

Summary
Faecal pollution contains a rich and diverse community of bacteria derived from animals and humans, many of which might serve as alternatives to the traditional enterococci and Escherichia coli faecal indicators. We used massively parallel sequencing (MPS) of the 16S rRNA gene to characterize microbial communities from wastewater treatment plant (WWTP) influent sewage from 12 cities geographically distributed across the USA. We examined members of the Clostridiales, which included the families Clostridiaceae, Lachnospiraceae and Ruminococcaceae for their potential as sewage indicators. Lachnospiraceae was one of the most abundant groups of faecal bacteria in sewage, and several Lachnospiraceae high-abundance sewage pyrotags occurred in at least 46 of 48 human faecal samples. Clone libraries targeting Clostridium coccoides (C. coccoides) in sewage samples demonstrated that Lachnospiraceae-annotated V6 pyrotags encompassed the previously reported C. coccoides group. We used oligotyping to profile the genus Blautia within Lachnospiraceae and found oligotypes comprised of 24 entropy components that showed patterns of host specificity. These findings suggest that indicators based on Blautia might have the capacity to discriminate between different faecal pollution sources. Development of source-specific alternative indicators would enhance water quality assessments, which leads to improved ecosystem health and reduced human health risk due to waterborne disease.
Background
Faecal pollution contains a broad array of microorganisms from animals and humans, the majority of which are faecal anaerobes (Franks et al., 1998; Eckburg et al., 2005; Ley et al., 2008). However, water quality surveillance relies upon a small subset of easily culturable facultative anaerobes, such as Escherichia coli (E. coli) or enterococci. These bacteria commonly occur in both animals and humans, thereby providing no information as to the source of faecal pollution. Faecal pollution remains a major source of water quality impairment of rivers, streams and coastal waters (USEPA, 2009). Receiving waters in watersheds often collect inputs from upstream rural and agricultural land and downstream urbanized regions, making it difficult to estimate relative contributions of various faecal pollution sources (sewage, agricultural animals, wildlife, etc.). Despite ambiguity about their source, the detection of faecal indicator bacteria commonly leads to advisories or closures of coastal beaches (USEPA, 2012).
Previous studies demonstrate that diet, host factors and host–microbe co-evolution shape the composition of gut microbiota (Ley et al., 2008; Sekelja et al., 2011; Shanks et al., 2011). If these processes have major influences on community structure, then characterization of host-associated microbiota might identify organisms that can serve as host-specific indicators of faecal pollution. Within the order Bacteroidales, traditional molecular methods have identified host-specific species and phylotypes. Terminal restriction fragment length polymorphism (TRFLP) and/or family-specific cloning and sequencing of nearly full-length 16 S rRNA genes by Sanger technologies have identified diagnostic phylotypes for humans, cows, pigs, dogs, etc. (Bernhard and Field, 2000a; Dick et al., 2005; Fogarty and Voytek, 2005; Kildare et al., 2007; Fremaux et al., 2009). Two established human-specific Bacteroides assays (Bernhard and Field, 2000b; Kildare et al., 2007) target the V2 region of very closely related phylotypes. Subtractive hybridization and genomic enrichment of the metagenome have identified candidate alternative indicators for host-specific assays, including a human-specific assay that targets an unidentified enzyme of a Bacteroides spp. (Shanks et al., 2007). While Bacteroidales is perhaps the most studied taxonomic group for the development of alternative indicators, a few indicators have also been described for Bifidobacteriaceae (Bonjoch et al., 2004; Gomez-Donate et al., 2012), and recently Lachnospiraceae (Newton et al., 2011).
In recent years the Human Microbiome Project (HMP) has generated large molecular data sets that provide new information about the complexity of microbial community composition in humans. Bacteroidales and Clostridiales are among the most abundant faecal anaerobes and the most diverse (Robinson et al., 2010). These studies demonstrate large interpersonal variation (Turnbaugh et al., 2009; Robinson et al., 2010; Lozupone et al., 2012), which makes it difficult to identify the most common and abundant host-specific microbes across human populations. Only a few studies have used analysis of treated or untreated sewage to search for novel indicators of human faecal pollution (McLellan et al., 2010; Wery et al., 2010). Sewage effectively represents a random, composite sampling of tens of thousands to millions of individuals, which circumvents the issue of individual variability in identifying common microorganisms in the human population.
Constancy of Bacteroidales and Clostridiales microbial communities has been reported across different waste-water treatment plants (WWTPs) (McLellan et al., 2010; Wery et al., 2010) and over a 2-year period in a single plant (McLellan et al., 2010). This paper examines the population structure of Clostridiales, and in particular Lachnospiraceae, a robust group of organisms that commonly occur in the gut of humans and other animals. Here, we characterize 38 sewage samples collected over a 4-year period from one city and 11 sewage samples collected from diverse geographic regions to demonstrate that surveys of sewage allow us to describe microbial community structure in the human population. We identified a complex array of unique Lachnospiraceae V6 pyrotags that appear to represent abundant, human-specific microbial populations. These Lachnospiraceae taxa have the potential to serve as alternative indicators of sewage that can differentiate between human and non-human faecal pollution sources.
Results
Distribution of Clostridiales in sewage, human, cattle and chickens
Table 1 lists the sewage and host samples that we included in this study. The family level composition of Clostridiales in sewage influent samples generally was similar to a composite data set of 48 human faecal samples; however, sewage reflected a higher relative abundance of Peptostreptococcaceae and Veillonellaceae (Fig. 1). Sequences annotated as Lachnospiraceae made up the majority of Clostridiales sequences in both the sewage and human faecal samples. Lachnospiraceae comprised 5.6% of the total microbial community in Milwaukee sewage samples and, on average, 6.2% of the total microbial community in sewage from multiple cities. Cattle faecal samples contained significantly higher Ruminococcaceae compared with sewage or humans (P < 0.05). Chickens had a low relative abundance of Clostridiales, most of which mapped to Lachnospiraceae.
Table 1.
Sewage, human, cattle and chicken data sets used in this study.
| Source | Collection timeframe | No. of samples | No. of high-quality bacterial reads | No. of high-quality Clostridiales reads | References |
|---|---|---|---|---|---|
| South Shore (SS) WWTP Milwaukee, WI | Apr. 2005–Aug. 2009 | 19 | 594 997 | 70 455 | McLellan et al. (2010); VandeWalle et al. (2012) |
| Jones Island (JI) WWTP Milwaukee, WI | Apr. 2005–May 2009 | 19 | 493 890 | 48 889 | McLellan et al. (2010); VandeWalle et al. (2012) |
| WWTPs in cities geographically distributeda | May 2006–Oct. 2007 | 11 | 323 070 | 46 816 | This study |
| Humans faecal samples | Mar. 2007–Oct. 2008 | 33 | 766 658 | 593 480 | Turnbaugh et al. (2009) |
| Humans faecal samples | Mar. 2005–Jan. 2006 | 15 | 462 690 | 244 155 | Dethlefsen et al. (2008) |
| Cattle faecal samples | 2008 | 30 | 633 877 | 374 566 | Shanks et al. (2011) |
| Chicken faecal sample | 2008 | 5 | 34 100 | 4 023 | This study |
The city and states included Seattle (WA), Duluth (MN), Rutland (VT), Albany (NY), Crystal Lake (IL), Clarksburg (WV), Elk Grove (CA), Tulsa (OK), Las Vegas (NV), Tallahassee (FL) and Kihei (HI).
Fig. 1.

Family level comparison of Clostridiales in untreated sewage, humans, cows and chickens. Non-faecal bacteria dominated sewage populations while faecal bacteria comprised approximately 15% of the community, which accounts for the lower relative abundance of Clostridiales in sewage compared with the individual faecal samples.
High-abundance Clostridiales pyrotags in sewage and humans
More than half of the top 30 most abundant Clostridiales pyrotags in sewage represented the family Lachnospiraceae, and abundance patterns were similar to the human faecal data set (Fig. 2). These abundant Lachnospiraceae pyrotags were rarely present in the cow or chicken faecal data sets. Ruminococcaceae, specifically pyrotags classified as Faecalibacterium sp., were also among the most abundant in the sewage and human faecal data sets, but many of these pyrotags were also present in the cow faecal data set. The top two most abundant V6 pyrotags in sewage resolved to Lachnospiraceae and matched with 100% identity to uncultured Lachnospiraceae A1-86 (pyrotags annotated as Roseburia) and Blautia wexlerae (pyrotags annotated as Blautia) respectively (Fig. 2). The majority of abundant pyrotags in the sewage data set, but not present in the human data set, were from the Clostridiaceae, Veillonellaceae and Peptostreptococcaceae families.
Fig. 2.

The relative abundance of pyrotags assigned as Clostridiales within each composite dataset is depicted with a heatmap. The inferred phylogenetic tree represents full-length reference sequences that had 100% identity to the 30 most abundant Clostridiales V6 pyrotags in sewage, the 30 most abundant Clostridiales V6 pyrotags in human faecal samples, the 10 most abundant pyrotags across all datasets in the family Clostridiaceae and type strains from each major phylogenetic group. GenBank accession numbers are shown in parentheses. A green circle indicates a sequence with 100% identity to the represented pyrotag was obtained from a C. coccoides specific clone library.
Relating Lachnospiraceae pyrotags to the Clostridium coccoides group
We generated 2018 near full-length 16S rRNA gene sequences from a subset of sewage samples using a primer set targeting the C. coccoides group. A total of 307 sequences were unique and 305 of those classified to the family Lachnospiraceae, mapping to unclassified Lachnospiraceae (33.1%), Blautia (26%) and Lachnospiraceae Incerte sedis (24.0%). Only two clones were not classified as Lachnospiraceae, instead mapping to Veillonellaceae, genus Anaeroarcus. All of the 30 most abundant Lachnospiraceae pyrotags in the sewage and human faecal data sets matched (had 100% identity) to at least one of the cloned sequences (Fig. 2). The great majority of cloned sequences (92%) matched a Lachnospiraceae pyrotag, but only 44% of the unique pyrotags matched one of our cloned sequences. Given the depth of sequencing (62 092 Lachnospiraceae annotated pyrotags compared with 2018 cloned sequences), we expected that lower-abundance pyrotags would not be represented in the cloned library. Given an equal sequencing depth for the two sequencing methods, we estimate that, 87% of the Lachnospiraceae pyrotags data set would be represented by the clone library.
Identification of core Lachnospiraceae in humans and sewage
The top three most abundant Lachnospiraceae sewage pyrotags (representing 15.5% of sewage Lachnospiraceae pyrotags) occurred in all 48 human faecal samples (Fig. 3), and the top eight most abundant Lachnospiraceae sewage pyrotags (representing 27.1% of sewage Lachnospiraceae pyrotags) occurred in at least 46 of 48 individuals, although the relative abundance of these pyrotags in any individual human faecal sample was highly variable (Table S1). For example, the most abundant Lachnospiraceae sewage pyrotag, which corresponded to a Roseburia (Fig. 2) and accounted for 6.1% of the Lachnospiraceae pyrotags in sewage, represented the most abundant Lachnospiraceae pyrotag in 29 of the 48 human faecal samples, averaging 13% of the Lachnospiraceae. However, this pyrotag was not among the top Lachnospiraceae in seven of the individuals and averaged less than 0.3% of the Lachnospiraceae recovered. The second most abundant Lachnospiraceae sewage pyrotag mapped to the genus Blautia (Fig. 2) and was highly abundant in 19 of 48 individuals with distribution patterns among human faecal samples that paralleled the Roseburia-classified pyrotag.
Fig. 3.

Plot of the 3000 most abundant sewage pyrotags vs. the number of human faecal samples (individuals) in which each pyrotag was found. The top eight pyrotags in sewage were found in at least 46 of 48 individuals. Not all data points are visible because of the overlap of data points for low-abundance sewage pyrotags found in < 35 individuals.
In Milwaukee sewage, the top five most abundant Lachnospiraceae pyrotags exhibited stable rank abundance patterns across multiple samples collected over multiple years (2005 and 2007–2009) (Fig. 4). Pyrotags further down the rank abundance distribution exhibited more rank variability over temporal scales, but were always relatively dominant in sewage influent. We also compared pyrotags from Milwaukee sewage (averaged across the 38 samples) with 11 sewage samples from across the USA. The rank abundance of Lachnospiraceae pyrotags in Milwaukee sewage was highly correlated with the rank abundance in 11 other cities (rho = 0.8441, P < 0.001). Only one of 11 cities (Tulsa) had a disparate pattern, but the correlation was still significant (rho = 0.4103, P < 0.001). Table S2 summarizes individual WWTP correlations based upon comparisons of ranked abundance of Lachnospiraceae V6 pyrotag sequences.
Fig. 4.
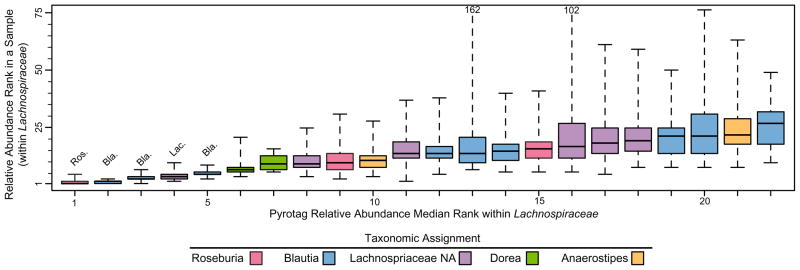
Box plot of ranked relative abundance for the 22 most abundant individual Lachnospiraceae pyrotags in sewage influent samples collected over 4 years from Milwaukee, WI wastewater treatment plants. Box vertices represent the 25% and 75% rank values and whiskers represent the maximum and minimum rank values. For visualization purposes, maximum rank values are listed on the plot for two pyrotags whose values greatly exceeded the other pyrotags. The taxonomic assignment is listed for the first five ranked pyrotags; abbreviations are as follows: Ros., Roseburia; Bla., Blautia; Lac., Lachnospiraceae NA.
Network analysis of Clostridiales
Network analysis was used to examine the distribution of the 400 most abundant Clostridiales V6 pyrotags from four faecal sources: humans, sewage, cattle and chickens (Fig. 5). When considering all Clostridiales taxa in sewage, we found Lachnospiraceae was the family with the most pyrotags shared between humans and sewage and these pyrotags were rarely present in the cow and chicken data sets. Overall 31.0% of Lachnospiraceae pyrotags in sewage overlapped with humans, 10.4% overlapped with cattle and 0.5% overlapped with chickens (Table 2). Ruminococcaceae was less specific; 18.5% of the sewage pyrotags overlapped with cattle. The number of unique Clostridiaceae pyrotags was approximately 10-fold lower than what was found with Lachnospiraceae, and the vast majority (94.5%) were unique to sewage. Further, there was more overlap between sewage Clostridiaceae pyrotags and cows and chickens than there was with the human faecal samples.
Fig. 5.

Network representation from a spring-embedded edge-weighted model of the 400 most abundant Clostridiales from each of four sources: Milwaukee sewage influent (red, composite data set of 38 samples), humans (yellow, composite data set of 48 samples), cattle (green, composite data set of 30 samples) and chickens (blue, composite data set five samples). See Table 1 for sample details. Composite samples are represented by a single point (circle), from which many lines radiate. Each pyrotag is represented by a small white circle. Lines connecting a pyrotag to the composite sample indicate the pyrotag was present in that sample set. Line thickness indicates relative abundance of the pyrotag in the connected sample set.
Table 2.
Clostridiales sewage pyrotags shared with humans, cattle and chickens.
| Family (n unique pyrotags in sewage) | Sewage pyrotags shared with other sources
|
||
|---|---|---|---|
| Humans | Cattle | Chickens | |
| Lachnospiraceae (n = 8933) | 31.0% | 10.4% | 0.5% |
| Ruminococcaceae (n = 5156) | 25.9% | 18.5% | 1.1% |
| Clostridiaceae (n = 770) | 5.5% | 8.7% | 1.8% |
Oligotypes within Blautia
To explore the level of host specificity within the genus Blautia we used oligotyping, a supervised computational method that can detect very subtle nucleotide variations among closely related taxa, thereby facilitating the identification of closely related but distinct organisms that may not be detected by taxonomic classification or de facto 3% clustering methods (Eren et al., 2011). Oligotyping analysis of 152 730 V6 reads that mapped to Blautia from our four source data sets revealed a total of 108 oligotypes. Figure 6 shows the distribution of oligotypes among samples. Some oligotypes exhibited remarkable host specificity. These oligotypes occurred only in chicken, only in cattle or only in human and sewage samples, indicating that V6 pyrotags could be used for faecal source identification. Figure 7 illustrates eight of the host-specific oligotypes and their abundance in individual samples. Table 3 lists the total counts and their parent V6 reads.
Fig. 6.

Stacked bar chart of oligotypes among groups of samples. Each bar in the figure represents a sample and each colour denotes a different oligotype. Bars at the bottom indicate sample sources.
Fig. 7.

Relative abundance representations among sample sources for eight Blautia host-specific oligotypes. Each dot in these panels represents the relative abundance of the listed oligotype (above each plot) among all Blautia assigned sequences in a faecal sample collected from a CHK = chicken (blue), CAT = cattle (green), HUM = human (orange) or a SEW = sewage influent sample (red). Box plots next to the dots provide the 25% and 75% and median relative abundance values for the samples. Whiskers represent 1.5 times the 25% and 75% quartile range.
Table 3.
Full-length V6 reads for oligotypes shown in Fig. 7.
| Host | Oligotype | V6 tag | Count |
|---|---|---|---|
| Chicken | AGGCACTACCACGGTGTTGAGGCA | AAGTCTTGACATCTGCCTGACCGTACCTTAACCGGTGCTTTCCTTCGGGACAGGCAAGAC | 1 814 |
| Human/sewage | AGGCACATCCACGGATTTGAGGCA | AAGTCTTGACATCCGCCTGACCGATCCTTAACCGGATCTTTCCTTCGGGACAGGCGAGAC | 35 140 |
| Human/sewage | AACTACGTCTATGGACTCAGGAGT | AAATCTTGACATCCCTCTGACCGGTCTTTAATCGGACCTTCTCTTCGGAGCAGAGGTGAC | 20 438 |
| Human/sewage | AGTCACGTCCACGGACTTGAGGAA | AAGTCTTGACATCCTCCTGACCGGTCCTTAACCGGACCTTTCCTTCGGGACAGGAGAGAC | 4 223 |
| Cattle | AGCGACGAACACGTTCTTAATCGT | AAGTCTTGACATCCCGATGACCGGAACTTAACCGTTCCTTTTCTTCGGAACATCGGTGAC | 126 |
| Cattle | AGCGACGTACACGTACTCAGTCGT | AAGTCTTGACATCCCGATGACCGGTACGTAACGGTACCTTCTCTTCGGAGCATCGGTGAC | 95 |
| Cattle | AGCGACTTCCACGGAATTAATCGA | AAGTCTTGACATCCCGATGACCGTTCCTTAACCGGAACTTTTCTTCGGAACATCGGAGAC | 77 |
| Cattle | AACTACGCTCATGAGTTTGAGAGT | AAATCTTGACATCCCTCTGACCGGCTCTTAATCGAGTCTTTCCTTCGGGACAGAGGTGAC | 165 |
Count column shows the total number of reads represented by a given oligotype.
Discussion
Lachnospiraceae are candidates for alternative indicators
Traditional faecal indicators can detect faecal pollution, but they fall short for identifying causes of poor water quality (Field and Samadpour, 2007; Stewart et al., 2008). Yet the efficient use of our limited resources for mitigation and, ultimately, reduction of human health risks requires information about the specific sources of faecal pollution. With the rapid advances in sequencing technologies, it is now possible to characterize microbial communities and their structure in great depth and interrogate these data sets to identify new host-specific indicators of faecal pollution. Ideally, host-specific indicators will: (i) represent abundant taxa in the host of interest, thereby maximizing sensitivity for detection, (ii) not occur in other hosts and thus provide specificity and (iii) prove to be robust over a large geographic region (NRC, 2004).
Clostridiales, a major group within the human gut microbiome, has been largely underexplored for identification of human-specific indicators (McLellan et al., 2010; Wery et al., 2010). In humans, the major families within Clostridiales include Lachnospiraceae, Ruminococcaceae and, to a lesser extent, Clostridiaceae (Eckburg et al., 2005). Lachnospiraceae is estimated to comprise 19% to 50% of faecal microbiota (Hayashi et al., 2002; 2006; Hold et al., 2002; Matsuki et al., 2004; Rajilic-Stojanovic et al., 2009; Gosalbes et al., 2011). Initial investigations of Lachnospiraceae suggest this group contains many organisms that could be used as faecal indicators (McLellan et al., 2010; Newton et al., 2011). Our profiling of untreated sewage using V6 sequencing revealed Lachnospiraceae was one of the most numerically dominant taxonomic groups, despite the overprinting of non-faecal bacteria that comprised nearly 85% of the total community (McLellan et al., 2010). From these data, we developed a qPCR assay that targets the second most abundant Lachnospiraceae (matching Blautia wexlerae) in our Milwaukee sewage samples and designated this gene sequence as Lachno2. We then demonstrated that the Lachno2 qPCR assay correlated to a previously described qPCR assay targeting a human Bacteroides sp. and provided evidence of chronic human faecal pollution in surface waters (Newton et al., 2011).
The evolving nomenclature for the Clostridiales makes it difficult to relate current and past studies of community structure and diversity. Within Clostridiales, Collins and colleagues (1994) proposed Clostridium clusters I to XIX. The C. coccoides group is analogous to Clostridium cluster XIVa (Collins et al., 1994; Matsuki et al., 2002), which consists of up to 20 different genera including Anaerostipes, Butyrivibrio, Clostridium, Roseburia and Ruminococcus to name a few (Hayashi et al., 2006; Liu et al., 2008). Several of these named genera have been reclassified into the recently described genus Blautia (Liu et al., 2008). Cloning with the previously described primer set targeting the C. coccoides group (Matsuki et al., 2002) generated clones representing much of the diversity in our Lachnospiraceae-annotated V6 pyrotags, including the most dominant pyrotags present in sewage and humans (Fig. 2). The general abundance patterns between clones and pyrotags were very similar, and the sequencing depth of the clone library appeared to be the largest factor limiting our ability to capture the Lachnospiraceae diversity identified in the pyrotags. Overall, our results suggest that the Lachnospiraceae pyrotags described in this study are analogous to the previously reported C. coccoides group and Clostridium cluster XIVa and include unclassified Lachnospiraceae not previously described.
Abundant sewage Lachnospiraceae pyrotags represent core human faecal microbiota
The high individual diversity but consistent metabolic pathways of the human gut microbiota suggests the presence of a microbiota functional core rather than a phylogenetic core (Eckburg et al., 2005; Turnbaugh et al., 2009; Robinson et al., 2010; Lozupone et al., 2012). Multiple studies have identified genera, phylogroups or phylotypes (represented by sequences or OTUs) that consistently occur in humans (Rajilic-Stojanovic et al., 2009; Tap et al., 2009; Qin et al., 2010; Turnbaugh et al., 2010; Sekelja et al., 2011). However, the prevailing thought is that it is unlikely that a core set (present in all humans) of microbial species exists for the human gut (Lozupone et al., 2012). Sewage represents faecal microbiota from thousands to millions of people, and represents the phylotypes, OTUs, etc. that are the most common among a human population. Therefore, the most abundant and most consistently present faecal microbes in sewage are likely to represent what could be considered a core gut community. Members of Lachnospiraceae (i.e. Clostridium cluster XIVa) are among the most frequently identified as ‘core’ gut microbes (Sekelja et al., 2011; Lozupone et al., 2012).
In sewage, we found the same Lachnospiraceae high-abundance pyrotags in all 12 cities’ WWTP influents, which suggests the presence of a cosmopolitan distribution for some gut bacteria in the human population of the USA. The most abundant of these shared Lachnospiraceae pyrotags were particularly stable in terms of rank relative abundance (i.e. most abundant, second most abundant, etc.) over a 4-year period in Milwaukee’s sewage influent. Instead of representing a binary relationship (either stable or not stable), there was a continuum of decreasing stability, where increasingly lower ranked pyrotags (i.e. lower overall relative abundance among Lachnospiraceae in sewage) showed increasing variability in rank abundance (Fig. 4). This pattern suggests to us that some pyrotags are present in a large percentage of Milwaukee’s population and could be considered core for this city, while other pyrotags occur in a smaller percentage of people, which leads to their increased variability. Sekelja and colleagues (2011) found that ‘core’ phylogroups were more stable over time than non-core, which supports our hypothesis that the highest abundance sewage pyrotags in sewage represent core microbiota.
Surveying sewage for abundant and specific indicators is useful, but such characterizations could have much broader applications. Sewage represents trends in a particular human population that cannot be readily observed by sampling a limited number of individuals. We have found that WWTP influent displays a more consistent pyrotag profile than that of individual human faecal samples (Fig. 6; Newton et al., 2011). In the present study, the relative abundance of the eight most abundant Clostridiales pyrotags in sewage was highly variable in 48 human faecal samples, which is consistent with other reports describing the abundance patterns of ‘core’ members (Qin et al., 2010; Turnbaugh et al., 2010; Sekelja et al., 2011). Interestingly, individuals whose microbiota was dominated by Bacteroides were more likely to have only minor representation of high-abundance sewage Lachnospiraceae (Table S1). Rather, the predominant Lachnospiraceae present in these individuals were those that were more rare across the averaged human population represented in sewage. This result suggests sewage profiles provide a benchmark for norms in a human population. Simple averaging of highly diverse individuals would not be sufficient to establish this same benchmark. We suggest sewage may be used to observe microbial community patterns in the human population that are linked to population level statistics such as age, health or dietary habit.
The role of Lachnospiraceae in humans and host-specific patterns
We focused our efforts on Lachnospiraceae because of its diversity and high abundance in sewage and humans. Further, a large number of pyrotags within the family Lachnospiraceae were found in both sewage and human faecal samples, but not in cattle or chicken faeces, suggesting that Lachnospiraceae might serve important functional roles specific to humans. Ongoing research suggests this may be the case. It is thought that Lachnospiraceae taxa have an important role in maintaining gut homeostasis (Frank et al., 2007) and are involved in human metabolism as butyrate producers (Sekelja et al., 2011). Lachnospiraceae taxa also appear to be important for the exclusion of pathogens (Reeves et al., 2012). Given the depth of Lachnospiraceae diversity observed in this study and by others, it remains unclear to what extent cultured strains represent the functional diversity of this group (Hayashi et al., 2002), particularly in relation to traits accounting for host specificity. To understand the functional role fulfilled by Lachnospiraceae, and whether or not these roles contribute to host specificity, cultured organisms that represent the range of diversity in the natural population are needed.
In contrast to Lachnospiraceae, the Ruminococcaceae and Clostridiaceae families did not show as much promise as groups that harboured large numbers of indicator organisms that could be used to detect human faecal pollution. Humans and cows and, to a lesser extent, chickens shared many Ruminococcaceae pyrotags including several high-abundance pyrotags present in the sewage data set (Fig. 2). In a previous survey of farm animals and humans, the Clostridium leptum group (i.e. Clostridium group IV, encompassed within Ruminococcaceae) was constantly present and exhibited low variability in abundance between humans and a number of animals including rabbits, goats, horses, sheep, cows and pigs (Furet et al., 2009). This same study demonstrated C. coccoides levels distinguished humans from the majority of these same sources, but not pigs (Furet et al., 2009). The genus Clostridium and other Clostridiaceae occurred at relatively low abundance in the human, cow and chicken data sets (Fig. 1). There were, however, four abundant sewage pyrotags that did not occur in the human, cattle or chicken data sets, suggesting Clostridiaceae or Clostridium sp. may serve as indicators for animals not tested in this study. Alternatively, these pyrotags may represent non-host-associated, free-living organisms. Additional examination of the occurrence in humans and analysis of more animal samples would clarify the range of host specificity among these Clostridiales taxa.
Small sequence variations in the 16S rRNA gene indicate host specificity
Large data sets of short-read sequences generated by massively parallel sequencing (MPS) show promise for identification of indicators to track faecal pollution sources; however, sensitive approaches are needed to discriminate among organisms with closely related 16S rRNA gene sequences but different ecological characteristics. Aggregating sequences into groups (OTUs) can reduce resolution to the point that the full suite of potential candidates cannot be identified. In this study and others by our lab we found unique V6 pyrotags that represented ecologically meaningful populations. For example, a single base pair change in pyrotags within the genus Acinetobacter mapped to two populations whose relative abundance fluctuated seasonally but inversely in urban sewer infrastructure (VandeWalle et al., 2012). In this study, we found several pyrotags (i.e. unique V6 sequences) that appeared in sewage and humans but not in chickens or cows, while a pyrotag with only a slight sequence variation to the human-specific pyrotag was present in one of the animal faecal data sets (Table 3). The host distribution, abundance patterns and relationship to phylogenetically distinct 16S rRNA genes (Figs 2 and 4) suggest that the pyrotags represent ecologically relevant phylotypes. Host-associated phylotypes, distinguished by small variations in 16S rRNA gene sequences, also have been observed in the studies of Bacteroidales (Dick et al., 2005; Jeter et al., 2009).
In this study, oligotyping was a useful tool for systematically identifying small sequence changes corresponding to within-genus sequence-based groupings that differentiated host organism microbial communities. By utilizing Shannon entropy to identify nucleotide locations of high variation among very closely related taxa, oligo-typing can elaborate the differences among samples with respect to the chosen taxon. Patterns of host-associated sequence types may not be easily inferred from phylogenies, as many of our identified host associations were represented by only a few nucleotide substitutions and thus would be represented by divergence only at the tips of phylogenetic representations of communities. In this study we targeted Blautia for oligotyping specifically because the second most abundant Lachnospiraceae pyrotag (Lachno2) appeared specific to humans and was classified to this genus (Newton et al., 2011). The great diversity that is concealed within Blautia but revealed by oligotyping suggests that there may be other genera in the Lachnospiraceae family that can be used for further identification of host-specific indicators. Oligotyping shows great promise for being able to confidently distinguish a large range of hosts’ microbes typically found in environmental samples.
Clustering of MPS data (i.e. OTUs) is frequently used to mitigate artefacts caused by sequencing errors (Huse et al., 2010). In this study, we did not use clustered sequence data, instead we chose the more stringent criterion of unique sequence groups for all analyses. Primarily sequence errors affect MPS data by artificially increasing the sequence types present in a sample. Since our primary objective was to identify common pyrotags or oligotypes across multiple samples and in this case many different sequencing runs, the confounding issue of random errors increasing sample diversity is not likely to have had a large effect on our analysis. Instead, by examining exact sequences and through oligotyping, we identified important patterns related to small sequence variations that would have been missed if cluster analysis had been used.
Applications for detecting sources of faecal pollution
In previous source tracking studies that focused on the C. coccoides group (i.e. Lachnospiraceae) as an indicator of human faecal pollution, either PCR was used as a faecal detection method (Bonkosky et al., 2009), or abundance patterns among faecal groups were used to distinguish humans from farm animals (Furet et al., 2009). In this study, MPS provided a higher resolution of the Clostridiales, particularly Lachnospiraceae, community structure leading to the identification of hundreds of candidate host-specific indicators. However, only a limited range of host animals was examined; therefore, more rigorous validation is needed to determine the extent of candidate indicator host specificity. In addition, these results only reflect microbiota data collected from humans, sewage and animals within the USA and thus, may or may not reflect sewage signature potential in other countries. Identification of potential candidates, or candidate genera, as in the case of Blautia, will streamline this process so that a combination of MPS approaches and targeted qPCR could be used to validate these alternative indicators.
Conclusions
As a first tier assessment of faecal pollution sources, distinguishing human sources from non-human sources is important because human faeces is a major reservoir for human pathogens. We found Lachnospiraceae to be the most promising group for identification of human host-specific indicators among Clostridiales. Multiple pyrotag and oligotype sequences identified in this study appear to be ecologically distinct and warrant further investigation. In contrast to Lachnospiraceae, Ruminococcaceae pyrotags were more commonly shared among host sources, particularly between humans and cattle, reducing specificity. Clostridium was neither commonly abundant nor frequently human-specific, which would impair sensitivity and specificity. Applications using a MPS approach have provided unprecedented insights into the population structure of human faecal communities and have documented high diversity among common taxa. Due to transient colonization of multiple hosts and equivalent niches among hosts, it is unlikely that a single indicator will be exclusively specific to a single host source, and have appropriate sensitivity for quick detection methods. Rather, future studies should consider using a suite of indicators, which are more likely to provide the specificity and sensitivity needed to profile contaminated water samples (Wu et al., 2010; Newton et al., 2013). The ecology of indicator organisms post release into the studied environment and the use of different taxonomic groups (e.g. Gram-positive vs. Gram-negative organisms) covering a range of persistence times in that environment should be considered and could be used to discern recent and past contamination events. As technology advances, such approaches will move from the research arena to improved tools for water quality assessments that are necessary for more efficiently addressing pollution concerns. We suggest a particularly powerful approach would be to identify and then incorporate a suite of general and host-specific phylotypes into platforms such as phylochips (Wu et al., 2010) or other rapid sequence profiliers that can characterize the community of impacted surface waters.
Experimental procedures
Analysis of 454 pyrosequencing data from sewage, humans, cattle and chickens
Three previously published data sets and one new data set with five chicken faecal samples were used to assess Clostridiales population structure. A total of 132 samples were used for analysis; their sources and previous studies are shown in Table 1. The chickens were from the same farm collected in Athens, GA at the US Department of Agriculture research facility. After collection, samples were frozen immediately and shipped on ice to the EPA. Upon arrival in the lab, the samples were stored at −80°C until time of DNA isolation (< 6 months). The DNA from five chicken faecal samples was sequenced as described previously (Shanks et al., 2011). Briefly, we amplified the V6 hypervariable region of the 16S rRNA coding region using a mixture of five fused primers at the 5′ end of the V6 region (E. coli positions 967–985) and four primers at the 3′ end (E. coli positions 1046–1028) to capture the breadth of diversity of rRNA sequences represented in molecular databases (Sogin et al., 2006; Huber et al., 2007). We amplified libraries from at least three independent PCR cocktails for each sample to minimize the impact of potential early-round PCR errors. Amplicons were prepared and sequenced using the Roche Genome Sequencer GS-FLX according to the Roche standard protocols.
The relative abundance of Clostridiales and the family composition in the different sources (sewage, humans, cattle and chickens) was determined using normalized (to the smallest) data sets of all bacterial tags. Approximately 1.38 M Clostridiales pyrotags were parsed from these data sets and used to assess the population structure and phylogenetic relationships of matched reference sequences and cloned representatives. For comparisons of pyrotags distributed among samples for network analysis, data were normalized to the amount of Clostridiales in each sample.
Phylogenetic tree reconstruction and heatmap
The 30 most abundant Clostridiales in sewage and the 30 most abundant in human faecal samples (12 sequences were shared between both sources) were chosen for construction of a heatmap. Only one representative in the family Clostridiaceae was among the 30 most abundant in sewage or humans, so nine additional pyrotags representing the most abundant Clostridiaceae were added to the heatmap data set. Peptostreptococcaceae and Veillonellaceae are not commonly found in humans and their abundance patterns in sewage suggest some members of these families are free-living (McLellan et al., 2010); therefore, Peptostreptococcaceae was not included in the tree but Veillonellaceae was included for a point of reference in the phylogenetic analyses.
A reference ARB database of all near full-length 16S rRNA gene sequences representing the families Lachnospiraceae, Clostridiaceae, Ruminococcaceae and Veillonellaceae was downloaded from the Silva database (Pruesse et al., 2007) (May 2010). The subset of abundant pyrotags used in the heatmap was added and aligned to the ARB database using the FAST_ALIGNER tool (Ludwig et al., 2004), before the sequences were heuristically adjusted using the rRNA secondary structure as a guide. Near full-length sequences that were identical to the tag sequences were identified and used for phylogenetic analysis. A mask trimming sequences to an equal length was applied before we used the ARB neighbour-joining algorithm for phylogenetic reconstruction.
Network analysis
A network analysis was implemented to visualize the relationship of the top 400 most abundant Clostridiales pyrotags from the average of 38 Milwaukee sewage influent samples. The network was generated with Cytoscape version 2.7 (Shannon et al., 2003) by implementing an edge-weighted spring-embedded model (Eades, 1984). Sample sources: Milwaukee sewage influent, human faeces, cow faeces and chicken faeces are represented by averages of the samples included in each data set. See Table 1 for data set details. Lines in the network indicate a pyrotag was present in the connected data set, where the thickness of the line represents the relative abundance of the pyrotag in that data set.
Identification of core microbiota in humans
To assess if abundant sewage pyrotags represented core phylotypes in humans, Lachnospiraceae-annotated V6 pyrotags were selected from the normalized total bacterial data set and the number of individuals in which a particular pyrotag occurred was counted and plotted against the abundance in sewage. We also compared the abundance rank to determine if the high-abundance sewage pyrotags were also most abundant in individuals. We defined the top eight ranked pyrotags as ‘high abundance’ because this cut-off encompassed 27.1% of the total Lachnospiraceae pyrotag sewage data set and these pyrotags were found in at least 46 of 48 individuals. The eight top ranked pyrotags in humans corresponded to 30% of the total Lachnospiraceae pyrotags in the composite data set of individuals.
Clostridium coccoides clone libraries
Clone libraries were constructed in order to generate longer sequences (~ 900 bp) from the family Lachnospiraceae that could be compared with V6 pyrotag data (~ 60 bp). Lachnospiraceae was PCR amplified from five sewage influent samples from two Milwaukee WWTPs (SS and JI) on three separate dates (18 August 2008, 19 November 2008 and 22 April 2009) using a combination of two forward primers, the previously published group-specific primer g-CcocF (Matsuki et al., 2002) and a second primer with two base pair changes that included unclassified Lachnospiraceae, designated BF-063 with the sequence 5′ AAGTGACGGTACCTGAATAA 3′. These forward primers in a 3:1 ratio, respectively, were paired with the universal reverse primer (1492R) so that the amplification product included the V6 region. PCR product was purified using QIAGEN PCR purification kit (Qiagen, Valencia, CA). Products were cloned into pCR2.1 vector using the TOPO TA cloning kit (Invitrogen, Carlsbad, CA). Plasmid DNA was isolated using a manual method adapted to a 96-well microtitre plate format (Sambrook and Russell, 2001). Sequencing was carried out from the M13R primer using the ABI Big Dye Terminator Kit (Applied Biosystems, Foster City, CA) on an ABI Prism 3700xi (Applied Biosystems, Foster City, CA), which generated on average 800 bp reads. Sequences were trimmed for quality using PHRED (Ewing and Green, 1998), vector sequence was removed and sequences less than 500 bp were removed from further analyses. A total of 2070 sequences were generated from the five clone libraries with 2018 high-quality sequences used for comparisons after quality filtering and removal of chimeras identified by Mallard (Ashelford et al., 2006). Sequences flagged by Mallard were analysed using Chimera Check (Cole et al., 2003) to verify. Clones were then blasted against the pyrotags to determine what percentage of the Lachnospiraceae family represented by the pyrotags was also represented by our clones.
Oligotyping analysis
For oligotyping analysis we used 152 730 quality-controlled V6 reads from 132 samples that were identified as Blautia by GAST (Huse et al., 2008). Reads were aligned with PyNAST (DOI 10.1093/bioinformatics/btp636) using the GreenGenes (DeSantis et al., 2006) gold standard 16S rRNA gene sequence templates for Blautia. Following the entropy analysis oligotyping was performed with 24 components using the version 0.6 of the oligotyping pipeline (available from http://oligotyping.org). To reduce noise, we imposed requirements that each oligotype must: (i) appear in at least three samples, (ii) occur in more than 1% of the reads for at least one sample and (iii) have a most abundant unique sequence to occur at a minimum of 30 reads. After removal of oligotypes that did not meet these criteria, the analysis retained 140 804 reads (92.19% of the original reads). Oligotyping analysis identified 108 oligotypes, 93 of which perfectly matched sequences in NCBI’s nr database over their entire length.
Statistical analysis
Student’s t-test was used to assess significance of family abundance differences among host sources. Standard Spearman rank correlations were carried out using the R package (R Development Core Team, 2012).
Sequence data submission
Clostridium coccoides cloned sequences from libraries are deposited under GenBank Accession Numbers JX228967–JX230954. Other sequences already published in Newton and colleagues (2011) from these same libraries are deposited under JF826248 to JF826279. Pyrotag sequences from chickens and sewage from geographically dispersed cities, as well as previously published data for sewage samples, cattle and human faecal samples (Table 1), are available through VAMPS (http://www.vamps.mbl.edu).
Supplementary Material
Table S1. Number of high-abundance sewage phylotypes in individual samples.
Table S2. Correlation of ranked abundance between sewage samples from Milwaukee and other geographically distributed cities.
Acknowledgments
This work was supported by the grants 1R21AI076970-02 and 1R01AI091829-01A1 to S. L. M. and NSF/BDI 0960626 to S. M. H. We would like to thank Giles Goetz for bioinformatics support in the clone library comparisons and Morgan Schroeder for assistance with network analysis. Information has been subjected to the US EPA’s peer and administrative review and has been approved for external publication. Any opinions expressed in this paper are those of the author(s) and do not necessarily reflect the official positions and policies of the US EPA. Any mention of trade names or commercial products does not constitute endorsement or recommendation for use.
Footnotes
Additional Supporting Information may be found in the online version of this article at the publisher’s web-site:
References
- Ashelford KE, Chuzhanova NA, Fry JC, Jones AJ, Weightman AJ. New screening software shows that most recent large 16S rRNA gene clone libraries contain chimeras. Appl Environ Microbiol. 2006;72:5734–5741. doi: 10.1128/AEM.00556-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bernhard AE, Field KG. Identification of non-point sources of fecal pollution in coastal waters by using host-specific 16S ribosomal DNA genetic markers from fecal anaerobes. Appl Environ Microbiol. 2000a;66:1587–1594. doi: 10.1128/aem.66.4.1587-1594.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bernhard AE, Field KG. A PCR assay to discriminate human and ruminant feces on the basis of host differences in Bacteroides–Prevotella genes encoding 16S rRNA. Appl Environ Microbiol. 2000b;66:4571–4574. doi: 10.1128/aem.66.10.4571-4574.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bonjoch X, Balleste E, Blanch AR. Multiplex PCR with 16S rRNA gene-targeted primers of Bifidobacterium spp. to identify sources of fecal pollution. Appl Environ Microbiol. 2004;70:3171–3175. doi: 10.1128/AEM.70.5.3171-3175.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bonkosky M, Hernandez-Delgado EA, Sandoz B, Robledo IE, Norat-Ramirez J, Mattei H. Detection of spatial fluctuations of non-point source fecal pollution in coral reef surrounding waters in southwestern Puerto Rico using PCR-based assays. Mar Pollut Bull. 2009;58:45–54. doi: 10.1016/j.marpolbul.2008.09.008. [DOI] [PubMed] [Google Scholar]
- Cole JR, Chai B, Marsh TL, Farris RJ, Wang Q, Kulam SA, et al. The Ribosomal Database Project (RDP-II): previewing a new autoaligner that allows regular updates and the new prokaryotic taxonomy. Nucleic Acids Res. 2003;31:442–443. doi: 10.1093/nar/gkg039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Collins MD, Lawson PA, Willems A, Cordoba JJ, Fernandezgarayzabal J, Garcia P, et al. The phylogeny of the genus Clostridium – proposal of 5 new genera and 11 new species combinations. Int J Syst Bacteriol. 1994;44:812–826. doi: 10.1099/00207713-44-4-812. [DOI] [PubMed] [Google Scholar]
- DeSantis TZ, Hugenholtz P, Larsen N, Rojas M, Brodie EL, Keller K, et al. Greengenes, a chimera-checked 16S rRNA gene database and workbench compatible with ARB. Appl Environ Microbiol. 2006;72:5069–5072. doi: 10.1128/AEM.03006-05. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dethlefsen L, Huse S, Sogin ML, Relman DA. The pervasive effects of an antibiotic on the human gut microbiota, as revealed by deep 16S rRNA sequencing. PLoS Biol. 2008;6:2283–2400. doi: 10.1371/journal.pbio.0060280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dick LK, Bernhard AE, Brodeur TJ, Santo Domingo JW, Simpson JM, Walters SP, Field KG. Host distributions of uncultivated fecal Bacteroidales bacteria reveal genetic markers for fecal source identification. Appl Environ Microbiol. 2005;71:3184–3191. doi: 10.1128/AEM.71.6.3184-3191.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eades P. A heuristic for graph drawing. Congr Numer. 1984;42:149–160. [Google Scholar]
- Eckburg PB, Bik EM, Bernstein CN, Purdom E, Dethlefsen L, Sargent M, et al. Diversity of the human intestinal microbial flora. Science. 2005;308:1635–1638. doi: 10.1126/science.1110591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eren AM, Zozaya M, Taylor CM, Dowd SE, Martin DH, Ferris MJ. Exploring the diversity of Gardnerella vaginalis in the genitourinary tract microbiota of monogamous couples through subtle nucleotide variation. PLoS ONE. 2011;6:e26732. doi: 10.1371/journal.pone.0026732. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ewing B, Green P. Base-calling of automated sequencer traces using phred. II. Error probabilities. Genome Res. 1998;8:186–194. [PubMed] [Google Scholar]
- Field KG, Samadpour M. Fecal source tracking, the indicator paradigm, and managing water quality. Water Res. 2007;41:3517–3538. doi: 10.1016/j.watres.2007.06.056. [DOI] [PubMed] [Google Scholar]
- Fogarty LR, Voytek MA. Comparison of Bacteroides–Prevotella 16S rRNA genetic markers for fecal samples from different animal species. Appl Environ Microbiol. 2005;71:5999–6007. doi: 10.1128/AEM.71.10.5999-6007.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frank DN, St Amand AL, Feldman RA, Boedeker EC, Harpaz N, Pace NR. Molecular-phylogenetic characterization of microbial community imbalances in human inflammatory bowel diseases. Proc Natl Acad Sci USA. 2007;104:13780–13785. doi: 10.1073/pnas.0706625104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Franks AH, Harmsen HJ, Raangs GC, Jansen GJ, Schut F, Welling GW. Variations of bacterial populations in human feces measured by fluorescent in situ hybridization with group-specific 16S rRNA-targeted oligonucleotide probes. Appl Environ Microbiol. 1998;64:3336–3345. doi: 10.1128/aem.64.9.3336-3345.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fremaux B, Gritzfeld J, Boa T, Yost CK. Evaluation of host-specific Bacteroidales 16S rRNA gene markers as a complementary tool for detecting fecal pollution in a prairie watershed. Water Res. 2009;43:4838–4849. doi: 10.1016/j.watres.2009.06.045. [DOI] [PubMed] [Google Scholar]
- Furet JP, Firmesse O, Gourmelon M, Bridonneau C, Tap J, Mondot S, et al. Comparative assessment of human and farm animal faecal microbiota using real-time quantitative PCR. FEMS Microbiol Ecol. 2009;68:351–362. doi: 10.1111/j.1574-6941.2009.00671.x. [DOI] [PubMed] [Google Scholar]
- Gomez-Donate M, Balleste E, Muniesa M, Blanch AR. New molecular quantitative PCR assay for detection of host-specific Bifidobacteriaceae suitable for microbial source tracking. Appl Environ Microbiol. 2012;78:5788–5795. doi: 10.1128/AEM.00895-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gosalbes MJ, Durban A, Pignatelli M, Abellan JJ, Jimenez-Hernandez N, Perez-Cobas AE, et al. Metatranscriptomic approach to analyze the functional human gut microbiota. PLoS ONE. 2011;6:e17447. doi: 10.1371/journal.pone.0017447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hayashi H, Sakamoto M, Benno Y. Phylogenetic analysis of the human gut microbiota using 16S rDNA clone libraries and strictly anaerobic culture-based methods. Microbiol Immunol. 2002;46:535–548. doi: 10.1111/j.1348-0421.2002.tb02731.x. [DOI] [PubMed] [Google Scholar]
- Hayashi H, Sakamoto M, Kitahara M, Benno Y. Diversity of the Clostridium coccoides group in human fecal microbiota as determined by 16S rRNA gene library. FEMS Microbiol Lett. 2006;257:202–207. doi: 10.1111/j.1574-6968.2006.00171.x. [DOI] [PubMed] [Google Scholar]
- Hold GL, Pryde SE, Russell VJ, Furrie E, Flint HJ. Assessment of microbial diversity in human colonic samples by 16S rDNA sequence analysis. FEMS Microbiol Ecol. 2002;39:33–39. doi: 10.1111/j.1574-6941.2002.tb00904.x. [DOI] [PubMed] [Google Scholar]
- Huber JA, Welch DB, Morrison HG, Huse SM, Neal PR, Butterfield DA, Sogin ML. Microbial population structures in the deep marine biosphere. Science. 2007;318:97–100. doi: 10.1126/science.1146689. [DOI] [PubMed] [Google Scholar]
- Huse SM, Dethlefsen L, Huber JA, Welch DM, Relman DA, Sogin ML. Exploring microbial diversity and taxonomy using SSU rRNA hypervariable tag sequencing. PLoS Genet. 2008;4:e1000255. doi: 10.1371/journal.pgen.1000255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huse SM, Welch DM, Morrison HG, Sogin ML. Ironing out the wrinkles in the rare biosphere through improved OTU clustering. Environ Microbiol. 2010;12:1889–1898. doi: 10.1111/j.1462-2920.2010.02193.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jeter SN, McDermott CM, Bower PA, Kinzelman JL, Bootsma MJ, Goetz GW, McLellan SL. Bacteroidales diversity in ring-billed gulls (Laurus delawarensis) residing at Lake Michigan beaches. Appl Environ Microbiol. 2009;75:1525–1533. doi: 10.1128/AEM.02261-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kildare BJ, Leutenegger CM, McSwain BS, Bambic DG, Rajal VB, Wuertz S. 16S rRNA-based assays for quantitative detection of universal, human-, cow-, and dog-specific fecal Bacteroidales: a Bayesian approach. Water Res. 2007;41:3701–3715. doi: 10.1016/j.watres.2007.06.037. [DOI] [PubMed] [Google Scholar]
- Ley RE, Hamady M, Lozupone C, Turnbaugh PJ, Ramey RR, Bircher JS, et al. Evolution of mammals and their gut microbes. Science. 2008;320:1647–1651. doi: 10.1126/science.1155725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu CX, Finegold SM, Song YJ, Lawson PA. Reclassification of Clostridium coccoides, Ruminococcus hansenii, Ruminococcus hydrogenotrophicus, Ruminococcus luti, Ruminococcus productus and Ruminococcus schinkii as Blautia coccoides gen. nov., comb. nov., Blautia hansenii comb. nov., Blautia hydrogenotrophica comb. nov., Blautia luti comb. nov., Blautia producta comb. nov., Blautia schinkii comb. nov and description of Blautia wexlerae sp nov., isolated from human faeces. Int J Syst Evol Microbiol. 2008;58:1896–1902. doi: 10.1099/ijs.0.65208-0. [DOI] [PubMed] [Google Scholar]
- Lozupone CA, Stombaugh JI, Gordon JI, Jansson JK, Knight R. Diversity, stability and resilience of the human gut microbiota. Nature. 2012;489:220–230. doi: 10.1038/nature11550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ludwig W, Strunk O, Westram R, Richter L, Meier H, et al. ARB: a software environment for sequence data. Nucleic Acids Res. 2004;32:1363–1371. doi: 10.1093/nar/gkh293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McLellan SL, Huse SM, Mueller-Spitz SR, Andreishcheva EN, Sogin ML. Diversity and population structure of sewage-derived microorganisms in wastewater treatment plant influent. Environ Microbiol. 2010;12:378–392. doi: 10.1111/j.1462-2920.2009.02075.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matsuki T, Watanabe K, Fujimoto J, Miyamoto Y, Takada T, Matsumoto K, et al. Development of 16S rRNA-gene-targeted group-specific primers for the detection and identification of predominant bacteria in human feces. Appl Environ Microbiol. 2002;68:5445–5451. doi: 10.1128/AEM.68.11.5445-5451.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matsuki T, Watanabe K, Fujimoto J, Takada T, Tanaka R. Use of 16S rRNA gene-targeted group-specific primers for real-time PCR analysis of predominant bacteria in human feces. Appl Environ Microbiol. 2004;70:7220–7228. doi: 10.1128/AEM.70.12.7220-7228.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Newton RJ, Vandewalle JL, Borchardt MA, Gorelick MH, McLellan SL. Lachnospiraceae and Bacteroidales alternative fecal indicators reveal chronic human sewage contamination in an urban harbor. Appl Environ Microbiol. 2011;77:6972–6981. doi: 10.1128/AEM.05480-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Newton RJ, Bootsma MJ, Morrison HG, Sogin ML, McLellan SL. Microbial signatures of fecal pollution in the waters off an urbanized coast of Lake Michigan. Microb Ecol. 2013 doi: 10.1007/s00248-013-0200-9. (in press) [DOI] [PMC free article] [PubMed] [Google Scholar]
- NRC. Indicators for Waterborne Pathogens. Washington, DC, USA: National Research Council of the National Academies; 2004. [Google Scholar]
- Pruesse E, Quast C, Knittel K, Fuchs BM, Ludwig W, Peplies J, Glockner FO. SILVA: a comprehensive online resource for quality checked and aligned ribosomal RNA sequence data compatible with ARB. Nucleic Acids Res. 2007;35:7188–7196. doi: 10.1093/nar/gkm864. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qin JJ, Li RQ, Raes J, Arumugam M, Burgdorf KS, Manichanh C, et al. A human gut microbial gene catalogue established by metagenomic sequencing. Nature. 2010;464:59–65. doi: 10.1038/nature08821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- R Development Core Team. R: A Language and Environment for Statistical Computing. Vienna, Austria: R Foundation for Statistical Computing; 2012. [WWW document]. URL http://www.R-project.org. [Google Scholar]
- Rajilic-Stojanovic M, Heilig HGHJ, Molenaar D, Kajander K, Surakka A, Smidt H, de Vos WM. Development and application of the human intestinal tract chip, a phylogenetic microarray: analysis of universally conserved phylotypes in the abundant microbiota of young and elderly adults. Environ Microbiol. 2009;11:1736–1751. doi: 10.1111/j.1462-2920.2009.01900.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reeves AE, Koenigsknecht MJ, Bergin IL, Young VB. Suppression of Clostridium difficile in the gastrointestinal tract of germ-free mice inoculated with a murine Lachnospiraceae isolate. Infect Immun. 2012 doi: 10.1128/IAI.00647-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robinson CJ, Bohannan BJM, Young VB. From structure to function: the ecology of host-associated microbial communities. Microbiol Mol Biol Rev. 2010;74:453–476. doi: 10.1128/MMBR.00014-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sambrook J, Russell D. Molecular Cloning: A Laboratory Manual. Cold Spring Harbor, NY, USA: Cold Spring Harbor Laboratory Press; 2001. [Google Scholar]
- Sekelja M, Berget I, Naes T, Rudi K. Unveiling an abundant core microbiota in the human adult colon by a phylogroup-independent searching approach. ISME J. 2011;5:519–531. doi: 10.1038/ismej.2010.129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shanks OC, Domingo JW, Lu J, Kelty CA, Graham JE. Identification of bacterial DNA markers for the detection of human fecal pollution in water. Appl Environ Microbiol. 2007;73:2416–2422. doi: 10.1128/AEM.02474-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shanks OC, Kelty CA, Archibeque S, Jenkins M, Newton RJ, McLellan SL, et al. Community structures of fecal bacteria in cattle from different animal feeding operations. Appl Environ Microbiol. 2011;77:2992–3001. doi: 10.1128/AEM.02988-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shannon P, Markiel A, Ozier O, Baliga NS, Wang JT, Ramage D, et al. Cytoscape: a software environment for integrated models of biomolecular interaction networks. Genome Res. 2003;13:2498–2504. doi: 10.1101/gr.1239303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sogin ML, Morrison HG, Huber JA, Welch DM, Huse SM, Neal PR, et al. Microbial diversity in the deep sea and the underexplored ‘rare biosphere’. Proc Natl Acad Sci USA. 2006;103:12115–12120. doi: 10.1073/pnas.0605127103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stewart JR, Gast RJ, Fujioka RS, Solo-Gabriele HM, Meschke JS, Amaral-Zettler LA, et al. The coastal environment and human health: microbial indicators, pathogens, sentinels and reservoirs. Environ Health. 2008;7 (Suppl 2):S3. doi: 10.1186/1476-069X-7-S2-S3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tap J, Mondot S, Levenez F, Pelletier E, Caron C, Furet JP, et al. Towards the human intestinal microbiota phylogenetic core. Environ Microbiol. 2009;11:2574–2584. doi: 10.1111/j.1462-2920.2009.01982.x. [DOI] [PubMed] [Google Scholar]
- Turnbaugh PJ, Hamady M, Yatsunenko T, Cantarel BL, Duncan A, Ley RE, et al. A core gut microbiome in obese and lean twins. Nature. 2009;457:480–484. doi: 10.1038/nature07540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Turnbaugh PJ, Quince C, Faith JJ, McHardy AC, Yatsunenko T, Niazi F, et al. Organismal, genetic, and transcriptional variation in the deeply sequenced gut microbiomes of identical twins. Proc Natl Acad Sci USA. 2010;107:7503–7508. doi: 10.1073/pnas.1002355107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- USEPA. National Water Quality Inventory: Report to Congress. Washington, DC, USA: Office of Water; 2009. [Google Scholar]
- USEPA. EPA BEACH Report: 2011 Swimming Season. Washington, DC, USA: USEPA; 2012. [Google Scholar]
- VandeWalle JL, Goetz GW, Huse SM, Morrison HG, Sogin ML, Hoffmann RG, et al. Acinetobacter, Aeromonas and Trichococcus populations dominate the microbial community within urban sewer infrastructure. Environ Microbiol. 2012;14:2538–2552. doi: 10.1111/j.1462-2920.2012.02757.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wery N, Monteil C, Pourcher AM, Godon JJ. Human-specific fecal bacteria in wastewater treatment plant effluents. Water Res. 2010;44:1873–1883. doi: 10.1016/j.watres.2009.11.027. [DOI] [PubMed] [Google Scholar]
- Wu CH, Sercu B, Van de Werfhorst LC, Wong J, DeSantis TZ, Brodie EL, et al. Characterization of coastal urban watershed bacterial communities leads to alternative community-based indicators. PLoS ONE. 2010;5:e11285. doi: 10.1371/journal.pone.0011285. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Table S1. Number of high-abundance sewage phylotypes in individual samples.
Table S2. Correlation of ranked abundance between sewage samples from Milwaukee and other geographically distributed cities.