

Abstract
Purpose: Retinal ischemic diseases primarily lead to damage of the inner retinal neurons. Electrophysiological studies also suggest impairment of the inner retinal neurons. Our recent studies with acute ocular hypertensive rats confirmed damage predominantly in the inner retinal layer along with the ganglion cell layer, changes that are ameliorated by the calpain inhibitor SNJ-1945. However, we do not know which specific neuronal cells in the inner retinal layer are damaged by calpains. Thus, the purpose of the present study was to identify specific calpain-damaged neuronal cells in the inner retina from acute ocular hypertensive rats.
Methods: Intraocular pressure was elevated to 110 mm Hg for 40 min. One hour after ocular hypertension (OH), SNJ-1945 was administrated as a single oral dose of 50 mg/kg. Retinal function was assessed by scotopic electroretinography (ERG). Histological degeneration was evaluated by hematoxylin and eosin, terminal deoxynucleotidyl transferase (TdT)-mediated dUTP nick-end-labeling (TUNEL), and immunostaining in thin sections and flat mounts of the retina. Calpain activation was determined by proteolysis of the calpain substrate α-spectrin.
Results: OH caused calpain activation, increased TUNEL-positive staining, decreased thickness of the inner nuclear layer (INL), and decreased amplitudes of the ERG a- and b-waves and oscillatory potentials (OPs). SNJ-1945 significantly inhibited calpain activation and the decrease in ERG values. Interestingly, the changes in the b-wave and OPs amplitudes were significantly correlated to changes in the thickness of the INL. In the inner retinal layer, the numbers of rod bipolar, cone-ON bipolar, and amacrine cells were decreased after OH. SNJ-1945 suppressed the loss of cone-ON bipolar and amacrine cells, but did not inhibit the loss of rod bipolar cells. We also observed increased glial fibrillary acid protein-positive staining in the Müller cells after OH and the treatment with SNJ-1945.
Conclusions: Calpains may contribute to ischemic retinal dysfunction by causing the loss of cone-ON bipolar and amacrine cells and causing the activation of Müller cells. Calpain inhibitor SNJ-1945 may be a candidate compound for treatment of retinal ischemic disease.
Introduction
Retinal ischemia is associated with a variety of ocular pathologies. Examples include optic neuropathy, retinal and choroidal vessel occlusion, diabetic retinopathy, and acute angle closure glaucoma.1 These conditions lead to visual impairment and, in severe cases, blindness. Severe acute hypertension lasting over 60 min in rats leads to retinal ischemia, loss of ganglion cells, and injury to the inner retinal layers.2,3 A milder hypertensive model lasting less than 45 min was recently reported and is probably closer to the human clinical condition.4
The underlying molecular mechanisms for degeneration of the inner retina in ischemia are not completely understood. Histological degeneration in rat inner retina is associated with loss of the electroretinography (ERG) b-waves, suggesting dysfunction of the ON bipolar and Müller cells.5 Calcium overload, glutamate toxicity and oxidative stress are also suggested mechanisms for the pathogenesis of retinal ischemia.1
Calcium overload activates a family of proteases called calpains (EC 3.4.22.17). Fifteen genes in mammals code for these cytoplasmic, cysteine proteases.6 Calpain activation is involved in the degeneration of the ganglion cell layer (GCL) after optic nerve crush,7 after acute elevation of intraocular pressure (IOP),8 and in light-induced photoreceptor degeneration.9 Activation of calpains, proteolysis of calpain substrates, apoptosis, retinal neuronal cell degeneration, and loss of ERG waves also occur in ocular hypertensive rats.10,11 The orally available calpain inhibitor SNJ-1945 suppresses proteolysis of calpain substrates and the degeneration of the ganglion cell and inner retinal layers.11 Studies in rats with chronic, ocular hypertension (OH) also suggest that calpain activation is associated with degeneration of the inner retina.12 However, these studies do not specifically identify which neuronal cells (e.g., photoreceptor, bipolar, horizontal, amacrine, and/or ganglion) are damaged by calpains.
The purposes of the present experiments were to (1) identify neuronal cells in the inner retinal layer that suffer degeneration and electrophysiological dysfunction during OH in rats and (2) document involvement of calpains and efficacy of the calpain inhibitor SNJ-1945 in preventing retinal cell pathology. The resulting data provide a novel mechanism to explain loss of specific retinal neuronal cells and function during retinal ischemia.
Methods
Experimental animals
Male Sprague-Dawley rats at 11–12 weeks of age were obtained from Charles River (Yokohama, Japan). Experimental animals were handled in accordance with the ARVO Statement for the Use of Animals in Ophthalmic and Vision Research and with the Guiding Principles in the Care and Use of Animals (DHEW Publication, NIH 80-23). All experiments were designed to minimize stress, number of animals used, and suffering.
Rat model of acute OH
Acute OH was produced in rats according to a modification of previously reported methods.11 Briefly, the rats were anesthetized with 3.5% sevoflurane/O2 gas. Rats were laid on a regulated heating pad maintaining body temperature at 37°C during anesthesia. For local anesthesia, 0.4% oxybuprocaine hydrochloride solution (Benoxil; Santen, Osaka, Japan) was instilled into the right eye. A 30-gauge steel cannula connected to a saline bag was inserted through the peripheral cornea into the anterior chamber, and the IOP was elevated to 110 mm Hg. The IOP was continuously measured with a calibrated pressure transducer (ADInstruments, Colorado Springs, CO), and data were compiled in LabChart 7.0 software with a PowerLab analog/digital interface (ADInstruments). After 30–50 min, the pressure was lowered to normal IOP, the cannula was removed, and an antibiotic ointment was applied. No procedure was performed on the left eye, since cannulation of sham-operated eyes does not cause retinal dysfunction or degeneration.10
Treatment with calpain inhibitor SNJ-1945
SNJ-1945 was suspended at 1.25% or 0.65% (w/v) in distilled water containing 0.5% carboxymethyl cellulose. One hour after OH, a single oral dose of 25 or 50 mg SNJ-1945/kg body weight was administrated with a feeding needle. The control group received the same volume of carboxymethyl cellulose vehicle solution without SNJ-1945.
Measurement of electroretinograms
Full-field scotopic ERGs were recorded before cannulation and at 1 and 7 days after OH according to a modification of methods previously reported.10 Briefly, the rats were dark-adapted overnight, and all further procedures were performed under dim red illumination. The rats were anesthetized by a 1.5 mL/kg intramuscular injection of a 4:1mixture of Ketalar (5% ketamine hydrochloride; Sankyo, Tokyo, Japan) and Celactal (2% xylazine hydrochloride; Bayer, Leverkusen, Germany)/kg body weight. The pupils had been dilated in advance with Mydrin-P (Santen). A small amount of hydroxyethylcellulose solution (Senju, Osaka, Japan) was applied to the corneal surface, and a contact lens electrode equipped with a light-emitting diode was positioned on the cornea. A wire electrode placed in the mouth served as the reference electrode, and a grounding needle electrode was subcutaneously inserted above the femur. The ERG response was amplified using a low-pass filter setting of 0.3 Hz and a high-pass filter setting of 500 Hz, and the ERG response was recorded with the PuREC system (Mayo, Inazawa, Japan). Five to ten, successive, white, stroboscopic flashes at 3 cds/m2 were presented to the cornea, and the maximal ERG response was used (Fig. 1A). The a-wave was measured as the difference between the amplitudes at the onset and the trough of the negative deflection. The b-wave amplitude was measured from the trough of the a-wave to the peak of the b-wave. The implicit times of a-and b-waves were measured from onset to the trough of the a-wave, and onset to the peak of the b-wave, respectively. The oscillatory potentials (OPs) were extracted from the above ERG data using 65–285 Hz band-pass filters and the PuREC 1.0 software (Mayo). The amplitude of each OP wavelet was measured from the baseline drawn between the troughs of successive wavelets to their peaks.13 Because the first of 5 wavelets are contaminated with the a-wave,14 the amplitudes of second to fifth wavelet were combined as the final adjusted OPs amplitude. Baselines were also recorded a few days before OH. All ERG analyses were performed using a masking procedure.
FIG. 1.

Representative ERGs (A) and quantitative measurement of a-waves (B), b-waves (C), and OPs amplitudes (D), showing significant decrease in amplitudes of a- and b-waves and OPs 7 days after OH, and the protective effect of oral 50 mg/kg SNJ-1945. Data are expressed as mean±SEM (n=16 each). #P<0.05 relative to normal (paired Wilcoxon's rank sum test). *P<0.05 relative to vehicle (Student's t-test). ERGs, electroretinographies; OPs, oscillatory potentials.
Quantitative measurement of cells in the ganglion cell layer and the thickness of the inner retinal layer
Immediately after recording the ERG at 7 days of OH, the rats were asphyxiated with CO2 gas, and the eyes were enucleated. Enucleated right globes were fixed overnight in 10% neutral-buffer formalin containing glutaraldehyde and embedded in paraffin. Sections (3 μm) of the retina through the optic disc were stained with hematoxylin and eosin as previously reported.11 The retinal sections were observed with an optical microscope and then digital images were captured with a CCD camera (DP72; Olympus, Tokyo, Japan) and e-Tiling 3.7 software (Mitani, Fukui, Japan). Cell nuclei in the ganglion cell layer (GCL) were counted in two, 0.8 mm-wide regions located 0.5–2.5 mm in both the right and left sides from the center of the optic disk and expressed as the mean of these four regions.10
The thicknesses of the inner plexiform layer (IPL) and the inner nuclear layer (INL), which included bipolar, amacrine, and horizontal cells, were measured in the same regions used above for GCL counting. Mean thickness values from the entire 0.8 mm-wide regions were computed with Image-Pro Plus 6.3 software (Media Cybernetics, Bethesda, MD). The mean thickness measurements were then calculated from the four regions in each section.15 The thicknesses of the outer nuclear layer (ONL) and the outer segment (OS), which include the photoreceptor cells, were measured using the same method as for the IPL and INL described above.
Immunohistochemistry and cell counting in retinal thin section and flat mount
Retinal thin section
Animals were deeply anesthetized and euthanized by inhalation of air-saturated sevoflurane. Partial blood washout in tissues was performed by intracardial perfusion with saline. The globes were enucleated, fixed for 2 h in 4% paraformaldehyde (PFA) in 0.1 M phosphate buffer (PB, pH 7.4), cryoprotected for 1 h in PB containing 20% sucrose, and embedded in OCT compound (Sakura Finetech, Tokyo, Japan) in liquid nitrogen. Cryosections were prepared at 10 μm thickness. The sections were immersed in 10 mM phosphate-buffered saline (PBS: pH 7.4) for 20 min at room temperature. The sections were then incubated overnight at 4°C with primary antibodies for protein kinase Cα (PKCα, 1:100; Cell Signaling Technologies, Beverly, MA) for visualization of rod bipolar cells and amacrine cells; Goα (1:50; Chemicon/Millipore, CA) for cone-ON bipolar and rod bipolar cells; calretinin (1:1,000; Santa Cruz Technologies, Santa Cruz, CA) for AII amacrine cells; calbindin (1:1,000; Swant, Bellinzona, Switzerland) for horizontal cells; and glial fibrillary acid protein (GFAP) (1:1,000. Dako, Glostrup, Denmark) for astrocytes and Müller cells.16,17 After washing thrice with PBS for 5 min each, the sections were incubated for 45 min with secondary antibodies conjugated to Alexa Fluor 488 or 568 (Invitrogen, Carlsbad, CA). The sections were washed 3 more times with PBS and embedded with Fluoromount (Diagnostic BioSystems, Pleasanton, CA) containing 0.01% TOPRO-3 (Invitrogen). Immunofluorescence images were acquired using a laser scanning confocal microscope (LSM710; Carl Zeiss, Hallbergmoos, Germany), and images were digitalized with ZEN2008 software. Immuno-positive cells were counted in the inner retinal layer in the same regions and using the same procedures as described above for the GCL.
Retinal flat mounts
After transcardiac perfusion, the globes were enucleated, fixed overnight at 4°C in 4% PFA/0.1 M PB. Posterior eyecups were prepared and the remaining vitreous was removed. To allow penetration of the antibody into the retina, eyecups were treated with the following modifications.18 The eyecups were incubated overnight at −30°C in dimethyl sulfoxide (DMSO)/methanol (1:4). After rinsing twice with 100% methanol for 30 min each, the eyecups were subjected to 3 cycles of freezing and thawing between−80°C and room temperature to fragment plasma membranes. The eyecups were then rehydrated in 70%, 50%, and 15% methanol, and PBS for 30 min each. The tissues were free-rotated for 1 h at 4°C in PBS blocking solution containing 2% skim milk, 5% DMSO, and 0.1% Triton X-100. The tissues were then incubated overnight with primary monoclonal antibody for Brn-3a (MAB1585, 1:100; Chemicon/Millipore), a transcription factor specifically expressed in the nuclei of ganglion cells.19 After washing 3 times with blocking solution for 90 min, the tissues were incubated overnight with secondary antibody conjugated to Alexa Fluor 488 (1:200). After washing 3 times for 90 min in PBS with 5% DMSO, whole retinas were isolated. Four radial incisions toward the optic disk were performed, and the retinas with the GCL up were flat-mounted in Vectashield (Vector, Burlingame, CA) onto glass slides. Immunofluorescence images of Brn-3a-positive RGC soma were acquired using a laser scanning confocal microscope (FV1000; Olympus) and digitalized with FV10-ASW software. Using Image-Pro Plus, RGCs were counted in two square areas (0.36 mm2) located 1 and 2 mm from the center of the optic disk in each retinal quadrant. For each retina, the number of RGCs was averaged over the eight square areas.
TUNEL staining
Cleavage of DNA was visualized in situ with Alexa Fluor 488 after marking the free 3′-H ends of DNA with terminal deoxynucleotidyl transferase (TdT)-mediated dUTP nick-end-labeling (TUNEL, Click-iT TUNEL Assay Kit; Invitrogen). TUNEL-positive cells were assumed to be undergoing apoptosis.
Immunoblotting
Immunoblotting was performed in rat retinas according to a modification of previously reported methods.11 Briefly, 4 h after OH, the retina from a right eye was homogenized together in buffer containing 20 mM Tris (pH 7.5), 5 mM EGTA, 5 mM EDTA, and 2 mM dithioerythritol. The soluble proteins were obtained by centrifugation at 13,000 g for 20 min at 4°C. Protein concentrations were measured using the BCA assay (Pierce, Rockford, IL) with bovine serum albumin as the standard. All steps were performed at 4°C.
SDS-PAGE of retinal soluble proteins (20 μg/lane) was performed on 4%–12% Bis-Tris gels (NuPAGE; Invitrogen) with the MOPS buffer system (Invitrogen). Immunoblotting was performed by electrotransferring the proteins from NuPAGE gels onto polyvinylidene fluoride membranes (Millipore, Bedford, MA). The membranes were probed with mouse monoclonal antibody to α-spectrin (nonerythroid, clone AA6; Affiniti Research Product, Exeter, United Kingdom) diluted to 1:1,000. Immunoreactivity was visualized with alkaline phosphatase conjugated to secondary antibody and BCIP/NBT (AP Conjugate Substrate Kit; Bio-Rad, Hercules, CA).
Statistical analysis
Statistical analysis was performed using JMP 9.0 software (SAS Institute, Cary, NC). The ERG a- and b-wave amplitudes, the sum of the OPs amplitudes, and the a- and b-wave implicit times 7 days after OH were expressed as the relative percentage of the baseline values before hypertension. Differences between means from baseline and hypertension groups were tested using the paired Wilcoxon's rank sum-test. The effect of SNJ-1945 was then analyzed with Student's t-test. Other data were analyzed using Tukey's test. Pearson's linear correlation analysis was used to determine the correlation between INL thickness and relative b-wave or OPs amplitudes. For all analyses, P<0.05 was considered statistically significant.
Results
OH depressed ERG waves
As in our previous studies,10 1 day after OH (110 mm Hg, 50 min), a-waves were almost entirely lost, and b-waves and OPs were completely depressed in the present study. These changes persisted for 7 days. Decreases in amplitudes were similar when hypertension was applied for 40 min on day 1, but a- and b-wave amplitudes and OPs partially recovered by 7 days. Thirty minutes of hypertension caused less attenuation on day 1 and more recovery by 7 days (data not shown). Therefore, for all studies below, we elevated IOP to 110 mm Hg for 40 min to produce maximal retinal dysfunction in minimal time.
OH activated calpains and apoptotic cell death
Compared to retina from nontreated normal rats (Fig. 2A), all retinal layers were histologically similar 1 day after OH (Fig. 2B). Thicknesses of the IPL in nontreated normal rats and ocular hypertensive rats were similar at 27.7±3.1 μm and 25.0±3.5 μm, respectively. The INL thicknesses in nontreated normal rats and hypertensive rats were also similar at 19.2±2.4 μm and 20.1±2.9 μm, respectively.
FIG. 2.

Degeneration in retina after OH in rats. Representative retinal sections stained with H&E from normal rats (A) and from rats 1 day (B) or 7 days (C) after OH at 110 mm Hg for 40 min. Representative TUNEL staining in normal retina (D) and 1 day after OH (E), showing TUNEL-positive nuclei (arrows) in INL and ONL. Cell nuclei were stained with TOPRO-3 (blue). Retinal sections stained with PKCα (F), Goα (G), and TUNEL (H) were merged (I). The area with an asterisk in (I) was magnified (J). Rod bipolar cells (J, arrows) staining Goα-positive (purple) and PKCα-positive (green), stained positive for TUNEL. Cone-ON bipolar cells (arrowheads), staining Goα-positive and PKCα-negative, likewise stained positive for TUNEL (J). Retinal section stained with calretinin (K) or with calbindin (N) were merged with TUNEL staining (L, O). AII amacrine cells (arrows), staining positive for calretinin (green), stained positive for TUNEL (M, merged K, L). Horizontal cells (N), staining positive for calbindin (green) were TUNEL-negative (O), are shown merged in (P). Scale bar=50 μm (A), 20 μm (D), and 10 μm (F, K, N). OH, ocular hypertension; H&E, hematoxylin and eosin; GCL, ganglion cell layer; IPL, inner plexiform layer; INL, inner nuclear layer; ONL, outer nuclear layer; OS, outer segment; TUNEL, terminal deoxynucleotidyl transferase (TdT)-mediated dUTP nick-end-labeling.
The number of cells in the GCL from ocular hypertensive rats was 66±3/mm, which was similar to that in nontreated rats at 65±3/mm. Seven days after OH, obvious degeneration and cell loss were observed in the GCL and the INL (Figs. 2C, 3A, 3C, and 4C), but these changes were not observed in the ONL and OS (Figs. 2C and 3A).
FIG. 3.

Photomicrographs of sections from rat retina stained with H&E (A) and measurement of thickness in IPL (B) and INL (C), showing significant a decrease in thickness of the IPL and INL 7 days after OH and the protective effect of a single oral dose of 50 mg SNJ-1945/kg. Scale bar=50 μm. Data are mean±SEM (n=16 each). #P<0.05 relative to normal and *P<0.05 relative to Vehicle (Tukey's test). Correlation between relative b-wave (D) or OPs (E) amplitudes and thickness of the INL in individual eyes from vehicle-treated (closed circles) and 50 mg/kg SNJ-1945-treated (open circles) rats 7 days after OH. When using all data, statistically significant, positive correlations were observed (D): % b-wave=3.86×(μm INL)−49.40, Pearson's linear correlation coefficient r=0.54, P<0.05 (n=16); and (E): % OPs=2.44×(μm IPL)−17.41, Pearson's linear correlation coefficient r=0.45, P<0.05 (n=16).
FIG. 4.

(A) Representative immunoblot of lysate from whole retina showing intact α-spectrin at 280 kDa (closed arrowhead) and SBDPs at 145 and 150 kDa (open arrowheads) 4 hrs after OH and inhibition of these changes by SNJ-1945. (B) Densitometric image analysis showing increased calpain-specific SBDP at 145 kDa and inhibition by oral SNJ-1945 at 50 mg/kg. Data are expressed as mean±SEM (n=8–10). #P<0.05 relative to normal and *P<0.05 relative to vehicle (Tukey's test). Dose-dependent inhibition of decreased cell numbers in the GCL (C) and decreased ganglion cells (D) by administration of oral SNJ-1945 after OH. Data are expressed as mean±SEM (n=27–29 (C), n=11–12 (D)). #P<0.05 relative to normal and *P<0.05 relative to vehicle (Tukey's test). SBDPs, α-spectrin breakdown products.
No TUNEL-positive cells were observed in normal retina (Fig. 2D). Although there were no obvious histological changes in retina at 1 day, OH produced numerous TUNEL-positive cells in the INL (Fig. 2E, arrows), with only minimal TUNEL-positive cells in the ONL. Rod bipolar cells (Fig. 2F, G, arrows) and cone-ON bipolar cells (Fig. 2G, arrowhead) in the INL stained positive for TUNEL (Fig. 2H–J) 1 day after OH. AII amacrine cells (Fig. 2K, arrow) also showed TUNEL-positive staining (Fig. 2L, M). No TUNEL-positive staining was observed in the horizontal cells in INL (Fig. 2N–P). These data suggested that ocular apoptosis of specific cell types in the inner retinal layer were related to the loss of ERG waves reported above.
Calpain inhibitor SNJ-1945 attenuated degeneration and dysfunction of the inner retinal layers
Amplitudes of the a- and b-waves and OPs in the control preoperative eyes were: −420±17, 1,011±40, and 613±39 (μV, mean±SEM), respectively. As performed in previous studies,20 our ERG results for experimental groups were expressed as percent of these control values. Seven days after OH, amplitudes of the a- and b-waves and OPs were significantly decreased to 74, 29, and 27%, respectively (Fig. 1, Pre vs. Vehicle). Oral 50 mg/kg SNJ-1945 significantly improved amplitudes of the a- and b- waves and OPs to 89%, 53%, and 51% of preoperated eye, respectively (Fig. 1B–D, SNJ-1945). Seven days after OH, the b-wave implicit time was significantly [preoperated; 73±1.83, 7 days after OH; 85±3.29 (ms, mean±SEM) P<0.05, paired Wilcoxon's rank sum test] prolonged to 117% of preoperated eyes. Oral 50 mg/kg SNJ-1945 significantly restored the b-wave implicit time back to the level of preoperated eyes [SNJ; 72±3.29 (ms, mean±SEM) P<0.05, Student's t-test]. In contrast, the a-wave implicit time was not obviously affected by OH [preoperated; 16.5±0.28, 7 days after OH; 16.8±0.43(ms, mean±SEM)].
OH decreased the thickness of IPL and INL to 64% and 79% of normal after 7 days, respectively (Fig. 3A–C, Normal vs. Vehicle). SNJ-1945 at 50 mg/kg significantly improved IPL and INL thickness to 81% and 92%, respectively (Fig. 3A–C, SNJ-1945 vs. Vehicle). The amplitudes of b-waves (Fig. 3D) and OPs (Fig. 3E) were significantly directly correlated to INL thickness. No changes were observed in the thickness of ONL and OS (data not shown).
An intact α-spectrin band at 280 kDa was observed in retina from normal rats (Fig. 4A, Nor). Proteolysis of α-spectrin is a marker for neuronal cell death, and breakdown products from α-spectrin appeared at 150 and 145 kDa 4 h after OH (Fig. 4A, OH). The 145 kDa fragment is produced only by calpains, while the 150 kDa fragment is produced by calpains and caspase-3.21 The caspase-3-specific α-spectrin breakdown product at 120 kDa was not observed, as previously observed.11 Oral administration of SNJ-1945 at 50 mg/kg significantly inhibited proteolysis of α-spectrin (Fig. 4A, B, SNJ).
Effect of SNJ-1945 on specific retinal neurons after OH
The b-waves are derived from the ON bipolar and Müller cells, and a-waves from the photoreceptor cells. OPs are mainly derived from amacrine cells and represent feedback neural pathways for the inner retina.22,23
Cells in GCL
The number of cells in the GCL from ocular hypertensive rats decreased to 72% of nontreated normal rats (Fig. 4C, Normal vs. Vehicle). Oral administration of SNJ-1945 significantly inhibited this loss of GCL cells in a dose-dependent manner (Fig. 4C, SNJ-1945 vs. Vehicle). The cell numbers in animals treated with SNJ-1945 at 50 and 25 mg/kg were 92% and 81% of normal, respectively. This suggested involvement of calpains in the loss of cells in the GCL. Since the RGL contains several cell types, the specific number of ganglion cells was measured. The number of ganglion cells in the ocular hypertensive rats significantly decreased compared to the nontreated normal rats (Fig. 4D, Normal vs. Vehicle). Oral administration of SNJ-1945 significantly inhibited this loss of ganglion cells in a dose-dependent manner (Fig. 4D, SNJ-1945 vs. Vehicle).
Bipolar cells
In normal retina, the cell bodies of rod bipolar cells were observed in the outer border of INL (Fig. 5A–C, arrows). Cone-ON bipolar cells were located in the inner side of the INL (Fig. 5A, C, arrowheads). Seven days after OH, rod and cone-ON bipolar cells were significantly decreased (Fig. 5D–F) to 85% and 68% of normal, respectively (Fig. 5J, K). While 50 mg/kg SNJ-1945 did not inhibit loss of rod bipolar cells after OH (Fig. 5K), SNJ-1945 significantly attenuates loss of cone-ON bipolar cells back to 91% of normal (Fig. 5G–I, J). Cone-OFF bipolar cells were not measured, since cone-OFF bipolar cells are not major participants in production of the b-waves.1
FIG. 5.

Immunohistochemistry for rod bipolar cells (arrows), staining Goα-positive (purple) (A, D, G) and PKCα-positive (green) (B, E, H); and cone-ON bipolar cells (arrowheads), staining Goα-positive (purple) and PKCα-negative. Quantitative measurement of cell numbers (J, K), showed loss of bipolar cells 7 days after OH (D–F) and protection of cone ON-bipolar cells by 50 mg/kg SNJ-1945 (G–I). Area with an asterisk in normal retina was magnified and inserted in panel (C). Scale bar=20 μm. Data are expressed as mean±SEM (n=3 each). *P<0.05 (Tukey's test). PKCα, protein kinase Cα.
AII amacrine cells
In normal retina, AII amacrine cells were present in the GCL, in the innermost side of the INL, and as 3 distinct layers in the IPL formed by the synaptic termini of AII amacrine cells (Fig. 6A, Normal). Seven days after OH, AII amacrine cells and their synaptic termini were significantly decreased (Fig. 6A, Vehicle) to 55% of normal INL levels (Fig. 6B). Oral 50 mg/kg SNJ-1945 allowed significant retention of AII amacrine cells and their synaptic termini (Fig. 6A, SNJ-1945) at 81% of the normal INL levels (Fig. 6B).
FIG. 6.

Immunohistochemistry of AII amacrine cells, staining (green) for calretinin-positive (arrows) (A), and quantitative measurement of cell numbers (B). AII amacrine cells were lost after 7 days of OH and protected by 50 mg/kg SNJ-1945. Scale bar=20 μm. Data are expressed as mean±SEM (n=3 each). *P<0.05 (Tukey's test).
Horizontal cells
OH caused no observable changes in horizontal cells (data not shown), agreeing with previously reported results.24
Müller cells
In normal retina, positive staining for GFAP was localized only in the GCL (Fig. 7, Normal). This staining originates from the astrocytes.20 Seven days after OH, GFAP-positive staining mainly increased in the GCL, but stained axons extended from the GCL to the outer retinal layer (Fig. 7, Vehicle). This new staining originated from the Müller cells because the staining pattern overlapped the published localization pattern for Müller cells, and GFAP is increased when Müller cells were activated following ischemia.20 Oral 50 mg/kg SNJ-1945 inhibited the increase in GFAP-positive staining (activation of Müller cells) (Fig. 7, SNJ-1945).
FIG. 7.
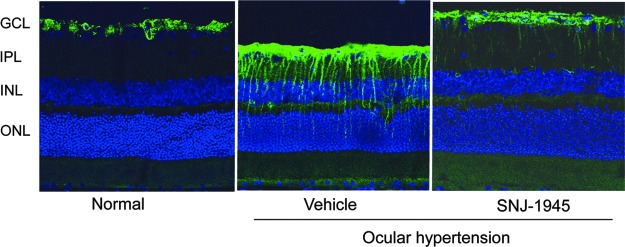
Immunohistochemistry of astrocytes and Müller cells, staining (green) for glial fibrillary acid protein.
Discussion
The major findings of the present study were that (1) OH induced activation of calpains and subsequent apoptosis of ON bipolar and amacrine cells. This led to degeneration of the INL and decreased ERG amplitudes. (2) Calpain inhibitor SNJ-1945 attenuated the loss of cone-ON bipolar and amacrine cells, leading to rescue of INL morphology and improvement of electrophysiologic function.
Activation of calpains, degeneration, and ERG dysfunction in retina
Our previous studies in ocular hypertensive rats using severe conditions (60 min increased IOP) showed increased retinal calcium, activation of calpains, proteolysis of calpain substrates, degeneration of retinal layers, and loss of ERG; suggesting involvement of calpains in retinal degeneration and dysfunction.10 The present experiments with milder OH (40 min) further showed that OH caused hydrolysis of intracellular α-spectrin to a calpain-specific breakdown product, TUNEL-positive retinal cells, and loss of specific inner retinal cells associated with degradation of ERG signals. In retina, acute OH -induced retinal ischemia/reperfusion is known to deplete intracellular ATP followed by the failure of the Na/K-ATPase pump, membrane depolarization, and accumulation of cytoplasmic sodium and calcium ions.1 Increased intracellular Ca2+ influx is required for the activation of calpains.10 The localization of calpains to specific retinal cell types is largely unknown. However, a complete calpain system, consisting of mRNA for calpains, calpain proteins, and the endogenous inhibitor calpastatin is known to be present and active in whole rat retina.25
Cells in the inner retinal layer damaged by calpain during OH
The cone-ON bipolar, rod bipolar, and amacrine cells, but not the horizontal cells in the inner retinal layer were apoptotic 1 day after OH (Fig. 2). These apoptotic cells were then lost on day 7 (Figs. 5 and 6). Bipolar cells, are second-order retinal neurons, and relay signals from photoreceptor cells to RGCs. The relay amacrine neurons vertically connect these bipolar cells to the ganglion cells. ON bipolar cells, including cone-ON and rod bipolar cells are the major sources of the electrical impulses producing b-waves, while the AII amacrine cells produce the OPs signals.26,27 The layers containing these connecting neurons showed apoptotic cells that were associated with significant decreases in the b-wave and OPs signals 1 day after OH. In our experiments, cone-ON bipolar and AII amacrine seem to be more vulnerable to OH than rod bipolar cells (Figs. 5 and 6), and this agrees with a previous report.19 Thus, hypertension-induced apoptosis and loss of electrophysiological function occurs before histologic degeneration of the inner retinal layers.
Although TUNEL-positive cells have been reported in the GCL 1 day after OH,28 we did not observe such cells, and a possible reason is that our model is less severe. Alternatively, apoptotic cells may have disappeared in our model 1 day after OH since the number of apoptotic cells peak at 6 h and then decrease.29
OH provoked a significant alternation in Müller cells, as shown by an increase in GFAP immunoreactivity 7 days after OH. This suggests that activation of Müller cells contributed to the observed decrease in the b-wave amplitudes.1 Oral SNJ-1945 decreased GFAP immunoreactivity in Müller cells. Possibly activated calpain causes the activation of Müller cells, leading to the decrease b-wave amplitudes. This would be an additional advantage of SNJ-1945 to improve electrophysiologic function by regulation of ocular glial cells.
Also of potential clinical significance, SNJ-1945 suppressed degeneration of inner retina and the decreases in b-waves and OPs. SNJ-1945 inhibited loss of cone-ON bipolar cells (Fig. 5), but not rod bipolar cells, suggesting that cone-ON bipolar cell death might be mediated by calpain activation. Although we do not know yet why SNJ-1945 did not inhibit rod bipolar cell death, localization of calpains and calpastatin to specific cells types may be a reason. For example, in the central nervous system, calpain 2 mRNA is higher in some localized neuronal populations, and this may explain the pathogenesis of certain regional neurological degenerative diseases.30 Activation of executioner caspases may be another explanation since caspases are not inhibited by SNJ-1945. In an acute model of OH, caspases were involved in apoptosis in retinal neuronal cells,31 although obvious caspase-specific α-spectrin breakdown product was not observed in present study. In addition to apoptosis and necrosis, another type of cell death, autophagy was recently observed in retina during acute OH.32,33 Autophagy involves lysosomal degradation of long-lived cytoplasmic proteins, and it has been observed during differentiation, starvation, and stress conditions such as oxidation.34 Activation of calpains and caspases are involved in autophagic cell death.32,33 Beclin-1, an essential protein for autophagic regulation, is proteolyzed by calpains.32 Future experiments are needed to determine which of the 3 specific mechanisms (apoptosis, necrosis, or autophagy) cause death of the rod bipolar cells.
SNJ-1945 also inhibited loss of AII amacrine cells (Fig. 6). Amacrine cells express ionotropic glutamate receptors (NMDA, AMPA, and kainate types), and over-agonized NMDA receptors cause entry of Ca2+ and activation of calpains leading to cell death. A large increase in extracellular glutamate is also observed in an acute model of OH.13 Activation of NMDA receptors causes influx of intracellular Ca2+, activation of calpains, and death of amacrine cells.35
After a single oral administration of 10 mg SNJ-1945/kg, concentrations of SNJ-1945 in retina reach a maximum at 15 min and then gradually decrease over 8 h.36 Four hours after administration, the concentration of SNJ-1945 in retina still exceeded the IC50's against calpains 1 and 2. Rapid penetration and long retention may contribute to the effect of SNJ-1945, since calpain was highly activated 1 h after reperfusion.32 SNJ-1945 also inhibits other cysteine proteases such as cathepsin B and L.37 The activation of cathepsin B is involved in retinal ganglion cell death after optic nerve injury.38 Therefore, inhibition of activated cathepsin B by SNJ-1945 may be another mechanism for ameliorating retinal cell death, in addition to inhibition by calpains.
Relevance to clinical findings
In the present rat experiments, cells in the GCL were decreased by OH, and SNJ-1945 inhibited loss of these cells, suggesting activation of calpain as in previous experiments.11 Further, a lower dose of SNJ-1945 (25 mg/kg) was effective for rescuing ganglion cells in the present experiments using our milder model. Synapses of the cone-ON bipolar cells directly connect to ganglion cell dendrites. In contrast, rod bipolar cells connect to AII amacrine dendrites, whose synapses are connected to cone-ON bipolar cells, modulating cone-mediated visual function. The human macula is largely dependent upon cone-mediated photopic vision, and cone-mediated visual dysfunction is reported in patients with retinal vessel occlusion and diabetic retinopathy.39,40 Calpain inhibitor rescued both cone-bipolar and AII amacrine cells important for cone-mediated visual function, suggesting that SNJ-1945 might be useful as a therapeutic medication for sustaining the visual field in patients retina ischemia.
Author Disclosure Statement
Dr. Shearer has significant financial interests (research contract and consulting fee) in Senju Pharmaceutical Co., Ltd., and Dr. Azuma is an employee of Senju Pharmaceutical Co., Ltd., a company that may have a commercial interest in the results of this research and technology. These potential conflicts of interest have been reviewed and managed by the OHSU Conflict of Interest in Research Committee.
References
- 1.Osborne N.N., Casson R.J., Wood J.P., Chidlow G., Graham M., and Melena J.Retinal ischemia: mechanisms of damage and potential therapeutic strategies. Prog. Retin. Eye Res. 23:91–147, 2004 [DOI] [PubMed] [Google Scholar]
- 2.Büchi E.R., Suivaizdis I., and Fu J.Pressure-induced retinal ischemia in rats: an experimental model for quantitative study. Ophthalmologica. 203:138–147, 1991 [DOI] [PubMed] [Google Scholar]
- 3.Büchi E.R.Cell death in the rat retina after a pressure-induced ischemia-reperfusion insult: an electron microscopic study. I. ganglion cell layer and inner nuclear layer. Exp. Eye Res. 55:605–613, 1992 [DOI] [PubMed] [Google Scholar]
- 4.Jehle T., Wingert K., Dimitriu C., Meschede W., Lasseck J., et al. Quantification of ischemic damage in the rat retina: a comparative study using evoked potentials, electroretinography, and histology. Invest. Ophthalmol. Vis. Sci. 49:1056–1064, 2008 [DOI] [PubMed] [Google Scholar]
- 5.Mukaida Y., Machida S., Masuda T., and Tazawa Y.Correlation of retinal function with retinal histopathology following ischemia-reperfusion in rat eyes. Curr. Eye Res. 28:381–389, 2004 [DOI] [PubMed] [Google Scholar]
- 6.Branca D.Calpain-related diseases. Biochem. Biophys. Res. Commun. 322:1098–1104, 2004 [DOI] [PubMed] [Google Scholar]
- 7.Ryu M., Yasuda M., Shi D., Shanab A.Y., Watanabe R., et al. Critical role of calpain in axonal damage-induced retinal ganglion cell death. J. Neurosci. Res. 90:802–815, 2012 [DOI] [PubMed] [Google Scholar]
- 8.Russo R., Cavaliere F., Berliocchi L., Nucci C., Gliozzi M., et al. Modulation of pro-survival and death-associated pathways under retinal ischemia/reperfusion: effects of NMDA receptor blockade. J. Neurochem. 107:1347–1357, 2008 [DOI] [PubMed] [Google Scholar]
- 9.Imai S., Shimazawa M., Nakanishi T., Tsuruma K., and Hara H.Calpain inhibitor protects cells against light-induced retinal degeneration. J. Pharmacol. Exp. Ther. 335:645–652, 2010 [DOI] [PubMed] [Google Scholar]
- 10.Oka T., Tamada Y., Nakajima E., Shearer T.R., and Azuma M.Presence of calpain-induced proteolysis in retinal degeneration and dysfunction in a rat model of acute ocular hypertension. J. Neurosci. Res. 83:1342–1351, 2006 [DOI] [PubMed] [Google Scholar]
- 11.Oka T., Walkup D., Tamada Y., Nakajima E., Tochigi A., et al. Amelioration of retinal degeneration and proteolysis in acute ocular hypertensive rats by calpain inhibitor ((1S)-1-((((1S)-1-benzyl-3-cyclopropylamino-2, 3-di-oxiopropyl)amino) carbonyl)-3-methylbutyl)carbamic acid 5-methoxy-3-oxapentyl ester. Neuroscience. 141:2139–2145, 2006 [DOI] [PubMed] [Google Scholar]
- 12.Hughes E., Spry P., and Diamond J.24-hour monitoring of intraocular pressure in glaucoma management: a retrospective review. J. Glaucoma 12:232–236, 2003 [DOI] [PubMed] [Google Scholar]
- 13.Fernandez D.C., Chianelli M.S., and Rosenstein R.E.Involvement of glutamate in retinal protection against ischemia/reperfusion damage induced by post-conditioning. J. Neurochem. 111:488–498, 2009 [DOI] [PubMed] [Google Scholar]
- 14.Akula J.D., Mocko J.A., Moskowitz A., Hansen R.M., and Fulton A.B.The Oscillatory potentials of the dark-adapted electroretinogram in retinopathy of prematurity. Invest. Ophthalmol. Vis. Sci. 48:5788–5797, 2007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Oka T., Nakajima T., Tamada Y., Shearer T.R., and Azuma M.Contribution of calpains to photoreceptor cell death in N-methyl-N-nitrosourea-treated rats. Exp. Neurol. 204:39–48, 2007 [DOI] [PubMed] [Google Scholar]
- 16.Hernandez M., Rodriguez F.D., Sharma S.C., and Vecino E.Immunohistochemical changes in rat retinas at various time periods of elevated intraocular pressure. Mol. Vis. 15:2696–2709, 2009 [PMC free article] [PubMed] [Google Scholar]
- 17.Sun D., Bui B.V., Vingrys A.J., and Kalloniatis M.Alterations in photoreceptor-bipolar cell signaling following ischemia/reperfusion in the rat retina. J. Comp. Neurol. 505:131–146, 2007 [DOI] [PubMed] [Google Scholar]
- 18.Yabuta C., Oka T., Kishimoto Y., Ohtori A., Yoshimatsu A., and Azuma M.Topical FK962 facilitates axonal regeneration and recovery of corneal sensitivity after flap surgery in rabbits. Am. J. Ophthalmol.153:651–660, 2012 [DOI] [PubMed] [Google Scholar]
- 19.Mojumder D.K., Sherry D.M., and Frishman L.J.Contribution of voltage-gated sodium channels to the b-wave of the mammalian flash electroretinogram. J Physiol. 586:2551–2580, 2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Osborne N.N., Ji D., Abdul Majid A.S., Fawcett R.J., Sparatore A., et al. ACS67, a hydrogen sulfide-releasing derivative of latanoprost acid, attenuates retinal ischemia and oxidative stress to RGC-5 cells in culture. Invest. Ophthalmol. Vis. Sci. 51:284–294, 2010 [DOI] [PubMed] [Google Scholar]
- 21.Nath R., Raser K.J., Stafford D., Hajimohammadreza I., Posner A., et al. Non-erythroid α-spectrin breakdown by calpain and interleukin 1β-converting-enzyme-like protease(s) in apoptotic cells: contributory roles of both protease families in neuronal apoptosis. Biochem. J. 319:683–690, 1996 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Dick E., and Miller R.F.Light-evoked potassium activity in mudpuppy retina: its relationship to the b-wave of the electroretinogram. Brain Res. 154:388–394, 1978 [DOI] [PubMed] [Google Scholar]
- 23.Kline R.P., Ripps H., and Dowling J.E.Generation of b-wave currents in the skate retina. Proc. Natl. Acad. Sci. U. S. A. 75:5727–5731, 1978 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Kim S.A., Jeon J.H., Son M.J., Cha J., Chun M.H., and Kim I.B.Changes in transcript and protein levels of calbindin D28k, calretinin and parvalbumin, and numbers of neuronal populations expressing these proteins in an ischemia model of rat retina. Anat. Cell Biol. 43:218–229, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Azuma M., Sakamoto-Mizutani K., Nakajima T., Kanaami-Daibo S., Tamada Y., and Shearer T.R.Involvement of calpain isoforms in retinal degeneration in WBN/Kob rats. Comp. Med. 54:533–542, 2004 [PubMed] [Google Scholar]
- 26.Wachtmeister L.Further studies of the chemical sensitivity of the oscillatory potentials of the electroretinogram (ERG) I. GABA- and glycine antagonists. Acta. Ophthalmol. (Copenh). 58:712–725, 1980 [DOI] [PubMed] [Google Scholar]
- 27.Wachtmeister L.Oscillatory potentials in the retina: what do they reveal. Prog. Retin. Eye Res. 17:485–521, 1998 [DOI] [PubMed] [Google Scholar]
- 28.Zhang X.Y., Xiao Y.Q., Zhang Y., and Ye W.Protective effect of pioglitazone on retinal ischemia/reperfusion injury in rats. Invest. Ophthalmol. Vis. Sci. 54:3912–3921, 2013 [DOI] [PubMed] [Google Scholar]
- 29.Lam T.T., Abler A.S., and Tso M.O.Apoptosis and caspases after ischemia-reperfusion injury in rat retina. Invest. Ophthalmol. Vis. Sci. 40:967–975, 1999 [PubMed] [Google Scholar]
- 30.Li J., Grynspan F., Berman S., Nixon R., and Bursztajn S.Regional differences in gene expression for calcium activated neutral proteases (calpains) and their endogenous inhibitor calpastatin in mouse brain and spinal cord. J. Neurobiol. 30:177–191, 1996 [DOI] [PubMed] [Google Scholar]
- 31.Singh M., Savitz S.I., Hoque R., Gupta G., Roth S., et al. Cell-specific caspase expression by different neuronal phenotypes in transient retinal ischemia. J. Neurochem. 77:466–475, 2001 [DOI] [PubMed] [Google Scholar]
- 32.Russo R., Berliocchi L., Adornetto A., Varano G.P., Cavaliere F., et al. Calpain-mediated cleavage of Beclin-1 and autophagy deregulation following retinal ischemic injury in vivo. Cell Death Dis. 2:e144, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Piras A., Gianetto D., Conte D., Bosone A., and Vercelli A.Activation of autophagy in a rat model of retinal ischemia following high intraocular pressure. PLoS One 6:e22514, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Shacka J.J., Roth K.A., and Zhang J.The autophagy-lysosomal degradation pathway: role in neurodegenerative disease and therapy. Front. Biosci. 13:718–736, 2008 [DOI] [PubMed] [Google Scholar]
- 35.Nakazawa T., Shimura M., Mourin R., Kondo M., Yokokura S., et al. Calpain-mediated degradation of G-substrate plays a critical role in retinal excitotoxicity for amacrine cells. J. Neurosci. Res. 87:1412–1423, 2009 [DOI] [PubMed] [Google Scholar]
- 36.Shirasaki Y., Yamaguchi M., and Miyashita H.Retinal penetration of calpain inhibitors in rats after oral administration. J. Ocul. Pharmacol. Ther. 22:417–424, 2006 [DOI] [PubMed] [Google Scholar]
- 37.Azuma M., and Shearer T.R.The role of calcium-activated protease calpain in experimental retinal pathology. Surv. Ophthalmol. 53:150–163, 2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Agudo M., Pérez-Marín M.C., Sobrado-Calvo P., Lönngren U., Salinas-Navarro M., et al. Immediate upregulation of proteins belonging to different branches of the apoptotic cascade in the retina after optic nerve transection and optic nerve crush. Invest. Ophthalmol. Vis. Sci. 50:424–431, 2009 [DOI] [PubMed] [Google Scholar]
- 39.Larsson J., Bauer B., and Andréasson S.The 30-Hz flicker cone ERG for monitoring the early course of central retinal vein occlusion. Acta. Ophthalmol. Scand. 78:187–190, 2000 [DOI] [PubMed] [Google Scholar]
- 40.Yamamoto S., Kamiyama M., Nitta K., Yamada T., and Hayasaka S.Selective reduction of the S cone electroretinogram in diabetes. Br. J. Ophthalmol. 80:973–975, 1996 [DOI] [PMC free article] [PubMed] [Google Scholar]