

Abstract
Anti-inflammatory strategies are often used to reduce muscle pain and soreness that can result from high-intensity muscular activity. However, studies indicate that components of the acute inflammatory response may be required for muscle repair and growth. The hypothesis of this study was that cyclooxygenase (COX)-2 activity is required for compensatory hypertrophy of skeletal muscle. We used the synergist ablation model of skeletal muscle hypertrophy, along with the specific COX-2 inhibitor NS-398, to investigate the role of COX-2 in overload-induced muscle growth in mice. COX-2 was expressed in plantaris muscles during compensatory hypertrophy and was localized mainly in or near muscle cell nuclei. Treatment with NS-398 blunted the increases in mass and protein content in overloaded muscles compared with vehicle-treated controls. Additionally, the COX-2 inhibitor decreased activity of the urokinase type plasminogen activator, macrophage accumulation, and cell proliferation, all of which are required for hypertrophy after synergist ablation. Expression of insulin-like growth factor-1 and phosphorylation of Akt, mammalian target of rapamycin, and p70S6K were increased following synergist ablation, but were not affected by NS-398. Additionally, expression of atrogin-1 was reduced during hypertrophy, but was also not affected by NS-398. These results demonstrate that COX-2 activity is required for skeletal muscle hypertrophy, possibly through facilitation of extracellular protease activity, macrophage accumulation, and cell proliferation.
Keywords: inflammation, nonsteroidal anti-inflammatory drug, macrophage, muscle growth
anti-inflammatory strategies are commonly used to reduce muscle pain or soreness that can result from exercise, especially high-intensity resistance training. However, studies indicate that acute inflammation may play important positive physiological roles in skeletal muscle repair and growth. Treatment with common nonsteroidal anti-inflammatory drugs (NSAIDs) can reduce postexercise muscle protein synthesis (46), inhibit muscle hypertrophy (42), and reduce force and torque production during long-term recovery from injury (32). Thus, the acute inflammatory response appears to be important in muscle remodeling. However, much remains to be learned about the role of the inflammatory response in muscle hypertrophy.
NSAIDs reduce inflammation in part by inhibiting the cyclooxygenase (COX) enzymes, which convert arachidonic acid to precursors of the prostaglandins and thromboxanes (33). COX-1 is constitutively expressed in most tissues, and is believed to have a homeostatic role (20). In contrast, COX-2 expression is increased by inflammatory stimuli, and COX-2-derived prostaglandins are typically considered proinflammatory (14). COX-2 metabolites are involved in regulating pain responses, blood flow, platelet activation, leukocyte adhesion, and tissue infiltration (14, 20). Selective COX-2 inhibitors effectively reduce pain, inflammation, and fever, without the gastrointestinal side effects associated with nonspecific NSAIDs (33). However, recent studies have demonstrated that COX-2 has important physiological functions in addition to its role as an inflammatory mediator. Indeed, potential side effects of COX-2 inhibitors include increased risk of hypertension and myocardial infarction (33).
COX-2 has been shown to influence a number of different pathways that may contribute to muscle hypertrophy. COX-2 activity is essential for efficient repair after muscle injury (8, 40), as well as recovery from atrophy (9). Inhibition of COX-2 during skeletal muscle regeneration results in increased fibrosis, elevated expression of transforming growth factor-β and myostatin, and reduced inflammatory cell accumulation (8, 40). In addition to anti-inflammatory effects, COX-2 inhibitors can affect muscle cells directly; cyclical stretch of cultured myoblasts causes an increase in COX-2 expression, and inhibition of COX-2 abolishes stretch-induced myoblast proliferation (34). COX-2-derived prostaglandins may also regulate activation, proliferation, migration, and fusion of cultured myoblasts (7, 9). Additionally, previous studies in pancreatic beta cells and liver have suggested that COX-2 activity increases insulin-like growth factor (IGF)-1 expression and signaling through Akt (17, 30), a pathway that promotes compensatory hypertrophy in muscle (5, 21). Finally, COX-2 is involved in regulation of the extracellular protease urokinase-type plasminogen activator (uPA) via production of the prostaglandin PGE2 (22, 35), and our previous study demonstrated that uPA is essential for muscle hypertrophy after synergist ablation (13). While these studies indicate that COX-2 is involved in the regulation of certain aspects of muscle remodeling, the role of COX-2 in skeletal muscle hypertrophy has not yet been established.
The hypothesis of the present study was that COX-2 activity is required for compensatory hypertrophy in skeletal muscle by promoting uPA activity, inflammatory cell infiltration, Akt signaling, and cell proliferation. To test this hypothesis, we used a specific COX-2 inhibitor, NS-398, and the synergist ablation model of mechanical overload. Our data indicate that COX-2 is expressed in the plantaris muscle during hypertrophy and is required for compensatory growth. Potential mechanisms by which COX-2 inhibition may block muscle hypertrophy include reduction of extracellular protease activity, inhibition of macrophage accumulation, and downregulation of cell proliferation.
METHODS
Mice and experimental groups.
Wild-type C57BL/6 mice were obtained from Harlan Laboratories. Mice were housed in groups of three to five mice at 22–24°C on a 12:12-h light-dark cycle. Food and water were provided ad libitum. All experiments were performed on 10- to 15-wk-old male mice and were approved by the Animal Care Committee of the University of Illinois.
For experiments on hypertrophy, mice were subjected to synergist ablation and treated daily with vehicle or NS-398, and plantaris muscles were collected at 14 days postablation (see Synergist ablation for details). For experiments on COX-2 localization, inflammatory cell accumulation, and cell proliferation, mice were subjected to synergist ablation and treated daily with vehicle or NS-398, and muscles were collected at 1, 3, 5 or 14 days postablation. For experiments on mRNA and protein analysis, mice were subjected to synergist ablation and treated daily with vehicle or NS-398, and muscles were collected at 5 days postablation. Additional control mice were not subjected to synergist ablation and were either left untreated or treated daily with NS-398 for 14 days.
Synergist ablation.
Bilateral synergist ablation was performed as previously described (13). Briefly, mice were anesthetized with intraperitoneal injection of ketamine (100 mg/kg) and xylazine (5 mg/kg). Under sterile conditions, a 1-cm incision was made on the lateral hind leg. Soleus and gastrocnemius muscles were removed, leaving the plantaris muscle and its neurovascular supply intact. The incision was closed with 7-0 nylon suture, and the procedure was repeated on the contralateral limb. NS-398 (10 mg·kg−1·day−1; Cayman Chemical) was administered by intraperitoneal injection 2 h prior to synergist ablation and at the same time daily until muscle collection. Controls were treated with vehicle (33% vol/vol dimethyl sulfoxide in physiological saline). For analysis of cell proliferation, mice received 30 mg/kg 5-bromo-2′-deoxyuridine (BrdU) by intraperitoneal injection 1 h prior to muscle collection. Mice were euthanized by cervical dislocation while anesthetized. Plantaris muscles were removed and weighed, and then mounted in tissue freezing medium and frozen in isopentane chilled with dry ice for histological analysis or flash-frozen in liquid nitrogen for biochemical assays.
Protein content.
Plantaris muscles (n = 6 each for untreated and NS-398-treated controls, n = 9 each for vehicle and NS-398-treated overloaded muscles) were homogenized with a dounce homogenizer in sample buffer (50 mM Tris, pH 6.8, 100 mM dithiothreitol, 2% SDS, 1 mM glycerol) supplemented with protease inhibitors (5 mM EDTA, 1 mM PMSF, 1 μM leupeptin, and 0.3 μM aprotinin). Samples were centrifuged at 16,000 g for 10 min at 4°C, and protein concentrations in the supernatant were determined (31). Total protein content for each muscle was determined by multiplying the protein concentration by the volume of the homogenate.
COX-2 localization.
Cryosections of 10-μm thickness were cut from the midbelly of four muscles per time point, air-dried, fixed in cold acetone, washed with PBS, and blocked with buffer containing 3% BSA. Sections were then incubated overnight with primary antibodies for COX-2 (1:50, rabbit polyclonal; Cayman Chemical), CD31 (1:100, rat monoclonal; BD Biosciences), monocyte/macrophage-2 antibody (MOMA-2; 1:200 rat monoclonal; Serotec), and/or desmin (1:50, mouse monoclonal; Novocastra). Sections were then washed with PBS and incubated with FITC-conjugated anti-rabbit secondary antibody, or with TRITC-conjugated anti-rabbit and FITC-conjugated anti-rat or anti-mouse secondary antibodies (1:200; Jackson ImmunoResearch). To visualize nuclei, slides were mounted with medium containing 4′,6 diamidino-2-phenylindole (Vector Laboratories). Images were acquired and merged using a Nikon Labphot-2 and SPOT software.
Inflammatory cell accumulation.
Immunohistochemical analysis was performed as described (13) for four to eight muscles per group and time point after ablation. Cryosections were air dried, fixed in cold acetone, washed with PBS, quenched with 0.3% hydrogen peroxide, and washed with PBS. Sections were blocked with buffer containing 3% BSA and then incubated overnight with F4/80 antibody to label macrophages (1:100; Serotec), or Ly6G antibody to label neutrophils (1:100; BD Biosciences). Sections were then washed with PBS and incubated with biotinylated anti-rat secondary antibody (1:200; Vector Laboratories). After washing with PBS, sections were incubated with avidin d-horseradish peroxidase (1:1,000) and developed with a 3-amino-9-ethylcarbazole kit (Vector Laboratories). The number of labeled cells was counted in six images (magnification, ×20; Labphot-2, Nikon; SPOT software, Diagnostic Instruments) per muscle, and then normalized to volume of muscle sampled (area of fields × section thickness).
Cell proliferation.
Cell proliferation was assessed by staining for BrdU, which is incorporated into newly synthesized DNA (13), in muscles collected at 5 days postablation (n = 5 for vehicle, n = 7 for NS-398 treated). Cryosections were air-dried, fixed in cold acetone, washed with PBS, incubated in 2 N HCl, washed with basic PBS (pH 8.5), and then washed with neutral PBS (pH 7.6). Sections were then incubated in 0.1% IGEPAL and then placed in blocking buffer containing 3% BSA. Proliferating cells were labeled with a BrdU antibody (1:10, Roche) for 1 h. Sections were washed with PBS and then incubated with FITC-conjugated anti-mouse secondary antibody (1:200; Jackson ImmunoResearch). The number of labeled cells was counted in six images (magnification, ×20) per muscle, and then normalized to volume of muscle sampled.
uPA activity.
uPA activity was assessed in soluble fractions of homogenates using zymography (23). Muscles (n = 4 per group) were homogenized in buffer [50 mM Tris-HCl (pH 7.6), 150 mM NaCl, and 0.5% Triton X-100] supplemented with protease inhibitors (5 mM EDTA, 1 mM PMSF, 1 μM leupeptin, and 0.3 μM aprotinin). Samples were centrifuged, and the soluble fraction was collected, mixed with loading buffer, and then separated on SDS-PAGE gels containing α-casein (4 mg/ml) and human Glu-plasminogen (20 μg/ml). Gels were washed in 1% Tween 20 in PBS and incubated in 0.1% Tween 20 in PBS overnight at 37°C. Finally, gels were stained with Coomassie blue dye and destained overnight. uPA activity was identified by the position of its lytic band (45 kDa) and quantified by densitometry.
Real-time PCR.
Total RNA was isolated from seven to eight muscles per group using the RNeasy kit (Qiagen) following manufacturer's instructions. RNA quantity was determined by UV absorption at 260 nm, and quality was verified by the 260/280 nm absorption ratio. cDNA was synthesized from 1 μg RNA using the Thermoscript RT-PCR System (Invitrogen). Real-time PCR was performed in a 7500 Fast System (Applied Biosystems) using TaqMan Universal PCR Master Mix and TaqMan Gene Expression Assay primer/probe sets (Applied Biosystems) for mouse IGF-1, uPA, plasminogen activator inhibitor (PAI)-1, atrogin-1, or GAPDH. The IGF-1 primers did not differentiate between muscle and endocrine IGF-1 transcripts. All reactions were performed in triplicate, and cycle threshold (CT) values were averaged over triplicates. Relative gene expression was determined using the 2−ΔΔCT method, as previously described (25), with nonablated muscle as baseline and GAPDH as endogenous control gene. Primer efficiency was verified using serial dilutions of sample cDNA.
Signaling.
Western blots were used to analyze signaling pathways previously associated with muscle growth (21). Muscles (n = 4 per group) were homogenized in buffer containing 40 mM Tris (pH 7.5), 1 mM EDTA, 5 mM EGTA, 0.5% Triton X-100, 25 mM β-glycerophosphate, 25 mM NaF, 1 mM Na3VO4, 10 μg/ml leupeptin, and 1 mM PMSF. After homogenates had been centrifuged, the supernatant was mixed with concentrated reducing SDS buffer and separated on SDS-PAGE gels. Proteins were transferred to a polyvinylidene difluoride membrane, and membranes were blocked with 5% powdered milk and then incubated overnight at 4°C with phospho-specific primary antibodies against Akt (473), mammalian target of rapamycin (mTOR; 2481), p70 (389), or p70 (421/424) (Upstate Biotechnology). Membranes were washed and then probed with horseradish peroxidase-conjugated secondary antibody. Following another wash, blots were developed using an enhanced chemiluminescence kit (Amersham). Membranes were then stained with Coomassie blue to verify equal loading in all lanes. Densitometric measurements were carried out using the public domain National Institutes of Health Image program (ImageJ).
Statistics.
Values are reported as means ± SD. For comparisons of muscle mass and protein, protein phosphorylation levels, and mRNA levels, data were compared across treatment groups using one-way ANOVA. For comparisons of inflammatory cell accumulation, data were compared across treatment groups and time points using two-way ANOVA. The Student-Newman-Keuls post hoc test was used when ANOVAs demonstrated significance. For groups that did not pass tests of normality and equal variance, the nonparametric Kruskal-Wallis ANOVA on ranks was used with Dunn's post hoc method. Comparisons of cell proliferation and uPA activity were made between treatment groups using t-tests. Differences between groups were considered significant if P ≤ 0.05.
RESULTS
COX-2 inhibition reduces compensatory hypertrophy.
As expected, synergist ablation resulted in compensatory hypertrophy of the plantaris muscle. In vehicle-treated mice, synergist ablation resulted in an ∼80% increase in plantaris mass on day 14 postablation relative to nonablated controls (Fig. 1A). Total protein content increased by ∼50% (Fig. 1B), indicating that the increase in mass was not solely due to edema. Plantaris mass and protein content on day 14 postablation did not differ between untreated mice and those given a daily dose of vehicle (muscle mass and protein = 32.0 ± 2.6 mg and 3,674 ± 349 μg for untreated, 32.6 ± 2.5 mg and 3,582 ± 211 μg for vehicle treated).
Fig. 1.

Cyclooxygenase (COX)-2 inhibition reduces compensatory hypertrophy. Plantaris mass (A) and total protein (B) were quantified for muscles from nonablated control mice, nonablated mice treated with NS-398 for 14 days, vehicle-treated mice at 14 days postablation, and NS-398-treated mice at 14 days postablation. NS-398 treatment did not significantly affect mass or protein content of muscles from nonablated mice. In vehicle-treated mice, synergist ablation resulted in increased plantaris mass and protein content. Treatment with NS-398 blunted this response. Data are presented as means ± SD; n = 6–9 muscles/group. *Significantly different from nonablated control (P ≤ 0.05). ‡Significantly different from vehicle-ablated mice (P ≤ 0.05).
In NS-398-treated mice, hypertrophy of the plantaris muscle was reduced by ∼75% compared with vehicle-treated controls (Fig. 1A). This resulted in plantaris mass that was ∼33% lower than in mice treated with vehicle at 14 days, and only ∼20% higher than in nonablated controls. Protein content in NS-398-treated muscles at 14 days was also reduced relative to vehicle, and did not differ significantly from nonablated controls (Fig. 1B). In mice not subjected to synergist ablation, treatment with NS-398 for 14 days did not alter plantaris mass or protein content (Fig. 1A–B). In addition, body weight did not differ between vehicle-treated and NS-398-treated mice at 14 days postablation (25.0 ± 1.1 g vs. 25.5 ± 1.5 g).
COX-2 is expressed during compensatory hypertrophy.
Immunofluorescence analysis was used to localize expression of COX-2 during compensatory hypertrophy. In muscle of nonablated control mice, COX-2 staining was sparsely distributed at the periphery of muscle fibers. COX-2-stained cells appeared to increase in number during compensatory hypertrophy (Fig. 2A). Dual fluorescence labeling was used to determine which cell types expressed COX-2 during hypertrophy. COX-2 expression was most often seen in or near nuclei, within or closely associated with muscle fibers or myogenic precursor cells, as evidenced by colocalization of COX-2 with 4′,6 diamidino-2-phenylindole and desmin (Fig. 2B). We observed minimal COX-2 colocalization with the monocyte/macrophage marker MOMA-2 or with the endothelial cell marker CD31 (Fig. 2C). Negative control sections stained with secondary antibody only showed no specific staining (Fig. 2D).
Fig. 2.
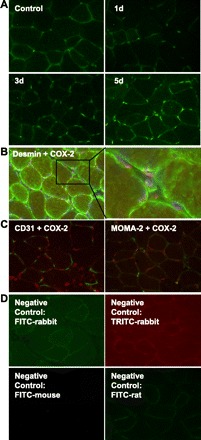
COX-2 is expressed in muscle during compensatory hypertrophy. A: COX-2 expression in plantaris was visualized by immunofluorescence in control muscle and at 1, 3, and 5 days postablation. B: localization of COX-2 expression at 5 days postablation was determined using immunofluorescence for COX-2 (red) and desmin (green). Nuclei were counterstained with 4′,6 diamidino-2-phenylindole (blue). COX-2 fluorescence was most often associated with nuclei within or near desmin-positive cells. C: immunofluorescence for COX-2 (red) and CD31 (green, left) or MOMA-2 (green, right) at 5 days postablation. COX-2 exhibited little colocalization with either the endothelial cell marker CD31 or the monocyte/macrophage marker MOMA-2. D: representative images of negative controls for muscles at 5 days postablation. Negative controls at other time points were similar. All images in Fig. 2 are representative of at least 4 muscles per condition.
COX-2 inhibition reduces macrophage accumulation.
Immunohistochemistry was used to assess macrophage and neutrophil accumulation after synergist ablation. As expected, control muscles contained relatively few macrophages and neutrophils (80 ± 149 per mm3 and 2,960 ± 2,488 per mm3, respectively). In vehicle-treated mice, macrophage accumulation was significantly increased compared with control values on days 1, 3, and 5 postablation, with a peak at day 5, before returning to control levels at day 14. NS-398 treatment reduced the macrophage number by ∼80% at day 5 postablation (Fig. 3A). In vehicle-treated mice, neutrophil accumulation peaked at day 1 postablation, and remained significantly above control levels until day 14 (Fig. 3B). Treatment with NS-398 reduced neutrophil accumulation only on day 5 postablation, when neutrophil number had already been reduced to near control levels.
Fig. 3.

Inflammatory cell accumulation during compensatory hypertrophy. Macrophages (A) and neutrophils (B) were quantified as number of cells stained positively for F4/80 or Ly6G antigen, respectively. Peak macrophage accumulation was markedly reduced by NS-398 treatment. NS-398 treatment did not affect peak neutrophil accumulation and only reduced neutrophil number at day 5, when neutrophil content had already declined to near control values. Data are presented as means ± SD; n = 4–8 muscles/group. ‡Significantly different from vehicle at same time point (P ≤ 0.05).
COX-2 inhibition reduces cell proliferation.
Incorporation of BrdU into newly synthesized DNA was used to assess cell proliferation at day 5 postablation, the time point at which we previously observed a peak in proliferation (13). In vehicle-treated mice, a substantial number of BrdU-positive cells were observed in the plantaris muscle, indicating an abundance of proliferating cells. Treatment with NS-398 reduced the number of BrdU-positive cells by ∼20% (Fig. 4). As expected, very few proliferating cells were observed in the plantaris muscles of unoperated (24 ± 48 per mm3) and sham-operated animals at 5 days after sham surgery (36 ± 55 per mm3), and these values were not significantly different from zero.
Fig. 4.

COX-2 inhibition reduces cell proliferation at 5 days postablation. 5-Bromo-2′-deoxyuridine (BrdU) was injected into mice 1 h prior to tissue collection, and cell proliferation was quantified by counting the number of cells positively labeled for BrdU. Treatment with NS-398 significantly reduced the number of proliferating cells at 5 days postablation. Data are presented as means ± SD; n = 5–7 muscles/group. ‡Significantly different from vehicle at same time-point (P ≤ 0.05).
COX-2 inhibition reduces uPA activity with no change in mRNA expression.
To determine whether inhibition of COX-2 results in altered uPA activity during muscle hypertrophy, casein zymograms were generated to evaluate uPA activity in the plantaris muscle at day 5 postablation, the time point at which maximum uPA activity was previously observed (13). uPA activity was reduced by NS-398 treatment compared with vehicle (Fig. 5A), indicating that COX-2 inhibition did indeed reduce uPA activity following synergist ablation. We also assessed expression of uPA and its inhibitor PAI-1 at the mRNA level using real-time PCR. While synergist ablation resulted in increased mRNA expression of both uPA and PAI-1, treatment with NS-398 had no effect on expression of either gene (Fig. 5, B and C).
Fig. 5.

COX-2 inhibition reduces urokinase-type plasminogen activator (uPA) activity but does not alter mRNA expression of uPA or plasminogen activator inhibitor (PAI)-1. A: uPA activity was assessed using casein zymograms and homogenates of muscles collected at 5 days postablation from vehicle- and NS-398-treated mice. Treatment with NS-398 resulted in reduced uPA activity. Zymograms shown are representative of n = 4 muscles per group. Means ± SD are displayed below respective images. B and C: real-time PCR was used to determine mRNA expression of uPA and PAI-1 in muscles from nonablated control mice and from vehicle- and NS-398-treated mice at 5 days postablation. Expression of both uPA and PAI-1 was increased after ablation in both vehicle- and NS-398-treated mice, but was not altered by NS-398 compared with vehicle. Data are presented as means ± SD; n = 7–8 muscles/group. *Significantly different from nonablated control (P ≤ 0.05).
COX-2 inhibition does not alter expression of IGF-1 or phosphorylation of Akt, mTOR, or p70S6K during hypertrophy.
We used real-time PCR to quantify IGF-1 expression during compensatory hypertrophy. Synergist ablation produced a ∼24-fold elevation in IGF-1 mRNA expression at day 5 postablation compared with unoperated controls. However, IGF-1 expression was not altered by treatment with NS-398 (Fig. 6A). Additionally, Western blots with phospho-specific antibodies were used to analyze the activity of the Akt, mTOR, and p70S6K signaling molecules. Phosphorylation of Akt, mTOR, and p70S6K increased at day 5 postablation in both vehicle- and NS-398-treated muscles (Fig. 6, B and C). However, NS-398 treatment did not significantly alter phosphorylation of any of these signaling molecules.
Fig. 6.

Insulin-like growth factor (IGF)-1 expression and phosphorylation of Akt, mammalian target of rapamycin (mTOR), and p70S6K during hypertrophy are not altered by COX-2 inhibition. A: IGF-1 mRNA was quantified by real-time PCR in muscles from nonablated control mice and from vehicle- and NS-398-treated mice at 5 days postablation. IGF-1 expression increased after synergist ablation, but was not affected by NS-398 treatment. Data are presented as mean ± SD; n = 7–8 muscles/group. *Significantly different from nonablated control (P ≤ 0.05). B: phosphorylation of Akt (473), mTOR (2481), p70 (389), and p70 (421/424) was determined by Western blot. Blots shown are representative of n = 4 muscles per condition. C: phosphorylation was quantified by densitometric analysis on Western blots. Phosphorylation of Akt, mTOR, and p70S6K was increased after synergist ablation, but was not significantly affected by NS-398 treatment. Data are presented as means ± SD; n = 4 muscles/group. *Significantly different from nonablated control (P ≤ 0.05).
Ubiquitin ligase expression decreases during compensatory hypertrophy.
We used real-time PCR to quantify expression of the E3 ubiquitin ligase atrogin-1 following synergist ablation. Atrogin-1 expression was reduced by ∼95% on day 5 postablation, but was unaffected by treatment with NS-398 (Fig. 7), suggesting that COX-2 inhibition does not alter this component of the proteasomal degradation pathway.
Fig. 7.

Atrogin-1 expression decreases after synergist ablation. Real-time PCR was used to determine mRNA expression of the E3 ubiquitin ligase atrogin-1 in muscles from nonablated control mice and from vehicle- and NS-398-treated mice at 5 days postablation. In both vehicle- and NS-398-treated mice, synergist ablation resulted in decreased expression of atrogin-1 relative to nonablated control. NS-398 did not alter atrogin-1 expression relative to vehicle. Data are displayed on a log scale to facilitate comparison of vehicle vs. NS-398-treated groups. Data are presented as means ± SD; n = 3–4 muscles/group. *Significantly different from nonablated control (P ≤ 0.05).
DISCUSSION
Previous studies have shown that components of the acute inflammatory response may be required for skeletal muscle remodeling following increased mechanical loading or injury (8, 9, 10, 13, 40, 42). Therefore, we hypothesized that COX-2 activity is necessary for compensatory muscle hypertrophy. The major findings of this study were that inhibition of COX-2 with NS-398 blunts muscle hypertrophy following synergist ablation. Associated with this impaired hypertrophy were decreases in uPA activity, macrophage accumulation, and cell proliferation, processes which have been previously shown to be required for muscle growth (1, 13). The impairment of hypertrophy occurred despite no differences in phosphorylation of Akt, mTOR, and p70S6K, which is thought to promote muscle growth (4, 21). In short, these findings indicate that COX-2 activity is required for compensatory muscle hypertrophy, possibly through its effects on extracellular protease activity, macrophage accumulation, and cell proliferation.
Recent studies have shown that COX-2 is expressed in muscle during regeneration and during recovery from atrophy (8, 9). Our results demonstrate that COX-2 is also expressed during compensatory hypertrophy. Concordant with a study on recovery from atrophy (9), COX-2 expression during hypertrophy was most prominent in or near nuclei within or closely associated with muscle cells. COX-2 colocalized strongly with the muscle cell marker desmin, but not with the monocyte/macrophage marker MOMA-2 or the endothelial marker CD31, suggesting that muscle fibers and/or myogenic precursor cells are the primary source of COX-2 during hypertrophy.
One potential mechanism by which COX-2 inhibition may blunt muscle hypertrophy is through a reduction in uPA activity. Our present data indicate that COX-2 inhibition results in decreased uPA activity during muscle overloading, and we previously demonstrated that uPA is essential for skeletal muscle hypertrophy (13). uPA could promote muscle hypertrophy through its influence on extracellular matrix remodeling (12), regulation of growth factor bioactivity (44, 48, 49), modulation of the inflammatory response (10, 13), and direct effects on myogenic precursor cells (6, 15). Consistent with our current results, inhibition of COX-2 reduced uPA activity in corneal organ culture (35). Based on another recent study in which uPA mRNA was reduced by COX-2 inhibition in gastric cancer cells (22), we also expected that uPA mRNA expression would be modulated by COX-2 during skeletal muscle hypertrophy. However, we found that mRNA expression of uPA and its inhibitor PAI-1, while elevated during hypertrophy, were not altered by COX-2 inhibition. This suggests that, during skeletal muscle hypertrophy, COX-2 upregulates uPA activity at a posttranscriptional level. uPA is secreted as a zymogen, and is activated by proteolytic processing by plasmin (2). In addition, uPA undergoes several posttranslational modifications, including glycosylation, phosphorylation, and fucosylation (2). Whether COX-2 influences uPA activity via any of these processes remains to be determined.
A second mechanism by which COX-2 inhibition may decrease muscle hypertrophy is through a reduction in macrophage accumulation; COX-2 inhibition in the present study dramatically reduced peak macrophage accumulation following synergist ablation, and our previous study demonstrated that macrophage accumulation is required for compensatory hypertrophy (13). Macrophages may assist in muscle remodeling by phagocytosing damaged tissue (29), releasing soluble factors that promote proliferation of myogenic precursor cells (11), and inhibiting apoptosis of myoblasts and myotubes (43). In addition, macrophages produce factors such as IGF-1 that could increase protein synthesis and hypertrophy in mature muscle fibers (13). Peak neutrophil accumulation during compensatory hypertrophy was unaffected by COX-2 inhibition, indicating that the reduction in macrophage number was specific and not due to a general defect in inflammatory cell responses. Recent studies have suggested that COX-2-derived prostaglandins promote macrophage chemotaxis (36, 45). In addition, COX-2 metabolites influence vascular responses that could also affect macrophage accumulation (20). Furthermore, our previous studies have demonstrated that uPA is required for macrophage chemotaxis (10) and for macrophage accumulation during muscle regeneration (10) and compensatory hypertrophy (13). Thus, reduction of uPA activity by COX-2 inhibition may have contributed to the decrease in macrophage accumulation observed in the present study.
A third mechanism by which COX-2 inhibition may result in reduced muscle hypertrophy is through impairment of cell proliferation; COX-2 inhibition in the present study reduced cell proliferation during compensatory hypertrophy. These proliferating cells could be myoblasts, macrophages, fibroblasts, or other cells, all of which may be important during hypertrophy. For example, addition of nuclei to muscle fibers during hypertrophy requires activation of muscle satellite cells, which then proliferate and fuse with mature fibers (19), and previous studies have shown that COX-2-derived prostaglandins promote proliferation of myogenic cells in vitro (9, 34). Interestingly, irradiation of the rat hindlimb largely prevented such nuclear addition and subsequent compensatory hypertrophy (1). This occurred despite increased expression of IGF-1 related molecules and changes in cellular signaling that were not different from those in nonirradiated muscle. These findings parallel those of the present study, in which COX-2 inhibition resulted in a reduction in hypertrophy despite a lack of effect on cell signaling. Finally, our previous study demonstrated that uPA deficiency or macrophage depletion reduces cell proliferation during hypertrophy (13). Since our present data indicate that COX-2 inhibition reduces uPA activity and macrophage accumulation, either of these mechanisms may contribute to the reduction in cell proliferation.
Mechanical stimuli, nutrients, or growth factors, such as IGF-1, can activate signaling through Akt, mTOR, and p70S6K (21), which promotes protein synthesis and skeletal muscle growth (4). Since COX-2 promotes IGF-1 expression and Akt signaling in other cell types (17, 30), we hypothesized that COX-2 inhibition would reduce IGF-1 expression and downstream signaling during hypertrophy, and this could be responsible for the observed reductions in muscle mass and protein content. However, our data indicate that COX-2 inhibition did not alter mRNA expression of IGF-1 and did not produce any significant alterations in phosphorylation of Akt, mTOR, or p70S6K at 5 days postablation. While COX-2 inhibition does reduce skeletal muscle hypertrophy, the mechanism does not seem to involve altered IGF-1 expression or signaling through Akt, mTOR or p70S6K.
The increase in muscle protein content during compensatory hypertrophy is likely due to a combination of increased protein synthesis and decreased protein degradation (39), the latter partly via the ubiquitin-proteasome pathway. The E3 ubiquitin ligase atrogin-1 is upregulated during atrophy (16) and after burn injury (24), and is downregulated by IGF-1 through Akt and mTOR (16, 24, 39). Our data indicate that atrogin-1 mRNA expression is markedly decreased during compensatory hypertrophy. This may result in decreased proteasome-mediated protein degradation and may contribute to increased total muscle protein content (26). In addition, a previous study has suggested a role for COX-2 in the regulation of atrogin-1 expression in muscle during experimental arthritis (17). However, in the present study, COX-2 inhibition did not affect atrogin-1 expression, despite reductions in total muscle protein content. Thus COX-2 inhibition may have caused a decrease in protein synthesis, or may have increased protein degradation via another pathway. The prostaglandins PGF2α and PGE2 have previously been implicated in regulation of muscle protein synthesis (37, 41) and degradation (37, 38), respectively, although conflicting reports have been generated (3, 18, 28). Ibuprofen, which inhibits both COX-1 and COX-2, reduces muscle PGF2α content (47) and protein synthesis (46) after exercise, so we suspect that protein synthesis was also reduced in the presence of the specific COX-2 inhibitor NS-398.
A limitation of this study is potential nonspecific effects of the COX-2 inhibitor NS-398 during compensatory hypertrophy. However, in vivo experiments with rats demonstrated that NS-398 blocked COX-2-mediated prostaglandin production, but did not block COX-1-mediated prostaglandins (27). In addition, NS-398 has been used to block prostaglandin production in cultured muscle cells and impair muscle regeneration in vivo (40). Nonetheless, the potential for nonspecific effects of NS-398 remains. Future experiments to determine whether administration of prostaglandins rescues compensatory hypertrophy in NS-398-treated mice would provide strong evidence on the specificity of NS-398 on COX-2 during hypertrophy.
Perspectives and Significance
Our data show that COX-2 activity is required for skeletal muscle hypertrophy, possibly through facilitation of uPA activity, macrophage accumulation, and cell proliferation. Inhibition of COX-2 reduced hypertrophy despite the absence of an effect on signaling associated with hypertrophy. These data indicate that nonmuscle cell types and extracellular processes contribute to the hypertrophic process. From a practical standpoint, anti-inflammatory strategies are commonly used to treat muscle pain and soreness that can result from exercise. Although much remains to be learned about the role of inflammatory processes in muscle hypertrophy, especially in humans, reducing COX-2 activity and acute inflammation has potential for hampering the very adaptations at which exercise training is aimed.
Footnotes
The costs of publication of this article were defrayed in part by the payment of page charges. The article must therefore be hereby marked “advertisement” in accordance with 18 U.S.C. Section 1734 solely to indicate this fact.
REFERENCES
- 1.Adams GR, Caiozzo VJ, Haddad F, Baldwin KM. Cellular and molecular responses to increased skeletal muscle loading after irradiation. Am J Physiol Cell Physiol 283: C1182–C1195, 2002 [DOI] [PubMed] [Google Scholar]
- 2.Alfano D, Franco P, Vocca I, Gambi N, Pisa V, Mancini A, Caputi M, Carriero MV, Iaccarino I, Stoppelli MP. The urokinase plasminogen activator and its receptor. Role in cell growth and apoptosis. Thromb Haemost 93: 205–211, 2005 [DOI] [PubMed] [Google Scholar]
- 3.Barnett JG, Ellis S. Prostaglandin-E2 and the regulation of protein degradation in skeletal muscle. Muscle Nerve 10: 556–559, 1987 [DOI] [PubMed] [Google Scholar]
- 4.Bodine SC. mTOR signaling and the molecular adaptation to resistance exercise. Med Sci Sports Exerc 38: 1950–1957, 2006 [DOI] [PubMed] [Google Scholar]
- 5.Bodine SC, Stitt TN, Gonzalez M, Kline WO, Stover GL, Bauerlein R, Zlotchenko E, Scrimgeour A, Lawrence JC, Glass DJ, Yancopoulos GD. Akt/mTOR pathway is a crucial regulator of skeletal muscle hypertrophy and can prevent muscle atrophy in vivo. Nat Cell Biol 3: 1014–1019, 2001 [DOI] [PubMed] [Google Scholar]
- 6.Bonavaud S, Charriere-Bertrand C, Rey C, Leibovitch MP, Pedersen N, Frisdal E, Planus E, Blasi F, Gherardi R, Barlovatz-Meimon G. Evidence of a non-conventional role for the urokinase tripartite complex (uPAR/uPA/PAI-1) in myogenic cell fusion. J Cell Sci 110: 1083–1089, 1997 [DOI] [PubMed] [Google Scholar]
- 7.Bondesen BA, Jones KA, Glasgow WC, Pavlath GK. Inhibition of myoblast migration by prostacyclin is associated with enhanced cell fusion. FASEB J 21: 3338–3345, 2007 [DOI] [PubMed] [Google Scholar]
- 8.Bondesen BA, Mills ST, Kegley KM, Pavlath GK. The COX-2 pathway is essential during early stages of skeletal muscle regeneration. Am J Physiol Cell Physiol 287: C475–C483, 2004 [DOI] [PubMed] [Google Scholar]
- 9.Bondesen BA, Mills ST, Pavlath GK. The COX-2 pathway regulates growth of atrophied muscle via multiple mechanisms. Am J Physiol Cell Physiol 290: C1651–C1659, 2006 [DOI] [PubMed] [Google Scholar]
- 10.Bryer SC, Fantuzzi G, Van Rooijen N, Koh TJ. Urokinase-type plasminogen activator plays essential roles in macrophage chemotaxis and skeletal muscle regeneration. J Immunol 180: 1179–1188, 2008 [DOI] [PubMed] [Google Scholar]
- 11.Cantini M, Giurisato E, Radu C, Tiozzo S, Pampinella F, Senigaglia D, Zaniolo G, Mazzoleni F, Vitiello L. Macrophage-secreted myogenic factors: a promising tool for greatly enhancing the proliferative capacity of myoblasts in vitro and in vivo. Neurol Sci 23: 189–194, 2002 [DOI] [PubMed] [Google Scholar]
- 12.Chapman HA, Reilly JJ, Kobzik L. Role of plasminogen-activator in degradation of extracellular-matrix protein by live human alveolar macrophages. Am Rev Respir Dis 137: 412–419, 1988 [DOI] [PubMed] [Google Scholar]
- 13.DiPasquale DM, Cheng M, Billich W, Huang SA, van Rooijen N, Hornberger TA, Koh TJ. Urokinase-type plasminogen activator and macrophages are required for skeletal muscle hypertrophy in mice. Am J Physiol Cell Physiol 293: C1278–C1285, 2007 [DOI] [PubMed] [Google Scholar]
- 14.DuBois RN, Abramson SB, Crofford L, Gupta RA, Simon LS, Van De Putte L, Lipsky PE. Cyclooxygenase in biology and disease. FASEB J 12: 1063–1073, 1998 [PubMed] [Google Scholar]
- 15.Fibbi G, Barletta E, Dini G, Del Rosso A, Pucci M, Cerletti M, Del Rosso M. Cell invasion is affected by differential expression of the urokinase plasminogen activated/urokinase plasminogen activator receptor system in muscle satellite cells from normal and dystrophic patients. Lab Invest 81: 27–39, 2001 [DOI] [PubMed] [Google Scholar]
- 16.Glass DJ. Skeletal muscle hypertrophy and atrophy signaling pathways. Int J Biochem Cell Biol 37: 1974–1984, 2005 [DOI] [PubMed] [Google Scholar]
- 17.Granado M, Martin AI, Villanua MA, Lopez-Calderon A. Experimental arthritis inhibits the insulin-like growth factor-I axis and induces muscle wasting through cyclooxygenase-2 activation. Am J Physiol Endocrinol Metab 292: E1656–E1665, 2007 [DOI] [PubMed] [Google Scholar]
- 18.Hasselgren PO, Zamir O, James JH, Fischer JE. Prostaglandin-E2 does not regulate total or myofibrillar protein breakdown in incubated skeletal-muscle from normal or septic rats. Biochem J 270: 45–50, 1990 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Hawke TJ, Garry DJ. Myogenic satellite cells: physiology to molecular biology. J Appl Physiol 91: 534–551, 2001 [DOI] [PubMed] [Google Scholar]
- 20.Hinz B, Brune K. Cyclooxygenase-2—10 years later. J Pharmacol Exp Ther 300: 367–375, 2002 [DOI] [PubMed] [Google Scholar]
- 21.Hornberger TA, Sukhija KB, Chien S. Regulation of mTOR by mechanically induced signaling events in skeletal muscle. Cell Cycle 5: 1391–1396, 2006 [DOI] [PubMed] [Google Scholar]
- 22.Iwamoto J, Mizokami Y, Takahashi K, Matsuoka T, Matsuzaki Y. The effects of cyclooxygenase2-prostaglandin E2 pathway on Helicobacter pylori-induced urokinase-type plasminogen activator system in the gastric cancer cells. Helicobacter 13: 174–182, 2008 [DOI] [PubMed] [Google Scholar]
- 23.Koh TJ, Bryer SC, Pucci AM, Sisson TH. Mice deficient in plasminogen activator inhibitor-1 have improved skeletal muscle regeneration. Am J Physiol Cell Physiol 289: C217–C223, 2005 [DOI] [PubMed] [Google Scholar]
- 24.Lang CH, Huber D, Frost RA. Burn-induced increase in atrogin-1 and MuRF-1 in skeletal muscle is glucocorticoid independent but downregulated by IGF-I. Am J Physiol Regul Integr Comp Physiol 292: R328–R336, 2007 [DOI] [PubMed] [Google Scholar]
- 25.Livak KJ, Schmittgen TD. Analysis of relative gene expression data using real-time quantitative PCR and the 2−ΔΔ CT method. Methods 25: 402–408, 2001 [DOI] [PubMed] [Google Scholar]
- 26.Marino JS, Tausch BJ, Dearth CL, Manacci MV, McLoughlin TJ, Rakyta SJ, Linsenmayer MP, Pizza FX. β2-integrins contribute to skeletal muscle hypertrophy in mice. Am J Physiol Cell Physiol 295: C1026–C1036, 2008 [DOI] [PubMed] [Google Scholar]
- 27.Masferrer JL, Zweifel BS, Manning PT, Hauser SD, Leahy KM, Smith WG, Isakson PC, Siebert K. Selective inhibition of inducible cyclooxygenase 2 in vivo is anti-inflammatory and nonulcerogenic. Proc Natl Acad Sci USA 91: 3228–3232, 1994 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.McElligott MA, Chaung LY, Baracos V, Gulve EA. Prostaglandin production in myotube cultures–influence on protein-turnover. Biochem J 253: 745–749, 1988 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.McLennan IS. Degenerating and regenerating skeletal muscles contain several subpopulations of macrophages with distinct spatial and temporal distributions. J Anat 188: 17–28, 1996 [PMC free article] [PubMed] [Google Scholar]
- 30.Meng ZX, Sun JX, Ling JJ, Lv JH, Zhu DY, Chen Q, Sun YJ, Han X. Prostaglandin E-2 regulates Foxo activity via the Akt pathway: implications for pancreatic islet beta cell dysfunction. Diabetologia 49: 2959–2968, 2006 [DOI] [PubMed] [Google Scholar]
- 31.Minamide LS, Bamburg JR. A filter-paper dye-binding assay for quantitative determination of protein without interference from reducing agents or detergents. Anal Biochem 190: 66–70, 1990 [DOI] [PubMed] [Google Scholar]
- 32.Mishra DK, Friden J, Schimitz MC, Lieber RL. Anti-inflammatory medication after muscle injury. A treatment resulting in short-term improvement but subsequent loss of muscle function. J Bone Joint Surg Am 77: 1510–1519, 1995 [DOI] [PubMed] [Google Scholar]
- 33.Mitchell JA, Warner TD. COX isoforms in the cardiovascular system: understanding the activities of non-steroidal anti-inflammatory drugs. Nat Rev Drug Discov 5: 75–85, 2006 [DOI] [PubMed] [Google Scholar]
- 34.Otis JS, Burkholder TJ, Pavlath GK. Stretch-induced myoblast proliferation is dependent on the COX2 pathway. Exp Cell Res 310: 417–425, 2005 [DOI] [PubMed] [Google Scholar]
- 35.Ottino P, Bazan H. Corneal stimulation of MMP-1,-9 and uPA by platelet-activating factor is mediated by cyclooxygenase-2 metabolites. Curr Eye Res 23: 77–85, 2001 [DOI] [PubMed] [Google Scholar]
- 36.Reding T, Bimmler D, Perren A, Sun LK, Fortunato F, Storni F, Graf R. A selective COX-2 inhibitor suppresses chronic pancreatitis in an animal model (WBN/Kob rats): significant reduction of macrophage infiltration and fibrosis. Gut 55: 1165–1173, 2006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Rodemann HP, Goldberg AL. Arachidonic-acid, prostaglandin E2 and F2-α influence rates of protein turnover in skeletal and cardiac muscle. J Biol Chem 257: 1632–1638, 1982 [PubMed] [Google Scholar]
- 38.Rodemann HP, Waxman L, Goldberg AL. The stimulation of protein degradation in muscle by Ca2+ is mediated by prostaglandin E2 and does not require the calcium-activated protease. J Biol Chem 257: 8716–8723, 1982 [PubMed] [Google Scholar]
- 39.Sandri M. Signaling in muscle atrophy and hypertrophy. Physiology (Bethesda) 23: 160–170, 2008 [DOI] [PubMed] [Google Scholar]
- 40.Shen W, Li Y, Tang Y, Cummins J, Huard J. NS-398, a cyclooxygenase-2-specific inhibitor, delays skeletal muscle healing by decreasing regeneration and promoting fibrosis. Am J Pathol 167: 1105–1117, 2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Smith RH, Palmer RM, Reeds PJ. Protein synthesis in isolated rabbit forelimb muscles–the possible role of metabolites of arachidonic acid in the response to intermittent stretching. Biochem J 214: 153–161, 1983 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Soltow QA, Betters JL, Sellman JE, Lira VA, Long J, Criswell DS. Ibuprofen inhibits skeletal muscle hypertrophy in rats. Med Sci Sports Exerc 38: 840–846, 2006 [DOI] [PubMed] [Google Scholar]
- 43.Sonnet C, Lafuste P, Arnold L, Brigitte M, Poron F, Authier FJ, Chretien F, Gherardi RK, Chazaud B. Human macrophages rescue myoblasts and myotubes from apoptosis through a set of adhesion molecular systems. J Cell Sci 119: 2497–2507, 2006 [DOI] [PubMed] [Google Scholar]
- 44.Taipale J, Keski-Oja J. Growth factors in the extracellular matrix. FASEB J 11: 51–59, 1997 [DOI] [PubMed] [Google Scholar]
- 45.Tajima T, Murata T, Aritake K, Urade Y, Hirai H, Nakamura M, Ozaki H, Hori M. Lipopolysaccharide induces macrophage migration via prostaglandin D-2 and prostaglandin E-2. J Pharmacol Exp Ther 326: 493–501, 2008 [DOI] [PubMed] [Google Scholar]
- 46.Trappe RA, White F, Lambert CP, Cesar D, Hellerstein M, Evans WJ. Effect of ibuprofen and acetaminophen on postexercise muscle protein synthesis. Am J Physiol Endocrinol Metab 282: E551–E556, 2002 [DOI] [PubMed] [Google Scholar]
- 47.Trappe TA, Fluckey JD, White F, Lambert CP, Evans WJ. Skeletal muscle PGF2α and PGE2 in response to eccentric resistance exercise: influence of ibuprofen and acetaminophen. J Clin Endocrinol Metab 86: 5067–5070, 2001 [DOI] [PubMed] [Google Scholar]
- 48.Yamada S, Buffinger N, DiMario J, Strohman RC. Fibroblast growth factor is stored in fiber extracellular matrix and plays a role in regulating muscle hypertrophy. Med Sci Sports Exerc 21: S173–S180, 1989 [PubMed] [Google Scholar]
- 49.Yamaguchi A, Ishii H, Morita I, Oota I, Takeda H. mRNA expression of fibroblast growth factors and hepatocyte growth factor in rat plantaris muscle following denervation and compensatory overload. Pflügers Arch 448: 539–546, 2004 [DOI] [PubMed] [Google Scholar]