

Abstract
Engineering artificial protein hydrogels for medical applications requires precise control over their mechanical properties, including stiffness, toughness, extensibility and stability in the physiological environment. Here we demonstrate topological entanglement as an effective strategy to robustly increase the mechanical tunability of a transient hydrogel network based on coiled-coil interactions. Chain extension and entanglement are achieved by coupling the cysteine residues near the N- and C- termini, and the resulting chain distribution is found to agree with the Jacobson-Stockmayer theory. By exploiting the reversible nature of the disulfide bonds, the entanglement effect can be switched on and off by redox stimuli. With the presence of entanglements, hydrogels exhibit a 7.2-fold enhanced creep resistance and a suppressed erosion rate by a factor of 5.8, making the gels more mechanically stable in a physiologically relevant open system. While hardly affecting material stiffness (only resulting in a 1.5-fold increase in the plateau modulus), the entanglements remarkably lead to hydrogels with a toughness of 65,000 J m-3 and extensibility to approximately 3,000% engineering strain, which enables the preparation of tough yet soft tissue simulants. This improvement in mechanical properties resembles that from double-network hydrogels, but is achieved with the use of a single associating network and topological entanglement. Therefore, redox-triggered chain entanglement offers an effective approach for constructing mechanically enhanced and responsive injectable hydrogels.
INTRODUCTION
Artificially engineered protein hydrogels have been widely investigated for regenerative medicine, tissue engineering and other biomedical applications.1-3 Advances in molecular biology and protein biosyntheses allow precise control of the protein structure, enabling bottom-up design of the gel mechanical properties. These mechanical properties play an important role in controlling cell-material interactions. For example, the differentiation of stem cells is greatly influenced by matrix elasticity,4, 5 and an abnormal stiffness of the matrix can alter the biological responses of cells.6 By incorporating different modular building blocks such as coiled-coils or elastin-like domains, engineered protein hydrogels can achieve a variety of mechanical properties, including elasticity, toughness and resilience.7-9 In particular, physical hydrogels with coiled-coil associating domains show shear-banding flow followed by remarkably rapid self-healing, which enables their use as injectable cell-encapsulated materials with high cell viability post-injection.10 Moreover, the binding affinity of coiled-coils can be changed by pH, ionic strength and temperature, allowing the mechanical properties of hydrogels to be tuned by external stimuli.9, 11, 12
Coiled-coil proteins have well-defined molecular structures, including monodisperse primary chains and sequence-defined coiled-coil domains that make them interesting model systems where the structure-property relationships, including thermodynamics (such as equilibrium modulus) and kinetics (such as stress relaxation) of gels can be compared with a broad spectrum of polymer physics theories.13-18 The physics of transient networks has attracted great interest since 1946, when Green and Tobolsky first proposed a kinetic model to capture the relaxation dynamics of transient networks.15 Tanaka and Edwards generalized the Green-Tobolsky theory by realizing that the deconstruction and recreation rates of the network junctions can be different, depending on the end-to-end distance of the network strand.16, 19-21 Annable et al. examined the rheology of hydrophobic ethoxylated urethane associative thickeners (HEUR-AT) in aqueous solution, further demonstrating that the complex network topologies (such as superbridges) impart the concentration dependence of the rheological behaviors to associating polymers.17 Leibler, Rubinstein, Colby and Semenov proposed the sticky Rouse and sticky reptation theories to describe the relaxation dynamics of polymers with pendent associating groups.22-24 They find that many material properties, such as the relaxation time and the zero-shear-rate viscosity, have a strong dependence on the concentration. The sticky Rouse and sticky reptation models have found the success in explaining some of the viscoelastic behaviors of many supramolecular polymer networks.25-27 In addition to these studies of polymers with associating groups covalently bonded along the main chain, several authors have investigated polymers with dimeric associating groups localized only at the chain ends, capable of chain extension but not network formation. Cates proposed a living polymer model to study polymers with end-functionalized dimeric associating domains.28 The model reveals several relaxation mechanisms, depending on the ratio of the lifetime of the associating groups to the reptation time, and ratio of the entanglement length to the average length of polymers. Many experimental studies confirm Cates’ theory, especially those investigating polymers with self-complementary H-bonding motifs.29, 30
Understanding the structure-property relationship of associating polymers as well as reversible transient networks enables preparation of physical hydrogels with versatile mechanical properties; however, there are several drawbacks of physically crosslinked injectable hydrogels that limit their practical applications. These gels generally undergo rapid erosion (dominated by surface erosion) when physical interactions governing the transient network relax due to thermal fluctuations.7, 31 In addition, a relatively low yield stress makes these hydrogels susceptible to both creep and mechanical failure if they are stressed during use.10 Finally, physically crosslinked hydrogels are usually brittle, exhibiting low toughness and extensibility. Although in certain cases, many of these shortcomings can be addressed by chemically crosslinking the protein strands,32-34 the injectable flow behavior is lost.
While polymer hydrogels are traditionally brittle, high molecular weight polymer melts are tough due to the presence of physical entanglements between polymer chains. These entanglements impart elastic recoil35 and strain hardening behavior36, 37 to polymer liquids. The low polymer concentration in gels requires a very high polymer molecular weight to achieve chain entanglements, and a few authors have reported chain entanglement in chemically crosslinked synthetic polymer gels, where the molecular weight of network strand exceeds the entanglement molecular weight.38, 39 However, it has not been realized in biocompatible and mechanically tunable engineered protein hydrogels. The molecular weight of engineered proteins achievable in high yield by biosyntheses is limited; therefore, an alternative relies on efficient coupling reactions to link protein molecules to produce long chains.40-43 Disulfide formation via thiol oxidation is a protein conjugation strategy that takes advantages of natural cysteine residues, does not require additional (potentially toxic) chemical modifiers, and can be driven to high yield with simple synthetic procedures. In addition, the disulfide bonds can break down in a reducing environment, providing an opportunity to control the molecular architecture with redox stimuli, and producing oxidatively responsive materials. Recently, responsive disulfide formation has been used to stabilize self-assembled nanofibers from peptide amphiphiles44 and to chemically crosslink coiled-coil domains in artificially engineered protein hydrogels.45 Although high molecular weight synthetic copolymers have been prepared by oxidation of thiols on the polymer chain ends,46 to the best of our knowledge, this strategy has not been applied to synthesize high molecular weight protein gels.
Herein, we demonstrate oxidatively-triggered disulfide bond formation as a method to prepare high molecular weight artificially engineered protein hydrogels containing coiled-coil associations. Under appropriate processing conditions these gels may be prepared in a physically entangled state. The physical entanglements lead to enhanced toughness, similar to that of double network hydrogels, with a single network. Unlike other approaches to gel toughening, large improvements in toughness and extensibility are demonstrated with only a small change in the hydrogel stiffness. Finally, it is shown that the oxidative trigger may be used to turn the entanglements on and off, leading to responsive transitions between a brittle and tough state that enable entanglements and the resulting mechanical property improvements to be incorporated into injectable biomaterials.
EXPERIMENTAL SECTION
Protein Synthesis and Purification
The genes encoding for Cys-P4-Cys and P4 were prepared and transformed into the SG13009 strain of Escherichia coli for protein expression by Glassman et al.47. Proteins were expressed at 37 °C in 1 L TB culture in a shaker flask. The expression was induced by 1.0 mM isopropyl β-D-1-thiogalactopyranoside (IPTG) at an optical density of 0.9 – 1.0. Six hours after induction, the cells were harvested by centrifugation at 4 °C and 6,000 RPM for 10 minutes, and the cell pellets were stored at -80 °C overnight. The cells were resuspended in lysis buffer (10 mM Tris, 1 mM EDTA, 100 mM NaCl, 5 mM MgCl2, pH 7.5). Lysozyme (100 mg) was added and the mixture was incubated at 4 °C for 2 hours, followed by sonication for 30 minutes. The lysate was centrifuged at 4 °C and 9,000 RPM for 30 minutes. DNAse I and RNAse A (2 mg each) were added to the clarified lysate and the mixture was incubated at 37 °C for 2 hours. The lysate was further denatured by 8 M urea, buffered with 100 mM phosphate, and 20 mM β-mercaptoethanol. Protein was first purified by precipitation in 20% – 30% ammonium sulfate and was further purified by anionic exchange using QAE-Sephadex A-50 resin (GE Healthcare) in 6 M urea, 20 mM Tris and 20 mM β-mercaptoethanol as wash buffer. Impurities were eluted using up to 100 mM NaCl and protein was eluted at 1 M NaCl. The purity was assessed by SDS-PAGE (Figure S1). A typical expression yield was 150 mg L-1.
Oxidation of Cysteine Residues to Trigger Chain Extension
Protein was dissolved in a buffer with 6 M urea, 20 mM Tris and pH 8.0 to a final concentration of 10 (w/v)%. To reduce the existing disulfide bonds during protein purification, a two-fold excess TCEP was added. The mixture was allowed to stir for a week at 4 °C. Oxidized protein was dialyzed and lyophilized. Oxidation can also be triggered by other methods (Figure S2): (1) Small amount of hydrogen peroxide was added to the reduced hydrogel, followed by mechanical mixing to ensure homogeneous distribution of hydrogen peroxide; (2) Chain extended protein can be reduced by the addition of a five-fold excess of DTT, following by periodic mechanical mixing to allow air oxidation.
Sodium Dodecyl Sulfate-Polyacrylamide Gel Electrophoresis (SDS-PAGE) Analysis of Chain Extension Reaction
The molecular weight distribution of the product mixture after chain extension reaction was analyzed by SDS-PAGE (Mini-PROTEAN® TGX™ gels, 4-15%, BioRad) under non-reducing condition at 150 V. The linear and ring species were assumed to have the same electrophoretic mobility. The weight fraction of species was estimated using densitometry with BioRad Image Lab software.
Circular Dichroism (CD)
Proteins were dissolved in phosphate buffer to a final concentration of 20 μM. Protein solutions were further filtered through 0.2 μm PES filters. The protein concentrations after filtering were determined spectrophotometrically. CD spectra were recorded on an Aviv Model 202 Circular Dichroism Spectrometer (Figure S3). Experiments were performed in a rectangular cell with a path length of 1 mm. Measurement was taken from 260 nm to 195 nm with 1 nm resolution and an averaging time of 10 s.
Phosphate Buffer Preparation
The phosphate buffer was prepared by dissolving 13 mmol NaH2PO4·H2O, 87 mmol Na2HPO4·7H2O in 1L MilliQ water to reach a final pH of 7.6, and was further filtered through 0.2 μm PES filters for sterilization.
Rheology
Frequency sweep and creep experiments were performed on an Anton Paar MCR 301 rheometer. A cone-plate geometry (25 mm and 1° cone) was used with TruGap® accessories. Protein samples were hydrated in phosphate buffer to a desired concentration for two days before the experiment. Samples were centrifuged to remove bubbles before loading onto the rheometer, and the edge of hydrogel sample was coated with mineral oil to minimize water evaporation. To eliminate thermal and shear history, samples were heated to 90 °C and cooled to 25 °C at 5 °C min-1. This brief thermal treatment unfolds the coiled-coil domains upon heating above 52 °C,9 enabling rapid stress relaxation in the protein network. Frequency sweep experiments were performed from 100 rad s-1 to 0.001 rad s-1 at 1% strain, which was confirmed to be in the linear viscoelastic regime (LVE) using strain sweep experiments. In creep experiments, samples were subjected to a constant load for 2 hours, after which the load was subsequently removed to examine the recovery behavior for 2 hours. Four different loading conditions (25 Pa, 50 Pa, 75 Pa and 100 Pa) were tested to ensure that the test was in the linear regime.
Surface Erosion
Protein hydrogel samples were hydrated in phosphate buffer to a final concentration of 15 (w/v)% for two days before the erosion experiment. Approximately 50 μL of protein hydrogel samples were loaded into a cylinder well with 6.4 mm in diameter and 8.5 mm in height in a 96-well plate. Hydrogels were centrifuged for 2 hours at 4,000 g to flatten the hydrogel surface and the thickness of each gel sample was 1.50 ± 0.12 mm. Extra phosphate buffer (250 uL) was added into each well. The erosion experiment was performed at 35 °C without mechanical agitation. The protein concentration in the supernatant was measured spectrophotometrically, with each measurement performed in triplicate.
Uniaxial Compression
Hydrogel specimens were pressed into cylindrical Teflon molds of 8 mm diameter and 8 mm height. Samples were annealed in a humidified chamber at 37 °C for 3 hours and stored in fridge at 4 °C for 2 days to allow stress relaxation. No visible defects were observed within the samples. Uniaxial compression experiments were performed on a Zwick/Roell Z2.5/TS1S materials testing machine and TestXpert V10.1 master software (Ulm, Germany) with a 20 N load cell. All tests were performed at room temperature, 24 ± 1 °C. A layer of soapy water was used to provide lubrication during compression. A preload of approximately 0.005 – 0.01 N was applied to samples to initiate contact. Two test procedures were used in this study. (A) Compression experiments were performed at three different nominal strain rates (0.1% s-1, 1% s-1 and 10% s-1) to a final true strain of 160%, followed by unloading to 0% strain at a nominal strain rate of 10% s-1. (B) Interval loading at 1% s-1 nominal strain rate to 160% true strain, with partial unloading (10% s-1 nominal strain rate) segments at 40%, 80%, 120% to probe the relaxation behavior.
Uniaxial Tension
For better visualization, hydrogels were hydrated in phosphate buffer supplemented with 0.1 mg mL-1 fluoresceinamine (isomer I, Sigma). Hydrogel specimens were prepared in rectangular Teflon molds 20 mm in width, 28 mm in length and 2 mm in thickness. Samples were annealed in a humidified chamber at 37 °C for 3 hours and stored in fridge at 4 °C for 2 days to allow stress relaxation. A custom designed cutter was used to obtain a dogbone shaped tensile specimen with a neck width of 4 mm (Figure S4). No visible defects were observed after cutting. Uniaxial tension experiments were performed on a Zwick/Roell Z2.5/TS1S materials testing machine and TestXpert V10.1 master software (Ulm, Germany) with a 20 N load cell. All tests were performed at room temperature, 24 ± 1 °C. Sandpaper was glued to the grips to provide friction and to ensure that the sample did not slip. The ramp-relaxation testing protocol consisted of loading ramps alternated with relaxation intervals of 600 seconds, until specimen failure. During the loading ramps the specimens were stretched at an average true strain rate at 0.06 s-1 to increasing levels of engineering strain (625%, 1,250% and 2,500%, 3,750%) until mechanical failure. Three independent experiments were performed and the error bars reported in the text are the standard deviations of the measurement. To study the effect of the wait period on the mechanical performance of gels, a one minute wait time was used as control (Figure S7).
RESULTS AND DISCUSSION
Oxidatively Responsive Topological Entanglements for Mechanical Reinforcement
Chain extension by thiol coupling in concentrated protein hydrogels leads to topological entanglement as the molecular weight is increased during macromolecular polycondensation, providing a basis for oxidatively responsive changes in gel mechanics. The artificial proteins Cys-P4-Cys and P4 (Figure S1) are chosen as model systems to study the chain extension effect: both proteins contain four coiled-coil domains (P) separated by flexible polyelectrolyte blocks (C10) (Figure 1A), but cysteine residues are engineered into Cys-P4-Cys near the N- and C-termini. Hydrophobic interactions and salt bridging drive the coiled-coil domains to form pentameric bundles, providing physical crosslinks in the hydrogel. The chain extension reaction by disulfide bond formation leads to the formation of high molecular weight products. Densitometry analysis using SDS-PAGE of the oxidation reaction mixture provides an estimation of the molecular weight distribution, which is expected to follow the Jacobson-Stockmayer distribution.48 This theory assumes that the chain distribution of the linear species obeys the Flory-Schulz distribution, and it additionally accounts for the effect of intramolecular polycondensation (ring formation). A nonlinear regression fit to this model gives an overall end-group conversion of p = 0.712 ± 0.061 and a ring weight fraction of wr = 0.262 ± 0.148. The weight average degrees of polymerization of the chain fraction, the ring fraction, and the system are X̄cw = 4.13 ± 1.17, X̄rw = 1.42 ± 0.13, and X̄w = 3.43 ± 0.95, corresponding to weight average molar mass of M̄cw = 261.0 ± 73.9 kDa, M̄rw = 89.7 ± 8.2 kDa, and M̄w = 216.8 ± 60.0 kDa (Figure 1B, see Supporting Information for calculation details). Despite the difficulty inherent in achieving high conversion by coupling high molar mass proteins, a significant fraction of high molecular weight chain entangled products was formed. Compared to the theoretical distribution, the monomeric protein has a smaller fraction of loops. This discrepancy may be due in part to approximations in the Jacobson-Stockmayer theory (Gaussian chain statistics and equal reactivity of endgroups) or also due to a fraction of cysteine residues that are unreactive (i.e. due to oxidation to sulfones). Circular dichroism (CD) experiments reveal that chain extension does not affect the folded structure of coiled-coils (Figure S3), suggesting that the folded coiled-coil junctions are unperturbed by chain extension.
Figure 1.
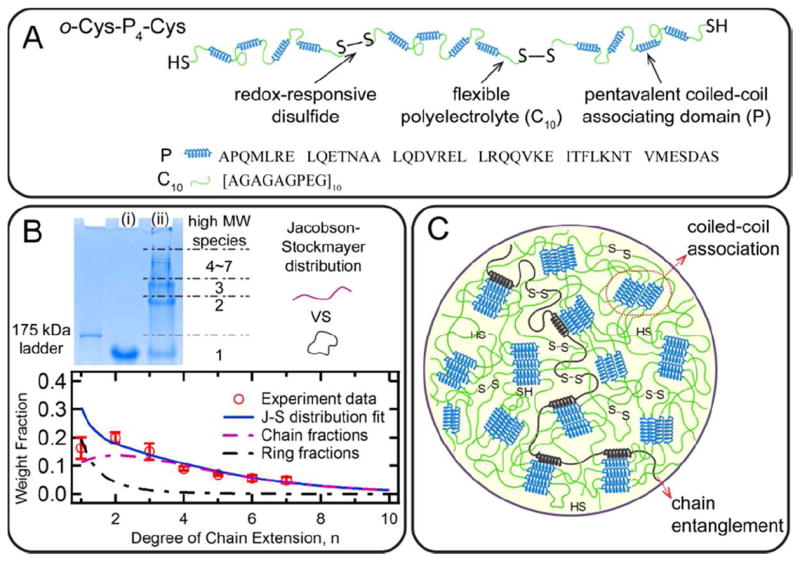
Illustration of the hydrogel design. (A) Molecular design of oxidatively-responsive chain extended artificially engineered proteins. (B) SDS-PAGE characterizing the molecular weight and its distribution of extended chains: (i) Cys-P4-Cys before oxidation reaction, and (ii) chain extended protein mixture. The molecular weight distribution is analyzed by densitometry and further fit to the Jacobson-Stockmayer distribution that accounts for the intramolecular looping. Error bars represent the standard deviations of three separate experiments. (C) Schematic illustration of networks combining coiled-coil association and chain entanglement in protein hydrogels.
Chain entanglements manifest in the linear viscoelastic response of the protein gels through a small increase in the high frequency plateau of the storage modulus and the appearance of a second plateau at low frequency (Figure 2A). At frequencies from 10 rad s-1 to 100 rad s-1 in small amplitude oscillatory shear (SAOS), both P4 and o-Cys-P4-Cys (oxidized, chain extended Cys-P4-Cys) display a plateau storage modulus (G’∞), at 10,500 ± 120 Pa and 15,300 ± 650 Pa, respectively. However, for P4, a crossover in G’ and G” is observed at approximately 0.35 rad s-1; below this frequency, G’ and G” approach to the terminal relaxation behavior (G’ ~ ω2 and G” ~ ω). This qualitative behavior is typical for physical gels.10, 49 A noticeable deviation from the Rouse-like behavior in this low frequency regime (starting from 0.01 rad s-1) might be due to the effect of coiled-coil association on the Rouse relaxation, resembling unentangled transient polymer networks with blocky H-bonding domains.27 In contrast, the simple oxidative chain extension of Cys-P4-Cys yields a gel where G’ is larger than G” over the entire frequency window, and a second plateau modulus (~ 900 Pa) is observed at low frequencies down to 10-3 rad s-1. This behavior is qualitatively similar to entangled polymer solutions and melts.50
Figure 2.
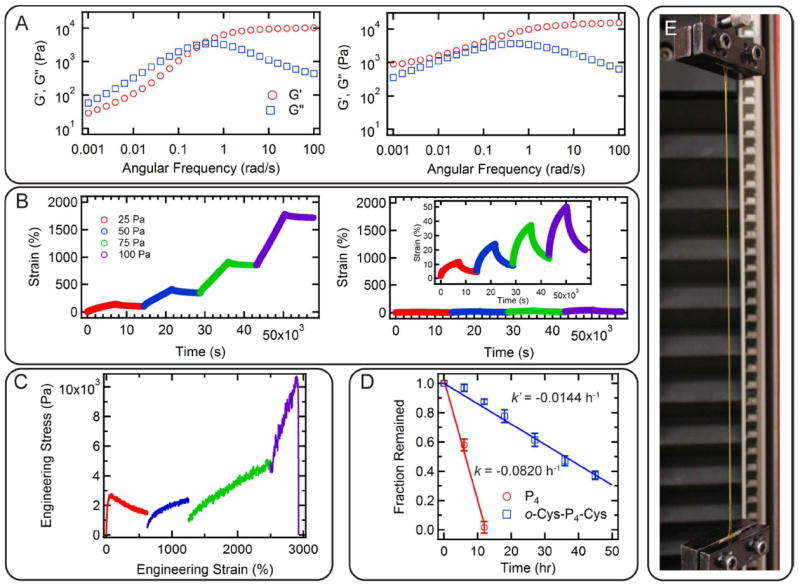
Comparison of the mechanical response of unextended gels and chain extended gels. (A) The effect of chain entanglement is demonstrated in the linear viscoelastic domain: frequency sweeps of hydrogel P4 (left) and o-Cys-P4-Cys (right) at 15 (w/v)% at 25 °C. (B) Compared to P4 (left), the entangled o-Cys-P4-Cys hydrogel (right) shows significant creep resistance and elastic recovery. The inset in the right panel shows the same data magnified to enhance detail. Measurements were performed on 15 (w/v)% samples at 25 °C. (C) Uniaxial tensile experiment (25 °C) on o-Cys-P4-Cys at 15 (w/v)% showing remarkable improvement on the hydrogel’s toughness and extensibility upon introducing entanglement. (D) Surface erosion of hydrogel P4 and o-Cys-P4-Cys at 15 (w/v)% at 35 °C. The erosion rate was estimated from a linear fit of the data. Error bars represent the standard deviations of three separate experiments. (E) A stretched dogbone gel specimen o-Cys-P4-Cys at 15 (w/v)% during tensile loading at 3,100% engineering strain.
To prove that this low frequency plateau modulus results from the formation of topological chain entanglements due to an increase in the molecular weight of o-Cys-P4-Cys, two simple tests are performed. First, chain extension is shown to be reversible, which provides the opportunity to turn entanglement on and off by redox stimuli. The addition of mild reducing agents such as TCEP to o-Cys-P4-Cys chain extended hydrogels causes the entanglement effects on mechanical properties to disappear as the molecular weight of the protein is reduced (Figure S2A). The term “mild” indicates that the added reagents do not chemically alter amino acid functional groups (except cysteines) or the protein structure. The robustness of thiol oxidation as a method to introduce entanglements via chain extension is also demonstrated by the use of other oxidation methods (Figure S2B). It is shown that the entanglement forms by adding a mild oxidant H2O2 to the reduced Cys-P4-Cys hydrogel, as evidenced by the similar properties observed in SAOS experiment. Second, the low frequency plateau modulus is shown to weakly depend on temperature. The entanglement plateau becomes more obvious as the frequency sweep spectrum shifts to the high frequency regime at elevated temperature (Figure S5); however, the physical associations among coiled-coils show a strongly temperature-dependent relaxation (Figure S6). The coiled-coil relaxation time is estimated from the frequency where a maximum in G” occurs in the high frequency regime, τc= 2π/ωh.22, 51 In general, the coiled-coil relaxation time decreases with increasing temperature according to the Arrhenius law, τc ~ exp(1/T), suggesting that the hydrogel properties are predominantly governed by association-disassociation dynamics in the high frequency regime.52
The presence of chain entanglements in these protein gels creates many of the same enhancements observed in double network hydrogels,47, 53-55 but with the use of a single network with topological entanglement. While hydrogel P4 is so brittle that its toughness and extensibility could not be measured in tensile loading, the entangled gels show enhanced toughness of 65,000 ± 24,500 J m-3 due to their dramatic increase in extensibility under a designed test profile, with failure engineering strain of 2,970 ± 860% (Figure 2C, the relationship between the engineering stress and strain and the true stress and strain can be found in Supporting Information). It is also particularly noteworthy that the presence of these topological entanglements leads to large increases in toughness and extensibility without a large increase in the small-strain modulus, which provides an effective approach to prepare tough yet soft hydrogels. Chain entanglements also lead to a decrease in the irrecoverable creep compliance by a factor of 7.2, from 0.227 Pa-1 for P4 to 1.63 Pa-1 for o-Cys-P4-Cys (Figure 2B), as well as a decrease in the surface erosion rate by a factor of 5.8 (Figure 2D), making the gels significantly more robust under sustained load and in open systems, where physical hydrogels have traditionally had a performance disadvantage relative to other materials.31, 45, 56
While the P4 hydrogel creeps readily under all examined loads (from 25 Pa to 100 Pa) and reaches a final strain of ~ 1,700% after four steps of loading-unloading, hydrogel o-Cys-P4-Cys only strains to 20% under the same conditions, almost two orders of magnitudes lower than that of P4. Elastic recovery after loading in the linear regime is 80% ~ 90%, comparable to some synthetic shape-memory elastomers57, 58 due to the elastic recoil of the entangled chains. Interestingly, unlike many double network hydrogels with chemical crosslinks, the chain extended hydrogel is still hand-injectable through a 26-gauge needle even with the presence of chain entanglements (Figure 3). To investigate the flow behavior during injection, the gel is subjected to steady shear for 50 strain units at 1 s-1 strain rate. The rupture of the transient network occurs within less than 10 s, with its shear modulus dropping from 11,200 Pa to 380 Pa. Further shear leads to a continuous slow decrease in the modulus. The recovery behavior after shear is monitored under SAOS at 1 rad s-1 for 1,000 s. It is found that the mechanical strength of gels rapidly recovers to more than 99% of its original modulus within several seconds. Such nearly-instantaneous recovery kinetics is consistent with observation in other protein hydrogels with the same associating coiled-coil domains.10
Figure 3.
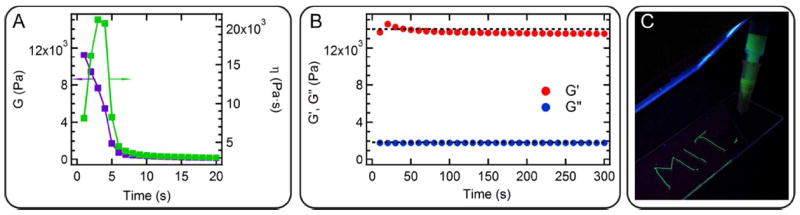
Rapid shear and recovery behavior of the hydrogel o-Cys-P4-Cys at 15 (w/v)%. (A) Fast drop in shear modulus and viscosity at the start-up of steady shear at 1 s-1 strain rate for 50 s. (For clarity, only data from the first 20 s is shown.) (B) Nearly instantaneous recovery of the mechanical properties of gels after shear, examined under SAOS at 1 rad s-1 and 1 % stain for 1,000 s. (For clarity, only data from the first 300 s is shown.) Dash lines in black showing the original values of G’ and G”. (C) Demonstration of the hand-injectability of the chain extended hydrogel through a 26-gauge needle. The concentration of fluorescein is at 0.1 mg mL-1 in the gels.
From Chain Extension to Topological Chain Entanglement
Chain extension leads to topological entanglement when the protein concentration is above the critical entanglement concentration, as is the case for entangled polymer solutions and melts. The contribution of chain extension/entanglement to the network modulus is examined by comparing the frequency spectra of both o-Cys-P4-Cys and P4 hydrogels at different protein concentrations and temperatures (Figure S5 and Figure S6, respectively). At 10.0 and 12.5 (w/v)%, the entanglement plateau signature is absent in the low-frequency regime, while the entanglement plateau is observed for concentrations of 15.0 (w/v)% and above. The observed critical entanglement concentration is close to that estimated using the sticky reptation theory (see calculation in Supporting Information). The quantitative relationship between the entanglement plateau modulus and the protein concentration is shown in Figure 4A. For entangled polymer solutions,51 the plateau modulus scales as Ge ~ φα, where φ is the volume fraction of the protein in gels. For the o-Cys-P4-Cys gels linear regression yields a concentration scaling exponent of α = 7.0. Compared to synthetic polymers that have a typical value of α in the range of 2.0 ~ 2.3, the entanglement plateau modulus of o-Cys-P4-Cys hydrogels depends strongly on the concentration. Two effects may contribute to this stronger than expected concentration dependence. First, interchain coiled-coil interactions may enhance the long range topological constraints between entangled chains, effectively decreasing the entanglement molecular weight (Me). Me in gels at different concentrations can be estimated from the entanglement plateau modulus (Figure 4A).59 According to the tube theory,60 the dynamic topological interaction can only be observed when the system has a molecular weight larger than Me. For hydrogels at low protein concentrations, the predicted Me is larger than 500 kg mol-1. The lack of entanglement comes from the fact that Me is much larger than the experimentally measured molecular weight for chain extended proteins. Second, topological rearrangement of the coiled-coil network at increasing concentration leads to an increased degree of entanglement in hydrogels. This effect is evidenced by changes in the plateau modulus of the gels. The modulus contributed by the coiled-coil associations is obtained by subtracting the entanglement plateau modulus from the high frequency modulus (Figure 4B), and the corresponding number density of the elastically effective chains is calculated based on the affine network theory (Figure 4C). It is found that chain extension and entanglement results in an increase in the plateau modulus at high frequency by a factor of 1.32 to 1.69, higher than the theoretical prediction of 1.23 (see Supporting Information for calculation details). Although a relatively high ratio is observed at low protein volume fractions, this measurement is in the vicinity of the critical gelation concentration where a small shift in gelation concentration due to chain extension effectively shifts the phase separation boundary, causing the ratio in moduli to increase. At higher concentrations, the increase in the number density of elastically effective chains suggests that chain extension and entanglement restructure the associative protein network. The transformation from intramolecular coiled-coil association to intermolecular association gives the strong concentration dependence of the entanglement plateau, consistent with the sticky reptation theory.24
Figure 4.
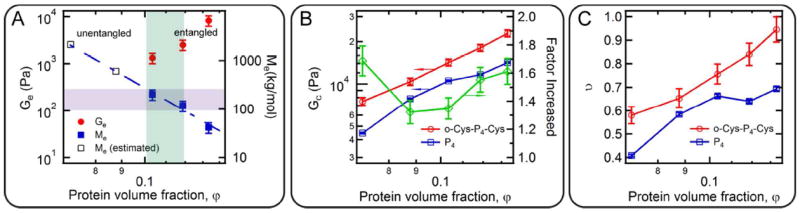
The contribution of chain entanglement and coiled-coil association to network mechanics. (A) Entanglement plateau modulus and the entanglement molecular weight as a function of protein concentration. The purple shaded area highlights the estimated molecular weight (from the lowest Mn to the highest Mw) from the analysis based on the Jacobson-Stockmayer theory. The green shaded area marks the experimentally observed onset of chain entanglement. (B) Comparison of plateau modulus contributed by coiled-coil association as a function of protein concentration at 25 °C (red for hydrogel o-Cys-P4-Cys, and blue for hydrogel P4). Data in green showing the ratio of the two moduli at the same concentration. (C) Comparison of the fractions of elastically effective chains in o-Cys-P4-Cys and P4. Error bars represent the standard deviations of the measurement. (N = 3)
Interactions between Topological Entanglement and Coiled-coil Association
The presence of chain entanglements in the protein gel significantly slows the relaxation of coiled-coil associations, as seen in frequency sweep in SAOS (Figure 5A, Figure S5 and Figure S6). At low protein concentration (10.0 and 12.5 (w/v)%), the relaxation times of coiled-coils in both P4 and o-Cys-P4-Cys hydrogels are very similar, as determined by τc = 2π/ωh. On the contrary, when the protein concentration exceeds the critical entanglement concentration, the coiled-coil relaxation time in the chain extended hydrogel is two to three times higher than that in the lower molecular weight gel. Although the association-disassociation dynamics of the coiled-coil domain localizes on a small length scale (the length scale of an alpha helix, ~5 nm), we hypothesize entanglements on a larger length scale (the length scale of an entangled segment) slow diffusion of the extended protein chains and effectively constrain thermal fluctuation of the coiled-coils, as is predicted by the sticky reptation theory.22 While the microscopic coiled-coil association and dissociation rates may remain unchanged, the slowed diffusion reduces the ability of the coiled-coil to exchange binding partners. According to sticky reptation theory, it is reasoned that a coiled-coil typically returns to its original cluster after an unsuccessful search to bind to a new cluster, effectively coupling stress relaxation due to coiled-coil strand exchange to the slower reptation dynamics.24 In addition to entanglements, the slowing of coiled-coil dynamics is likely responsible for some of the observed decrease in creep compliance and erosion rate (Figure 2B and Figure 2C).
Figure 5.

The interplay between the coiled-coil association and chain entanglement. (A) Coiled-coil relaxation time as a function of concentration and temperature, determined from the frequency where G” reaches its maximum at the high frequency regime in SAOS. Lines connecting data points are used to guide the eye. (B) Zero-shear rate viscosity of o-Cys-P4-Cys as a function of temperature and concentration, showing that coiled-coil association introduces a very strong concentration dependence of viscosity. Error bars represent the standard deviations of the measurement (N = 3).
The associating coiled-coil domains, in turn, have effects on reptation. The zero-shear-rate viscosities (η0) of o-Cys-P4-Cys at different temperatures and concentrations, obtained from the inverse of the slope in creep at steady-state,51 are compared in Figure 5B. Below the entanglement concentration, η0 shows strong dependence on temperature, following Arrhenius law, consistent with the previous argument that coiled-coil association is the exclusive contribution to the network properties in this unentangled regime.52 On the contrary, in the entangled regime, η0 shows a stronger dependence on concentration and a weak dependence on temperature (the zero-shear-rate viscosity should be linearly proportional to the temperature in this regime24). Over the narrow range of concentration investigated, a power law fit to η0 ~ φβ gives an average exponent β = 19, which is a significantly larger value than the exponent of 3.4 predicted by the tube theory of reptation.60 In addition, the sticky reptation theory predicts the exponents in the regimes φe < φ < φren, φren < φ < φs and φs < φ < φle are 6.8, 8.5 and 3.75, respectively.24 Although the range of concentration studied is not large enough to clearly establish a power law regime, and the average exponent is significantly larger than the ones predicted by the sticky reptation theory, both the experiment and theory demonstrate the strong concentration dependence upon chain entanglement. Furthermore, a key difference between our experimental result and the sticky reptation theory is that coiled-coils form pentameric bundles instead of the dimeric associating domains treated in the theory. The relaxation dynamics of these systems is in contrast to end-associating polymers28 where bond relaxation leads to changes in the effective chain molecular weight and can relax entanglements.
Mechanical Enhancement in Axial Loading
Topological entanglement significantly affects the rate-dependence and nonlinearity of the large-strain mechanical response of protein gels. The tensile responses of hydrogels o-Cys-P4-Cys and P4 cannot be directly compared, as P4 is extremely fragile and the tensile specimens immediately fail at the grips. The mechanical properties of two hydrogels are therefore compared in uniaxial compression at 0.1, 1 and 10% s-1 nominal strain rates (Figure 6A and 6B, the definition of the true stress and true strain can be found in Supporting Information). The use of true stress-strain enables direct comparison between compression and tension data, and it also reveals the strain hardening behavior at large strains under stretching. The entangled gels display higher initial moduli and flow stress, as well as enhanced entropic stiffening at larger strains. The differences are more significant at low strain rates, consistent with the observations in shear rheology. At the highest strain rate of 10% s-1, the fragility of the P4 gel leads to partial failure/splitting around the periphery of the specimen at larger strains, as reflected in the stress-strain response. Conversely, at the 10% s-1 strain rate, the o-Cys-P4-Cys gel does not fail and exhibits enhanced elastic recovery during unloading due to the entropic recoil of the entangled molecular network. This finding again suggests the presence of chain entanglement with a characteristically long relaxation time associated with chain reptation. At low strain rates, both gels show similar unloading behaviors, as the imposed time scale is larger than the longest network relaxation time. Indeed, the unloading slopes of the stress-strain response during interval loading/partial unloading cycles at 1% s-1 cannot be readily distinguished (Figure 6C). In summary, the main effects of entanglements at these intermediate levels of deformation (up to a true strain of 1.6) are increasing the (small strain) modulus through the increase in the number of elastically effective chains and increasing the resistance to viscous flow, especially at low strain rates.
Figure 6.
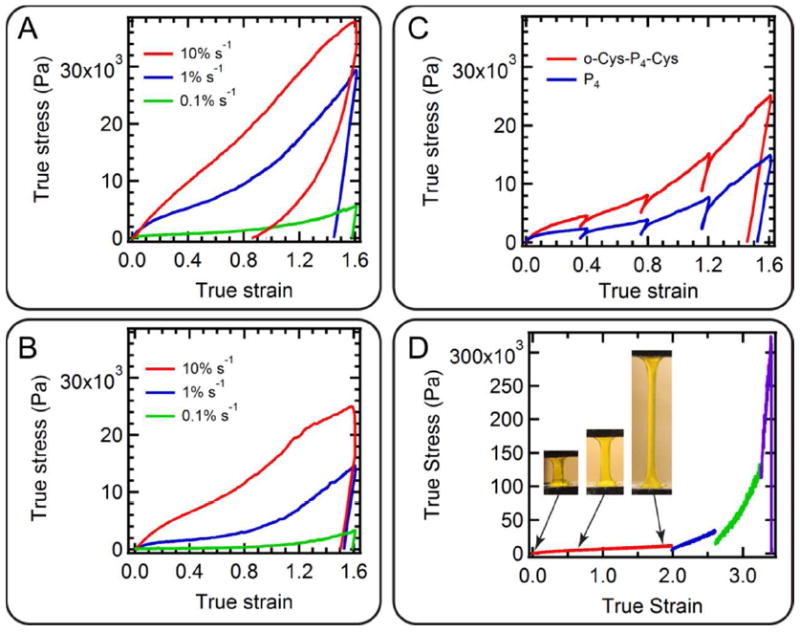
Mechanical enhancement in axial loading. Uniaxial compression of o-Cys-P4-Cys (A) and P4 (B) showing the rate-dependence and nonlinearity of the stress response of gels to 80% nominal strain. (C) Uniaxial compression at 1% s-1 nominal strain rate showing that chain entanglement does not affect the relaxation behavior at small deformation. (D) True stress-strain curve of topologically entangled gel reveals the strain hardening behavior during tensile loading. The photographs in the inset illustrate the neck stabilization and propagation, and the stable drawing of the specimen. All hydrogels are prepared at 15 (w/v)% concentration.
The entanglement effect becomes more pronounced at very large strain that is well characterized under tensile loading. Dramatic stiffening of the true stress-strain response is found to occur at true strains over 200%. Although less apparent, the early stiffening associated with stretch of the entangled network is sufficient to stabilize the tensile response of the o-Cys-P4-Cys gel specimen. This leads to neck propagation and drawing, with engineering strains at failure reaching approximately 3,000% (Figure 2C, Figure 2E and Fig 6D). The selected ramp-relaxation tensile protocol also demonstrates significant load relaxation at intermediate deformations, possibly owing to the fast coiled-coil association-disassociation dynamics, in agreement with the compression experiment results. Interestingly, at large deformations, the stress level is maintained even during relaxation. This effect might be ascribed to the strain-induced alignment of the molecular network, with entangled protein chains undergoing a coil-stretch transition that leads to remarkable resistance to strain localization and mechanical failure. Sufficient relaxation time intervals in the ramp-relaxation loading protocol are found to be critical to reach this extremely high extensibility. If the relaxation interval is shortened from ten minutes to one minute, the ultimate tensile strain is reduced to 1,250% engineering strain (Figure S7). Further studies are underway to study the structural transformation during tensile loading.
CONCLUSIONS
Topological entanglements are introduced into a coiled-coil protein hydrogel, achieving a dramatic enhancement in many mechanical properties. The interplay between reptation and coiled-coil association significantly suppresses the network relaxation, making hydrogels more resistant to creep and erosion in an open system. Chain entanglement promotes a mechanical transition from a highly brittle state to a tough and highly extensible state, enabling artificial protein hydrogels to be stretched up to approximately 3,000% engineering strain. Based on the reversible nature of the disulfide linkages, the improvement in network properties can be triggered by different redox stimuli. This network enhancement strategy can potentially be generalized to other hydrogel systems, and the presented redox responsive network toughening approach shows promise of synthetizing hydrogels that can mimic the high toughness of native tissues such as tendon and cartilage.
Supplementary Material
Acknowledgments
This research was supported by the U.S. Army Research Office through the Institute of Soldier Nanotechnologies under contract W911NF-07-D-0004. The authors would like to thank Biophysical Instrumentation Facility at MIT (NSF-0070319) for the use of the CD spectrophotometer. M.J.G. was supported by an NIH Interdepartmental Biotechnology Training Program (2-T32-GM08334) and S.L. was supported by a Summer Undergraduate Research Fellowship (SURF) from Caltech Student-Faculty Programs.
Footnotes
Supporting Information. Details for calculating protein volume fractions in hydrogels, molecular weight and distribution of chain extended proteins, tensile stress and strain, entanglement molecular weight, and density of elastically effective chains; SDS-PAGE of proteins; circular dichroism spectrum; additional rheology and tensile experiment data. This information is available free of charge via the Internet at http://pubs.acs.org/.
The authors declare no competing financial interest.
References
- 1.Shen W. Engineered Polypeptides for Tissue Engineering. In: Burdick J, Mauck R, editors. Biomaterials for Tissue Engineering Applications. Springer; Vienna: 2011. pp. 243–275. [Google Scholar]
- 2.Gomes S, Leonor IB, Mano JF, Reis RL, Kaplan DL. Progress in Polymer Science. 2012;37(1):1–17. doi: 10.1016/j.progpolymsci.2011.07.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.DiMarco RL, Heilshorn SC. Adv Mater. 2012;24(29):3923–3940. doi: 10.1002/adma.201200051. [DOI] [PubMed] [Google Scholar]
- 4.Engler AJ, Sen S, Sweeney HL, Discher DE. Cell. 2006;126(4):677–689. doi: 10.1016/j.cell.2006.06.044. [DOI] [PubMed] [Google Scholar]
- 5.Mammoto A, Ingber DE. Current Opinion in Cell Biology. 2009;21(6):864–870. doi: 10.1016/j.ceb.2009.08.001. [DOI] [PubMed] [Google Scholar]
- 6.Erickson IE, Huang AH, Sengupta S, Kestle S, Burdick JA, Mauck RL. Osteoarthritis and Cartilage. 2009;17(12):1639–1648. doi: 10.1016/j.joca.2009.07.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Kim M, Tang S, Olsen BD. Journal of Polymer Science Part B: Polymer Physics. 2013;51(7):587–601. [Google Scholar]
- 8.Urry DW. Angewandte Chemie International Edition in English. 1993;32(6):819–841. [Google Scholar]
- 9.Petka WA, Harden JL, McGrath KP, Wirtz D, Tirrell DA. Science. 1998;281(5375):389–392. doi: 10.1126/science.281.5375.389. [DOI] [PubMed] [Google Scholar]
- 10.Olsen BD, Kornfield JA, Tirrell DA. Macromolecules. 2010;43(21):9094–9099. doi: 10.1021/ma101434a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Banta S, Wheeldon IR, Blenner M. Annual Review of Biomedical Engineering. 2010;12(1):167–186. doi: 10.1146/annurev-bioeng-070909-105334. [DOI] [PubMed] [Google Scholar]
- 12.Kopeček J, Yang J. Angewandte Chemie International Edition. 2012;51(30):7396–7417. doi: 10.1002/anie.201201040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Semenov AN, Rubinstein M. Macromolecules. 1998;31(4):1373–1385. [Google Scholar]
- 14.Dobrynin AV. Macromolecules. 2004;37(10):3881–3893. [Google Scholar]
- 15.Green MS, Tobolsky AV. The Journal of Chemical Physics. 1946;14(2):80–92. [Google Scholar]
- 16.Tanaka F, Edwards SF. Macromolecules. 1992;25(5):1516–1523. [Google Scholar]
- 17.Annable T, Buscall R, Ettelaie R, Whittlestone D. Journal of Rheology. 1993;37(4):695–726. [Google Scholar]
- 18.Tripathi A, Tam KC, McKinley GH. Macromolecules. 2006;39(5):1981–1999. [Google Scholar]
- 19.Tanaka F, Edwards SF. Journal of Non-Newtonian Fluid Mechanics. 1992;43(2–3):247–271. [Google Scholar]
- 20.Tanaka F, Edwards SF. Journal of Non-Newtonian Fluid Mechanics. 1992;43(2–3):273–288. [Google Scholar]
- 21.Tanaka F, Edwards SF. Journal of Non-Newtonian Fluid Mechanics. 1992;43(2–3):289–309. [Google Scholar]
- 22.Leibler L, Rubinstein M, Colby RH. Macromolecules. 1991;24(16):4701–4707. [Google Scholar]
- 23.Rubinstein M, Semenov AN. Macromolecules. 1998;31(4):1386–1397. [Google Scholar]
- 24.Rubinstein M, Semenov AN. Macromolecules. 2001;34(4):1058–1068. [Google Scholar]
- 25.Xu D, Craig SL. Macromolecules. 2011;44(13):5465–5472. doi: 10.1021/ma200096s. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Hackelbusch S, Rossow T, van Assenbergh P, Seiffert S. Macromolecules. 2013;46(15):6273–6286. [Google Scholar]
- 27.Feldman KE, Kade MJ, Meijer EW, Hawker CJ, Kramer EJ. Macromolecules. 2009;42(22):9072–9081. [Google Scholar]
- 28.Cates ME. Macromolecules. 1987;20(9):2289–2296. [Google Scholar]
- 29.Sijbesma RP, Beijer FH, Brunsveld L, Folmer BJB, Hirschberg JHKK, Lange RFM, Lowe JKL, Meijer EW. Science. 1997;278(5343):1601–1604. doi: 10.1126/science.278.5343.1601. [DOI] [PubMed] [Google Scholar]
- 30.Folmer BJB, Sijbesma RP, Versteegen RM, van der Rijt JAJ, Meijer EW. Adv Mater. 2000;12(12):874–878. [Google Scholar]
- 31.Shen W, Zhang K, Kornfield JA, Tirrell DA. Nat Mater. 2006;5(2):153–158. doi: 10.1038/nmat1573. [DOI] [PubMed] [Google Scholar]
- 32.Welsh ER, Tirrell DA. Biomacromolecules. 2000;1(1):23–30. doi: 10.1021/bm0002914. [DOI] [PubMed] [Google Scholar]
- 33.Charati MB, Ifkovits JL, Burdick JA, Linhardt JG, Kiick KL. Soft Matter. 2009;5(18):3412–3416. doi: 10.1039/b910980c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Asai D, Xu D, Liu W, Garcia Quiroz F, Callahan DJ, Zalutsky MR, Craig SL, Chilkoti A. Biomaterials. 2012;33(21):5451–5458. doi: 10.1016/j.biomaterials.2012.03.083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Edwards SF, Vilgis T. Polymer. 1986;27(4):483–492. [Google Scholar]
- 36.Liu G, Sun H, Rangou S, Ntetsikas K, Avgeropoulos A, Wang S-Q. Journal of Rheology. 2013;57(1):89–104. [Google Scholar]
- 37.Donald AM, Kramer EJ. Journal of Polymer Science: Polymer Physics Edition. 1982;20(5):899–909. [Google Scholar]
- 38.Yang J, Wang X-P, Xie X-M. Soft Matter. 2012;8(4):1058–1063. [Google Scholar]
- 39.Bose RK, Lau KKS. Biomacromolecules. 2010;11(8):2116–2122. doi: 10.1021/bm100498a. [DOI] [PubMed] [Google Scholar]
- 40.Xiao J, Tolbert TJ. Organic Letters. 2009;11(18):4144–4147. doi: 10.1021/ol9016468. [DOI] [PubMed] [Google Scholar]
- 41.Fang G-M, Li Y-M, Shen F, Huang Y-C, Li J-B, Lin Y, Cui H-K, Liu L. Angewandte Chemie International Edition. 2011;50(33):7645–7649. doi: 10.1002/anie.201100996. [DOI] [PubMed] [Google Scholar]
- 42.Sahin E, Kiick KL. Biomacromolecules. 2009;10(10):2740–2749. doi: 10.1021/bm900474k. [DOI] [PubMed] [Google Scholar]
- 43.Vila-Perelló M, Liu Z, Shah NH, Willis JA, Idoyaga J, Muir TW. Journal of the American Chemical Society. 2012;135(1):286–292. doi: 10.1021/ja309126m. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Hartgerink JD, Beniash E, Stupp SI. Proceedings of the National Academy of Sciences. 2002;99(8):5133–5138. doi: 10.1073/pnas.072699999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Shen W, Lammertink RGH, Sakata JK, Kornfield JA, Tirrell DA. Macromolecules. 2005;38(9):3909–3916. [Google Scholar]
- 46.Zhang Q, Ye J, Lu Y, Nie T, Xie D, Song Q, Chen H, Zhang G, Tang Y, Wu C, Xie Z. Macromolecules. 2008;41(6):2228–2234. [Google Scholar]
- 47.Glassman MJ, Chan J, Olsen BD. Advanced Functional Materials. 2013;23(9):1182–1193. doi: 10.1002/adfm.201202034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Jacobson H, Stockmayer WH. The Journal of Chemical Physics. 1950;18(12):1600–1606. [Google Scholar]
- 49.Skrzeszewska PJ, de Wolf FA, Werten MWT, Moers APHA, Cohen Stuart MA, van der Gucht J. Soft Matter. 2009;5(10):2057–2062. [Google Scholar]
- 50.Ferry J. Viscoelastic properties of polymers. 3. Wiley; New York: 1980. p. xxiv.p. 641. [Google Scholar]
- 51.Graessley WW. Polymeric liquids and networks : dynamics and rheology. Garland Science; London; New York: 2008. p. xix.p. 801. [Google Scholar]
- 52.Séréro Y, Aznar R, Porte G, Berret JF, Calvet D, Collet A, Viguier M. Physical Review Letters. 1998;81(25):5584–5587. [Google Scholar]
- 53.Gong JP. Soft Matter. 2010;6(12):2583–2590. [Google Scholar]
- 54.Sun J-Y, Zhao X, Illeperuma WRK, Chaudhuri O, Oh KH, Mooney DJ, Vlassak JJ, Suo Z. Nature. 2012;489(7414):133–136. doi: 10.1038/nature11409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Glassman MJ, Olsen BD. Soft Matter. 2013;9(29):6814–6823. doi: 10.1039/C3SM00102D. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Betre H, Setton LA, Meyer DE, Chilkoti A. Biomacromolecules. 2002;3(5):910–916. doi: 10.1021/bm0255037. [DOI] [PubMed] [Google Scholar]
- 57.Li J, Viveros JA, Wrue MH, Anthamatten M. Adv Mater. 2007;19(19):2851–2855. [Google Scholar]
- 58.Chen Y, Kushner AM, Williams GA, Guan Z. Nat Chem. 2012;4(6):467–472. doi: 10.1038/nchem.1314. [DOI] [PubMed] [Google Scholar]
- 59.Larson RG, Sridhar T, Leal LG, McKinley GH, Likhtman AE, McLeish TCB. Journal of Rheology. 2003;47(3):809–818. [Google Scholar]
- 60.McLeish TCB. Adv Phys. 2002;51(6):1379–1527. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.