

Abstract
Pd-catalyzed allylic relay Suzuki cross-coupling reactions of secondary alkyl tosylates, featuring a sterically-hindered oxidative addition and precise control of β-hydride elimination, are reported. The identification of a linear free energy relationship between the relative rates of substrate consumption and the electronic nature of the substrate alkene suggests that the oxidative addition requires direct alkene involvement. A study of the effect of chain length on the reaction outcome supports a chelation-controlled oxidative addition.
Introduction
The Pd-catalyzed Suzuki cross-coupling is among the most widely-used transition metal-catalyzed carbon–carbon bond-forming reactions because of the ease with which a vast array of coupling partners can be combined from commercially-available reactants under mild conditions.1 Despite the diversity of coupling partners commonly employed in these reactions, including many sp2 and sp3 nucleophiles and electrophiles, secondary sp3 electrophiles remain difficult to utilize.2,3 This is chiefly a result of the SN2-type oxidative addition of secondary halides and -pseudohalides to Pd(0), which is severely limited through presumed steric interactions.4
We recently reported an unusual example of such an oxidative addition in the context of exploring the reaction depicted in Scheme 1A.5 In this report, primary homoallylic tosylates undergo oxidative addition followed by the relay of Pd to the allylic position via a proposed sequential β-hydride elimination and migratory insertion.6 The resultant stabilized Pd-allyl intermediate then undergoes cross-coupling to an aryl boronic acid to afford difficult-to-access secondary allylic Suzuki products3 with generally good selectivity (>10:1) over the less-hindered linear homoallylic cross-coupling product. Through two mechanistic experiments employing secondary homostyrenyl tosylates, oxidative addition was found to proceed through a stereoinvertive, SN2-type mechanism – a rare example of a Pd-catalyzed oxidative addition of an unactivated secondary electrophile.7 Herein, we present an improved procedure for Pd-catalyzed relay Suzuki reactions of this challenging electrophile class (1, Scheme 1B), as well as preliminary mechanistic evidence as to why such secondary electrophiles are capable of undergoing oxidative addition.
Scheme 1.

A Previous relay Suzuki cross-coupling of primary homostyrenyl tosylates. B. Mechanistic rationale to account for three potential products resulting from the reaction of a secondary homostyrenyl tosylate under Pd(quinox) catalysis.
Since mixtures of allylic (relay) and homoallylic (non-relay) cross-coupling products were routinely observed in reactions of primary homostyrenyl tosylates (Scheme 1A),5 secondary homostyrenyl tosylates such as 1 were anticipated to afford mixtures of three cross-coupling products (Scheme 1B). Mechanistically, these products could result from divergent reaction following oxidative addition. Specifically, from Pd-alkyl B, transmetallation of a boronic acid and subsequent reductive elimination could give homoallylic cross-coupling product 3; alternatively, β-hydride elimination/migratory insertion could transpose the Pd atom to either the stabilized allylic position, A, or to the terminal alkyl position, C, which, after transmetallation and reductive elimination, could afford either the allylic cross-coupling product 2 or the bishomoallylic cross-coupling product 4, respectively. Using Pd(quinox) as catalyst, we now report that relay Suzuki reactions of secondary homostyrenyl tosylates afford the allylic cross-coupling product, 2, exclusively, thus showcasing the control of β-hydride elimination in addition to the surprising oxidative addition of a secondary electrophile.
Results and Discussion
Our previous relay Suzuki cross-coupling conditions, which employed KF·2H2O as base in isopropanol (Scheme 1A), necessitated the use of 10–15 mol % of Pd(quinox)Cl2 to achieve sufficient conversion of secondary homostyrenyl tosylates for mechanistic studies.5 In an effort to decrease the precatalyst load for secondary tosylates, we identified Cs2CO3 and THF/H2O to be a suitable base/solvent combination, allowing the reaction to be carried out at ambient temperature with just 5 mol % load of Pd(quinox)Cl2.8 Under these conditions, the scope of boronic acid coupling partners in the relay Suzuki cross-coupling of 1 was probed using three equivalents of boronic acid (Table 1). Unsubstituted (2a) and 4-alkoxy-substituted phenylboronic acids (2b–2d) were found to afford good yields of the corresponding desired products. On the other hand, the presence of electron-deficient substituents on the arylboronic acid results in noticeably attenuated yields. For example, 3-fluoro-4-isopropoxyphenylboronic acid gives 2e in just 36% yield, 4-chlorophenylboronic acid couples to give 42% yield of 2f, and 3-carbomethoxyphenylboronic acid affords 41% yield of the corresponding relay product 2g (although the methyl ester may be sensitive to the hydrolytic reaction conditions). Interestingly, 3-methoxyphenylboronic acid, which also possesses a relatively electron-deficient boron-bearing carbon atom, is coupled in good yield (2h). Electron-rich 3-ethylphenylboronic acid also affords a very good yield of the corresponding relay product 2i. Polycyclic heteroaromatic boronic acids were also investigated for their propensity to undergo relay cross-coupling. To this end, ortho-substituted 2-dibenzofuranyl boronic acid gave 2j in reasonable yield, and indole-incorporated relay product 2k was accessed in 60% yield.
Table 1.
Scope of aryl boronic acids in the relay Suzuki reaction[a]
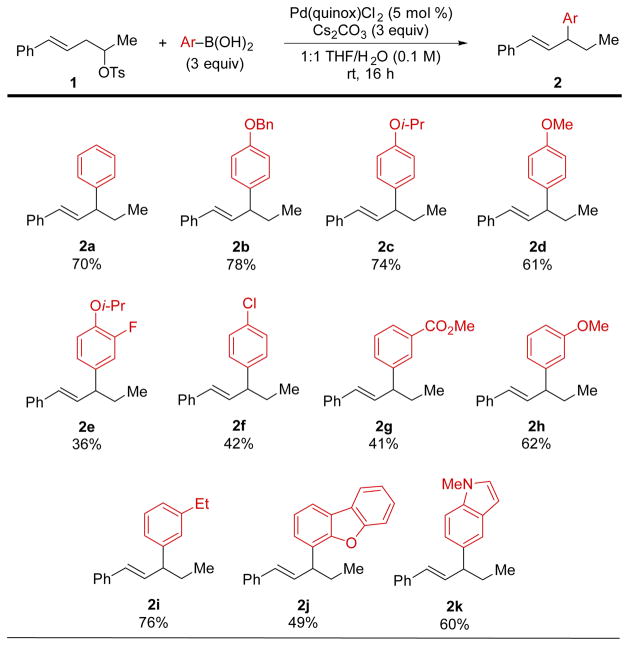
|
Isolated yields are reported.
An important observation based on crude NMR analyses of each of these reactions is that the yield is generally comparable to substrate conversion. That is to say, substrate 1 is not fully consumed in these reactions, even upon doubling the amount of reactants, reagents, and catalyst.8,9 The conversion is not improved substantially by the addition of a second batch of catalyst after 16 h, which suggests that boronic acid decomposition, rather than catalyst decomposition, prevents substrate conversion under these conditions.10 This may also explain the lower yields observed in relay cross-couplings of electron-deficient boronic acids as shown in Table 1.11
Turning our attention towards substrate scope, we next evaluated a series of electronically-disparate alkene substrates through the preparation of para-substituted secondary homostyrenyl tosylates (5a–5f, Table 2). Isolated yields were fair for trifluoromethyl-, chloro-, fluoro-, and methyl- containing products (6a–6d), while the 4-methoxy-substituted variant resisted reaction almost entirely, resulting in less than 5% yield of product 6e by NMR analysis. Finally, substrate 5f, containing an aryl bromide functional group, afforded 20% yield of 6f. In this reaction, both 1 and 2a were observed as byproducts, thus illustrating that aryl bromides are prone to oxidative addition to Pd(0) under these conditions, while aryl chlorides are not (cf., 6b).
Table 2.
Scope of substrates containing para-substituents.[a]
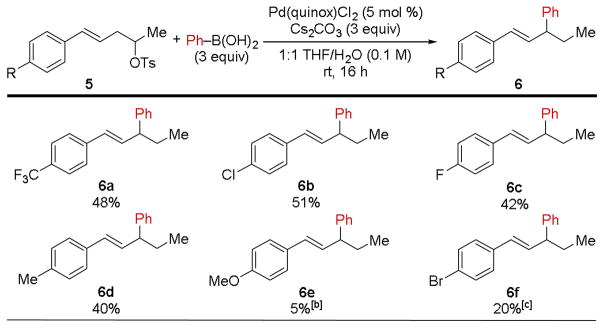
|
Isolated yields are reported.
Yield determined by 1H NMR spectroscopy using CH2Br2 as an ex situ internal standard.
In addition, 1 and 2a were isolated in 10% and 23% yield, respectively.
Based on the observed sensitivity of the reaction to the electronics of the alkene, we became intrigued by the possibility that the styrenyl portion of the substrate may facilitate the oxidative addition of the hindered tosylate. To probe this hypothesis, a relative rate experiment was designed to compare the rate of conversion of homostyrenyl tosylates 5a–5e to that of 1. This revealed that the relative rates correlate to the Hammett σ values12 of the arene substituent (Scheme 2). The positive ρ value observed implies a buildup of negative charge in the transition state of the conversion-influencing step, which suggests an inverse-electron-demand ligand substitution of the alkene onto Pd(0) that would be enabled by a more electron-deficient alkene.13,14 This evidence supports the hypothesis that the substrate alkene is required for the oxidative addition step in relay Suzuki cross-coupling reactions.15
Scheme 2.

Hammett correlation to the relative rates of consumption of para-unsubstituted- and para-substituted secondary homostyrenyl tosylates.
A proposed mechanism for the relay Suzuki cross-coupling reaction of secondary homostyrenyl tosylates is summarized in Scheme 3. Initially, the alkene of 1 coordinates Pd(0) (7) and brings Pd into proximity to the tosylate for the SN2-type oxidative addition, leading to destabilized Pd-alkyl 8. Then, β-hydride elimination may generate the diene–Pd–H intermediate 9, which undergoes migratory insertion to afford the stabilized allyl-Pd intermediate 10.16 Boronic acid transmetallation and reductive elimination may then give rise to the cross-coupling product 2a.
Scheme 3.

Proposed mechanism for the relay Suzuki cross-coupling of a secondary homostyrenyl tosylate to phenylboronic acid.
Based on this newfound insight regarding the alkene-dependent oxidative addition, we sought to investigate the influence that the number of carbon atoms separating the alkene from the tosylate might have on oxidative addition. This was accomplished by first subjecting bishomostyrenyl tosylate 12 to the optimized conditions (Scheme 4). This resulted in reasonable substrate conversion, and all three of the possible cross-coupling products (2a, 3a, and 4a) were observed despite having only observed the allylic relay product (2) in the aforementioned reactions of homostyrenyl tosylates 1 and 5a– 5f. Interestingly, under our previously-reported reaction conditions, the major reaction product is not the allylic relay product 2a, but rather the linear arylation product 4a.5 This selectivity reversal signifies the sensitivity of relay Suzuki cross-couplings to reaction conditions.
Scheme 4.
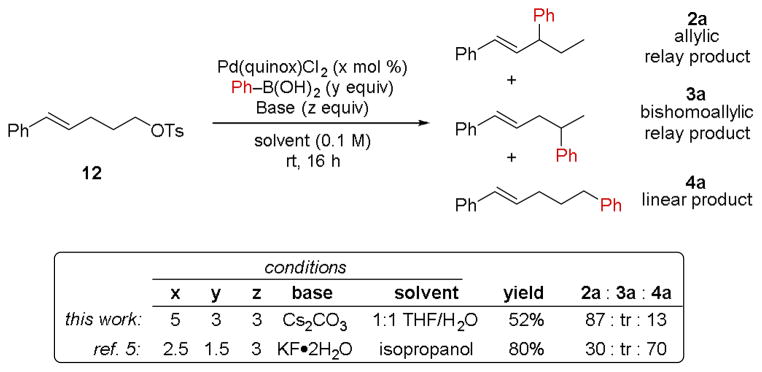
Investigation of the relay cross-coupling of bishomostyrenyl tosylate.
Incorporating an additional carbon between the alkene and tosylate, as in substrate 13, resulted in no reaction (Eq 1). This implies that the olefin and tosylate are not able to bind to the metal simultaneously in order for oxidative addition to be achieved. This may be due to excessive strain in the oxidative addition transition state arising from the increased length of the aliphatic chain, which could prohibit access to the requisite proposed transition state 14.
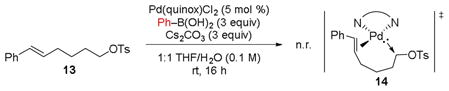 |
(1) |
Conclusions
We have developed an allyl-selective relay Suzuki cross-coupling reaction of secondary homostyrenyl tosylates at ambient temperature. Linear free energy relationship analysis is consistent with a mechanism whereby the substrate alkene coordinates prior to oxidative addition. Systematic evaluation of the distance between the alkene and the electrophile reveals that the alkene must be within three carbon atoms of the oxidant. Thus, homostyrenyl and bishomostyrenyl substituents may now be considered useful functional handles for oxidative additions of unactivated alkyl electrophiles to Pd(0). New cross-coupling reactions exploiting alkene-assisted oxidative addition of traditionally inert alkyl electrophiles are currently being explored.
Supplementary Material
Acknowledgments
The research reported in this publication was supported by the National Institute of General Medical Science of the National Institutes of Health under award numbers R01GM063540 and F32GM099254 (B.J.S.), as well as a UROP undergraduate assistantship from the University of Utah (A.J.B.).
Footnotes
Electronic Supplementary Information (ESI) available: Detailed experimental procedures, reaction optimization data, and spectroscopic data of all new compounds. See DOI: 10.1039/b000000x/
Notes and references
- 1.For reviews on transition metal-catalyzed cross-coupling reactions, see: Diederich F, Stang PJ, editors. Metal-Catalyzed Cross-Coupling Reactions. Wiley-VCH; Weinheim: 1998. de Meijere A, Diederich F, editors. Metal-Catalyzed Cross-Coupling Reactions. Wiley-VCH; Weinheim: 2004. 2nd Completely Revised and Enlarged ed.
- 2.For reviews on cross-coupling reactions of alkyl halides, see: Netherton MR, Fu GC. Top Organomet Chem. 2005;14:85.Frisch AC, Beller M. Angew Chem Int Ed. 2005;44:674. doi: 10.1002/anie.200461432.Glorius F. Angew Chem Int Ed. 2008;47:8347. doi: 10.1002/anie.200803509.Rudolph A, Lautens M. Angew Chem Int Ed. 2009;48:2656. doi: 10.1002/anie.200803611.Nakamura M, Ito S. Modern Arylation Methods. Wiley-VCH Verlag GmbH & Co. KGaA; 2009. pp. 155–181.Kambe N, Iwasaki T, Terao J. Chem Soc Rev. 2011;40:4937. doi: 10.1039/c1cs15129k.
- 3.Secondary allylic electrophiles are a particularly challenging substrate for Pd-catalyzed Suzuki cross-couplings. For leading reports, see: Li C, Xing J, Zhao J, Huynh P, Zhang W, Jiang P, Zhang YJ. Org Lett. 2012;14:390. doi: 10.1021/ol203154j.Zhao J, Ye J, Zhang YJ. Adv Synth Catal. 2013;355:491.
- 4.(a) Pearson RG, Figdore PE. J Am Chem Soc. 1980;102:1541. [Google Scholar]; (b) Hills ID, Netherton MR, Fu GC. Angew Chem Int Ed. 2003;42:5749. doi: 10.1002/anie.200352858. [DOI] [PubMed] [Google Scholar]; (c) Ariafard A, Lin Z. Organometallics. 2006;25:4030. [Google Scholar]
- 5.Stokes BJ, Opra SM, Sigman MS. J Am Chem Soc. 2012;134:11408. doi: 10.1021/ja305403s. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.For recent reports on selective catalytic C–C bond-forming reactions involving unstabilized Pd-alkyl intermediates, see: DeLuca RJ, Sigman MS. J Am Chem Soc. 2011;133:11454. doi: 10.1021/ja204080s.Saini V, Sigman MS. J Am Chem Soc. 2012;134:11372. doi: 10.1021/ja304344h.Aspin S, Goutierre AS, Larini P, Jazzar R, Baudoin O. Angew Chem Int Ed. 2012;51:10808. doi: 10.1002/anie.201206237.Werner EW, Mei TS, Burckle AJ, Sigman MS. Science. 2012;338:1455. doi: 10.1126/science.1229208.DeLuca RJ, Sigman MS. Org Lett. 2012;15:92. doi: 10.1021/ol303129p.Shang Y, Jie X, Zhou J, Hu P, Huang S, Su W. Angew Chem Int Ed. 2013;52:1299. doi: 10.1002/anie.201208627.Aspin S, López-Suárez L, Larini P, Goutierre AS, Jazzar R, Baudoin O. Org Lett. 2013;15:5056. doi: 10.1021/ol402398s.Millet A, Larini P, Clot E, Baudoin O. Chem Sci. 2013;4:2241.Mei TS, Werner EW, Burckle AJ, Sigman MS. J Am Chem Soc. 2013;135:6830. doi: 10.1021/ja402916z.Saini V, Liao L, Wang Q, Jana R, Sigman MS. Org Lett. 2013;15:5008. doi: 10.1021/ol4023358.
- 7.Xiao B, Liu ZJ, Liu L, Fu Y. J Am Chem Soc. 2013;135:616. doi: 10.1021/ja3113752. [DOI] [PubMed] [Google Scholar]
- 8.For complete optimization data, please refer to the Supporting Information.
- 9.The secondary tosylates were not observed to undergo hydrolysis under the reaction conditions.
- 10.Substantial quantities of boronic acid homocoupling and protodeboronated products are observed in the crude reaction mixtures.
- 11.Aqueous basic conditions such as these promote boronic acid protodeboronation, especially of electron-deficient arylboronic acids. For a review, see: Hall DG. Boronic Acids. Wiley-VCH Verlag GmbH & Co. KGaA; 2011. pp. 1–133.
- 12.Hansch C, Leo A, Taft RW. Chem Rev. 1991;91:165. [Google Scholar]
- 13.While Pd0(quinox)–alkene complexes have not previously been implicated as reactive intermediates, PdII(quinox) and PdII(pyrox) catalysts are well-known to engage alkenes. For mechanistic studies, see: Michel BW, Steffens LD, Sigman MS. J Am Chem Soc. 2011;133:8317. doi: 10.1021/ja2017043.Weinstein AB, Stahl SS. Angew Chem Int Ed. 2012;51:11505. doi: 10.1002/anie.201206702.Holder JC, Zou L, Marziale AN, Liu P, Lan Y, Gatti M, Kikushima K, Houk KN, Stoltz BM. J Am Chem Soc. 2013;135:14996. doi: 10.1021/ja401713g.
- 14.For leading reports on inverse-electron-demand alkene ligand substitution on Pd(0) complexes, see: Stahl SS, Thorman JL, de Silva N, Guzei IA, Clark RW. J Am Chem Soc. 2002;125:12. doi: 10.1021/ja028738z.Popp BV, Thorman JL, Morales CM, Landis CR, Stahl SS. J Am Chem Soc. 2004;126:14832. doi: 10.1021/ja0459734.
- 15.For alkene involvement in Ni(0)-catalyzed Negishi cross-coupling reactions, see: Devasagayaraj A, Stüdemann T, Knochel P. Angew Chem, Int Ed Engl. 1995;34:2723.Nielsen DK, Huang CY, Doyle AG. J Am Chem Soc. 2013;135:13605. doi: 10.1021/ja4076716.
- 16.Liao L, Sigman MS. J Am Chem Soc. 2010;132:10209. doi: 10.1021/ja105010t. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.