

Abstract
Background
Combination antiretroviral regimens including nelfinavir (NFV) are commonly used in pregnancy. We studied the safety, antiviral effect, and pharmacokinetics of NFV and its M8 metabolite with two dosing regimens in combination with zidovudine (ZDV) and lamivudine (3TC) in HIV-infected pregnant women.
Method
HIV-infected pregnant women between 14 and 34 weeks gestation received NFV (Cohort 1: 750 mg tid, n = 10; Cohort 2: 1250 mg bid, n = 23) with ZDV and 3TC. Serial blood sampling for NFV concentrations was performed antepartum (AP) and 6 weeks postpartum (PP). Maternal and cord blood samples were also obtained at delivery. NFV and M8 levels were determined by high-performance liquid chromatography. The pharmacokinetic (PK) target was an extrapolated NFV AUC0–24 > 30 μg · h/mL. Mothers were followed frequently for potential clinical and laboratory toxicity.
Results
Overall, NFV in combination with ZDV and 3TC was well tolerated. The PK target was met in 3/8 AP and 5/7 PP in Cohort 1 and 17/21 AP and 16/17 PP in Cohort 2. When Cohort 2 NFV PK parameters AP and PP were compared, median Cmax (3.90 μg/mL vs. 5.01 μg/mL, p < .05) and AUC0–24 (56.6 vs. 86.8 μg · h/mL, p < .05) were increased PP and oral clearance (Cl/F; 44.2 vs. 28.8 L/h, p < .05) was decreased PP. The average M8/NFV ratio was increased PP compared to AP (0.085 vs. 0.29, p < .001). Placental transfer of NFV was low with a median cord blood:maternal plasma ratio at delivery of 0.05. Maternal mean CD4+ T cell counts increased significantly and plasma HIV-1 RNA levels decreased from entry to delivery and 6 to 12 weeks postpartum.
Conclusion
NFV used in combination with ZDV and 3TC was well tolerated in pregnant HIV-infected women and produced a significant improvement in HIV disease parameters. NFV drug exposure is inadequate in most pregnant women receiving 750 mg tid but is much improved with 1250 mg bid. NFV crosses the placenta poorly. The AP increase in NFV oral clearance and decrease in M8/NFV ratio suggest that CYP3A activity increases relative to CYP2C19 activity during pregnancy.
Keywords: HIV, nelfinavir, pregnant women
Current antiretroviral guidelines recommending triple combination highly active antiretroviral therapy (HAART) during pregnancy for both treatment of the mother and prevention of mother-to-child transmission of HIV have resulted in rates of perinatal transmission below 2%.1 Treatment with the protease inhibitor (PI) nelfinavir (NFV) in combination with zidovudine (ZDV) and lamivudine (3TC) is a currently preferred HAART regimen during pregnancy.1,2 In addition, this combination has been proposed for both prophylaxis and early treatment of HIV-exposed and -infected newborns. The recent report of excess liver toxicity associated with use of a nevirapine-based regimen in women with CD4+ lymphocyte counts over 250 cells/mm3 emphasizes the importance of studying the safety of antiretrovirals in pregnant women.3,4 The physiologic changes of pregnancy may have a large impact on drug absorption, distribution, metabolism, and excretion, making it crucial to study the pharmacokinetics of antiretrovirals during pregnancy.5,6 It is also important to know whether antiretroviral agents cross the placenta, because placental passage has implications for prevention of maternal–infant HIV transmission and for potential effects on the fetus.6 This study was initiated to evaluate the pharmacokinetics and safety of two dosing regimens of NFV when used in combination with ZDV and 3TC in HIV-positive mothers during pregnancy and postpartum.
STUDY DESIGN
The Pediatric AIDS Clinical Trials Group (PACTG) Protocol 353 was a multicenter phase 1 study of the safety, tolerance, and pharmacokinetics of NFV co-administered with ZDV and 3TC in HIV-infected pregnant women and their infants. The data on infants have been reported separately.7
Population
HIV-infected pregnant women between 14 and 34 weeks of gestation with singleton pregnancies were eligible for entry after providing informed consent. Women and infants were enrolled between 1997 and 2002, and all women were treated in accordance with the standard of care. In Cohort 1, women had no prior treatment with any PI and less than 3 weeks of treatment with ZDV. In Cohort 2, women could be drug naïve or have received either less than 3 weeks of NFV or another PI in combination with nucleosides or more than 3 weeks of therapy if within the 3 months before enrollment the CD4+ T-cell count was stable and HIV-1 RNA levels were below 400 copies/mL. Women could have no serious medical conditions, prior history of preterm labor, or current pregnancy complications. They had to have a normal detailed fetal ultrasound with a normal amniotic fluid volume at entry.
Drug Regimen
The study was planned to have a dose escalation depending on the pharmacokinetic targets and tolerance results in the first cohort. Cohort 1 mothers received NFV 750 mg (three 250 mg tablets) three times daily with Combivir (ZDV 300 mg/3TC 150 mg) twice daily from entry through 12 weeks postpartum. Cohort 2 mothers received NFV 1250 mg (five 250 mg tablets) twice daily with Combivir twice daily. During labor, women in both groups received intravenous ZDV and oral 3TC and NFV. Women were instructed to take their medications with food or Ensure liquid. During labor, NFV was given with liquid dietary supplements to enhance absorption. Baseline studies included T-cell subsets and plasma HIV-1 RNA by Roche Amplicor 1.5 (Roche Diagnostic Systems, Inc., Branchburg, New Jersey, USA). The ultra-sensitive HIV-1 RNA assay was performed on plasma samples with <400 copies/mL. Follow-up samples were collected every 4 weeks, at delivery, and at 2, 4, 6, and 12 weeks postpartum. Complete blood count (CBC) with differential and platelets and chemistry studies were performed at baseline and follow-up at the same time points. Mothers were followed for clinical and laboratory toxicities, and adverse events were graded according to the National Institute of Allergy and Infectious Diseases Division of AIDS Toxicity grading table. Toxicities were classified as follows: mild, grade 1; moderate, grade 2; severe, grade 3; and life threatening, grade 4. The site investigators managed any adverse events and assessed their relationship to the study medications. The protocol team subsequently reviewed all adverse events on at least a monthly basis. Protocol-targeted adverse events included all new diagnoses, all signs and symptoms and laboratory abnormalities of grade 3 or 4, and all grade 2 levels of uric acid, SGOT (serum glutamic oxaloacetic transaminase), SGPT (serum glutamic pyruvic transaminase), and glucose. All study drugs (ZDV, 3TC, and NFV) were permanently discontinued for a confirmed grade 4 or persistent grade 3 toxicity.
Pharmacokinetic Samples
Serial blood samples were obtained over a dosing interval at 34 weeks gestation and again at 5–6 weeks postpartum. Samples were obtained before a morning dose and at 1, 2, 3, 4, 5, 6, and 8 hours after the dose from mothers in Cohort 1 and before a morning dose and at 2, 4, 6, 8, 10, and 12 hours after the dose from mothers in Cohort 2. Additional samples were collected at delivery from maternal blood and cord blood. Plasma concentrations of NFV and M8, its active hydroxyl-tert-butylamide metabolite, were measured by high-performance liquid chromatography (HPLC).
Samples were frozen at -20°C or colder, shipped to the central analytical lab (University of California, San Diego, USA), and assayed on average within 2 weeks of collection. Serum concentrations of NFV and its active metabolite M8 were determined simultaneously by HPLC. The method was derived from that described by Wu et al.8 with the following modifications. Proteins were precipitated from 0.1–0.3 mL serum samples (depending on the size of the available specimen) using acetonitrile, and 0.1 mL of the supernatant was injected directly onto a C18 reversed phase HPLC column (150 × 4.6 mm). The drugs were separated isocratically using 10 mM potassium phosphate buffer, pH 4.0 (62%), and acetonitrile (38%). Ultraviolet detection at 206 nm was used. The mean recovery was 97% for NFV and 89% for M8. The assay was linear from the lower limit of quantitation of 0.1 μg/mL to 10.34 μg/mL for NFV and from the lower limit of quantitation of 0.063 to 5 μg/mL for M8. The mean inter- and intra-assay coefficient of variation based on other controls was 10.56 ± 2.04% and 4.01 ± 2.38% for NFV and 11.6 ± 1.12% and 5.96 ± 1.69% for M8.
Noncompartmental pharmacokinetic parameters were calculated using two commercial computer programs (WinNonlin, Pharsight Corp., Palo Alto, California, USA, and Microsoft Excel, Redmond, Washington, USA). Predose concentration, peak concentration (Cmax), time of peak concentration (Tmax), and concentration at the end of sampling (C8hour or C12hour) were determined by inspection of concentration time curves. Area under the serum concentration-time curve (AUC) during the dosing interval (8-hour AUC or 12-hour AUC) was calculated by the trapezoidal rule. AUC values were extrapolated to a 24-hour AUC by multiplying 8-hour AUCs by 3- and 12-hour AUCs by 2. Oral clearance (Cl/F) was calculated from the formula Cl/F = dose/AUC. Terminal elimination half-lives (t1/2) for Cohort 2 were determined by weighted least-squares fitting. T1/2 could not be calculated for Cohort 1 due to prolonged absorption relative to the 8-hour dosing and sampling interval. The ratio of M8 to NFV was calculated for each plasma sample. These ratios were then averaged for each sampling period, excluding samples with NFV concentrations below 1.0 μg/mL.
The pharmacokinetic target for Cohorts 1 and 2 was an extrapolated 24-hour NFV AUC above 30 μg · h/mL, which is equivalent to the 10th percentile of the AUC in nonpregnant adults. AUCs observed after 8- and 12-hour dosing were extrapolated to 24 hours as described previously. After the data from Cohort 1 had been analyzed, the NFV dose for Cohort 2 was changed to 1250 mg bid.9
Statistical Analysis
The Wilcoxon test was used to compare antepartum and postpartum pharmacokinetic parameters. The paired t test was used to evaluate mean changes in CD4+ T-cell counts between baseline, delivery, and postpartum. McNemar’s test was used to compare the number of patients who had HIV-1 RNA levels <400 copies/mL at baseline, delivery, and postpartum.
RESULTS
The baseline characteristics of the 33 women (10 in Cohort 1 and 23 in Cohort 2) enrolled in the study are shown in Table 1. The median gestational age at entry was 27 weeks overall with median HIV-1 RNA level and CD4 T-cell count in Cohort 1 of 30,643 copies/mL and 220 CD4 T cells/mm3 versus 3,317 copies/mL and 338 CD4 T cells/mm3 in Cohort 2. Twenty-one women were naïve to PIs (all 10 in Cohort 1 and 11 [48%] in Cohort 2). The remaining women in Cohort 2 only received NFV during the current pregnancy. In Cohort 1, 8 of the 10 enrolled women completed the study, one woman had an elective pregnancy termination, and one mother had a fetal demise with obstetrical complications shortly after starting study medications. Both of these events were considered unrelated to study treatment. In Cohort 2, 21 of the 23 enrolled women completed the study, and 2 women withdrew from the study postpartum.
Table 1.
Baseline characteristics
| Number of mothers enrolled | Total (N = 33) | Cohort 1 (n = 10) | Cohort 2 (n = 23) |
|---|---|---|---|
| Median age, yrs (range) | 27 (18–37) | 28 (19–37) | 27 (18–35) |
| Race/ethnicity, n (%) | |||
| White, non-Hispanic | 4 (12%) | 3 (30%) | 1 (4%) |
| Black, non-Hispanic | 9 (27%) | 1 (10%) | 8 (35%) |
| Hispanic | 20 (61%) | 6 (60%) | 14 (61%) |
| Median gestational age at entry, wks (range) | 27 (14–37) | 25 (14–29) | 29 (15–37) |
| Median HIV-1 RNA, copies/mLa (range) | 5,754 (<400–347,000) | 30,643 (<400–282,000) | 3,317 (<400–347,000) |
| Median CD4 T-cell count, cells/mm3 (range) | 293 (46–1005) | 220 (46–836) | 338 (78–1005) |
| No. of patients who completed study | 29 | 8 | 21 |
| Pregnancy outcome | |||
| Elective termination | 1 | 1 | 0 |
| Fetal demise | 1 | 1 | 0 |
| Neonatal death | 1 | 1 | 0 |
| Live births | 31 | 8 | 23 |
Ten women (2 in Cohort 1 and 8 in Cohort 2) had HIV-1 RNA levels <400 copies/mL at baseline.
Maternal Pharmacokinetic Results
Pharmacokinetic sampling was successfully completed in 8 women antepartum and 7 women postpartum in Cohort 1 and 21 women antepartum and 17 women postpartum in Cohort 2. Pharmacokinetic parameters for the mothers in both cohorts are presented in Table 2. Only 3 of 8 of women in Cohort 1 met the 24-hour NFV AUC target of 30 μg · h/mL during pregnancy, while 5 of 7 met the target postpartum. In contrast, 17 of 21 women in Cohort 2 met the AUC target antepartum and 16 of 17 met the target postpartum. NFV concentration time curves for Cohort 1 are presented in Figure 1 and for Cohort 2 in Figure 2. Among the Cohort 2 women, median NFV AUC, predose concentration, peak concentration, and 12-hour postdose concentration were significantly higher postpartum than during pregnancy, whereas median Cl/F was significantly lower (p < .05 for all comparisons). The Cohort 2 mothers demonstrated significant diurnal variation in NFV concentrations, with higher median predose than 12-hour postdose concentrations both antepartum (1.45 μg/mL vs. 0.53 μg/mL, p=.02) and postpartum (2.77 μg/mL vs. 1.60 μg/mL, p = .003). The ratio of M8 to NFV plasma concentrations could be calculated for 21 women. The median M8 to NFV ratio was decreased antepartum compared to postpartum (0.085 [range, 0 to 0.29] vs. 0.21 [range, 0.02 to 0.41], p < .001).
Table 2.
Pharmacokinetic parameters for Cohorts 1 and 2
| Cohort 1 | Pregnant (n = 8) | Postpartum (n = 7) |
|---|---|---|
| Median (range) | Median (range) | |
| Predose concentration, μg/mL | 0.80 (0.29–2.69) | 0.85 (bql–1.87) |
| Cmax, μg/mL | 1.98 (0.80–3.07) | 2.66 (1.23–3.03) |
| Tmax, hours | 2.0 (0.0–8.0) | 2.3 (1.0–5.0) |
| 8-hour concentration, μg/mL | 0.53 (0.15–1.85) | 0.58 (0.23–2.09) |
| AUC0–24, μg · h/mL | 24.7 (7.2–48.2) | 38.1 (13.8–46.8) |
| Cl/F, L/h | 91.1 (46.7–312.5) | 59.1 (48.0–163.0) |
| Cl/F, L/h/kg | 1.06 (0.56–4.53) | 1.01 (0.59–2.48) |
| Pregnant (n = 21) | Postpartum (n = 17) | |
| Cohort 2 | Median (range) | Median (range) |
|
| ||
| Predose concentration, μg/mL | 1.45 (bql–5.50) | 2.77 (bql–9.59)* |
| Cmax, μg/mL | 3.90 (1.37–6.72) | 5.01 (1.84–10.17)* |
| Tmax, hours | 4.0 (0.0–8.0) | 4.0 (1.3–6.0) |
| 12-hour concentration, μg/mL | 0.53 (0.10–2.81) | 1.60 (0.24–4.42)* |
| AUC0–24, μg · h/mL | 56.6 (15.2–92.6) | 86.8 (21.7–186.7)* |
| t1/2, hours | 3.0 (1.9–12.3) | 3.9 (2.6–9.6) |
| Cl/F, L/h | 44.2 (27.0–164.0) | 28.8 (13.4–115.3)* |
| Cl/F, L/h/kg | 0.63 (0.30–1.88) | 0.46 (0.13–1.41) |
Note: bql = below quantitation limit of assay.
Pregnant vs. postpartum, p < .05.
Figure 1.
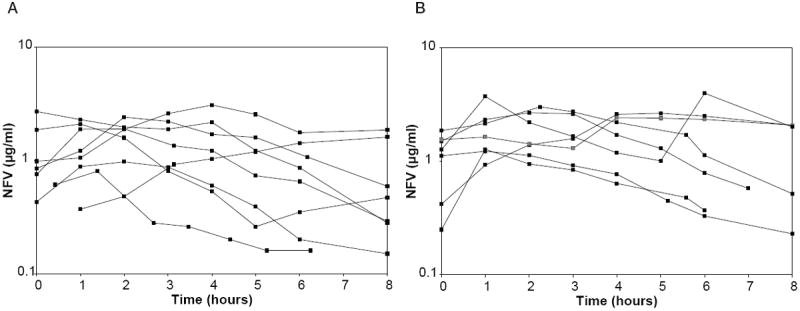
(A) Nelfinavir (NFV) concentration time plots for Cohort 1 antepartum. (B) NFV concentration time plots for Cohort 1 at 6 weeks postpartum.
Figure 2.
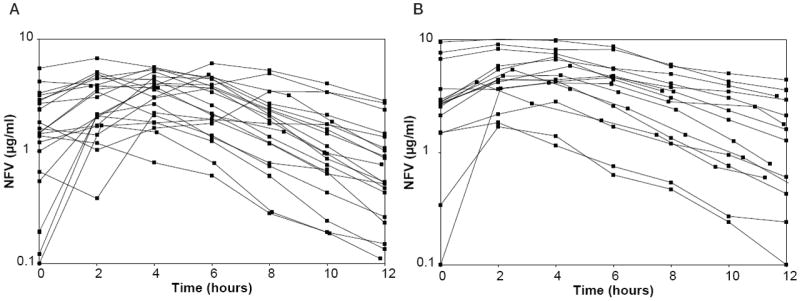
(A) Nelfinavir (NFV) concentration time plots for Cohort 2 antepartum. (B) NFV concentration time plots for Cohort 2 at 6 weeks postpartum.
Placental Transfer of NFV
Plasma from the mothers at the time of delivery and from cord blood was available from 25 mothers. Plasma NFV concentration was below the assay limit of detection in one mother. Median maternal plasma NFV concentration in the remaining 24 women at delivery was 1.12 μg/mL (range, 0.17 to 4.09 μg/mL). Median cord blood NFV concentration was 0.07 μg/mL, ranging from below the assay limit of detection in 12 infants to a maximum of 0.94 μg/mL. The median ratio of the NFV concentration in cord blood to that of maternal plasma at delivery was 0.05.
Virologic and CD4+ T-Cell Response to HAART During Gestation and Postpartum
CD4+ T-cell counts at baseline, delivery, and 12 weeks postpartum were available in 28 women.
As shown in Table 3, 27 women had CD4 count measurements at baseline, delivery, and postpartum. The mean CD4 T+ cell number in the two cohorts combined increased significantly from baseline to postpartum, from a mean of 402 CD4 T+ cells/mm3 to 458 cells/mm3 at delivery (p = .101) to 524 cells/mm3 at 12 weeks postpartum (p < .001). The mean changes from baseline to postpartum in Cohorts 1 and 2 were 102 cells/mm3 (n = 7, p = .200) and 129 cells/mm3 (n = 20, p < .001), respectively. Across the two cohorts combined, PI-naïve women also showed significant increase in CD4 T+ cells (p = .003) toward larger mean changes than PI-experienced women from baseline to 12 weeks postpartum (133 vs. 106 cells/mm3).
Table 3.
Maternal CD4 T-cell counts at baseline, delivery, and 12 weeks postpartum
| Mean CD4 cells/mm3 | N = 33 | Baseline | Delivery | pa | Postpartum | pb |
|---|---|---|---|---|---|---|
| Overall | 27c | 402 | 458 | .101 | 524 | .0003 |
| Cohort 1 | 7 | 343 | 432 | .201 | 446 | .200 |
| Cohort 2 | 20 | 422 | 467 | .271 | 551 | .0008 |
| Naïve | 16 | 320 | 405 | .022 | 454 | .003 |
| Not naïve | 11 | 521 | 535 | .826 | 626 | .063 |
Comparison of baseline with delivery.
Comparison of baseline with postpartum.
Number with all three values.
Table 4 shows the virologic response on treatment as measured by the number of women who achieved undetectable HIV-1 RNA levels in plasma (<400 HIV-1 RNA copies/mL). Over the two cohorts combined, most women (25/31 or 81%) achieved undetectable levels of virus at delivery and continued undetectable postpartum (15/29 or 52%). The rates showed a trend toward improvement in the proportion with undetectable HIV-1 RNA in Cohort 2 versus Cohort 1 at delivery (20/23 or 87% vs. 5/8 or 63%) but were more similar at 12 weeks postpartum (10/21 or 48% vs. 5/8 or 63%). In the PI-naïve group, 3/19 (16%) had undetectable HIV RNA at entry, 14/19 (74%) at delivery, and 8/18 (44%) at 12 weeks postpartum. One infant was infected in utero (positive DNA and RNA PCR at birth) despite an undetectable maternal plasma viral load at delivery; however the mother had not initiated treatment until 34 weeks gestation.
Table 4.
Maternal plasma RNA levels <400 copies/mL at baseline, delivery, and postpartum
| n | Baseline | Delivery | Postpartum (12 wks) | |
|---|---|---|---|---|
| Overall | 33 | 10/31 (32%) | 25/31 (81%) | 15/29 (52%) |
| Naïve | 21 | 3/19 (16%) | 14/19 (74%) | 8/18 (44%) |
| Not naïve | 12 | 7/12 (58%) | 11/12 (92%) | 7/11 (64%) |
Maternal Drug Tolerance and Adverse Events
NFV in combination with ZDV and 3TC was generally well tolerated by HIV-infected women during pregnancy, labor, and postpartum. No adverse obstetrical outcomes were judged to be definitely related to study drugs. One woman underwent an elective pregnancy termination, one fetal demise occurred due to obstetrical complications shortly after treatment was started, and one neonatal death occurred due to sepsis; all three events occurred among women enrolled in Cohort 1. The median gestational age of the infants at birth was 38.5 weeks with median birth weight of 3028 g. There were no preterm births. No major fetal anomalies or toxicities were noted at birth.
Table 5 summarizes protocol-targeted maternal adverse events possibly related to study medications, the timing, and outcome. Ten adverse events were reported including hypoglycemia, increased liver enzymes, anemia, vomiting, and neutropenia. Only one event, grade 4 neutropenia at 12 weeks postpartum, required permanent discontinuation of study drugs. Other laboratory abnormalities resolved, and treatment was continued.
Table 5.
Maternal adverse events judged to be possibly or probably treatment related
| Event | Grade | Timing (wks from delivery) | Treatment change |
|---|---|---|---|
| Clinical events | |||
| Vomiting/nausea | 3 | At delivery | No |
| Decreased amniotic fluid volume | 3 | At delivery | No |
| Laboratory abnormalities | |||
| Hypoglycemia | 2 | Antepartum (10 wks prior delivery) | No |
| Hypoglycemia | 2 | Antepartum (4 wks prior delivery) | No |
| Elevated AST (aspartate aminotransferase) | 2 | Antepartum (4 wks prior delivery) | No |
| Elevated GGT (gamma glutamyltransferase) | 3 | 5 wks postpartum | Temporarily held, restarted without incident |
| Hemoglobin | 3 | At delivery | No |
| Hemoglobin | 3 | At delivery | No |
| Total amylase | 3 | At delivery | No |
| Neutropenia | 4 | 12 wks postpartum | No |
| Hemoglobina | 4 | 2 wks postpartum | Permanently discontinued |
Normocytic normachromic anemia.
DISCUSSION
This study showed that the PI NFV at a dose of 1250 mg bid used in combination with ZDV and 3TC was generally well tolerated in a group of HIV-infected women during pregnancy and postpartum and produced significant plasma HIV suppression associated with an increase in maternal CD4+ T-cell numbers. PIs are potent inhibitors of HIV replication especially when used in combination with other antiretrovirals as part of a HAART regimen.2 NFV has become one of the most commonly used PIs during pregnancy because of its tolerability and potent antiviral effect. In a clinical trial (PACTG 316) that enrolled HIV-infected pregnant women from the United States, Europe, Brazil, and the Caribbean between 1997 and 2000, the use of PIs was common (41%), with the majority of these women (60%) receiving NFV.10
NFV pharmacokinetics in nonpregnant adults are characterized by variable absorption and diurnal variation in predose concentrations. The oral bioavailability of NFV ranges from 20% to 80% and increases two or three times when administered with food.2,11 NFV is highly protein bound (greater than 98%) and has a volume of distribution of 2 to 7 L/kg. NFV is extensively metabolized by enzymes of the cytochrome P-450 system. CY2C19 metabolizes NFV to its active metabolite, M8, and both NFV and M8 are eliminated via metabolism by CYP3A family enzymes.11,12 The elimination half-life of NFV in nonpregnant adults is 3 to 5 hours, consistent with the observed half-life of 3 hours during pregnancy and 3.9 hours postpartum in this study.
Only three women (37%) met the target AUC during pregnancy with 750 mg tid. NFV exposure was much improved in pregnant women who received 1250 mg bid, with only 4 (19%) of 21 pregnant women failing to meet the exposure target. When NFV pharmacokinetic parameters in these women were compared during pregnancy and at 6 weeks postpartum, Cl/F was significantly increased and Cmax, morning and evening predose concentrations, AUC, and M8:NFV ratio were significantly decreased during pregnancy.
Diurnal variation in NFV predose concentrations has been observed in previous studies in non-pregnant adults.9 We observed significant diurnal variation in NFV plasma concentrations during pregnancy and postpartum, and both morning and evening predose concentrations were higher postpartum than during pregnancy in the current study. It is unknown whether the diurnal variation in NFV concentrations and their change during pregnancy are due to differences in drug absorption or elimination or both.
Several previous studies have reported NFV concentrations in pregnant women receiving 1250 mg bid. One study found NFV concentrations were significantly lower in pregnant women compared to nonpregnant women, whereas another study showed too much variability in the pharmacokinetic parameters to see a pregnancy effect.13,14 Although some studies suggested that therapeutic drug monitoring might be an option for some women who did not meet the trough target set of 0.8 μg/mL, typical diurnal variation in NFV predose concentrations, with their average morning predose NFV concentration exceeding 1.0 μg/mL both during pregnancy and postpartum, suggest that many women may exceed the therapeutic target in the morning.15 A recent study showed pharmacokinetic results similar to our study when comparing AUC levels, which tend to be more reliable. Trough concentrations are more difficult to compare with NFV because of diurnal variation. Our median for morning trough was 1.45 μg/mL and only 6/21 were below 0.8 μg/mL, whereas the median for evening trough was 0.53 with 12/21 falling below 0.8 μg/mL.16 During clinical care, samples for therapeutic drug monitoring are most likely to be drawn in the morning and do not reflect the significantly lower evening concentrations. In nonpregnant adults receiving NFV 1250 mg bid, morning predose concentrations average 2.2 μg/mL whereas evening predose concentrations average 0.7 μg/mL, suggesting that most nonpregnant adults receiving 1250 mg bid would fail in the evening to meet the 0.8 μg/mL threshold for dosage modification.9 The value of therapeutic drug monitoring of NFV has not been established.17
Because we did not use intravenous NFV dosing or measure NFV protein binding, we could not determine the independent contributions of changes in protein binding, bioavailability, volume of distribution, and clearance on the differences observed in the pharmacokinetic parameters during pregnancy and postpartum. It was interesting to note that although the total daily dose with 1250 mg bid is only 10% greater than the 750 mg tid, there was a dramatic increase in the AUC with the twice-a-day dose, suggesting that a saturable process, perhaps affecting bioavailability, may play an important role. The antepartum increase in NFV oral clearance and decrease in M8:NFV ratio suggest that CYP3A activity increases relative to CYP2C19 activity during pregnancy.
Cord blood NFV concentrations were low compared to maternal NFV concentrations at the time of delivery, consistent with the growing evidence from other studies of poor placental transport of PIs.6 In contrast, both nucleoside and non-nucleoside reverse transcriptase inhibitors readily cross the placenta.10,18-20 Nucleoside inhibitors such as ZDV and stavudine (d4T) can be found at higher concentrations in amniotic fluid and have cord blood levels almost equivalent to maternal plasma levels.21,22 Poor placental transfer of PIs may limit fetal PI exposure in utero, which may protect the fetus from potential toxic or teratogenic effects of these agents. Over 1,600 NFV pregnancy exposures, including 496 in the first trimester, have been reported to the Antiretroviral Pregnancy Registry with no evidence of an increased risk of birth defects.23,24 The mechanism of action of the PIs in reducing mother-to-child transmission is most likely by the reduction of maternal virus load.25 Because PIs do not cross the placenta well, the administration of PIs to the mother during labor should not provide postexposure prophylaxis of the newborn at the time of birth, unlike NVP and nucleoside reverse transcriptase inhibitors.26,27 PIs could however be given directly to the infant following birth.
There was a significant increase in CD4+ T-cell number observed in both cohorts during gestation that continued into the postpartum period but was most notable (though not significantly so) in women who were drug naïve. The entry criteria for women who were not drug naïve included HIV RNA levels <400 copies/mL and stable CD4+ T cells for 3 months, which explains the higher CD4+ T-cell counts at entry; therefore immune reconstitution had most likely occurred prior to study enrollment.1,3,4
This antiretroviral combination reduced maternal viral plasma RNA to <400 HIV RNA copies/mL in the majority of the women, which is a major goal in the prevention of MTCT.25 We did identify one infant infected in utero despite the mother having an undetectable plasma viral load at delivery. However, she started HAART relatively late at 34 weeks gestation.
The combined regimen with NFV was well tolerated overall with no significant clinical adverse events attributed to the treatment. Studies in non-pregnant adults have reported the most common side effects of NFV to be gastrointestinal with diarrhea occurring in approximately 5% of patients, which was not significant in our study. We did not observe any preterm infants in this study, although other studies have shown controversial results.28-32 In PACTG 353, there were 29 infants and none were preterm.
This study, along with other recent reports of pharmacokinetics and safety of antiretrovirals in pregnancy, emphasizes the importance and necessity of ensuring adequate drug exposure and safety in pregnancy and in the newborn with new drugs as they become available. This study found the dose of 1250 mg bid to be superior to the three times a day dosage. Data describing perinatal pharmacokinetics, including placental transfer to the fetus and amniotic fluid, are critical not only for adequate treatment of the woman during pregnancy and postpartum but also for the design of future interventions to prevent mother-to-child HIV transmission.
Acknowledgments
Supported by the Pediatric AIDS Clinical Trials Group of the National Institute of Allergy and Infectious Diseases (U01 AI27550), funded by the National Center for Research Resources (including grant M01RR0533), and by the Pediatric/Perinatal HIV Clinical Trials Network of the National Institute of Child Health and Human Development. In addition, we would like to acknowledge the General Clinical Reserach Center grants (GCRC; NIH M01-RR00865, RR00533, and M01-RR00425) and the HIV Retrovirology Core Lab grant (NIH U01 AI-41089).
We wish to acknowledge the contributions of the following members of the PACTG 353 protocol team: Kimberly Hughes, Maureen Shannon, CNM, FNP, MS, and Bethann Cunningham.
The following investigators and institutions participated in this study: Boston Medical Center, Boston, MA: Laureen Kaye, RN, Anne Marie Regan, MEd, PNP, MSN, Ellen Cooper, MD, Stephen Pelton, MD; Children’s Hospital & Medical Center, Seattle, WA: Jane Hitti, MD, Deborah Goldman, ARNP, MPH, Michele Acker, Kathleen Mohan, ARNP, MN; San Juan City Hospital, San Juan, PR: Jorge Gandia, MD, Rodrigo Diaz-Velasco, MD, Milagros Gonzalez, MD; Bronx/Lebanon Hospital, Bronx, NY: Andrew Wiznia, MD, Wanda Biernick, RN, Karen Dorio, RN, Joanna Dobroszycki, MD; Los Angeles County Medical Center, Los Angeles, CA: Alice Stek, MD, Andrea Kovacs, MD, James Homans, MD; Howard University Hospital, Washington, DC: Sohail Rana, MD, Deepika Darbari, MD, Patricia Houston Yu, MS, Jhoanna Roa, MD; Harbor-UCLA Medical Center, Los Angeles, CA: Nasser Redjal, MD, Marie Beall, MD, Judy Hayes, RN; University of California Medical Center, Los Angeles, CA: Karin Nielsen, MD, MPH, Jaime Deville, MD, Maryanne Dillon, BSN, NP, Bonnie J. Ank (and Laura W. Sheehan for her assistance); University of California, San Diego, CA: Andrew, D. Hull, BMedSci, BMBS, Patricia Franklin, RN, NP, Stephen A. Spector, MD.
References
- 1.Centers for Disease Control and Prevention. US Public Health Service Task Force Recommendations for Use of Antiretroviral Drugs During Pregnancy for Maternal Health and Reduction of Perinatal Transmission of Human Immunodeficiency Virus Type 1 in the United States. MMWR Morb Mortal Wkly Rep. 1998:1–30. [Google Scholar]
- 2.Cooper ER, Charurat M, Mofenson L, Hanson IC, Pitt J, Diaz C, et al. Combination antiretroviral strategies for the treatment of pregnant HIV-1-infected women and prevention of perinatal HIV-1 transmission. J Acquir Immune Defic Syndr. 2002;29:484–494. doi: 10.1097/00126334-200204150-00009. [DOI] [PubMed] [Google Scholar]
- 3.Lyons F, Hopkins S, Kelleher B, McGeary A, Sheehan G, Geoghegan J, et al. Maternal hepatotoxicity with nevirapine as part of combination antiretroviral therapy in pregnancy. HIV Med. 2006;7:255–260. doi: 10.1111/j.1468-1293.2006.00369.x. [DOI] [PubMed] [Google Scholar]
- 4.Hitti J, Frenkel LM, Stek AM, Nachman SA, Baker D, Gonzalez-Garcia A, et al. Maternal toxicity with continuous nevirapine in pregnancy: results from PACTG 1022. J Acquir Immune Defic Syndr. 2004;36:772–776. doi: 10.1097/00126334-200407010-00002. [DOI] [PubMed] [Google Scholar]
- 5.Harris RZ, Benet LZ, Schwartz JB. Gender effects in pharmacokinetics and pharmacodynamics. Drugs. 1995;50:222–239. doi: 10.2165/00003495-199550020-00003. [DOI] [PubMed] [Google Scholar]
- 6.Mirochnick M, Dorenbaum A, Holland D, Cunningham-Schrader B, Cunningham C, Gelber R, et al. Concentrations of protease inhibitors in cord blood after in utero exposure. Pediatr Infect Dis J. 2002;21:835–838. doi: 10.1097/00006454-200209000-00010. [DOI] [PubMed] [Google Scholar]
- 7.Mirochnick M, Stek A, Acevedo M, Keller M, Holland D, Capparelli E, et al. Safety and pharmacokinetics of nelfinavir coadministered with zidovudine and lamivudine in infants during the first 6 weeks of life. J Acquir Immune Defic Syndr. 2005;39:189–194. [PubMed] [Google Scholar]
- 8.Wu EY, Wilkinson JM, 2nd, Naret DG, Daniels VL, Williams LJ, Khalil DA, et al. High-performance liquid chromatographic method for the determination of nelfinavir, a novel HIV-1 protease inhibitor, in human plasma. J Chromatogr B Biomed Sci Appl. 1997;695:373–380. doi: 10.1016/s0378-4347(97)00193-x. [DOI] [PubMed] [Google Scholar]
- 9.Viracept package insert. La Jolla, CA: Agouron Pharmaceuticals; 2004. [Google Scholar]
- 10.Dorenbaum A, Cunningham CK, Gelber RD, Culnane M, Mofenson L, Britto P, et al. Two-dose intrapartum/newborn nevirapine and standard antiretroviral therapy to reduce perinatal HIV transmission: a randomized trial. JAMA. 2002;288:189–198. doi: 10.1001/jama.288.2.189. [DOI] [PubMed] [Google Scholar]
- 11.Baede-van Dijk PA, Hugen PW, Verweij-van Wissen CP, Koopmans PP, Burger DM, Hekster YA. Analysis of variation in plasma concentrations of nelfinavir and its active metabolite M8 in HIV-positive patients. AIDS. 2001;15:991–998. doi: 10.1097/00002030-200105250-00007. [DOI] [PubMed] [Google Scholar]
- 12.Zhang KE, Wu E, Patick AK, Kerr B, Zorbas M, Lankford A, et al. Circulating metabolites of the human immunodeficiency virus protease inhibitor nelfinavir in humans: structural identification, levels in plasma, and antiviral activities. Antimicrob Agents Chemother. 2001;45:1086–1093. doi: 10.1128/AAC.45.4.1086-1093.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Kosel BW, Beckerman KP, Hayashi S, Homma M, Aweeka FT. Pharmacokinetics of nelfinavir and indinavir in HIV-1-infected pregnant women. AIDS. 2003;17:1195–1199. doi: 10.1097/00002030-200305230-00011. [DOI] [PubMed] [Google Scholar]
- 14.van Heeswijk RP, Veldkamp A, Mulder JW, Meenhorst PL, Lange JM, Beijnen JH, et al. Combination of protease inhibitors for the treatment of HIV-1-infected patients: a review of pharmacokinetics and clinical experience. Antivir Ther. 2001;6:201–229. [PubMed] [Google Scholar]
- 15.Pellegrin I, Breilh D, Montestruc F, Caumont A, Garrigue I, Morlat P, et al. Virologic response to nelfinavir-based regimens: pharmacokinetics and drug resistance mutations (VIRAPHAR study) AIDS. 2002;16:1331–1340. doi: 10.1097/00002030-200207050-00004. [DOI] [PubMed] [Google Scholar]
- 16.Villani P, Floridia M, Pirillo MF, Cusato M, Tamburrini E, Cavaliere AF, et al. Pharmacokinetics of nelfinavir in HIV-1-infected pregnant and nonpregnant women. Br J Clin Pharmacol. 2006;62:309–315. doi: 10.1111/j.1365-2125.2006.02669.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Angel JB, Khaliq Y, Monpetit ML, Cameron DW, Gallicano K. An argument for routine therapeutic drug monitoring of HIV-1 protease inhibitors during pregnancy. AIDS. 2001;15:417–419. doi: 10.1097/00002030-200102160-00017. [DOI] [PubMed] [Google Scholar]
- 18.Mirochnick M, Clarke DF, Dorenbaum A. Nevirapine: pharmacokinetic considerations in children and pregnant women. Clin Pharmacokinet. 2000;39:281–293. doi: 10.2165/00003088-200039040-00004. [DOI] [PubMed] [Google Scholar]
- 19.Wang Y, Livingston E, Patil S, McKinney RE, Bardeguez AD, Gandia J, et al. Pharmacokinetics of didanosine in antepartum and postpartum human immunodeficiency virus–infected pregnant women and their neonates: an AIDS clinical trials group study. J Infect Dis. 1999;180:1536–1541. doi: 10.1086/315067. [DOI] [PubMed] [Google Scholar]
- 20.Moodley J, Moodley D, Pillay K, Coovadia H, Saba J, van Leeuwen R, et al. Pharmacokinetics and antiretroviral activity of lamivudine alone or when coadministered with zidovudine in human immunodeficiency virus type 1-infected pregnant women and their offspring. J Infect Dis. 1998;178:1327–1333. doi: 10.1086/314431. [DOI] [PubMed] [Google Scholar]
- 21.O’Sullivan MJ, Boyer PJ, Scott GB, Parks WP, Weller S, Blum MR, et al. The pharmacokinetics and safety of zidovudine in the third trimester of pregnancy for women infected with human immunodeficiency virus and their infants: phase I acquired immunodeficiency syndrome clinical trials group study (protocol 082). Zidovudine Collaborative Working Group. Am J Obstet Gynecol. 1993;168:1510–1516. doi: 10.1016/s0002-9378(11)90791-1. [DOI] [PubMed] [Google Scholar]
- 22.Wade N, et al. Pharmacokinetics and safety of d4T in HIV-infected pregnant women and their infants: Pediatric AIDS Clinical Trials Group Protocol 332. J Infect Dis. doi: 10.1086/425903. In press. [DOI] [PubMed] [Google Scholar]
- 23.Covington DL, Conner SD, Doi PA, Swinson J, Daniels EM. Risk of birth defects associated with nelfinavir exposure during pregnancy. Obstet Gynecol. 2004;103:1181–1189. doi: 10.1097/01.AOG.0000127440.68730.23. [DOI] [PubMed] [Google Scholar]
- 24.Wilmington NC Registry Coordinating Center. The Antiretroviral Pregnancy Registry International Interim Report for January 1, 1989, through January 31, 2005. Accessible at: www.apregistry.com/forms/exec-summary.pdf.
- 25.Dickover RE, Garratty EM, Herman SA, Sim MS, Plaeger S, Boyer PJ, et al. Identification of levels of maternal HIV-1 RNA associated with risk of perinatal transmission. Effect of maternal zidovudine treatment on viral load. JAMA. 1996;275:599–605. [PubMed] [Google Scholar]
- 26.Connor EM, Sperling RS, Gelber R, Kiselev P, Scott G, O’Sullivan MJ, et al. Reduction of maternal-infant transmission of human immunodeficiency virus type 1 with zidovudine treatment. Pediatric AIDS Clinical Trials Group Protocol 076 Study Group. N Engl J Med. 1994;331:1173–1180. doi: 10.1056/NEJM199411033311801. [DOI] [PubMed] [Google Scholar]
- 27.Eshleman SH, Guay LA, Mwatha A, Cunningham SP, Brown ER, Musoke P, et al. Comparison of nevirapine (NVP) resistance in Ugandan women 7 days vs. 6-8 weeks after single-dose NVP prophylaxis: HIVNET 012. AIDS Res Hum Retroviruses. 2004;20:595–599. doi: 10.1089/0889222041217518. [DOI] [PubMed] [Google Scholar]
- 28.The European Collaborative Study and the Swiss Mother and Child HIV Cohort Study. Combination antiretroviral therapy and duration of pregnancy. AIDS. 2000;14:2913–2920. doi: 10.1097/00002030-200012220-00013. [DOI] [PubMed] [Google Scholar]
- 29.Lorenzi P, Spicher VM, Laubereau B, Hirschel B, Kind C, Rudin C, et al. Antiretroviral therapies in pregnancy: maternal, fetal and neonatal effects. Swiss HIV Cohort Study, the Swiss Collaborative HIV and Pregnancy Study, and the Swiss Neonatal HIV Study. AIDS. 1998;12:F241–247. doi: 10.1097/00002030-199818000-00002. [DOI] [PubMed] [Google Scholar]
- 30.Thorne C, Patel D, Newell ML. Increased risk of adverse pregnancy outcomes in HIV-infected women treated with highly active antiretroviral therapy in Europe. AIDS. 2004;18:2337–2339. doi: 10.1097/00002030-200411190-00019. [DOI] [PubMed] [Google Scholar]
- 31.Tuomala RE, Yawetz S. Protease inhibitor use during pregnancy: Is there an obstetrical risk? J Infect Dis. 2006;193:1191–1194. doi: 10.1086/503049. [DOI] [PubMed] [Google Scholar]
- 32.Cotter AM, Garcia AG, Duthely ML, Luke B, O’Sullivan MJ. Is antiretroviral therapy during pregnancy associated with an increased risk of preterm delivery, low birth weight, or stillbirth? J Infect Dis. 2006;193:1195–1201. doi: 10.1086/503045. [DOI] [PubMed] [Google Scholar]