associated with inhibiting cell growth [41] and therefore we believe that the growth inhibition observed in RKO colorectal cancer cells may be due in part to this epigenetic change. Our study showed that in addition to its HDAC inhibitory activity, NaB was able to down-regulate DNMT1 expression as well and the expression further decreased with the combination treatment. Data from other studies support decreased DNMT1 expression, at both the protein and mRNA levels, after HDAC inhibitor treatment [28, 42].
The common expressed biomarker signaling DNA damage is γ-H2AX. Class I, II, and III HDACs have been implicated in the DNA damage response, homologous recombination (HR), and chromatin integrity [43]. HDAC1 have been shown to assist with the DNA damage response by recruiting DNA repair proteins to the site of damage. Butyrate and trichostatin A (TSA) have been show to exert defects in the repair process. Studies with cells lacking HDAC1 have been shown to be hypersensitive to DNA damaging agents and exhibit sustaining DNA damaging signaling reflective of defects in double-strand break (DSB) repair [44]. The results from our study showed a significant decrease in HDAC1 levels in the combination treatment for all three cell lines. Further, an assessment of γ-H2AX levels in RKO cell lines showed a significant increase in the combination treatment significantly as compared to the control and individual treatments. The increase in γ-H2AX levels with the concomitant decrease in HDAC1 levels may account for increased DNA damage with decreased cell-replicative capacity. Colonogenic assays performed in RKO colon cells showed that the combination significantly reduced colony formation and a growth reduction potential was also observed. These findings support the hypothesis that the combination treatment induces DNA damage, arrests the cells in the cell cycle and prevents an increase in cell numbers (data not shown).
HDAC1 has been shown to repress the transcriptional activity of NF-κB-p65. Treatment of the RKO colorectal cancer cells to HDAC1 inhibitor NaB in our study increased NF-κB-p65 mRNA and protein, and the expression was higher with NaB treatment and in the combination treatment as compared to the control. Upregulation of NF-κB has been observed in response to DNA damage. HDAC1 has been reported to directly interact with the p65-subunit of NF-κB and affect its transactivation functions [45]. The inhibition of HDAC1 by NaB in our studies may provide an explanation for the observed increase in nuclear NF-κB-p65 levels where the protein is in its activated state and is associated with DNA damage. Studies have reported that the increase in NF-κB in response to DNA damage may allow for repair of the damaged DNA [46]; however, the repair process is mediated through a host of various other proteins and includes HDAC1. We propose that HDAC1 is a corepressor of NF-κB-p65 and that HDAC1 can directly associate with the p65 subunit of NF-κB. Despite the increase in NF-κB-p65 levels, the decreased levels of HDAC1 may hinder repair functions arresting cells in the cell cycle at the G2/M phase through the induction of p21 as was specifically observed in RKO colorectal cells. Thus, EGCG and NaB through their epigenetic modulating effects, in addition to their potent roles as regulators of chromatin structure, and through direct or indirect corepressor or coactivator functions can mediate cellular responses by upregulating genes necessary for cell cycle arrest favoring a therapeutic outcome.
Conclusions
In summary, we have shown that EGCG and NaB with different predominant epigenetic functions are a potent combination with in vitro anti-colorectal cancer properties by suppressing cell proliferation, inducing cell cycle arrest and promoting apoptosis. These results can form the basis of suitable clinical therapies for colorectal cancer.
Figure 9. Effect of EGCG and NaB on HDAC activity in all the three colon cancer cells and percent CpG methylation in RKO colon cancer cells.

The combination treatment (10 μM EGCG and 5 mM NaB) was significantly effective in decreasing HDAC activity (* < 0.05, ** < 0.01) in all three cell lines tested (A). Percent CpG methylation was analyzed in RKO colon cancer cells and the decrease observed in the combination treatment was significant as compared to the individual treatment (B). The increase in percent methylation of CpGs with NaB may be due to the compensatory effects of de novo methylating enzymes such as DNMT3B. Data are from three independent experiments and represent mean ± SEM.
Highlights.
EGCG and NaB as a combination inhibits colorectal cancer cell proliferation.
The combination treatment induces DNA damage, G2/M and G1 arrest and apoptosis.
Survivin is effectively down-regulated by the combination treatment.
p21 and p53 expression is induced by the combination treatment.
Epigenetic proteins DNMT1 and HDAC1 are effectively down-regulated by the treatment.
Acknowledgments
This work was supported in part by grants from the NCI (CA 129415) and the American Institute for Cancer Research. We would like to thank Dr. Karyn Scissum Gunn for the use of her laboratory space and various facilities at Alabama State University, Montgomery AL. We appreciate the efforts of Dr. Yuanyuan Li for organizing and helping with experimental supplies when needed.
Abbreviations
- EGCG
epigallocatechingallate
- NaB
sodium butyrate
- HDAC
histone deacetylase
- HAT
histone acetyltransferase
- DMSO
dimethylsulfoxide
- DNMT1
DNA methyltransferase 1
- DNMT3A
DNA methyltransferase 3A
- γ-H2AX
gamma-H2A histone family member X
- NF-κB
nuclear factor kappa B
- MTT
3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazoliumbromide
- PI
propidiumiodide
- PCR
polymerase chain reaction
- ChIP
chromatin immunoprecipitation assay
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Contributor Information
Sabita N. Saldanha, Email: ssaldanha@alasu.edu.
Rishabh Kala, Email: sabivan@uab.edu.
Trygve O. Tollefsbol, Email: trygve@uab.edu.
References
- 1.Zangemeister-Wittke U, Simon HU. An IAP in action: the multiple roles of survivin in differentiation, immunity and malignancy. Cell Cycle. 2004;3:1121–1123. [PubMed] [Google Scholar]
- 2.Kallio MJ, Nieminen M, Eriksson JE. Human inhibitor of apoptosis protein (IAP) survivin participates in regulation of chromosome segregation and mitotic exit. FASEB J. 2001;15:2721–2723. doi: 10.1096/fj.01-0280fje. [DOI] [PubMed] [Google Scholar]
- 3.Alper M, Cukur S, Belenli O, Suna M. Evaluation of the immunohistochemical stain patterns of survivin, Bak and Bag-1 in colorectal cancers and comparison with polyps situated in the colon. Hepatogastroenterology. 2008;55:1269–1273. [PubMed] [Google Scholar]
- 4.Kawasaki H, Altieri DC, Lu CD, Toyoda M, Tenjo T, Tanigawa N. Inhibition of apoptosis by survivin predicts shorter survival rates in colorectal cancer. Cancer Res. 1998;58:5071–5074. [PubMed] [Google Scholar]
- 5.Yako-Suketomo H, Marugame T. Comparison of time trends in colon, rectum and anus cancer incidence (1973–2002) in Asia, from ‘Cancer Incidence in Five Continents, Vols IV-IX’. Jpn J Clin Oncol. 2009;39:196–198. doi: 10.1093/jjco/hyp017. [DOI] [PubMed] [Google Scholar]
- 6.den Besten G, van Eunen K, Groen AK, Venema K, Reijngoud DJ, Bakker BM. The role of short-chain fatty acids in the interplay between diet, gut microbiota and host energy metabolism. J Lipid Res. 2013 doi: 10.1194/jlr.R036012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Hague A, Manning AM, Hanlon KA, Huschtscha LI, Hart D, Paraskeva C. Sodium butyrate induces apoptosis in human colonic tumour cell lines in a p53-independent pathway: implications for the possible role of dietary fibre in the prevention of large-bowel cancer. Int J Cancer. 1993;55:498–505. doi: 10.1002/ijc.2910550329. [DOI] [PubMed] [Google Scholar]
- 8.Davie JR. Inhibition of histone deacetylase activity by butyrate. J Nutr. 2003;133:2485S–2493S. doi: 10.1093/jn/133.7.2485S. [DOI] [PubMed] [Google Scholar]
- 9.Domokos M, Jakus J, Szeker K, Csizinszky R, Csiko G, Neogrady Z, Csordas A, Galfi P. Butyrate-induced cell death and differentiation are associated with distinct patterns of ROS in HT29-derived human colon cancer cells. Dig Dis Sci. 2010;55:920–930. doi: 10.1007/s10620-009-0820-6. [DOI] [PubMed] [Google Scholar]
- 10.Nor C, Sassi FA, de Farias CB, Schwartsmann G, Abujamra AL, Lenz G, Brunetto AL, Roesler R. The Histone Deacetylase Inhibitor Sodium Butyrate Promotes Cell Death and Differentiation and Reduces Neurosphere Formation in Human Medulloblastoma Cells. Mol Neurobiol. 2013 doi: 10.1007/s12035-013-8441-7. [DOI] [PubMed] [Google Scholar]
- 11.Lee WJ, Shim JY, Zhu BT. Mechanisms for the inhibition of DNA methyltransferases by tea catechins and bioflavonoids. Mol Pharmacol. 2005;68:1018–1030. doi: 10.1124/mol.104.008367. [DOI] [PubMed] [Google Scholar]
- 12.Saldanha SN, Tollefsbol TO. The role of nutraceuticals in chemoprevention and chemotherapy and their clinical outcomes. J Oncol. 2012;2012:192464. doi: 10.1155/2012/192464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Tang Y, Zhao DY, Elliott S, Zhao W, Curiel TJ, Beckman BS, Burow ME. Epigallocatechin-3 gallate induces growth inhibition and apoptosis in human breast cancer cells through survivin suppression. Int J Oncol. 2007;31:705–711. [PubMed] [Google Scholar]
- 14.Kim EH, Kim HS, Kim SU, Noh EJ, Lee JS, Choi KS. Sodium butyrate sensitizes human glioma cells to TRAIL-mediated apoptosis through inhibition of Cdc2 and the subsequent downregulation of survivin and XIAP. Oncogene. 2005;24:6877–6889. doi: 10.1038/sj.onc.1208851. [DOI] [PubMed] [Google Scholar]
- 15.Kim S, Lee MJ, Hong J, Li C, Smith TJ, Yang GY, Seril DN, Yang CS. Plasma and tissue levels of tea catechins in rats and mice during chronic consumption of green tea polyphenols. Nutr Cancer. 2000;37:41–48. doi: 10.1207/S15327914NC3701_5. [DOI] [PubMed] [Google Scholar]
- 16.Chen L, Lee MJ, Li H, Yang CS. Absorption, distribution, elimination of tea polyphenols in rats. Drug Metab Dispos. 1997;25:1045–1050. [PubMed] [Google Scholar]
- 17.Mortensen PB, Clausen MR. Short-chain fatty acids in the human colon: relation to gastrointestinal health and disease. Scand J Gastroenterol Suppl. 1996;216:132–148. doi: 10.3109/00365529609094568. [DOI] [PubMed] [Google Scholar]
- 18.Vogt JA, Wolever TM. Fecal acetate is inversely related to acetate absorption from the human rectum and distal colon. J Nutr. 2003;133:3145–3148. doi: 10.1093/jn/133.10.3145. [DOI] [PubMed] [Google Scholar]
- 19.Evers BM, Ko TC, Li J, Thompson EA. Cell cycle protein suppression and p21 induction in differentiating Caco-2 cells. Am J Physiol. 1996;271:G722–727. doi: 10.1152/ajpgi.1996.271.4.G722. [DOI] [PubMed] [Google Scholar]
- 20.Xiong J, Li YR, Tang ZM, Dou LF, Wang L, Hu LH. The effect of p21 on transcription of survivin in hepatocellular carcinoma HepG2 cells its regulation mechanism. Zhonghua Zhong Liu Za Zhi. 2008;30:583–587. [PubMed] [Google Scholar]
- 21.Chiu SJ, Chao JI, Lee YJ, Hsu TS. Regulation of gamma-H2AX and securin contribute to apoptosis by oxaliplatin via a p38 mitogen-activated protein kinase-dependent pathway in human colorectal cancer cells. Toxicol Lett. 2008;179:63–70. doi: 10.1016/j.toxlet.2008.04.004. [DOI] [PubMed] [Google Scholar]
- 22.Zhang B, Wang X, Wang Y. Altered gene expression and miRNA expression associated with cancerous IEC-6 cell transformed by MNNG. J Exp Clin Cancer Res. 2009;28:56. doi: 10.1186/1756-9966-28-56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Lambert JD, Lee MJ, Diamond L, Ju J, Hong J, Bose M, Newmark HL, Yang CS. Dose-dependent levels of epigallocatechin-3-gallate in human colon cancer cells and mouse plasma and tissues. Drug Metab Dispos. 2006;34:8–11. doi: 10.1124/dmd.104.003434. [DOI] [PubMed] [Google Scholar]
- 24.Lambert JD, Hong J, Kim DH, Mishin VM, Yang CS. Piperine enhances the bioavailability of the tea polyphenol (−)-epigallocatechin-3-gallate in mice. J Nutr. 2004;134:1948–1952. doi: 10.1093/jn/134.8.1948. [DOI] [PubMed] [Google Scholar]
- 25.De Preter V, Geboes KP, Bulteel V, Vandermeulen G, Suenaert P, Rutgeerts P, Verbeke K. Kinetics of butyrate metabolism in the normal colon and in ulcerative colitis: the effects of substrate concentration and carnitine on the beta-oxidation pathway. Aliment Pharmacol Ther. 2011;34:526–532. doi: 10.1111/j.1365-2036.2011.04757.x. [DOI] [PubMed] [Google Scholar]
- 26.Ahmed D, Eide PW, Eilertsen IA, Danielsen SA, Eknaes M, Hektoen M, Lind GE, Lothe RA. Epigenetic and genetic features of 24 colon cancer cell lines. Oncogenesis. 2013;2:e71. doi: 10.1038/oncsis.2013.35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Dangond F, Gullans SR. Differential expression of human histone deacetylase mRNAs in response to immune cell apoptosis induction by trichostatin A and butyrate. Biochem Biophys Res Commun. 1998;247:833–837. doi: 10.1006/bbrc.1998.8891. [DOI] [PubMed] [Google Scholar]
- 28.Sarkar S, Abujamra AL, Loew JE, Forman LW, Perrine SP, Faller DV. Histone deacetylase inhibitors reverse CpG methylation by regulating DNMT1 through ERK signaling. Anticancer Res. 2011;31:2723–2732. [PubMed] [Google Scholar]
- 29.Wilson AJ, Chueh AC, Togel L, Corner GA, Ahmed N, Goel S, Byun DS, Nasser S, Houston MA, Jhawer M, et al. Apoptotic sensitivity of colon cancer cells to histone deacetylase inhibitors is mediated by an Sp1/Sp3-activated transcriptional program involving immediate-early gene induction. Cancer Res. 2010;70:609–620. doi: 10.1158/0008-5472.CAN-09-2327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Hague A, Elder DJ, Hicks DJ, Paraskeva C. Apoptosis in colorectal tumour cells: induction by the short chain fatty acids butyrate, propionate and acetate and by the bile salt deoxycholate. Int J Cancer. 1995;60:400–406. doi: 10.1002/ijc.2910600322. [DOI] [PubMed] [Google Scholar]
- 31.Hague A, Butt AJ, Paraskeva C. The role of butyrate in human colonic epithelial cells: an energy source or inducer of differentiation and apoptosis? Proc Nutr Soc. 1996;55:937–943. doi: 10.1079/pns19960090. [DOI] [PubMed] [Google Scholar]
- 32.Archer SY, Johnson J, Kim HJ, Ma Q, Mou H, Daesety V, Meng S, Hodin RA. The histone deacetylase inhibitor butyrate downregulates cyclin B1 gene expression via a p21/WAF-1-dependent mechanism in human colon cancer cells. Am J Physiol Gastrointest Liver Physiol. 2005;289:G696–703. doi: 10.1152/ajpgi.00575.2004. [DOI] [PubMed] [Google Scholar]
- 33.Siavoshian S, Blottiere HM, Cherbut C, Galmiche JP. Butyrate stimulates cyclin D and p21 and inhibits cyclin-dependent kinase 2 expression in HT-29 colonic epithelial cells. Biochem Biophys Res Commun. 1997;232:169–172. doi: 10.1006/bbrc.1997.6255. [DOI] [PubMed] [Google Scholar]
- 34.Sanchez-Tena S, Vizan P, Dudeja PK, Centelles JJ, Cascante M. Green tea phenolics inhibit butyrate-induced differentiation of colon cancer cells by interacting with monocarboxylate transporter 1. Biochim Biophys Acta. 2013;1832:2264–2270. doi: 10.1016/j.bbadis.2013.08.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Afshari CA, Vojta PJ, Annab LA, Futreal PA, Willard TB, Barrett JC. Investigation of the role of G1/S cell cycle mediators in cellular senescence. Exp Cell Res. 1993;209:231–237. doi: 10.1006/excr.1993.1306. [DOI] [PubMed] [Google Scholar]
- 36.Samowitz WS. Genetic and epigenetic changes in colon cancer. Exp Mol Pathol. 2008;85:64–67. doi: 10.1016/j.yexmp.2008.03.008. [DOI] [PubMed] [Google Scholar]
- 37.Ajamian F, Salminen A, Reeben M. Selective regulation of class I and class II histone deacetylases expression by inhibitors of histone deacetylases in cultured mouse neural cells. Neurosci Lett. 2004;365:64–68. doi: 10.1016/j.neulet.2004.04.087. [DOI] [PubMed] [Google Scholar]
- 38.David G, Neptune MA, DePinho RA. SUMO-1 modification of histone deacetylase 1 (HDAC1) modulates its biological activities. J Biol Chem. 2002;277:23658–23663. doi: 10.1074/jbc.M203690200. [DOI] [PubMed] [Google Scholar]
- 39.de Haan JB, Gevers W, Parker MI. Effects of sodium butyrate on the synthesis and methylation of DNA in normal cells and their transformed counterparts. Cancer Res. 1986;46:713–716. [PubMed] [Google Scholar]
- 40.Cosgrove DE, Cox GS. Effects of sodium butyrate and 5-azacytidine on DNA methylation in human tumor cell lines: variable response to drug treatment and withdrawal. Biochim Biophys Acta. 1990;1087:80–86. doi: 10.1016/0167-4781(90)90124-k. [DOI] [PubMed] [Google Scholar]
- 41.Wu JT, Archer SY, Hinnebusch B, Meng S, Hodin RA. Transient vs. prolonged histone hyperacetylation: effects on colon cancer cell growth, differentiation, and apoptosis. Am J Physiol Gastrointest Liver Physiol. 2001;280:G482–490. doi: 10.1152/ajpgi.2001.280.3.G482. [DOI] [PubMed] [Google Scholar]
- 42.Zhou Q, Agoston AT, Atadja P, Nelson WG, Davidson NE. Inhibition of histone deacetylases promotes ubiquitin-dependent proteasomal degradation of DNA methyltransferase 1 in human breast cancer cells. Mol Cancer Res. 2008;6:873–883. doi: 10.1158/1541-7786.MCR-07-0330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Rajendran P, Ho E, Williams DE, Dashwood RH. Dietary phytochemicals, HDAC inhibition, and DNA damage/repair defects in cancer cells. Clin Epigenetics. 2011;3:4. doi: 10.1186/1868-7083-3-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Miller KM, Tjeertes JV, Coates J, Legube G, Polo SE, Britton S, Jackson SP. Human HDAC1 and HDAC2 function in the DNA-damage response to promote DNA nonhomologous end-joining. Nat Struct Mol Biol. 2010;17:1144–1151. doi: 10.1038/nsmb.1899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Ashburner BP, Westerheide SD, Baldwin AS., Jr The p65 (RelA) subunit of NF-kappaB interacts with the histone deacetylase (HDAC) corepressors HDAC1 and HDAC2 to negatively regulate gene expression. Mol Cell Biol. 2001;21:7065–7077. doi: 10.1128/MCB.21.20.7065-7077.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.McCool KW, Miyamoto S. DNA damage-dependent NF-kappaB activation: NEMO turns nuclear signaling inside out. Immunol Rev. 2012;246:311–326. doi: 10.1111/j.1600-065X.2012.01101.x. [DOI] [PMC free article] [PubMed] [Google Scholar]