

Abstract
Background
Substance P is a sensory nerve neuropeptide located near coronary vessels in the heart. Therefore, substance P may be one of the first mediators released in the heart in response to hypertension, and can contribute to adverse myocardial remodeling via interactions with the neurokinin-1 receptor. We asked: 1) whether substance P promoted cardiac hypertrophy, including the expression of fetal genes known to be re-expressed during pathological hypertrophy; and 2) the extent to which substance P regulated collagen production and fibrosis.
Methods and Results
Spontaneously hypertensive rats (SHR) were treated with the neurokinin-1 receptor antagonist L732138 (5 mg/kg/d) from 8 to 24 weeks of age. Age-matched WKY served as controls. The gene encoding substance P, TAC1, was up-regulated as blood pressure increased in SHR. Fetal gene expression by cardiomyocytes was increased in SHR and was prevented by L732138. Cardiac fibrosis also occurred in the SHR and was prevented by L732138. Endothelin-1 was up-regulated in the SHR and this was prevented by L732138. In isolated cardiac fibroblasts, substance P transiently up-regulated several genes related to cell-cell adhesion, cell-matrix adhesion, and extracellular matrix regulation, however, no changes in fibroblast function were observed.
Conclusions
Substance P activation of the neurokinin-1 receptor induced expression of fetal genes related to pathological hypertrophy in the hypertensive heart. Additionally, activation of the neurokinin-1 receptor was critical to the development of cardiac fibrosis. Since no functional changes were induced in isolated cardiac fibroblasts by substance P, we conclude that substance P mediates fibrosis via up-regulation of endothelin-1.
Keywords: Hypertrophy, fetal genes, fibrosis, substance P, cardiac fibroblast
INTRODUCTION
Substance P is an 11 amino acid member of the tachykinin family of neuropeptides and is encoded by the TAC1 gene. Its effects are manifested through neurokinin receptors with a several hundred fold greater affinity for the neurokinin-1 (NK-1) receptor over NK-2 and -3 receptors.(1) In the normal heart, substance P is found in sensory nerves that project to coronary arteries and arterioles, and in a small population of coronary endothelial cells.(2-5) This localization is of significance because it places substance P in the ideal location to be released in response to changes in coronary pressure/flow/homeostasis. While this may be advantageous initially since substance P is a potent vasodilator, long-term up-regulation of substance P may have detrimental effects. Only a few studies have investigated the role of substance P in long-term remodeling and function of the heart. In models of myocarditis, deletion of substance P protected mice from developing heart failure.(6;7) Similarly, we found that deletion of substance P protected mice from ventricular dilatation in a model of chronic cardiac volume overload.(8) In a rat model of chronic magnesium deficiency, Mak et al, (9) found that NK-1 receptor blockade improved E/A ratio and fractional shortening. However, no studies have investigated the role of substance P and the NK-1 receptor in adverse remodeling associated with the more prevalent condition of hypertension. Further, no studies have investigated the mechanisms by which substance P mediates adverse myocardial remodeling. Given the localization of substance P to the coronary vasculature, we hypothesized that substance P would be a key mediator in initiating adverse myocardial remodeling in response to hypertension. In the current study we report that: 1) TAC1 expression increased as blood pressure increased; 2) blockade of the NK-1 receptor prevented induction of fetal genes characteristic of pathological myocardial hypertrophy; 3) blockade of the NK-1 receptor prevented the development of cardiac fibrosis; 4) substance P regulated myocardial endothelin-1 (ET-1) levels; and 5) substance P transiently up-regulated specific genes related to extracellular matrix regulation and cellular adhesion in isolated cardiac fibroblasts.
METHODS
Animals
All animals were maintained according to a protocol approved by Institutional Animal Care and Use Committee at the Medical College of Wisconsin (Protocol Number AUA00002402) and conformed to the principles of the National Institutes of Health Guide for the Care and Use of Laboratory Animals. Rats were housed under standard environmental conditions and maintained on commercial rat chow and tap water ad libitum. Eight week-old SHR were randomly assigned to 2 groups: 1) untreated (n=8); and 2) those treated with the NK-1 receptor antagonist, L732138 (5 mg/kg/day, IP; n=8), with the experimental endpoint being at 24 weeks of age. These time-points were chosen so that drug treatment was initiated before the onset of hypertension and the study concluded during well-established hypertension and associated myocardial remodeling. This allowed us to determine the importance of substance P and the NK-1 receptor in initiating adverse myocardial remodeling. The dosage of L732138 was based on our previous studies.(8) Untreated WKY rats served as age-matched controls. Anesthesia at the experimental end point was affected by 2% isoflurane at which point blood pressure was measured via tail-cuff method before excision of the heart. Following euthanasia, the heart was removed and the atria and great vessels dissected away, with the left ventricle (LV) and right ventricle (RV) separated and weighed. A transverse mid-section of the LV was placed in formalin for fixation, and the apical portion of the LV snap frozen in liquid nitrogen and stored at -80°C. Lung weight was also obtained after the removal of the esophagus and trachea with the pleural surface blotted dry.
Additional information is available in the Methods section in the Online Data Supplement.
RESULTS
Biometric Parameters
There were no statistical differences in body weight between any of the groups at each time-point (Table 1). At 8 weeks of age there were no differences in blood pressure between SHR and WKY (Figure 1A). However, by 12 weeks of age the SHR had developed a statistically significant increase in blood pressure that continued to increase through to the final time-point of 24 weeks of age. Treatment with the NK-1 receptor antagonist, L732138, did not alter the increased blood pressure in the SHR at any time-point. Similar to blood pressure, LV mass was not different between SHR and WKY at 8 weeks of age (Table 1). However, by 12 weeks of age the SHR had developed a statistically significant increase in LV mass that continued to increase through to the final time-point of 24 weeks of age (Table 1). There was a trend towards a reduced LV mass in SHR treated with L732138, however, this did not reach statistical significance. There were no statistical differences in RV and lung weights between any of the groups (Table 1).
Table 1.
Biometric Data
| Weight (g) | LV/BW | RV/BW | Lung/BW | ||
|---|---|---|---|---|---|
| WKY | 8 weeks | 190±10 | 2.24±0.19 | 0.7±0.16 | 4.38±0.47 |
| 12 weeks | 300±11 | 2.18±0.10 | 0.58±0.07 | 3.47±0.22 | |
| 16 weeks | 309±11 | 2.17±0.08 | 0.58±0.05 | 3.43±0.17 | |
| 20 weeks | 325±16 | 2.13±0.08 | 0.56±0.11 | 3.29±0.24 | |
| 24 weeks | 399±31 | 2.13±0.06 | 0.55±0.04 | 3.30±0.20 | |
| SHR | 8 weeks | 213±5 | 2.44±0.08 | 0.56±0.03 | 4.39±0.19 |
| 12 weeks | 278±12 | 2.59±0.10* | 0.56±0.06 | 3.75±0.21 | |
| 16 weeks | 323±5 | 2.54±0.15* | 0.47±0.06 | 3.52±0.21 | |
| 20 weeks | 365±12 | 2.63±0.07* | 0.43±0.06 | 3.4±0.24 | |
| 24 weeks | 369±29 | 2.81±0.31* | 0.64±0.14 | 3.8±0.30 | |
| SHR + L732138 | 24 weeks | 356±36 | 2.6±0.20* | 0.54±0.06 | 3.88±0.25 |
Data is reported as mean values ± standard deviation. A 2-way ANOVA with a Bonferonni post-test was used to compare age-matched strains from 8 to 24 weeks of age.
p < 0.05.
FIGURE 1.
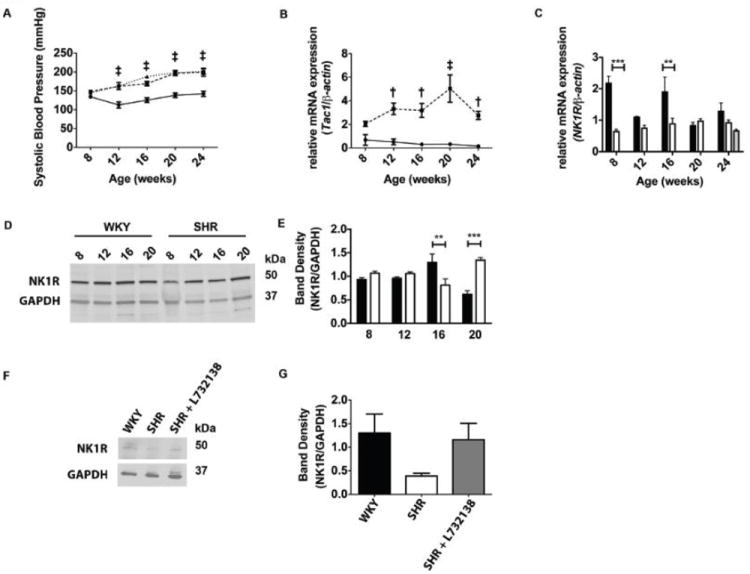
TAC1, but not NK-1 receptor expression corresponds with increased blood pressure in the SHR. A. Systolic blood pressure was measured every 4 weeks in WKY (circles with solid line, n=8), SHR (squares with dashed line, n=8), and SHR treated with L732138 (triangles with dotted line, n=8). Systolic blood pressure measurements are plotted against age of rat at time of measurement. Error bars represent SEM, ‡p < 0.005, 2-way ANOVA with a Bonferonni post-test for SHR and SHR+L732138 groups compared to the WKY. B. Relative fold expression of TAC1 mRNA was measured by qRT-PCR from left ventricular tissue in 8, 12, 16, 20, and 24-week old WKY (circles with solid line, n=5) and SHR (squares with dashed line, n=5). TAC1 Ct values were normalized to β-actin Ct values and fold expression were determined using the ΔΔCt method. Error bars represent SEM, †p < 0.01 and ‡p < 0.005 using a 2-way ANOVA between WKY and SHR with a Bonferonni post-test. C. Relative fold expression of Nk-1 receptor mRNA was measured by qRT-PCR and normalized to β-actin expression in left ventricular tissue isolated at 8, 12, 16, 20, and 24-weeks of age in WKY (black bars, n=5-8) and SHR (white bars, n=5-8). Nk-1 receptor mRNA was measured from the left ventricle of 24-week old SHR treated with L732138 (grey bar, n=5). Error bars represent SEM. **p < 0.01 and ***p < 0.005 using a 2-way ANOVA with a Bonferroni post-test. D. NK-1 receptor and GAPDH were detected by western blot from left ventricular tissue isolated at 8, 12, 16, and 20-week old WKY and SHR. E. Band densities from NK-1 receptor were normalized to band densities of GAPDH. The black bars represent WKY samples (n=4) and the white bars represent SHR samples (n=4). **p < 0.01 and ***p < 0.005 using a 2-way ANOVA with a Bonferonni post-test. F. NK-1 receptor and GAPDH expression were detected by western blot from left ventricular tissue isolated at 24-weeks from WKY, SHR, and SHR treated with L732138. G. Band densities from NK-1 receptor detection in F were normalized to GAPDH in WKY (black bar, n=5), SHR (white bar, n=5), and SHR treated with L732138 (grey bar, n=5).
Myocardial TAC1 Gene Expression
We asked whether myocardial expression of TAC1, the gene for substance P, was altered during the progression of hypertension in the SHR. TAC1 was not increased in the SHR compared to the WKY at 8 weeks of age (Figure 1B), before the onset of hypertension. However, from 12 weeks of age onwards expression was increased in the SHR as blood pressure increased.
Myocardial Neurokinin-1 Receptor mRNA and Protein Levels
Given the increased mRNA levels of TAC1, we sought to determine the mRNA and protein levels of the NK-1 receptor in SHR and WKY hearts. NK-1 receptor mRNA was increased in the WKY at 8 and 16 weeks of age, but was not different from SHR at the other time-points (Figure 1C). NK-1 receptor blockade with L732138 did not significantly alter NK-1 receptor mRNA at 24 weeks of age. NK-1 receptor protein levels were also somewhat cyclical in nature, but did not mirror mRNA. While there were no differences between SHR and WKY at 8 and 12 weeks of age, SHR levels fell below the WKY at 16 weeks of age before increasing above WKY at 20 weeks of age (Figure 1D & E). By 24 weeks of age, there was no statistically significant difference between WKY and SHR, although the trend was for decreased levels in the SHR (Figure F & G); NK-1 receptor blockade with L732138 did not cause any statistically significant change in receptor levels (Figure 1F & G).
Left Ventricular Structure and Function
Echocardiography was used to gain a greater understanding of the effects of NK-1 receptor blockade on LV structure and function. Echocardiography measurements revealed that the LV posterior wall during diastole (LVPWd) was thicker in the SHR beginning at 16 weeks of age through 24 weeks of age, compared to WKY (2.3+0.06 vs. 1.6+0.05 mm at 24 weeks of age; Figure 2A). L732138 tended to reduce LVPWd from 20 weeks onwards (2.0+0.07 mm at 24 weeks), however, this did not reach statistical significance. LVPWd was only statistically greater than the WKY in the SHR+L732138 group at 16 weeks of age. There were no significant differences between any groups for LV internal diameter in diastole (LVIDd), although chamber size tended to be smaller in the SHR (Figure 2B). As a result of the increased wall thickness, LV mass, determined by echocardiography was increased in the SHR from 16 weeks of age onwards compared to the WKY (Figure 2C). NK-1 receptor blockade did not prevent hypertrophy from occurring although there was a non-significant trend towards a decreased LV mass; this was consistent with the wet weight data in Table 1. The LV wall thickness-to-chamber ratio was significantly larger in the SHR at 16, 20, and 24 weeks of age compared to the WKY (Figure 2D). The SHR+L738132 group was increased compared to the WKY at 16 weeks of age, however, at 20 and 24 weeks of age, their wall thickness-to-chamber ratio was not statistically different from the WKY or SHR. Ventricular function was determined by ejection fraction and fractional shortening. Neither parameter was different between any of the groups at any time-point (Figure 2E & F). Figure 2G shows images of transverse LV sections representative of the findings described above.
FIGURE 2.
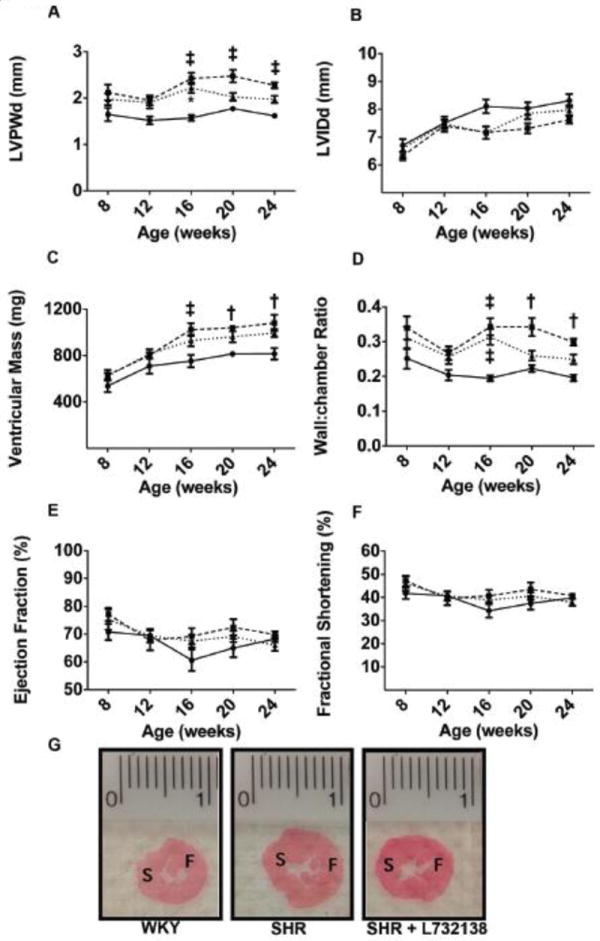
A. Left ventricular posterior wall thickness in diastole (LVPWd) was measured by echocardiogram in WKY (solid line, n=6-8), SHR (dashed line, n=6-8), and SHR treated with L732138 (dotted line, n=6-8) at 8, 12, 16, 20, and 24-weeks of age. ‡p < 0.005, 2-way ANOVA with a Bonferonni post-test. B. Left ventricular internal diameter in diastole (LVIDd) was measured in WKY, SHR, and SHR treated with L732138 as in A. C. Ventricular mass (mg) as determined by echocardiogram is plotted (y-axis) against age of rat (x-axis) for WKY (solid line, n=6-8), SHR (dashed line, n=6-8), and SHR treated with L732138 (dotted line, n=8). †p<0.01 and ‡p<0.005, 2-way ANOVA with a Bonferonni post-test. D. The ratio of ventricular wall thickness to chamber diameter in WKY (solid line, n=6-8), SHR (dashed line, n=6-8), and SHR treated with L732138 (dotted line, n=8) is plotted against the age of rat at the time of measurement. †p<0.01 and ‡p<0.005, 2-way ANOVA with a Bonferonni post-test. E. The percent ejection fraction was measured as in A-D and plotted against age of rat. F. The percent fractional shortening was measured as in A-E and plotted against age of rat. G. Left ventricular sections from 24-week old WKY, SHR, and SHR treated with L732138 stained with picrosirius red. Orientation is noted by S, septum, and F, free wall.
Myocardial Fetal Gene Expression
Since there was a trend for hypertrophy to be reduced by NK-1 receptor blockade, we examined the expression of four fetal genes, β-myosin heavy chain (β-MHC), skeletal α-actin (SkA), atrial natriuretic factor (ANF), and B-type natriuretic peptide (BNP), as markers of pathological hypertrophy (Figure 3). All four of these fetal genes showed significantly increased expression in the SHR hearts, indicative of pathological hypertrophy. Interestingly, expression of β-MHC, ANF, and BNP were all normalized by L732138; SkA was attenuated by L732138, but not normalized.
FIGURE 3.
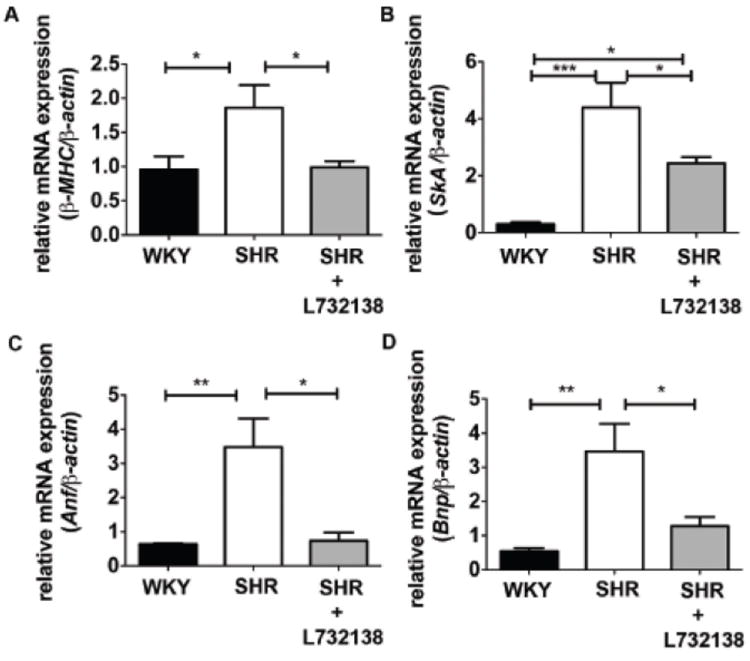
Fetal gene expression is enhanced in 24-week old SHR left ventricle and prevented by NK-1 receptor blockade. A – D. mRNA expression of β-MHC, SkA, Anf, and Bnp was measured by qRT-PCR and normalized to β-actin in 24-week old left ventricular tissue isolated from WKY (black bars, n=5), SHR (white bars, n=3-5), and SHR treated with L732138 (grey bars, n=4-5). Fold expression was determined using the ΔΔCt method. *p < 0.05, **p < 0.01, ***p < 0.001, 1-way ANOVA with the Tukey post-test.
Collagen Volume Fraction
Collagen content was determined in picrosirius red stained sections of LV from WKY, SHR, and SHR+L732138. The amount of collagen was increased in SHR hearts compared to WKY hearts, indicative of hypertension-induced cardiac fibrosis (Figure 4A & B). NK-1 receptor blockade prevented fibrosis, reducing collagen levels to a value slightly below that of the WKY.
FIGURE 4.
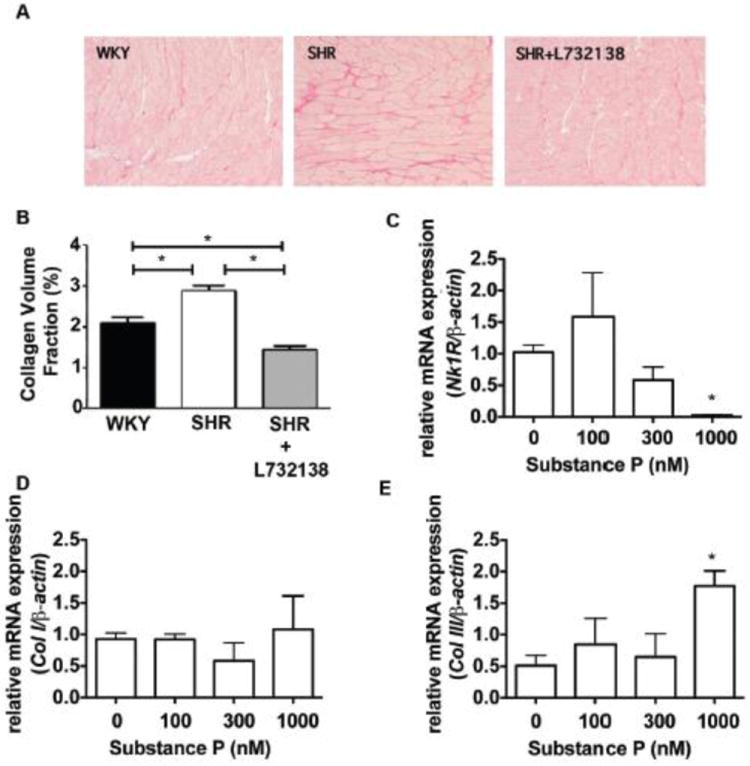
A. Collagen was stained with picrosirius red in 24-week old left ventricular tissue slices from WKY, SHR, and SHR treated with L732138. B. Collagen volume fraction was quantified from the picrosirius staining (A). n=8 for each group, WKY (black bar), SHR (white bar), and SHR treated with L732138 (grey bar). *p<0.05, 1-way ANOVA with a Tukey post-test. C. Nk-1 receptor mRNA was measured and expression was normalized to β-actin in cardiac fibroblasts incubated with increasing concentrations of substance P for 24 hours. Relative fold expression was calculated using the ΔΔCt method. *p<0.05, 1-way ANOVA with a Tukey post-test. D-E. Collagen I (D) and Collagen III (E) mRNA was measured in substance P treated cardiac fibroblasts after 24-hours. Fold expression was determined using the ΔΔCt method. *p<0.05, 1-way ANOVA with a Tukey post-test.
Isolated Cardiac Fibroblast mRNA Responses to Substance P
To examine mechanisms by which substance P may cause fibrosis, we performed a series of experiments using adult rat isolated cardiac fibroblasts. Firstly, we determined that cardiac fibroblasts possessed the NK-1 receptor using western blotting (Supplemental Figure 1). While increasing concentrations of substance P decreased mRNA expression for the NK-1 receptor (Figure 4C), this did not translate into changes at the protein level. We also examined additional genes related to extracellular matrix regulation, cell-cell adhesion and cell-matrix adhesion (Supplemental Table 1). We observed up-regulation of several genes related to cell-cell adhesion (cadherin 2 and cadherin 3), cell-matrix adhesion (integrin β1 and integrin α5), and extracellular matrix regulation (collagen III, membrane type 1-matrix metalloproteinase, matrix metalloproteinase-3, and laminin α4). We found that while increasing concentrations of substance P for 24 hours did not induce an up-regulation of collagen I mRNA, collagen III mRNA was increased by the highest concentration of substance P tested (1000 nM; Figure 4D & E). We also identified that membrane type 1-matrix metalloproteinase (MT1-MMP) and MMP-3 were significantly increased following a 24 hour incubation with substance P (Figure 5A & B). Peak up-regulation of MT1-MMP occurred at the 1000 nM concentration of substance P, while peak up-regulation of MMP-3 occurred at 300 nM. Up-regulation of MT1-MMP and MMP-3 at these concentrations was via the NK-1 receptor since L732138 blocked these responses (Figure 5C & D). However, up-regulation of these genes did not involve NF-κB as determined by reporter assay (Figure 5E). Interestingly, by 48 hours of treatment with substance P, collagen III, MT1-MMP, and MMP-3 gene expression had all returned to basal levels (Supplemental Figure 2).
FIGURE 5.
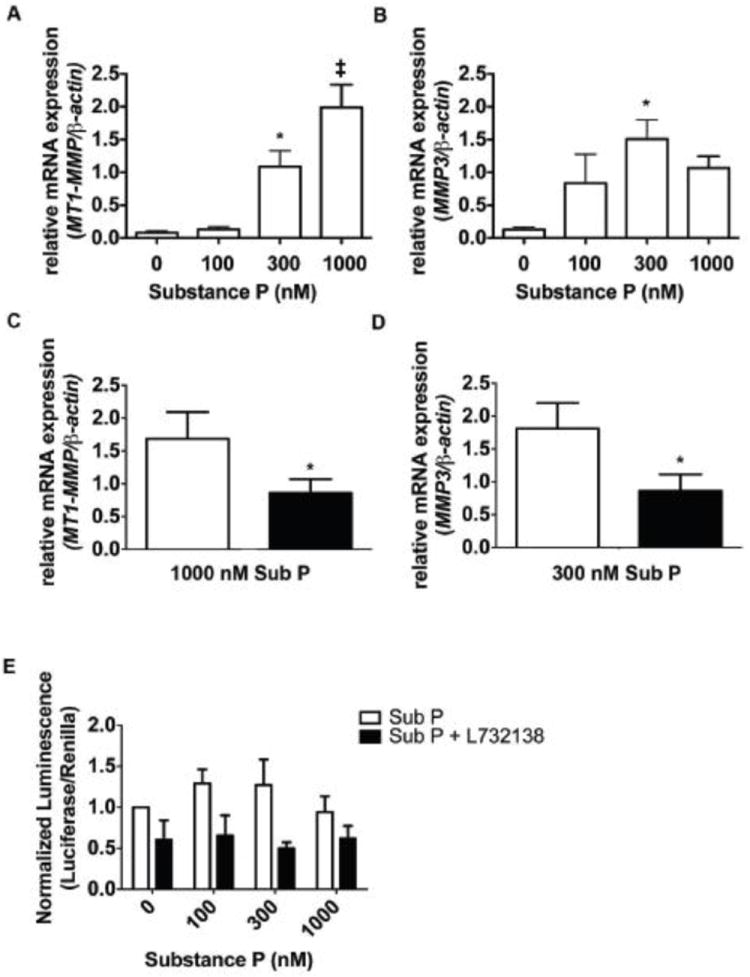
A-B. MT1-MMP and MMP3 mRNA expression was measured in cardiac fibroblasts incubated with increasing concentrations of substance P for 24 hours and normalized to β-actin. Fold expression was determined using the ΔΔCt method. *p<0.05 and ‡p<0.005 using a 1-way ANOVA with a Tukey post-test. C-D. MT1-MMP and MMP3 mRNA expression was measured in cardiac fibroblasts incubated with 1000 nM substance P (C) and 300 nM substance P (D) in the presence (black bars, n=4) and absence of L732138 (white bars, n=4) for 24 hours and normalized to β-actin. Relative fold expression was calculated using the ΔΔCt method. *p < 0.05 with an unpaired t-test. E. NF-κB transactivation was measured in cardiac fibroblasts incubated with increasing concentrations of substance P in the presence (black bars, n=4) and absence (white bars, n=3-4) of L732138 for 24-hours. Transcriptional activation as indicated by luciferase luminescence was normalized to renilla luminescence. *p<0.05, 2-way ANOVA with a Bonferonni post-test.
Isolated Cardiac Fibroblast Phenotype and Functional Responses to Substance P
Since we had observed up-regulation of specific genes in cardiac fibroblasts in response to 24 hours of substance P exposure, we performed experiments to determine if this resulted in changes to fibroblast phenotype and function. Following 24 hours of treatment with increasing concentrations of substance P, there was no increase in ED-A fibronectin, as an indicator of myofibroblast conversion (Supplemental Figure 3A). Further, there was no increase in collagen secretion as determined by hydroxyproline assay (Supplemental Figure 3B). There was also no change in migratory capacity in the presence of substance P (Supplemental Figure 3C).
Myocardial Endothelin-1is Mediated by Substance P and the NK-1 Receptor
We measured endothelin-1 (ET-1) levels from LV tissue isolated from 24 week old WKY, SHR and found a significant increase in SHR samples compared to WKY (Figure 6A). NK-1 receptor blockade with L732138 in SHR prevented the increase in ET-1.
FIGURE 6.
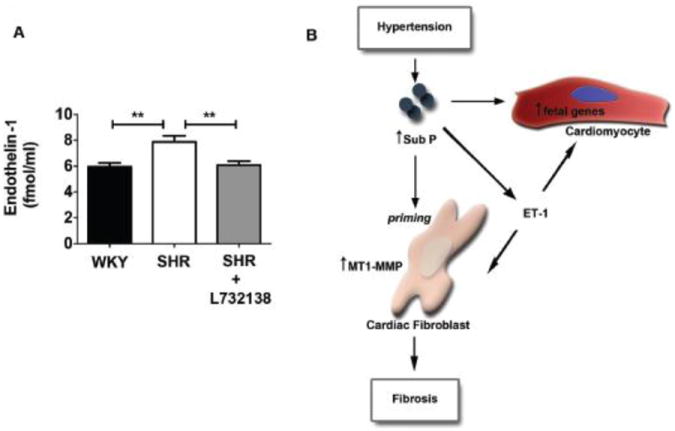
A. Endothelin-1 levels measured by ELISA from left ventricular tissue isolated from 24-week WKY (black bar, n=8), SHR (white bar, n=8), and SHR treated with L732138 (grey bar, n=7). **p < 0.01 using a 1-way ANOVA with a Tukey post-test. B. Schematic depicting the mechanisms by which substance P mediates adverse myocardial remodeling. Substance P is increased in response to hypertension, which leads to increased levels of endothelin-1 (ET-1). Substance P and/or ET-1 induce the expression of fetal genes in the cardiomyocyte. In response to substance P the cardiac fibroblasts are ‘primed’ to express numerous genes related including MT-1 MMP, with ET-1 providing a subsequent pro-fibrotic stimulus to these primed cells resulting in fibrosis.
DISCUSSION
Substance P is a sensory nerve neuropeptide that is encoded by the TAC1 gene, and primarily exerts its effects via the NK-1 receptor. Recently, substance P has been shown to have a role in adverse myocardial remodeling in the settings of myocarditis,(6;7) chronic volume overload,(8) and magnesium-deficiency.(9-11) It’s location in the heart in sensory nerves associated with the coronary vasculature, as well as in a small population of coronary endothelial cells, suggests that it may be released in response to changes in coronary perfusion pressure and/or flow. Therefore, substance P is likely released in the hypertensive heart where it may influence tissue remodeling. Of the handful of studies that have investigated substance P and adverse myocardial remodeling, none have investigated the hypertensive heart or performed in-depth analysis of the mechanisms by which substance P mediates remodeling. Accordingly, there are several major novel findings from the current study, including the following: 1) myocardial TAC1 gene expression increases as blood pressure increases; 2) substance P induces expression of fetal genes in the hypertensive heart; 3) substance P mediates cardiac fibrosis in the hypertensive heart; 4) substance P is a regulator of genes related to cell adhesion and regulation of the extracellular matrix in cardiac fibroblasts; and 5) substance P regulates myocardial ET-1 levels. The later four responses are mediated via the NK-1 receptor.
As SHR age they develop increased blood pressure and subsequent cardiac hypertrophy, which ultimately results in heart failure.(12) Part of this pathological hypertrophic response in the hypertensive or pressure overloaded heart is the re-induction of specific fetal genes that are usually quiescent in the adult heart.(13) An intriguing finding from this study is that NK-1 receptor blockade completely prevented the re-induction of ANP, BNP, and β-MHC gene expression, while attenuating SkA. This effect occurred even though NK-1 receptor blockade did not significantly attenuate overall myocardial hypertrophy as determined by wet weights and echocardiography. It is likely that hypertrophy in the NK-1 receptor antagonist group was still a necessary compensatory response, since blood pressure remained elevated in these animals. That fetal gene expression was prevented argues that the resultant hypertrophy was not pathological in nature. This is supported by our echocardiography observation that LV wall thickness to chamber size ratio was not different from the WKY in the SHR group treated with L732138. Thus, it appears that NK-1 receptor blockade may lead to a physiological form of hypertrophy. Cardiomyocytes possess the NK-1 receptor(14), which would suggest that substance P regulates fetal gene expression by direct effects on these cells. In fact, Church et al.(14) found that substance P did induce ANF release from cultured neonatal cardiomyocytes, suggesting the possibility of a direct effect. They did not investigate other fetal genes. Interestingly though, in the current study we also found that NK-1 receptor blockade prevented the increase in ET-1 observed in the untreated SHR. Since ET-1 is known to have effects on fetal genes in cardiomyocytes,(15) this may also represent an indirect mechanism by which substance P regulates fetal gene expression.
Our additional finding that NK-1 receptor blockade prevented fibrosis concomitant with the hypertrophy, would further indicate that the hypertrophic response was not pathological. In an attempt to identify mechanisms by which substance P regulated fibrosis, we conducted additional experiments in isolated adult rat cardiac fibroblasts. These cells were treated with increasing concentrations of substance P and assessed using real time PCR for a range of genes related to cell-cell adhesion, cell-matrix adhesion, and extra cellular matrix regulation. We identified several genes that were up-regulated in response to 24 hours of substance P (Supplemental Table 1), including MT1-MMP and MMP-3. MT1-MMP is increased in patients with LV pressure overload,(16) as well as pressure overloaded animals.(17;18) It is important in the development of fibrosis due to its role in processing latent TGF binding protein-1, resulting in the release of active TGFβ.(19-25) Additionally, it is important in processing latent MMP-2 to its active form, which is also important in fibrosis development.(26) The relevance of MMP-3 regulation is less clear since Spinale et al.(27) recently described MMP-3 as being either down-regulated or unchanged in LV pressure overload. NK-1 receptor blockade was able to prevent the up-regulation of both genes in cardiac fibroblasts, and the effects of substance P on these genes were not mediated by NF-κB since reporter assay showed no clear increase in activation. An interesting point is that these genes peaked in their expression at different concentrations of substance P; MT1-MMP at 1000 nM and MMP-3 at 300 nM, indicating that different genes may be expressed in the heart depending on the concentration of substance P present. Also of interest was that the up-regulation of MT1-MMP and MMP3 was a transient response in that expression returned to normal by 48 hours of treatment. To determine the extent to which this transient up-regulation had an effect on fibroblast phenotype and function, we performed a number of assays following 24 hours of substance P treatment. Surprisingly, we found no evidence of phenotype conversion to the more active myofibroblast, and no increase in cell migratory capacity. The lack of effect on migration was surprising given that α5 and β1 integrin sub-units were up-regulated at the gene level by substance P. We also found no increase in collagen synthesis despite collagen III mRNA being increased by substance P. Interestingly, it has been previously reported that tenocytes (tendon fibroblasts) stimulated with 100 nM of substance P also show up-regulation of collagen III and MMP-3 mRNA,(28) similar to our results. In that study, collagen III was up-regulated at 24 hours, which is equivalent to our findings, however, MMP-3 was up-regulated at 6 and 12 hours, but had returned to normal by 24 hours. Thus, tenocytes may have a slightly different temporal response to substance P for some genes, but still show evidence of a phasic gene response similar to what we have described herein for cardiac fibroblasts. Collagen secretion and MT1-MMP were not examined in the tenocyte study. Our findings would also seem consistent with another report in isolated cardiac fibroblasts where substance P increased proliferation, but did not increase collagen secretion.(29) Together with our findings, this suggests the interesting possibility that substance P induces fibroblast proliferation and up-regulation of select genes in anticipation of a pro-fibrotic action. However, this only proceeds if other pro-fibrotic stimuli follow. Substance P has been shown to have this type of ‘priming’ role in other settings. For example, substance P together with IGF-1 had a greater effect on migration of corneal epithelial cells than IGF-1 alone.(30) This was also the case for fibronectin and IL-6. Similarly, substance P augmented the release of histamine by rat peritoneal mast cells stimulated with anti-IgE.(31) Since we found that NK-1 receptor blockade normalized myocardial ET-1 levels in the SHR, ET-1 may represent an indirect mechanism by which substance P regulated fibrosis in the whole heart. If substance P does prime cardiac fibroblasts, up-regulation of ET-1 by substance P could then exert additional effects on the primed cells. Alternatively, substance P may regulate fibrosis in the hypertensive heart purely by up-regulating ET-1 without any priming type effect since ET-1 is well known as a pro-fibrotic stimuli in its own right.(32)
The effects of NK-1 receptor blockade that we observed on adverse myocardial remodeling were independent of blood pressure since L732138 did not reduce blood pressure in the SHR. However, TAC1 expression does appear to be closely related to blood pressure. This makes sense because hypertension involves increased systolic and diastolic blood pressure, with diastolic blood pressure providing the basis for coronary perfusion pressure. With substance P being located around the coronary vasculature, it is likely that expression is closely tied to perfusion pressure. Up-regulation of TAC1, and hence substance P, is likely an attempt to reduce perfusion pressure via vasodilation of coronary vessels since substance P is a potent coronary vasodilator.(33) This characteristic has been attributed to the beneficial effects of substance P in the acute period following ischemia reperfusion of the heart.(34-36) This acute beneficial effect likely also occurs in the hypertensive heart before the adverse effects of chronic up-regulation of substance P ensues. While TAC1 appears to be linked to blood pressure, this is not the case for either NK-1 receptor mRNA or protein levels. NK-1 receptor levels were observed to cycle up and down relative to the WKY across the 16 week study period. After binding substance P, the NK-1 receptor is rapidly phosphorylated, internalized, and recycled,(37) possibly explaining the results we observed. We also observed a decreased expression of NK-1 receptor mRNA in isolated fibroblasts treated with substance P, however, this did not manifest as changes at the protein level.
In the current study, we were interested in the ability of substance P to initiate adverse remodeling. As such, we investigated hearts in the earlier stages of remodeling that were well compensated and, thus, there were no differences in function as determined by fractional shortening and ejection fraction. Accordingly, follow-up studies where NK-1 blockade is maintained through to the point of expected heart failure, or pressure overload is induced in TAC1-/- mice and remodeling allowed to progress to expected failure are important to determine whether heart failure can be prevented. These studies would also more definitively determine the extent to which NK-1 receptor blockade induces physiological hypertrophy. Evidence from previous studies would suggest that prevention of heart failure is possible. Mouse models of viral(7) and parasitic(6) myocarditis are protected from heart failure by deletion of substance P. Further, we have reported that mice deficient in substance P were protected from ventricular dilatation in a model of chronic cardiac volume overload.(8) Mak et al.(9) also found that NK-1 receptor blockade improved E/A ratio and fractional shortening in a rat model of cardiac dysfunction due to magnesium-deficiency. Thus, it would seem possible that removing the effects of substance P could prevent heart failure from occurring in the hypertensive heart, however, these studies still need to be performed. Substance P is increased in the plasma of heart failure patients(38), indicating that it may remain influential in the decompensated heart.
In summary, we have demonstrated that blockade of the NK-1 receptor, the receptor for substance P, prevents adverse myocardial remodeling in the hypertensive rat heart. The mechanisms are summarized in Figure 6B. Part of this effect is the prevention of increased expression of specific fetal genes that are characteristic of pathological hypertrophy. These effects on fetal genes may be direct or may also be mediated by ET-1, which we showed to be regulated by the NK-1 receptor. Further, NK-1 receptor blockade prevented the development of cardiac fibrosis. The regulation of fibrosis by substance P in vivo likely involves ET-1 since we were unable to induce changes in cardiac fibroblast function in vitro with substance P, despite observing transient up-regulation of important genes related to the extracellular matrix. Since NK-1 receptor blockade was initiated before the onset of hypertension, hypertrophy, and TAC1 up-regulation, we conclude that NK-1 receptor activation is a critical factor in initiating the onset of pathological hypertrophy.
Supplementary Material
Acknowledgments
Sources of Funding: This work was supported by the NIH grants R00-HL093215 (S.P.L) and T32-HL007792 training grant (H.M.D).
Footnotes
This author takes responsibility for all aspects of the reliability and freedom from bias of the data presented and their discussed interpretation
Disclosures: None
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Reference List
- 1.Nakanishi S. Mammalian Tachykinin Receptors. Annual Review of Neuroscience. 1991;14:123–36. doi: 10.1146/annurev.ne.14.030191.001011. [DOI] [PubMed] [Google Scholar]
- 2.Reinecke M, Weihe E, Forssmann WG. Substance P-immunoreactive nerve fibers in the heart. Neurosci Lett. 1980;20:265–9. doi: 10.1016/0304-3940(80)90158-5. [DOI] [PubMed] [Google Scholar]
- 3.Wharton J, Polak JM, McGregor GP, Bishop AE, Bloom SR. The distribution of substrate P-like immunoreactive nerves in the guinea-pig heart. Neuroscience. 1981;6:2193–204. doi: 10.1016/0306-4522(81)90007-5. [DOI] [PubMed] [Google Scholar]
- 4.Dalsgaard CJ, Franco-Cereceda A, Saria A, et al. Distribution and origin of substance P- and neuropeptide Y-immunoreactive nerves in the guinea-pig heart. Cell Tissue Res. 1986;243:477–85. doi: 10.1007/BF00218054. [DOI] [PubMed] [Google Scholar]
- 5.Milner P, Ralevic V, Hopwood AM, et al. Ultrastructural localisation of substance P and choline acetyltransferase in endothelial cells of rat coronary artery and release of substance P and acetylcholine during hypoxia. Experientia. 1989;45:121–5. doi: 10.1007/BF01954843. [DOI] [PubMed] [Google Scholar]
- 6.D’Souza M, Garza MA, Xie M, et al. Substance P is associated with heart enlargement and apoptosis in murine dilated cardiomyopathy induced by Taenia crassiceps infection. J Parasitol. 2007;93:1121–7. doi: 10.1645/GE-596R1.1. [DOI] [PubMed] [Google Scholar]
- 7.Robinson P, Garza A, Moore J, et al. Substance P is required for the pathogenesis of EMCV infection in mice. Int J Clin Exp Med. 2009;2:76–86. [PMC free article] [PubMed] [Google Scholar]
- 8.Melendez GC, Li J, Law BA, et al. Substance P Induces Adverse Myocardial Remodeling via a Mechanism Involving Cardiac Mast Cells. Cardiovasc Res. 2011;92:420–9. doi: 10.1093/cvr/cvr244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Mak IT, Chmielinska JJ, Kramer JH, Spurney CF, Weglicki WB. Loss of neutral endopeptidase activity contributes to neutrophil activation and cardiac dysfunction during chronic hypomagnesemia: Protection by substance P receptor blockade. Exp Clin Cardiol. 2011;16:121–4. [PMC free article] [PubMed] [Google Scholar]
- 10.Kramer JH, Phillips TM, Weglicki WB. Magnesium-deficiency-enhanced post-ischemic myocardial injury is reduced by substance P receptor blockade. J Mol Cell Cardiol. 1997;29:97–110. doi: 10.1006/jmcc.1996.0255. [DOI] [PubMed] [Google Scholar]
- 11.Weglicki WB, Mak IT, Phillips TM. Blockade of cardiac inflammation in Mg2+ deficiency by substance P receptor inhibition. Circ Res. 1994;74:1009–13. doi: 10.1161/01.res.74.5.1009. [DOI] [PubMed] [Google Scholar]
- 12.Boluyt MO, Bing OH. Matrix gene expression and decompensated heart failure: the aged SHR model. Cardiovasc Res. 2000;46:239–49. doi: 10.1016/s0008-6363(00)00043-2. [DOI] [PubMed] [Google Scholar]
- 13.Wilkins BJ, Dai YS, Bueno OF, et al. Calcineurin/NFAT coupling participates in pathological, but not physiological, cardiac hypertrophy. Circ Res. 2004;94:110–8. doi: 10.1161/01.RES.0000109415.17511.18. [DOI] [PubMed] [Google Scholar]
- 14.Church DJ, Arkinstall SJ, Vallotton MB, et al. Stimulation of atrial natriuretic peptide release by neurokinins in neonatal rat ventricular cardiomyocytes. Am J Physiol. 1996;270:H935–H944. doi: 10.1152/ajpheart.1996.270.3.H935. [DOI] [PubMed] [Google Scholar]
- 15.Ehmke H, Faulhaber J, Munter K, Kirchengast M, Wiesner RJ. Chronic ETA receptor blockade attenuates cardiac hypertrophy independently of blood pressure effects in renovascular hypertensive rats. Hypertension. 1999;33:954–60. doi: 10.1161/01.hyp.33.4.954. [DOI] [PubMed] [Google Scholar]
- 16.Polyakova V, Hein S, Kostin S, Ziegelhoeffer T, Schaper J. Matrix metalloproteinases and their tissue inhibitors in pressure-overloaded human myocardium during heart failure progression. Journal of the American College of Cardiology. 2004;44:1609–18. doi: 10.1016/j.jacc.2004.07.023. [DOI] [PubMed] [Google Scholar]
- 17.Zile MR, Baicu CF, Stroud RE, et al. Pressure overload-dependent membrane type 1-matrix metalloproteinase induction: relationship to LV remodeling and fibrosis. Am J Physiol Heart Circ Physiol. 2012;302:H1429–H1437. doi: 10.1152/ajpheart.00580.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Yarbrough WM, Mukherjee R, Stroud RE, et al. Progressive induction of left ventricular pressure overload in a large animal model elicits myocardial remodeling and a unique matrix signature. J Thorac Cardiovasc Surg. 2012;143:215–23. doi: 10.1016/j.jtcvs.2011.09.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Koli K, Saharinen J, Hyytiainen M, Penttinen C, Keski-Oja J. Latency, activation, and binding proteins of TGF-beta. Microsc Res Tech. 2001;52:354–62. doi: 10.1002/1097-0029(20010215)52:4<354::AID-JEMT1020>3.0.CO;2-G. [DOI] [PubMed] [Google Scholar]
- 20.Dallas SL, Sivakumar P, Jones CJ, et al. Fibronectin regulates latent transforming growth factor-beta (TGF beta) by controlling matrix assembly of latent TGF beta-binding protein-1. J Biol Chem. 2005;280:18871–80. doi: 10.1074/jbc.M410762200. [DOI] [PubMed] [Google Scholar]
- 21.Neptune ER, Frischmeyer PA, Arking DE, et al. Dysregulation of TGF-beta activation contributes to pathogenesis in Marfan syndrome. Nat Genet. 2003;33:407–11. doi: 10.1038/ng1116. [DOI] [PubMed] [Google Scholar]
- 22.Rifkin DB. Latent transforming growth factor-beta (TGF-beta) binding proteins: orchestrators of TGF-beta availability. J Biol Chem. 2005;280:7409–12. doi: 10.1074/jbc.R400029200. [DOI] [PubMed] [Google Scholar]
- 23.Feng XH, Derynck R. Specificity and versatility in tgf-beta signaling through Smads. Annu Rev Cell Dev Biol. 2005;21:659–93. doi: 10.1146/annurev.cellbio.21.022404.142018. [DOI] [PubMed] [Google Scholar]
- 24.Bobik A. Transforming growth factor-betas and vascular disorders. Arterioscler Thromb Vasc Biol. 2006;26:1712–20. doi: 10.1161/01.ATV.0000225287.20034.2c. [DOI] [PubMed] [Google Scholar]
- 25.Ghosh AK. Factors involved in the regulation of type I collagen gene expression: implication in fibrosis. Exp Biol Med (Maywood) 2002;227:301–14. doi: 10.1177/153537020222700502. [DOI] [PubMed] [Google Scholar]
- 26.Matsusaka H, Ide T, Matsushima S, et al. Targeted deletion of matrix metalloproteinase 2 ameliorates myocardial remodeling in mice with chronic pressure overload. Hypertension. 2006;47:711–7. doi: 10.1161/01.HYP.0000208840.30778.00. [DOI] [PubMed] [Google Scholar]
- 27.Spinale FG, Janicki JS, Zile MR. Membrane-associated matrix proteolysis and heart failure. Circ Res. 2013;112:195–208. doi: 10.1161/CIRCRESAHA.112.266882. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Fong G, Backman LJ, Hart DA, et al. Substance P enhances collagen remodeling and MMP-3 expression by human tenocytes. J Orthop Res. 2013;31:91–8. doi: 10.1002/jor.22191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Kumaran C, Shivakumar K. Calcium- and superoxide anion-mediated mitogenic action of substance P on cardiac fibroblasts. Am J Physiol Heart Circ Physiol. 2002;282:H1855–H1862. doi: 10.1152/ajpheart.00747.2001. [DOI] [PubMed] [Google Scholar]
- 30.Yamada N, Yanai R, Inui M, Nishida T. Sensitizing effect of substance P on corneal epithelial migration induced by IGF-1, fibronectin, or interleukin-6. Invest Ophthalmol Vis Sci. 2005;46:833–9. doi: 10.1167/iovs.04-0775. [DOI] [PubMed] [Google Scholar]
- 31.Lau AHY, Chow SSM, Ng YS. Immunologically induced histamine release from rat peritoneal mast cells is enhanced by low levels of substance P. European Journal of Pharmacology. 2001;414:295–303. doi: 10.1016/s0014-2999(01)00805-6. [DOI] [PubMed] [Google Scholar]
- 32.Kong P, Christia P, Frangogiannis NG. The pathogenesis of cardiac fibrosis. Cell Mol Life Sci. 2013 doi: 10.1007/s00018-013-1349-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Hoover DB. Effects of substance P on rate and perfusion pressure in the isolated guinea pig heart. J Pharmacol Exp Ther. 1990;252:179–84. [PubMed] [Google Scholar]
- 34.Ustinova EE, Bergren D, Schultz HD. Neuropeptide depletion impairs postischemic recovery of the isolated rat heart: role of substance P. Cardiovasc Res. 1995;30:55–63. [PubMed] [Google Scholar]
- 35.Wang L, Wang DH. TRPV1 gene knockout impairs postischemic recovery in isolated perfused heart in mice. Circulation. 2005;112:3617–23. doi: 10.1161/CIRCULATIONAHA.105.556274. [DOI] [PubMed] [Google Scholar]
- 36.Zhong B, Wang DH. TRPV1 gene knockout impairs preconditioning protection against myocardial injury in isolated perfused hearts in mice. Am J Physiol Heart Circ Physiol. 2007;293:H1791–H1798. doi: 10.1152/ajpheart.00169.2007. [DOI] [PubMed] [Google Scholar]
- 37.Mantyh PW, Allen CJ, Ghilardi JR, et al. Rapid endocytosis of a G protein-coupled receptor: substance P evoked internalization of its receptor in the rat striatum in vivo. Proc Natl Acad Sci U S A. 1995;92:2622–6. doi: 10.1073/pnas.92.7.2622. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Valdemarsson S, Edvinsson L, Ekman R, Hedner P, Sjoholm A. Increased plasma level of substance P in patients with severe congestive heart failure treated with ACE inhibitors. J Intern Med. 1991;230:325–31. doi: 10.1111/j.1365-2796.1991.tb00452.x. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.