

Abstract
Cerebellar granule cell precursors (GCPs), which give rise to the most abundant neuronal type in the mammalian brain, arise from a restricted pool of primary progenitors in the rhombic lip (RL). Sonic hedgehog (Shh) secreted by developing Purkinje cells is essential for the expansion of GCPs and for cerebellar morphogenesis. Recent studies have shown that the primary cilium concentrates components of Shh signaling and that this structure is required for Shh signaling. GCPs have a primary cilium on their surface (Del Cerro and Snider, 1972). Here, we show that 1) this cilium can be conditionally ablated by crossing Kif3afl/- mice with hGFAP-Cre mice, 2) removal of Kif3a from GCPs disrupts cerebellar development, and 3) these defects are due to a drastic reduction in Shh-dependent expansion of GCPs. A similar phenotype is observed when Smoothened (Smo), an essential transducer of Shh signaling, is removed from the same population of GCPs. Interestingly, Kif3a-Smo double conditional mutants show that Kif3a is epistatic to Smo. This work shows that Kif3a is essential for Shh-dependent expansion of cerebellar progenitors. Dysfunctional cilia are associated with diverse human disorders including Bardet-Biedl and Joubert syndromes. Cerebellar abnormalities observed in these patients could be explained by defects in Shh-induced GCP expansion.
Keywords: Neurogenesis, Primary cilia, Kif3a, Sonic hedgehog, Cerebellar development, Joubert syndrome
Introduction
The most abundant neurons in the cerebellum, and in the entire brain, are granule neurons. Granule cell precursors (GCPs) arise from a restricted germinal center that develops in the anterior margins of the 4th ventricle known as the rhombic lip (RL) (Altman and Bayer, 1997). In the mouse, the GCPs leave the RL at around embryonic day (E) 13 and migrate rostrally over the dorsal surface of the cerebellar anlage to form the external granule cell layer (EGL). During the first two weeks after birth, cells in the EGL undergo extensive proliferation to produce the pool of GCPs required to generate the large number of granule neurons. Developing GCPs then exit the cell cycle, and migrate internally past the Purkinje cells to form the inner granule cell layer (IGL) (Sotelo, 2004).
Sonic hedgehog (Shh) is a secreted morphogen that plays essential roles during brain development. Binding of Shh to its receptor Patched1 (Ptc1) relieves inhibition of the signaling pathway activated by the transmembrane protein Smoothened (Smo), resulting in the activation of transcriptional targets by members of the Gli family of transcription factors (Ingham, 1998). In the spinal cord, Shh specifies a subpopulation of ventral cell types in a concentration-dependent manner (Chiang et al., 1996; Briscoe et al., 1999). In the limb, Shh is essential for proper morphogenesis and digit identity (Riddle et al., 1993; Litingtung et al., 2002). In the cerebellum, Shh is expressed in Purkinje cells and regulates the proliferation of GCPs (Dahmane and Ruiz i Altaba, 1999; Wechsler-Reya and Scott, 1999; Wallace, 1999). The cellular and molecular mechanisms through which Shh signaling regulates GCP proliferation are still under investigation. However, a number of recent studies suggested that one direct target of Shh signaling is the proto-oncogene N-myc1, which mediates GCP proliferation (Kenney et al., 2003; Knoepfler et al., 2002) via cyclin D1 and cyclin D2 (Kenney and Rowitch, 2000; Ciemerych et al., 2002).
The primary cilium is an antenna-like specialized structure that projects from the surface of many eukaryotic cells (Singla and Reiter, 2006). It has an axoneme with a 9+0 microtubule arrangement and is assembled and maintained by intraflagellar transport (IFT) (Rosenbaum and Witman, 2002). IFT is a specialized intracellular trafficking process that moves multimeric protein complexes bidirectionally along the ciliary axoneme. The anterograde and retrograde movements of proteins are mediated by kinesin-II and cytoplasmic dynein complex, respectively. Kinesin-II is composed of two different motor subunits (KIF3A and KIF3B) and one non-motor accessory subunit (KAP3). Genetic and localization experiments have shown that kinesin-II is essential for the construction and maintenance of cilia (for review see Marszalek and Goldstein, 2000). In the central nervous system (CNS), primary cilia are present in cells in the ventricular zone (VZ) during embryogenesis (Cohen et al, 1988; Huangfu and Anderson, 2005), in granule cell precursors in the EGL at early post-natal stages (Del Cerro and Snider, 1972), in neural progenitor cells (Alvarez-Buylla et al., 1998; Doetsch et al., 1999) and in neurons in the adult (Fuchs and Schwark, 2004; Whitfield, 2004). Recent studies demonstrated that primary cilia are required for the specification of ventral cell types in the spinal cord in response to Shh (Huangfu et al., 2003; Huangfu and Anderson, 2005). Moreover, essential components of Shh signaling, Smo (Corbit et al., 2005), the Gli transcription factors, Suppressor of fused (Sufu) (Haycraft et al., 2005), and Ptc1 (Rohatgi et al., 2007) have been shown to localize to primary cilia. However, little is known about the function of this intriguing cellular organelle in other cells in the developing CNS.
Here, we have investigated the function of primary cilia in the development of cerebellar GCPs and its contribution to Shh signaling in these cells. Conditional ablation of Kif3a in cells derived from Cre-expressing cells under the human glial fibrillary acidic protein (hGFAP) promoter resulted in GCPs without primary cilia. In these animals, GCPs are specified, but a severe defect in late embryonic and early postnatal expansion of GCPs results in atrophied cerebella. We show that the primary cilium is required for Shh-induced GCP proliferation via induction of Gli1 and cyclin D1 gene expression. Therefore, our results suggest that the primary cilium in GCPs is required for their Shh-induced expansion and cerebellar development.
Materials and Methods
Transgenic Mice
All animal care was in accordance with the guidelines of the National Institutes of Health, the University of California and European law. All the mice used in this study have been described previously: the hGFAP∷Cre mouse line containing 2.2 kb of the hGFAP promoter linked to the coding region of Cre recombinase (Zhuo et al., 2001), Kif3afl mice containing loxP sites flanking exon 2 of the Kif3a gene and kif3a-/+ mice containing the recombined allele (Marszalek et al., 1999), R26R mice (Soriano, 1999), Z/EG mice (Novak et al., 2000) and Smofl mice (Long et al., 2001). Mice used in this study were in C57Bl6/J or mixed backgrounds.
Immunostaining
Brains were fixed by immersion (embryonic brains) in 4% paraformaldehyde at 4°C for 16 hours or by perfusion (postnatal brains) in the same fixative and washed overnight at 4°C in PBS and cryoprotected in PBS containing 30% sucrose. Histological sagittal sections were cut at 10 or 12 μm on a cryostat and pre-blocked for ICC for 30 minutes to 1 hour in TBS or PBS with 0,1% Triton X-100 and 10% normal goat or horse serum. Sections were incubated overnight at 4°C with the primary antibodies. The following antibodies were used: rabbit polyclonal (pAb) anti-GFAP (1:200; Sigma), chicken pAb anti-GFP (1:500, Aves Labs inc.), rabbit pAb anti-GABAA receptor α6 subunit (α6, 1:1000, Chemicon), rabbit pAb anti-γ-tubulin (1:1000, Sigma), rabbit pAb anti-CaBP (1:5000, Swant), rabbit pAb anti-Pax6 (Osumi et al., 1997), mouse monoclonal (mAb) anti-Cre (1:500, Euromedex), mouse mAb anti-acetylated α–tubulin (1:1000, Sigma), rat monoclonal (rAb) anti-BrdU (1:200, Oxford Biotechnologies), goat monoclonal (gAb) anti-β–galactosidase (1:700, Biotrend), and species-specific secondary antibodies (Molecular Probes or Jackson ImmunoResearch). Sections were counterstained with DAPI (10 μg/ml, Sigma), mounted in Fluoromount and examined with a fluorescence microscope or a fluorescence confocal microscope (DM IRBE and SP2 Leica).
Isolation of cells enriched in GCPs and Purkinje cells, RNA preparation and quantitative real-time RT-PCR (qRT-PCR)
Cells enriched in GCPs and Purkinje cells were isolated from 2-4-day-old (P2-4) mutant and wild-type pups as described (Hatten, 1985; Baptista et al., 1994; Weschler-Reya and Scott, 1999). Cerebella were digested in solution containing 10 μg/ml trypsin and 0,5 mg/ml DNAse and triturated to obtain a cell suspension. This suspension was centrifuged through 35% and 60% Percoll (Pharmacia), and large and small cell fractions were harvested from the upper phase and the 35%/60% interface, respectively. Glial cells were removed from both fractions by sequential platings on poly-D Lysine coated dishes for 30 minutes at 37°C. To evaluate the purity of Purkinje cells in the large cell fraction, a small aliquot was immediately plated on a dish and immunostained with anti-CaBP antibody. This large cell fraction contains approximately 60% of Purkinje cells (not shown). RNAs from cells were extracted using Quiazol (Qiagen) according to the manufacturer’s protocol. Isolated RNA was quantified and analyzed for impurities by ultraviolet spectrometer. Total RNA was reverse transcribed using the SuperScript-II reverse transcriptase (Invitrogen), according to manufacturer’s instructions. qRT-PCR was performed with an MX3000P QPCR System (Stratagene). The PCR primers 5’-AAAGCTGACCCCTTTAGCCTA -3’ and 5’-TTCGGAGTTTCTTGTGATCTTC -3’ were designed to target Shh exon 1. The PCR primers 5’- TCGACCTGCAAACCGTAATCC -3’ and 5’-TCCTAAAGAAGGGCTCATGGTA -3’ targeted the splice junction between Gli1 exons 4 and 5. The PCR primers 5’- CAGAAGTGCGAAGAGGAGGTC-3’ and 5’-TCATCTTAGAGGCCACGAACAT-3’ targeted cyclin D1 exon 2. The housekeeping gene hprt was used to normalize the data. To compare the relative amount of Gli1, cyclinD1 and Shh in wt and hGFAP∷Cre; Kif3afl/- conditional mutant, the Ct values of these genes were divided by the Ct values of hprt in the same sample.
Cerebellar slice cultures
Cerebella from wild-type and mutants were removed aseptically, and cut into 300 μm thick parasagittal sections using a tissue chopper. Slices were cultured on Millicel CM culture inserts (Millipore, Bedford, MA) in 6-well plates containing serum-free medium supplemented with 6 mg/ml glucose, 25 mM potassium chloride in the presence or absence of a biologically active N-terminal fragment of Shh (Shh-N; R&D Systems; 3 μg/ml). After 48 hours, BrdU was added to the medium, and slices were cultured for 16 hours. Slices were then fixed with 4% paraformaldehyde, treated with 2 N HCl followed by 0.1 M borate, and stained overnight with anti-BrdU antibody. The next day, slices were washed and stained with alexa 488-anti-rat antibody, counterstained with DAPI and mounted in Fluoromount G. Staining was visualized and quantified using a fluorescent confocal microscope (DM IRBE, Leica, Nussloch, Germany).
Bromodeoxyuridine (BrdU) administrations
BrdU (Sigma, St. Louis, MO) at 50 μg/g body weight (dissolved in 0.9% saline) was injected 1 hour before sacrifice at E14.5, E16.5, E18.5 or P0. BrdU was injected intraperitoneally into the mother or subcutaneously into the young mice postnatally. For each animal analyzed, the total number of BrdU+ cells in the EGL in each of lobe I to IV and RL region was counted on 3 sagittal sections of comparable mediolateral levels (n=3) using a fluorescent microscope (Zeiss, Germany).
In situ hybridization
Antisense riboprobes were labeled as described previously (Spassky et al., 1998) by in vitro transcription of cDNAs encoding mouse Shh (gift from A. McMahon) or Gli1 (gift from A. Ruiz i Altaba). In situ hybridization was done as described (Spassky et al., 1998).
Terminal deoxynucleotidyl transferase-mediated biotinylated UTP nickend labeling (TUNEL)
Apoptosis was detected by TUNEL assay with the In Situ Cell Death Detection Kit (Roche), according to the manufacturer’s instructions.
Electron microscopy (EM)
P0 mice were killed by intracardial perfusion with 2% PFA and 2.5% glutaraldehyde. Brains were postfixed in 2.5% glutaraldehyde overnight at 4°C, cut sagittaly at 300 μm, and processed for EM as described previously (Doetsch et al., 1997). Briefly, sections were postfixed in 2% osmium tetroxide in 0.1M PB for 2h. Semithin sections, 1.5 μm thick, were cut with a diamond knife and stained with 1% toluidine blue. For the identification of primary cilia, 300 successive ultrathin (0.06 μm) sections were cut with a diamond knife, stained with lead citrate, and examined under a Jeol (Peabody, MA) 100CX electron microscope.
Results
The cerebellar granule cell lineage is derived from hGFAP∷Cre expressing precursors
Recent work has demonstrated that GFAP, an intermediate filament protein previously thought to be present in mature terminally differentiated astroglial cells (Bignami et al., 1972), is also expressed by neural stem cells (Doetsch et al., 1999; Laywell et al., 2000; Garcia et al., 2004). Developmental lineage tracing experiments using the mouse or human GFAP promoter also indicate that a subpopulation of embryonic neural progenitors (Malatesta et al., 2003; Casper and McCarthy, 2006), including precursor cells within the cerebellum (Marino et al., 2000; Zhuo et al., 2001, Kwon et al., 2001), express this intermediate filament protein. We thus tested whether GCPs undergo recombination in mice carrying hGFAP∷Cre. In hGFAP∷Cre mice, Cre immunoreactivity was detected at embryonic day 14.5 (E14.5) in proliferating progenitors in the cerebellar ventricular zone and in the RL (Fig. 1A-B), but not in the EGL. To visualize the progeny of these cells, we crossed the hGFAP∷Cre mice with the R26R or Z/EG reporter mice (Soriano, 1999; Novak et al., 2000). Similar patterns of expression of both reporters were observed at all developmental stages. In E16.5 hGFAP∷Cre; Z/EG embryos, GFP+ cells were found in the RL, EGL and dispersed in the cerebellum, suggesting that they were derived from Cre+ progenitors in the RL and ventricular zone, and migrated to the EGL (Fig. 1C). We confirmed that GFP+ cells in the EGL were GCPs by immunostaining with GCP marker Pax6 (Lin et al., 2001) (Fig. 1D). At all stages examined, GFP+ cells were not immunoreactive for the Purkinje cell marker calbindin D28k16, indicating that these cells are generated before the onset of Cre expression in the cerebellar ventricular zone (Sotelo, 2004; Fig. 1E and data not shown). At postnatal day 15 (P15) hGFAP∷Cre; R26R mice, β-galactosidase activity is restricted to Cre+ Bergmann glia in the Purkinje cell layer (PCL) and to α6+ granule neurons in the IGL (Fig. 1F and data not shown). Taken together, these results showed that hGFAP∷Cre can efficiently recombine genes in GCPs but not in Purkinje cells.
Fig. 1. The granule cell lineage is recombined by the hGFAP∷Cre transgene.
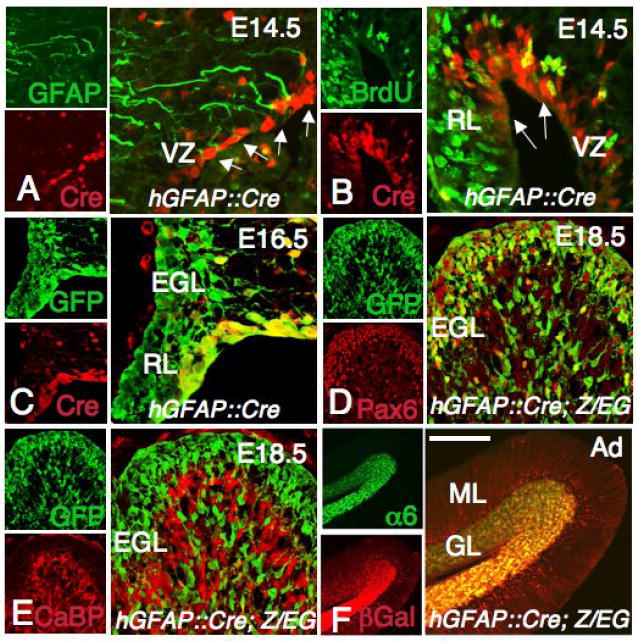
(A-F) Sagittal sections of the cerebellum from a hGFAP∷Cre (A-B), hGFAP∷Cre; Z/EG (C-E) and hGFAP∷Cre; R26R (F) mice at E14.5 (A-B), E16.5 (C), E18.5 (D-E) and adult (Ad, F) stained with the antibodies shown in each panel. (A, B) Cre immunoreactivity is detected in proliferating GFAP+ cells in the ventricular zone (VZ) and the RL (arrows in A, B). (C) At E16.5, Cre+ cells are still present in the ventricular zone and in the RL but are not observed in the EGL, which already contains progeny of Cre+ cells (GFP+). (D, E) At E18.5, Cells derived from Cre+ cells (GFP+) are localized in the EGL and inner layers, and express the GCP marker Pax6 (D), but not the Purkinje cell marker CaBP (E). (F) In the adult, Bergmann glial fibers in the ML and α6+ granule cells are β-galactosidase+. Scale bars: 50 μm (A, B), 30 μm (C), 100 μm (D, E) and 300 μm (F).
Loss of Kif3a in GCPs severely disrupts cerebellar development
During development, Shh expressed in Purkinje cells induce the proliferation of GCPs (Dahmane and Ruiz i Altaba, 1999; Wallace, 1999; Wechsler-Reya and Scott, 1999). Work from the ventral spinal cord and limb development suggests that the primary cilia plays essential roles in Shh signal transduction (Huangfu et al., 2003; Huangfu and Anderson, 2005, Liu et al., 2005; Haycraft et al., 2005; May et al., 2005). To test whether primary cilia in GCPs are required for cerebellar development, we generated knockouts of Kif3a in GCPs by using Cre/LoxP recombination. Kif3a encodes a subunit of kinesin-II, an anterograde IFT motor, which is essential for ciliogenesis (Marszalek et al., 1999). hGFAP∷Cre transgenic mice were crossed with Kif3afl/- and Kif3afl/fl mice to produce conditional Kif3a mutants: hGFAP∷Cre; Kif3afl/- and hGFAP∷Cre; Kif3afl/fl mice. Cre-mediated recombination between the loxP sites results in deletion of exon 2, which introduces a frameshift in the coding region resulting in premature termination of translation. PCR analysis of hGFAP∷Cre; Kif3afl/- progeny showed that the floxed allele was recombined in purified GCPs, resulting in cells with no functional KIF3A (Fig. 2A). Both hGFAP∷Cre; Kif3afl/- and hGFAP∷Cre; Kif3afl/fl mice showed the same phenotype. These mice will be called conditional Kif3a mutants throughout the manuscript.
Fig.2. Lack of Kif3a inhibits ciliogenesis in the EGL.
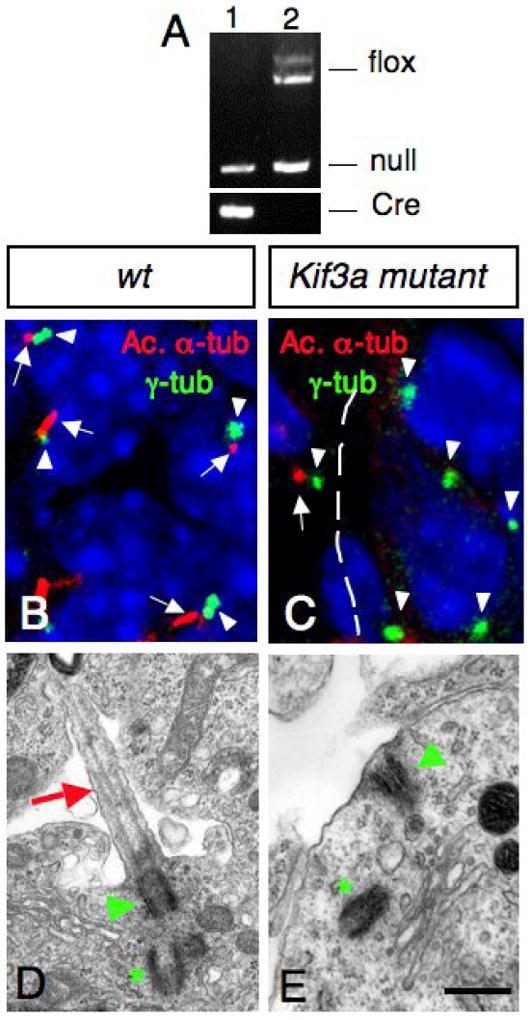
(A) PCR products obtained after amplification of genomic DNA of purified GCPs from hGFAP∷Cre; Kif3afl/- (lane 1) and Kif3afl/- (lane 2) cerebellum. Note that the LoxP band is absent in purified GCPs from hGFAP∷Cre+ mice (lane 1). (B, C) Confocal projections of double immunostaining with an anti-acetylated α-tubulin antibody (red, arrow) and an anti-γ-tubulin antibody (green, arrowhead) on sections of wild type (B) and conditional Kif3a mutant (C) at E18.5. In control animals, most GCPs extend a primary cilium (arrows) from a basal body (arrowheads). These cilia were absent in the EGL in the conditional Kif3a mutant. Basal bodies are still present in the mutant EGL (arrowhead). Note that the mutation does not affect meningeal cells since ciliary staining could still be observed in this structure (arrow in C). (D, E) Electron micrographs of GCPs from wild type and conditional Kif3a mutants at P0. (D) In wild type animals, a primary cilium (red arrow) extends from a basal body (green arrowhead) nearby a centriole (green asterisk). (E) In conditional Kif3a mutant a basal body (green arrowhead) is attached to the cell membrane nearby a centriole (green asterisk), but the primary cilium is absent. 5 μm (B-C), 0.5 μm (D-E).
To confirm that primary cilia were ablated in GCPs in these conditional Kif3a mutant animals, we performed transmission electron microscopy and immunofluorescence analysis using an anti-acetylated α-tubulin antibody to label microtubules within primary cilia, and an anti-γ-tubulin antibody to visualize the basal body. In wild type cerebellum, primary cilia extended from basal bodies in GCPs in the EGL (Supplementary Fig. 1; Fig. 2B, D). In contrast, conditional Kif3a mutants completely lacked primary cilia and only had basal bodies. Nine and ten immunofluorescence fields were sampled to detect primary cilia from wild-type and Kif3a conditional mutants, respectively. In wild-type, primary cilia were detected from all 9 fields and 77 out of 107 basal bodies were associated with a primary cilium at E18.5. In contrast, only 2 out of 102 basal bodies were associated with primary cilium in 10 fields from conditional Kif3a mutants. However, basal bodies were often observed apposed to the cell membrane as in wild type animals, suggesting that the mechanism of basal body anchorage is still functional in conditional Kif3a mutants (Fig. 2C, E; Supplementary Fig. 2). The loss of cilia was specific to the cell lineages expressing hGFAP∷Cre, since neighboring meningeal cells had cilia associated with the basal body (arrow in Fig. 2C). Consistent with hGFAP∷Cre expression pattern, Bergmann glia also lacked primary cilia in conditional Kif3a mutants (data not shown).
At birth, mutant and control pups were indistinguishable. However by P10, the mutants showed ataxia and tremor. Interestingly, these mice spent excessive time digging and grooming themselves. The vast majority of the mutant mice died around P25. In the forebrain, the hGFAP promoter is known to target Cre expression in a subset of radial glial cells from E13.5 (Malatesta et al., 2003). At postnatal stages, hydrocephalus develops in the forebrain of Kif3a mutants, probably due to defects in ependymal ciliary beating leading to a thinner cortex. Although the overall size of the forebrain in the mutant mice was not altered, the cerebellum of these animals was extremely small compared to control littermates (28±2% of wild type size, Fig. 3A-E). The ratio of the cerebellum to forebrain was 0.35 in control brain and 0.1 in conditional Kif3a mutants. A molecular layer (ML), PCL, and IGL were discernable in the severely atrophic P25 mutant cerebella, but there were significant abnormalities with foliation. Folia III, IV and VI, VII were fused (Fig. 3D-E). In the mutant, the total cerebellar area and cell number was significantly reduced. The ML was thinner and contained fewer cells, the PCL was frequently interrupted and Purkinje cell position was not limited to a single row of cells as in the controls (not shown). Instead, in the mutant animals, Purkinje cells formed a thick layer, which is probably a consequence of the reduction in cerebellar surface and reduced number of folia. Indeed, cerebellar growth and foliation are dependent on the expansion of GCPs (Corrales et al., 2006). Consistently, the most dramatic change in the mutant cerebellum from birth was a great reduction in the number of GCPs in the EGL. The defects were already present at birth and became obvious by P5 (Fig. 3F-I). At this age the EGL of mutant cerebellum was much thinner than that of wild-type cerebellum.
Fig. 3. Lack of Kif3a severely disrupts cerebellar development.
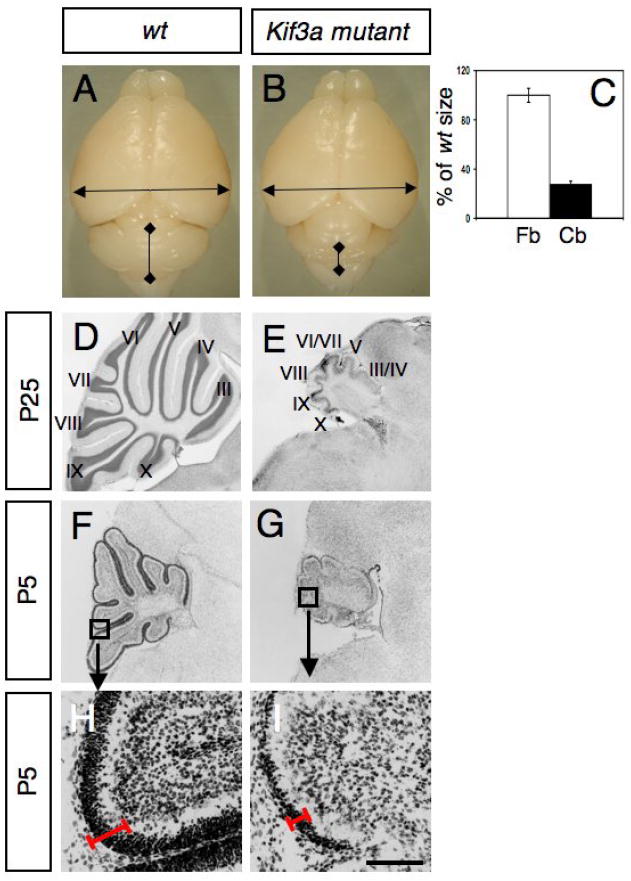
(A-B) Gross appearance of a brain from a wild-type littermate (A) and a conditional Kif3a mutant (B) at P25. (C) The size of the forebrain (Fb) and cerebellum (Cb) from wild-type and conditional Kif3a mutant were measured as indicated by arrows in A, B. Although the forebrain from conditional Kif3a mutants is not significantly different from wild-type, the size of the cerebellum is dramatically reduced. (D-I) Cresyl violet staining of sagittal sections of cerebellum from wild type (D, F, H) and conditional Kif3a mutant (E, G, I) cerebellum at P25 (D-E) and P5 (F-I). (F-I) Lack of Kif3a limits the expansion of the EGL. (H-I) High magnifications of the squares shown in (F,G) illustrate that EGL is thinner in the mutants (red line). Scale bars: 3mm (A, B), 800 μm (D-G), 130 μm (H, I).
This GFAP-driven Cre recombinase is also expressed in Bergmann glia (Fig. 1F and data not shown). Granule neurons migrate to the IGL along Bergmann glia fibers (Yacubova and Komuro, 2003), and Bergmann glia differentiation defects leads to layering defects including severe granule neuron migration defects and abnormal laminar formation (Yue et al., 2005). Although Bergmann glial fibers were slightly deformed and disorganized in conditional Kif3a mutants compared with the ordered and linear morphology of wild-type Bergmann glial fibers, they expressed the late marker GFAP and their glial fibers extend to the pial surface suggesting that Bergmann glia differentiation proceeds quite far (Supplementary Fig. 3). Moreover, no migratory defects of granule neurons or abnormal laminar formation were detected in conditional Kif3a mutants (Fig. 3), indicating that the phenotype observed is not due to Bergmann glia differentiation defects.
Loss of Kif3a results in GCP proliferation defects
The observed reduction of GCPs in conditional Kif3a mutants could be due to a decreased proliferation or an increase in the number of cells undergoing apoptosis. To test the latter possibility, we first analyzed P0 cerebellum using transmission EM. 300 ultrathin serial sections from control and mutant EGL were carefully studied and the number of picnotic cells counted. No significant increase in the number of cell death could be detected in the mutant compared to wild-type (less than 1% of cells in both animals, data not shown). To confirm these results, we stained sections from control and mutant E18 and P0 cerebellum for TUNEL and activated caspase 3. The number of TUNEL+ or activated caspase 3+ cells in the cerebellum at these developmental stages was very similar in controls and conditional Kif3a mice: 10±4 TUNEL+ (or activated caspase+) cells per section in controls (n=5) and 8±3 per section in conditional mutants (n=6) (Supplementary Fig. 4 and data not shown). This suggests that increased cell death cannot account for the severe reduction in the number of granule cells observed in the mutant mice.
To test if primary cilia are required for the early proliferation of GCPs, the number of cells incorporating BrdU was examined at E14.5 and 16.5 in control and conditional Kif3a mutant cerebella. BrdU+ cells were concentrated in the RL and the EGL and scattered in the inner layers of the cerebellum. Mutant mice showed no significant difference in the distribution or the number of BrdU+ cells at these early ages (Fig. 4A-C and data not shown). In contrast, at E18.5, conditional Kif3a mutant cerebella exhibited a striking reduction in the number of proliferating GCPs as determined by the extent of BrdU incorporation in the EGL (Fig. 4D-F). We counted the number of BrdU+ GCPs in five regions along the AP axis of conditional Kif3a mutants and control cerebellum (see Materials and Methods and Fig. 4D). In rostral areas indicated schematically by regions I and II in Fig. 4D-E, where Shh is first expressed (Corrales et al., 2004; Lewis et al., 2004), the number of BrdU+cells was significantly reduced in conditional Kif3a mutants. In more posterior regions (III, IV) of the cerebellum and in the RL the number of BrdU+ cells was only slightly reduced in conditional Kif3a mutant animals (Fig.4F) at this stage. At P0, when Shh is expressed in all rostrocaudal regions of the cerebellum, the number of BrdU+cells was significantly decreased in all regions of the EGL (data not shown). These results suggest that loss of Kif3a results in a dramatic reduction in the proliferation of GCPs and that it coincides in time and space with the pattern of Shh expression.
Fig. 4. Abnormal proliferation of GCPs in conditional Kif3a and Ftm mutant mice.
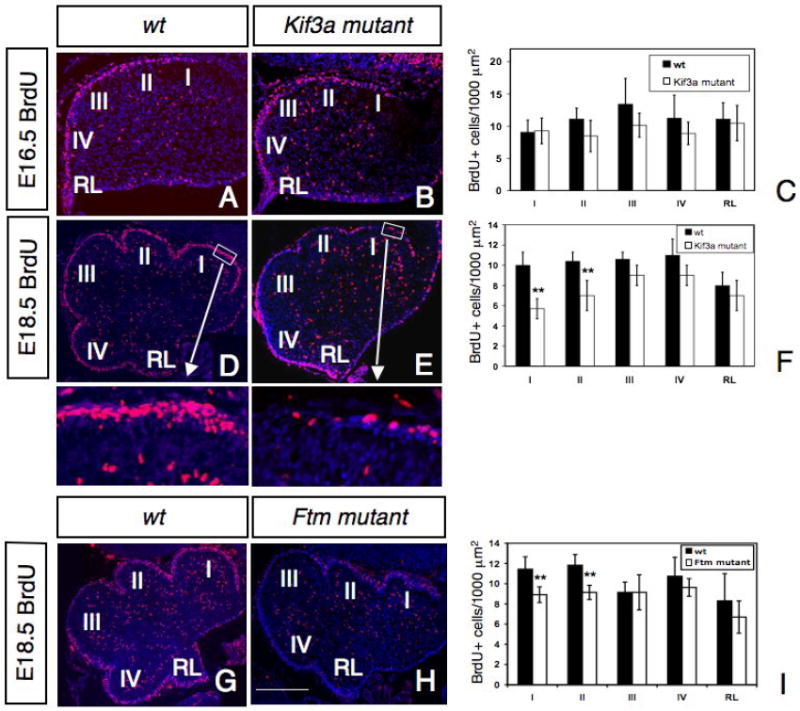
BrdU staining of a control (A, D, G), a conditional Kif3a mutant (B, E) and a Ftm mutant (H) cerebellum at E16.5 (A-B) and E18.5 (D-E, G-H). Rostral is to the right. Squares in D, E are shown at higher magnifications below. (C, F, I) Quantification of BrdU+ cells per 1000 μm2 in regions I to IV and in the RL. Note that at E18.5, the number of BrdU+ cells is much smaller in the rostral part of the cerebellum in conditional Kif3a and Ftm mutants compared to wild type, while no significant difference was observed in caudal regions of conditional Kif3a and Ftm mutants. The BrdU+ cells were counted from at least three sections from each mouse at comparable mediolateral levels. Data from three mice per group were pooled for statistical analysis with Student’s t-test. **: p<0.01. Scale bars: 100 μm (A, B), 200 μm (D-E, G-H).
Loss of Ftm results in GCP proliferation defects
To provide further evidence for the defective proliferation of GCPs in the conditional Kif3a mutants is due to the loss of cilia, we next analyzed the proliferation of GCPs in another ciliary mutant, Fantom (Ftm, also called nephrocystin-8 or Rpgrip1l) mutant mice. Ftm encodes a basal body protein and mutations in this gene cause ciliary disorders including Joubert and Meckel syndromes. Consistently, mutant mice for this gene show defective ciliogenesis and importantly, cerebellar hypoplasia (Delous et al., 2007; Vierkotten et al., 2007). At E18.5, BrdU proliferation was significantly reduced in rostral regions I and II (Fig. 4G-I). The reduced proliferation of GCPs observed in Ftm mutant mice in rostral regions of the cerebellum is consistent with our results from Kif3a mutant mice, suggesting that primary cilia play an important role in the proliferation of GCPs.
Loss of Kif3a in GCP disrupts the expression of Shh target genes
The above results suggested that loss of Kif3a in GCPs could cause defective Shh signaling in these cells. In order to examine how this pathway is affected, we investigated if the patterns and levels of expression of Shh and its target gene, Gli1, were altered using in situ hybridization and qRT-PCR. At P2, Shh was expressed in a deep layer corresponding to the presumptive PCL (Fig. 5A). This pattern of staining is similar to that described previously (Dahmane and Ruiz I Altaba, 1999; Corrales et al., 2004; Lewis et al., 2004). A similar pattern of expression was detected in the conditional Kif3a mutant cerebellum, although its expression seemed more pronounced (Fig. 5B). To quantify the expression of Shh mRNA in Purkinje cells at P2-4, qRT-PCR was performed using RNA prepared from a large cell fraction, which is enriched with Purkinje cells, isolated from control and conditional Kif3a mutant cerebella (see Materials and Methods). The level of Shh mRNA in a large cell fraction isolated from Kif3a mutant cerebellum was increased to 400-450% of that found in wild-type (Fig. 5C), indicating that the lack of cilia in GCPs increases the level of expression of Shh although it does not affect the pattern of expression of Shh in the developing cerebellum. Although approximately 60% of the large cell fraction is Purkinje cells, it was shown that Shh is not expressed by Bergmann glia and is restricted to Purkinje cells (Dahmane and Ruiz i Altaba, 1999; Corrales et al., 2004; Lewis et al., 2004), suggesting that the level of Shh mRNA expression in Purkinje cells in Kif3a mutant is increased compared to controls. To determine whether Shh signaling was disrupted in conditional Kif3a mutants, the expression of Gli1, a Shh downstream target gene, was analyzed in P2-4 conditional Kif3a mutants and control animals. As previously described, Gli1 was detected by in situ hybridization in the EGL and in some deeper cells in the anterior region of the control cerebellum, corresponding to developing Bergmann glia (Fig. 5D; Corrales et al., 2004). In contrast, very low or no expression of Gli1 could be detected in the mutant GCPs by in situ hybridization or qRT-PCR (Fig. 5E-F). Shh mediates GCP proliferation via cyclin D1, as cyclin D1 mRNA is also upregulated in response to Shh in GCPs (Kenney and Rowitch, 2000). We therefore compared the expression of cyclin D1 in the control and mutant GCPs using qRT-PCR. Although Shh was upregulated in Purkinje cells of the mutants, the level of cyclin D1 expression in mutant GCPs was reduced by 50% compared to that found in wild-type GCPs (Fig. 5G). These findings indicate that while Shh was expressed in the cerebellum at the right time and place, the downstream genes, Gli1 and cyclin D1, were dramatically down-regulated. These findings suggest that Shh target genes fail to be expressed in GCPs in the absence of primary cilia.
Fig. 5. Abnormal expression of Shh and Shh target genes, Gli1 and cyclin D1, in conditional Kif3a mutants.
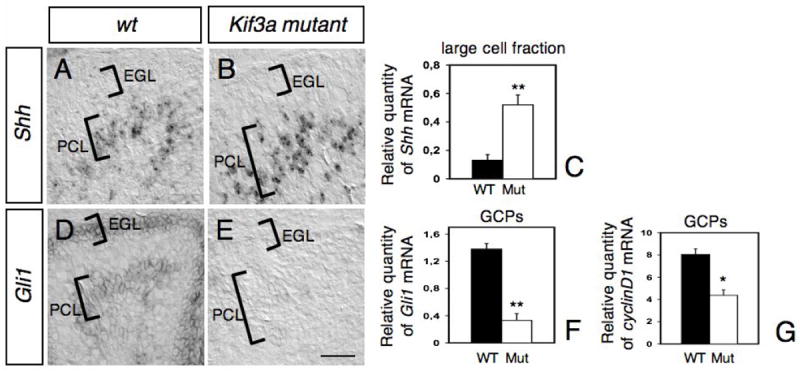
In situ hybridization on sagittal sections of wild type (A, D) and mutant (B, E) cerebellum at P2 with antisense riboprobes specific for Shh (A-B) and Gli1 (D-E). The location of the EGL and PCL are indicated on each panel. Note that whereas Shh expression is increased in the PCL of mutant compared to wild-type (A-C), Gli1 was no longer detected neither in the EGL nor the PCL in the mutant (E). (C, F-G) Levels of Shh mRNAs in the large cell fraction enriched in Purkinje cells (C), and levels of Gli1 (F) and cyclin D1 mRNAs in GCPs (G) at P2-4 were evaluated by semi-quantitative real-time RT-PCR and are shown as the relative quantities normalized to the level of hprt mRNA expression. The level of Shh mRNA in conditional Kif3a mutant is significantly higher than that in the wild-type, whereas the levels of Gli1 and cyclin D1 mRNAs in the mutant are significantly lower than those in the wild-type. Each column and the vertical line represent the mean± SEM. Data from three mice per group were pooled for statistical analysis with Student’s t-test. *: p<0.05; **: p<0.01. Scale bar: 80 μm.
Kif3a is required for Shh-induced proliferation of GCPs
The phenotype, timing of the defect and expression of Shh downstream genes, Gli1 and cyclin D1, in the conditional Kif3a mutant mice, suggest that these mice have a defect in Shh induced proliferation in GCPs. To test if primary cilia in GCPs are required for Shh-induced proliferation, we examined the effect of the biologically active N-terminal fragment of Shh (Shh-N) on cerebellar slice cultures (Weschler-Reya and Scott, 1999). Slices of P3 cerebellum from conditional Kif3a mutant and control littermates were cultured for 48 hours in the presence or absence of Shh-N. Proliferating cells in these slices were labeled with BrdU and quantified from single optical slices through the EGL using a confocal microscope by counting the percentage of BrdU+ cells per DAPI total cells. EGL boundaries were determined by BrdU and DAPI staining on each section. In control slices from wild type mice, exposure to Shh-N resulted in a 10-fold increase in the number of BrdU+ cells compared to non-treated control slices (from 8±6% to 87±5% of DAPI-positive cells after Shh-N exposure) (Fig. 6A, C, E). In contrast, no significant difference was observed in the number of BrdU+ cells after exposure of mutant slices to Shh-N (from 10±4% to 13±7% after Shh-N exposure) (Fig. 6B, D, E).
Fig. 6. Abnormal Shh-induced proliferation of GCPs in cerebellar cultures from conditional Kif3a mutants.
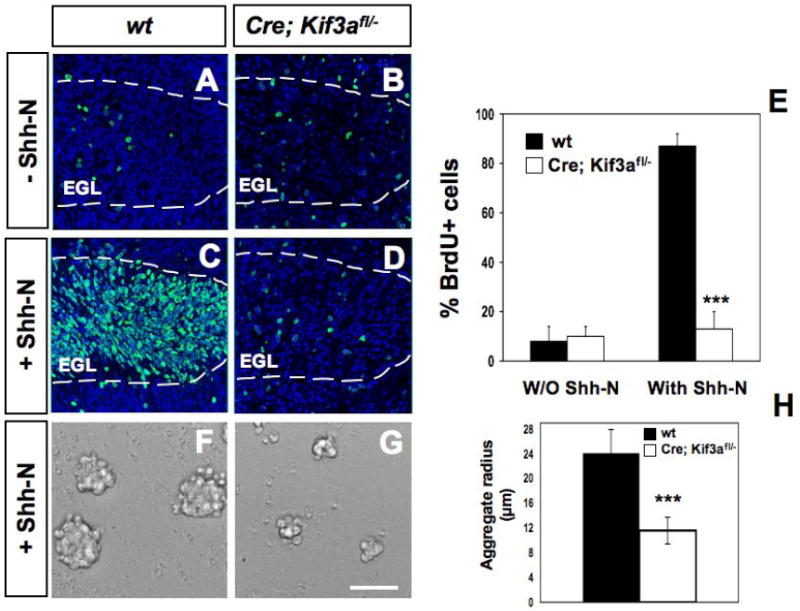
BrdU staining on cerebella sagittal slices from P3 wild type (A, C) and mutant mice (B, D) cultured with no stimulus (A-B) or 3 μg/ml Shh-N (C-D). BrdU staining (green) was counterstained with DAPI (blue) and visualized using confocal microscopy. (E) Quantification of the percentage of BrdU+ and DAPI+ nuclei in wild type and conditional Kif3a mutant slices with and without Shh-N. (F-G) Proliferation of isolated GCPs in aggregates obtained from P1 Kif3a mutants (G) compared to controls (F) with Shh-N for 48h. (H) Quantification of aggregate radius in wild type and conditional Kif3a mutants after 48h. Data from three mice per group were pooled for statistical analysis with Student’s t-test. ***: p<0.001. Scale bar: 100 μm.
To further test whether loss of Kif3a in GCPs cause cell-autonomous proliferation defects in response to Shh, GCPs were isolated from the cerebella of P3-5 Kif3a mutant and control mice (see Methods) and were allowed to form aggregates in vitro as previously described (Dahmane and Ruiz I Altaba, 1999). Addition of Shh to the serum-free culture media for 48h increased the size of the control aggregates by 2-fold on average, compared to Kif3a mutant (Fig. 6F-H). Thus, these observations strongly suggest that Kif3a is required for GCPs proliferation in response to Shh.
Loss of Smo in GCPs mimics the phenotype of Kif3a deficient mice
If the phenotype observed in the conditional Kif3a mutants is due to defective Shh signaling, we should expect a similar phenotype in conditional Smo mutants. hGFAP∷Cre transgenic mice were crossed to Smofl/fl mice (Long et al., 2001) to generate hGFAP∷Cre; Smofl/fl mutant mice (called conditional Smo mutants). Conditional Smo mutants showed ataxia and tremor similar to what we observed in the conditional Kif3a mutants. The overall size of P21 conditional Smo mutant brains was slightly smaller than wild-type brains (not shown). However, the cerebellum showed a pronounced reduction in size similar to that observed in the conditional Kif3a mutant. As the conditional Kif3a mice, the conditional Smo mutant mice had the basic layered cytoarchitecture of the cerebellum, but showed more severe defects in foliation (Fig. 7A, B). Although Shh signaling cascade was disrupted in conditional Smo mutant mice as no expression of Gli1 could be detected by in situ hybridization (not shown), primary cilia were still present in the GCPs (Fig. 7D). It has been shown that the extent of cerebellar foliation correlates with the level of Shh signaling (Corrales et al., 2006). Our results suggest that loss of Smo in GCPs results in a similar, but more severe cerebellar foliation defects compared to Kif3a mutation.
Fig.7. Abnormal proliferation of GCPs in conditional Smo mutant and conditional Kif3a and Smo double mutant mice.
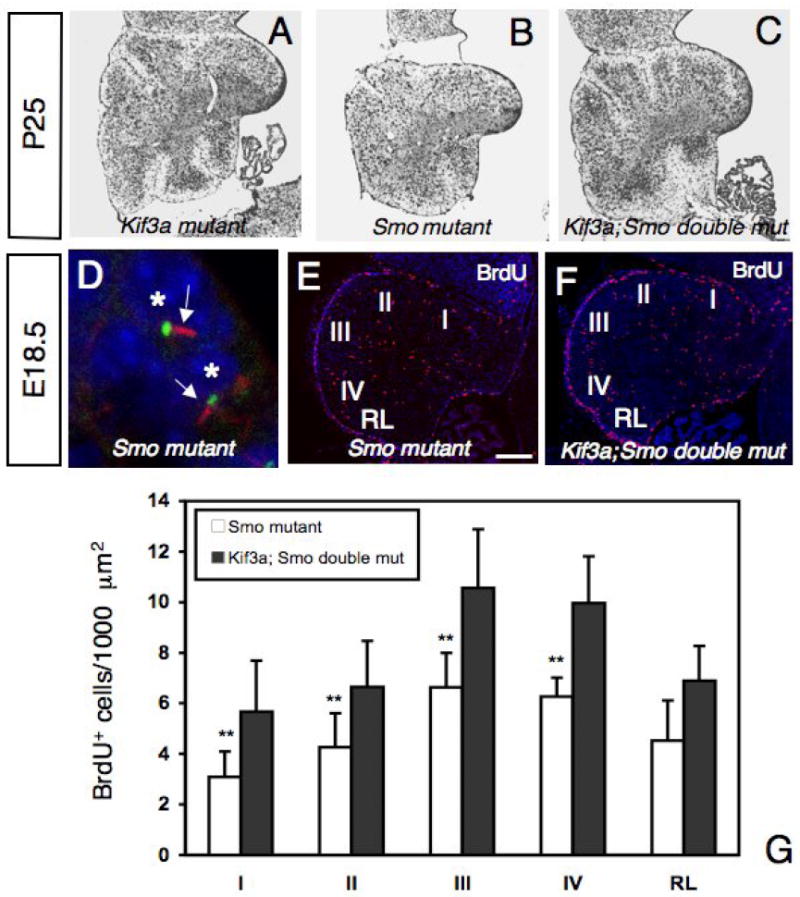
(A-C) Cresyl violet staining of sagittal sections of cerebellum from conditional Kif3a mutant (A), conditional Smo mutant (B), and conditional Kif3a and Smo double mutant cerebellum at P25. Rostral is to the right. (D) Double immunostaining with an anti-acetylated α-tubulin antibody (red) and an anti-γ-tubulin antibody (green) on sections of conditional Smo mutant at E18.5. In conditional Smo mutants, most GCPs extend a primary cilium (arrow) from a basal body. Each nucleus associated with a primary cilium is indicated by an asterisk. (E-F) BrdU staining of a conditional Smo mutant (E) and a conditional Kif3a and Smo double mutant (F) cerebellum at E18.5. Rostral is to the right. (G) Quantification of BrdU+ cells per 1000 μm2 in regions I to IV and in the RL. Note that the number of BrdU+ cells is much smaller in conditional Smo mutants than in conditional Kif3a and Smo double mutants, while no significant difference was observed between conditional Kif3a mutants and conditional Kif3a and Smo double mutants (compare with Fig.4F). The BrdU+ cells were counted from at least three sections from each mouse at comparable mediolateral levels. Data from three mice per group were pooled for statistical analysis with Student’s t-test. **: p<0.01. Scale bar: 230 μm (A-C), 2 μm (D) and 100 μm (E-F).
Kif3a is epistatic to Smo in cerebellar development
The more severe defects in the cerebellar development in conditional Smo mutants than in conditional Kif3a mutants suggested that some Shh signaling persisted in the absence of primary cilia. If this was the case, the removal of both Kif3a and Smo should result in a phenotype similar to that observed in conditional Smo mutants. However, at P25 the conditional Kif3a and Smo double mutant had a cerebellum strikingly similar to that of the conditional Kif3a mutant. In contrast to severe loss of foliation in conditional Smo mutants, the cerebellum of double mutants still retained rudimentary folia like in conditional Kif3a mutants (Fig. 7A-C). To further investigate the differences in the conditional Kif3a mutants, conditional Smo mutants and conditional Kif3a and Smo double mutant mice, we compared the proliferation of GCPs at E18.5. We counted the number of BrdU+ GCPs in five regions along the AP axis of mutant cerebellum. The number of BrdU+ cells in conditional Smo mutants was reduced in all five cerebellar regions studied. In contrast, in conditional Kif3a mutants the reduction was less severe in the posterior two lobes (compare Fig. 4E, F with Fig.7E, G). Consistent with the level of foliation, in the conditional Kif3a and Smo double mutants, the number of BrdU+ cells was significantly increased compared to conditional Smo mutants and comparable to the level observed in conditional Kif3a mutants (Compare Fig. 4E, F with Fig.7). Thus, Kif3a is epistatic to Smo in cerebellar development. Altogether, our study strongly suggests that IFT protein KIF3A is required for GCPs proliferation in response to Shh and acts downstream of Smo.
Discussion
In this study, we have examined the role of primary cilia in cerebellar development and specifically on GCP proliferation. We utilized Cre recombinase under the control of the hGFAP promoter to generate conditional mutant mice for Kif3a and/or Smo in GCPs. We observed a severe cerebellar defect due to reduced proliferation of neuronal progenitors in these conditional mutants. Interfering with intraflagellar transport in GCPs severely disrupts their Shh-induced proliferation. The defects were similar to those observed when Smo is inactivated in this same lineage. The study therefore suggests a critical role for primary cilia in cerebellar development and a possible explanation for the hypoplasia of cerebellar vermis found in Joubert and Bardet-Biedl syndromes associated with defective primary cilia (Louie and Gleeson, 2005).
The proliferation defect of GCPs is cell-autonomous
We report here that hGFAP-driven expression of Cre is detected at E14.5 in precursor cells located in the VZ and RL of the developing cerebellum. Similar results were obtained using the endogenous mouse GFAP promoter, suggesting that these precursor cells express GFAP very early during cerebellar development (Delaney et al., 1996; Marino et al., 2000; Kwon et al., 2001). We traced the fate of these cells using conditional reporter mice and found that progenitors expressing hGFAP-driven Cre produce granule neurons. The present report shows that the granule cell lineage can be effectively targeted using hGFAP∷Cre mice.
Primary cilia were absent in GCPs in conditional Kif3a mutants but were present in the neighboring meningeal cells, supporting the specific requirement of primary cilia in GCPs. In addition, we show that inhibition of ciliogenesis in GCPs from E14.5 does not lead to any cerebellar defects until the time between E16.5 and E18.5, suggesting that the initial specification of GCPs is not dependent on the presence of the primary cilia. In contrast, significantly less proliferating cells were counted in the EGL from E18.5, suggesting that GCPs require primary cilia for the extensive expansion to make the large number of cerebellar granule cells. Conditional ablation of Smo in the same population of hGFAP expressing cells showed similar phenotypes to Kif3a mutants, and cilia are present in GCPs of conditional Smo mutants, suggesting that the absence of cilia in Kif3a mutants is not secondary to the cerebellar defects itself. hGFAP promoter target gene expression to Bergmann glia in addition to GCPs. We therefore cannot exclude that the observed disruption of cerebellar development could be due in part to loss of Kif3a or Smo in these cells. However, the proliferation defects observed in isolated GCPs from Kif3a mutants compared to controls provide an additional argument in favor of a cell-autonomous proliferation defect of GCPs in Kif3a mutants. Recent work suggests that the kinesin-II complex may also have a function in intracellular trafficking and mitosis (Brown et al., 2005; Teng et al., 2005; Haraguchi et al., 2006). However, the distributions of cell-cell adhesion molecules such as N-cadherin and β-catenin appeared normal in GCPs from Kif3a mutants (Supplementary Fig. 5). Moreover, we did not observe an abnormal cytokinesis nor an increase in cell death at P0, which would occur as a consequence of defective mitosis (Supplementary Fig. 4). Instead, multiple experimental evidence strongly suggest that kinesin-II has a prominent function in ciliogenesis (Kozminski et al., 1995; Morris and Scholey, 1997; Nonaka et al., 1998; Takeda et al., 1999; Marszalek et al., 1999; Sarpal et al., 2003; Snow et al., 2004). Similarly, the GCPs in the conditional Kif3a mutants showed normal proliferation at E16.5 and defects begin to manifest at E18.5 coinciding with the initiation of Shh-dependent expansion of the GCPs. Our double mutant analyses also showed that Kif3a act downstream of Smo, which is not likely to occur if Kif3a is required for general cytokinesis or intracellular trafficking in GCPs. Furthermore, defective proliferation of GCP was also observed in other ciliary mutants, Ift88 (Chizikoff et al., 2007) and Ftm mutants (this study). Thus, the simplest interpretation of the phenotypes observed in Kif3a mutants is that Kif3a is required to make primary cilia to mediate Shh signaling in GCPs. Nonetheless, we cannot completely rule out the possibility that the cerebellar phenotype observed in Kif3a conditional mutants could be due to other possible roles of Kif3a since it is also shown to localize to axons (Kondo et al., 1994) and synapses (Muresan et al., 1999). It is also possible that signaling other than Shh signaling is also affected in our conditional Kif3a mutants since 5-HT, Sst3, and PDGF receptors were also shown to localize to primary cilia (for review, see Marshall and Nonaka, 2006) although the significance of this localization in vivo is still lacking.
Requirement of cilia for Shh signal transduction
It was recently demonstrated that primary cilia play an essential role in Shh signaling. Defective primary cilia cause diverse defects including neural tube patterning defects and polydactyly, which resemble those occur as a consequence of Shh signaling defects (Huangfu et al., 2003; Huangfu and Anderson, 2005; Liu et al., 2005; Haycraft et al., 2005; May et al., 2005). Consistently, Smo localizes to primary cilia in response to Shh, and IFT function and primary cilia are required for Smo activity (Corbit et al., 2005; May et al., 2005).
In this study, we provide the first evidence that the primary cilium is essential for Shh-induced proliferation of GCPs. Defects in proliferation were first detected in conditional Kif3a mutants around E18.5, which coincides with the first expression of Shh in Purkinje cells (Corrales et al., 2004; Lewis et al., 2004). Inhibition of ciliogenesis in GCP leads to a dramatic decrease in two positive readouts of Shh activity, the expression of Gli1 and cyclin D1 mRNA, in these cells. Consistently, although Shh elicited a proliferative response in isolated GCPs and GCPs in slices made from wild-type, it was unable to induce GCP proliferation from conditional Kif3a mutants. Interestingly, Shh expression was increased in the large cell fraction enriched in Purkinje cells in conditional Kif3a mutants compared with controls. Although the large cell fraction might contain other cell types like Bergmann glia or astrocytes, these cells do not express Shh (Dahmane and Ruiz I Altaba, 1999; Lewis et al., 2004). These results thus suggest that Shh expression by Purkinje cells might be in part regulated by GCPs. The transcription factors acting upstream in the pathway that control Shh expression in the cerebellum are unknown, but six Shh enhancers that control Shh expression in the ventral neural tube have recently been identified (Jeong et al., 2006). The level of Shh signaling is directly linked to the complexity of the cerebellar foliation pattern (Corrales et al., 2006). Furthermore, inappropriate Shh pathway activation in the cerebellum can cause medulloblastomas (Goodrich et al., 1997; Oliver et al., 2005) and the inhibition of the Shh signaling pathway can arrest the growth of a number of brain tumors (Romer et al., 2004; Sanchez and Ruiz i Altaba, 2005). For all these reasons it is crucial to better understand how Shh expression in Purkinje cells is regulated. Our observations suggest that Purkinje cells increase Shh production when the number of GCPs is reduced.
In the absence of Smo, the rate of proliferation of GCPs at E18.5 is reduced significantly more than that in conditional Kif3a mutants. The cerebellar foliation in conditional Smo mutants were also more severely disrupted than that in conditional Kif3a mutants. Previous studies in spinal cord and limb developments showed that both activator and repressor functions of Gli proteins require primary cilia (Haycraft et al., 2005; Huangfu et al., 2005; Liu et al., 2005; May et al., 2005). Thus, less Gli repressor activity in conditional Kif3a mutants than in conditional Smo mutants could result in less severe phenotype in conditional Kif3a mutants. It was recently shown that removing one copy of Gli3 in Shh null mutants partially rescued the structure of the cerebellum at E18.5 (Blaess et al., 2006). Alternatively, the less severe phenotype in conditional Kif3a mutants could be due to some Shh signaling occurring independently of primary cilia. In Drosophila, where sensory neuron and sperm are the only ciliated cells, mutations disrupting ciliogenesis do not affect Shh signaling (Han et al., 2003; Sarpal et al., 2003). We tested these two possibilities by examining the phenotype of the conditional Kif3a and Smo double mutant. This double mutant showed a very similar phenotype to that of the conditional Kif3a mutant, thus supporting the first possibility. We, therefore, suggest that in conditional Smo mutants, a cilia dependent-repressor activity (probably Gli3) remains, leading to a more severe proliferation defect, whereas in conditional Kif3a mutants or conditional Kif3a and Smo double mutants, the loss of repressor (Gli3) as well as activators (Gli1, 2) leads to a less severe proliferation defects. It would be interesting to directly address this question by comparing Gli3 processing in GCPs from Kif3a, Smo and Kif3a and Smo double mutants.
Connection between cilia and Joubert syndrome
Conditional Kif3a mutant mice display cerebellar vermis hypoplasia very similar to that observed in Joubert syndrome and in Bardet-Biedl syndrome (Louie and Gleeson, 2005). In these patients, a diminished density of granule neurons has been reported, which could be due to proliferation defects of GCPs (Joubert et al., 1969). Genetic analysis of these patients has identified mutations in 3 families of genes (nephrocystin, Bardet-Biedl syndrome and AHI1), several of which encodes proteins that have been localized to cilia and/or implicated in ciliary function and assembly. In addition, some of these genes are expressed in the cerebellum and are required for cerebellar development. First, the AHI1 gene encoding the Jouberin protein, whose function is still unknown, is expressed at a maximum level in the mouse cerebellum at birth (Dixon-Salazar et al., 2004). Structural studies of the Jouberin protein suggest that it may interact with nephrocystin-1 protein, which is localized to primary cilia (Otto et al., 2003). Second, the nephrocystin-6 gene is first expressed around E18 and is superimposable to the Gli1 expression pattern in the developing murine cerebellum (Valente et al., 2006). Moreover, nephrocystin-6 protein is localized to the centrosome and to the base of cilia, and abrogation of its function in zebrafish recapitulates the cerebellar phenotype of Joubert syndrome (Sayer et al., 2006; Valente et al., 2006). Finally, the nephrocystin-8 gene (also called Rpgrip1l or Fantom) has recently been identified to be mutated in Joubert syndrome (Delous et al., 2007; Arts et al., 2007). Most importantly, mutant mice in which nephrocystin-8 gene has been inactivated showed cerebellar hypoplasia and defects in GCPs proliferation similar to Kif3a mutants (Delous et al., 2007; this study).
These results are consistent with our findings that cilia play a crucial role in cerebellar development and specifically in Shh-induced proliferation of GCPs. To further understand the pathogenesis in Joubert syndrome, it would be interesting to test whether AHI1, nephrocystin-6 and nephrocystin-8 gene expression are controlled by Shh signaling and/or how these proteins interact with downstream effectors of Shh signaling pathway in primary cilia. It remains intriguing why key signaling processes, as those mediated by Shh, require this minute hair-like extension from the surface of the cells. Primary cilia may act as an antenna (Singla and Reiter, 2006) to concentrate relevant receptors on a small surface of the cell, thereby allowing signaling to reach the threshold required for activation of signal transduction. Placing receptors and signaling components in a thin protrusion at one point in the cell may help the cells extend its reach into the extracellular space, and orient the cellular response with respect to an external stimulus. Recent work highlights the importance of primary cilia in multiple processes including cell specification in CNS. The present work extends this view by showing how this primary cilium is required for the proliferation of an important neuronal progenitor and the development of an entire brain structure.
Supplementary Material
Low magnifications of confocal projections of double immunostaining with an anti-acetylated α-tubulin antibody (red) and an anti-γ-tubulin antibody (green) on sagittal sections of wild type (A, C) and conditional Kif3a mutant (B, D) at E18.5. White square in A, B shows the portion of the EGL that is magnified in C, D. White square in C, D shows which portion of the EGL is magnified in Fig. 2. Scale bar : 310 μm (A, B), 17 μm (C, D).
Serial sections of mutant GCPs at P0, which spans the entire thickness of a basal body, show the lack of primary cilia. The basal body (arrowhead) lacking the primary cilium is anchored to the cell membrane. A centriole (asterisk) is attached to the basal body. Scale bar: 0.2 μm.
GFAP immunostaining on cerebella sagittal cryosections from P5 wild type (A) and conditional Kif3a mutant (B) mice. Although glial fibers appear slightly disorganized in mutants compared to wild type, these cells are GFAP+ and their processes extend toward the pial surface. Scale bar: 20 μm.
Tunel staining of a control (A) and a conditional Kif3a mutant (B) on sagittal sections of cerebellum at P0. Scale bar: 200 μm.
Immunofluorescence of cerebellar sagittal sections from E18.5 wild type (A, C) and conditional Kif3a mutant (B, D) mice with anti-N-cadherin and anti-β-catenin antibodies. Scale bar: 20 μm.
Acknowledgments
This work was supported by NIH grant NS 028478 to A.A-B, by Human Frontier Science Program and Agence Nationale de la Recherche to N.S, and Mark Linder/American Brain Tumor Association Fellowship to Y.-G. H. SP2 confocal microscopy at DERC Microscopy and Imaging Core at UCSF was supported by NIH grant P30 DK063720. The authors would also like to acknowledge John and Frances Bowes for their support of this research. We are grateful to L.S. Goldstein for providing us with the Kif3a conditional mutant mice, A. Messing for providing us with the hGFAP-Cre mice and J. Reiter for providing us with the Smo conditional mutant mice. We thank Cynthia Yaschine for editorial comments and Rebecca Ihrie, Marion Wassef and Bernard Zalc for critical reading of the manuscript and support of this work. We also thank all members of Alvarez-Buylla laboratory for insightful and stimulating discussions.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Altman J, Bayer SA. Development of the Cerebellar system: In relation to its evolution, Structure, and Functions. New York, NY: CRC Press; 1997. [Google Scholar]
- Alvarez-Buylla A, Garcia-Verdugo JM, Mateo AS, Merchant-Larios H. Primary neural precursors and intermitotic nuclear migration in the ventricular zone of adult canaries. J Neurosci. 1998;18:1020–37. doi: 10.1523/JNEUROSCI.18-03-01020.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arts HH, Doherty D, van Beersum SE, Parisi MA, Letteboer SJ, Gorden NT, Peters TA, Marker T, Voesenek K, Kartono A, Ozyurek H, Farin FM, Kroes HY, Wolfrum U, Brunner HG, Cremers FP, Glass IA, Knoers NV, Roepman R. Mutations in the gene encoding the basal body protein RPGRIP1L, a nephrocystin-4 interactor, cause Joubert syndrome. Nat Genet. 2007;39:882–8. doi: 10.1038/ng2069. [DOI] [PubMed] [Google Scholar]
- Baptista CA, Hatten ME, Blazeski R, Mason CA. Cell-cell interactions influence survival and differentiation of purified Purkinje cells in vitro. Neuron. 1994;12(2):243–60. doi: 10.1016/0896-6273(94)90268-2. [DOI] [PubMed] [Google Scholar]
- Bignami A, Eng LF, Dahl D, Uyeda CT. Localization of the glial fibrillary acidic protein in astrocytes by immunofluorescence. Brain Res. 1972;43:429–35. doi: 10.1016/0006-8993(72)90398-8. [DOI] [PubMed] [Google Scholar]
- Blaess S, Corrales JD, Joyner AL. Sonic hedgehog regulates Gli activator and repressor functions with spatial and temporal precision in the mid/hindbrain region. Development. 2006;133:1799–1809. doi: 10.1242/dev.02339. [DOI] [PubMed] [Google Scholar]
- Briscoe J, Sussel L, Serup P, Hartigan-O’Connor D, Jessell TM, Rubenstein JL, Ericson J. Homeobox gene Nkx2.2 and specification of neuronal identity by graded Sonic hedgehog signalling. Nature. 1999;398:622–7. doi: 10.1038/19315. [DOI] [PubMed] [Google Scholar]
- Brown CL, Maier KC, Stauber T, Ginkel LM, Wordeman L, Vernos I, Schroer TA. Kinesin-2 is a motor for late endosomes and lysosomes. Traffic. 2005;6:1114–24. doi: 10.1111/j.1600-0854.2005.00347.x. [DOI] [PubMed] [Google Scholar]
- Casper KB, McCarthy KD. GFAP-positive progenitor cells produce neurons and oligodendrocytes throughout the CNS. Mol Cell Neurosci. 2006;31:676–84. doi: 10.1016/j.mcn.2005.12.006. [DOI] [PubMed] [Google Scholar]
- Chiang C, Litingtung Y, Lee E, Young KE, Corden JL, Westphal H, Beachy PA. Cyclopia and defective axial patterning in mice lacking Sonic hedgehog gene function. Nature. 1996;383:407–13. doi: 10.1038/383407a0. [DOI] [PubMed] [Google Scholar]
- Ciemerych MA, Kenney AM, Sicinska E, Kalaszczynska I, Bronson RT, Rowitch DH, Gardner H, Sicinski P. Development of mice expressing a single D-type cyclin. Genes Dev. 2002;16:3277–89. doi: 10.1101/gad.1023602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cohen E, Binet S, Meininger V. Ciliogenesis and centriole formation in the mouse embryonic nervous system. An ultrastructural analysis. Biol Cell. 1988;62:165–9. [PubMed] [Google Scholar]
- Corbit KC, Aanstad P, Singla V, Norman AR, Stainier DY, Reiter JF. Vertebrate Smoothened functions at the primary cilium. Nature. 2005;437:1018–21. doi: 10.1038/nature04117. [DOI] [PubMed] [Google Scholar]
- Corrales JD, Blaess S, Mahoney EM, Joyner AL. The level of sonic hedgehog signaling regulates the complexity of cerebellar foliation. Development. 2006;133:1811–21. doi: 10.1242/dev.02351. [DOI] [PubMed] [Google Scholar]
- Corrales JD, Rocco GL, Blaess S, Guo Q, Joyner AL. Spatial pattern of sonic hedgehog signaling through Gli genes during cerebellum development. Development. 2004;131:5581–90. doi: 10.1242/dev.01438. [DOI] [PubMed] [Google Scholar]
- Dahmane N, Ruiz i Altaba A. Sonic hedgehog regulates the growth and patterning of the cerebellum. Development. 1999;126:3089–100. doi: 10.1242/dev.126.14.3089. [DOI] [PubMed] [Google Scholar]
- Del Cerro MP, Snider RS. Studies on the developing cerebellum. II. The ultrastructure of the external granular layer. J Comp Neurol. 1972;144:131–64. doi: 10.1002/cne.901440202. [DOI] [PubMed] [Google Scholar]
- Delaney CL, Brenner M, Messing A. Conditional ablation of cerebellar astrocytes in postnatal transgenic mice. J Neurosci. 1996;16:6908–18. doi: 10.1523/JNEUROSCI.16-21-06908.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Delous M, Baala L, Salomon R, Laclef C, Vierkotten J, Tory K, Golzio C, Lacoste T, Besse L, Ozilou C, Moutkine I, Hellman NE, Anselme I, Silbermann F, Vesque C, Gerhardt C, Rattenberry E, Wolf MT, Gubler MC, Martinovic J, Encha-Razavi F, Boddaert N, Gonzales M, Macher MA, Nivet H, Champion G, Bertheleme JP, Niaudet P, McDonald F, Hildebrandt F, Johnson CA, Vekemans M, Antignac C, Ruther U, Schneider-Maunoury S, Attie-Bitach T, Saunier S. The ciliary gene RPGRIP1L is mutated in cerebello-oculorenal syndrome (Joubert syndrome type B) and Meckel syndrome. Nat Genet. 2007;39:875–81. doi: 10.1038/ng2039. [DOI] [PubMed] [Google Scholar]
- Dixon-Salazar T, Silhavy JL, Marsh SE, Louie CM, Scott LC, Gururaj A, Al-Gazali L, Al-Tawari AA, Kayserili H, Sztriha L, Gleeson JG. Mutations in the AHI1 gene, encoding jouberin, cause Joubert syndrome with cortical polymicrogyria. Am J Hum Genet. 2004;75:979–87. doi: 10.1086/425985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Doetsch F, Caille I, Lim DA, Garcia-Verdugo JM, Alvarez-Buylla A. Subventricular zone astrocytes are neural stem cells in the adult mammalian brain. Cell. 1999;97:703–16. doi: 10.1016/s0092-8674(00)80783-7. [DOI] [PubMed] [Google Scholar]
- Doetsch F, Garcia-Verdugo JM, Alvarez-Buylla A. Cellular composition and three-dimensional organization of the subventricular germinal zone in the adult mammalian brain. J Neurosci. 1997;17:5046–61. doi: 10.1523/JNEUROSCI.17-13-05046.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fuchs JL, Schwark HD. Neuronal primary cilia: a review. Cell Biol Int. 2004;28:111–8. doi: 10.1016/j.cellbi.2003.11.008. [DOI] [PubMed] [Google Scholar]
- Garcia AD, Doan NB, Imura T, Bush TG, Sofroniew MV. GFAP-expressing progenitors are the principal source of constitutive neurogenesis in adult mouse forebrain. Nat Neurosci. 2004;7:1233–41. doi: 10.1038/nn1340. [DOI] [PubMed] [Google Scholar]
- Goodrich LV, Milenkovic L, Higgins KM, Scott MP. Altered neural cell fates and medulloblastoma in mouse patched mutants. Science. 1997;277:1109–1113. doi: 10.1126/science.277.5329.1109. [DOI] [PubMed] [Google Scholar]
- Han YG, Kwok BH, Kernan MJ. Intraflagellar transport is required in Drosophila to differentiate sensory cilia but not sperm. Curr Biol. 2003;19:1679–86. doi: 10.1016/j.cub.2003.08.034. [DOI] [PubMed] [Google Scholar]
- Haraguchi K, Hayashi T, Jimbo T, Yamamoto T, Akiyama T. Role of the kinesin-2 family protein, KIF3, during mitosis. J Biol Chem. 2006;281:4094–9. doi: 10.1074/jbc.M507028200. [DOI] [PubMed] [Google Scholar]
- Hatten ME. Neuronal regulation of astroglial morphology and proliferation in vitro. J Cell Biol. 1985;100:384–96. doi: 10.1083/jcb.100.2.384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haycraft CJ, Banizs B, Aydin-Son Y, Zhang Q, Michaud EJ, Yoder BK. Gli2 and Gli3 localize to cilia and require the intraflagellar transport protein polaris for processing and function. PLoS Genet. 2005;1:e53. doi: 10.1371/journal.pgen.0010053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huangfu D, Anderson KV. Cilia and Hedgehog responsiveness in the mouse. Proc Natl Acad Sci U S A. 2005;102:11325–30. doi: 10.1073/pnas.0505328102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huangfu D, Liu A, Rakeman AS, Murcia NS, Niswander L, Anderson KV. Hedgehog signalling in the mouse requires intraflagellar transport proteins. Nature. 2003;426:83–7. doi: 10.1038/nature02061. [DOI] [PubMed] [Google Scholar]
- Ingham PW. Transducing Hedgehog: the story so far. Embo J. 1998;17:3505–11. doi: 10.1093/emboj/17.13.3505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jeong Y, El-Jaick K, Roessler E, Muenke M, Epstein DJ. A functional screen for sonic hedgehog regulatory elements across a 1 Mb interval identifies long-range ventral forebrain enhancers. Development. 2006;133:761–72. doi: 10.1242/dev.02239. [DOI] [PubMed] [Google Scholar]
- Joubert M, Eisenring JJ, Robb JP, Andermann F. Familial agenesis of the cerebellar vermis. A syndrome of episodic hyperpnea, abnormal eye movements, ataxia, and retardation. Neurology. 1969;19:813–25. doi: 10.1212/wnl.19.9.813. [DOI] [PubMed] [Google Scholar]
- Kenney AM, Cole MD, Rowitch DH. Nmyc upregulation by sonic hedgehog signaling promotes proliferation in developing cerebellar granule neuron precursors. Development. 2003;130:15–28. doi: 10.1242/dev.00182. [DOI] [PubMed] [Google Scholar]
- Kenney AM, Rowitch DH. Sonic hedgehog promotes G(1) cyclin expression and sustained cell cycle progression in mammalian neuronal precursors. Mol Cell Biol. 2000;20:9055–67. doi: 10.1128/mcb.20.23.9055-9067.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Knoepfler PS, Cheng PF, Eisenman RN. N-myc is essential during neurogenesis for the rapid expansion of progenitor cell populations and the inhibition of neuronal differentiation. Genes Dev. 2002;16:2699–712. doi: 10.1101/gad.1021202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kondo S, Sato-Yoshitake R, Noda Y, Aizawa H, Nakata T, Matsuura Y, Hirokawa N. KIF3A is a new microtubule-based anterograde motor in the nerve axon. J Cell Biol. 1994;125(5):1095–107. doi: 10.1083/jcb.125.5.1095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kozminski KG, Beech PL, Rosenbaum JL. The Chlamydomonas kinesin-like protein FLA10 is involved in motility associated with the flagellar membrane. J Cell Biol. 1995;131:1517–27. doi: 10.1083/jcb.131.6.1517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kwon CH, Zhu X, Zhang J, Knoop LL, Tharp R, Smeyne RJ, Eberhart CG, Burger PC, Baker SJ. Pten regulates neuronal soma size: a mouse model of Lhermitte-Duclos disease. Nat Genet. 2001;29:404–11. doi: 10.1038/ng781. [DOI] [PubMed] [Google Scholar]
- Laywell ED, Rakic P, Kukekov VG, Holland EC, Steindler DA. Identification of a multipotent astrocytic stem cell in the immature and adult mouse brain. Proc Natl Acad Sci U S A. 2000;97:13883–8. doi: 10.1073/pnas.250471697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lewis PM, Gritli-Linde A, Smeyne R, Kottmann A, McMahon AP. Sonic hedgehog signaling is required for expansion of granule neuron precursors and patterning of the mouse cerebellum. Dev Biol. 2004;270:393–410. doi: 10.1016/j.ydbio.2004.03.007. [DOI] [PubMed] [Google Scholar]
- Lin JC, Cai L, Cepko CL. The external granule layer of the developing chick cerebellum generates granule cells and cells of the isthmus and rostral hindbrain. J Neurosci. 2001;21:159–68. doi: 10.1523/JNEUROSCI.21-01-00159.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Litingtung Y, Dahn RD, Li Y, Fallon JF, Chiang C. Shh and Gli3 are dispensable for limb skeleton formation but regulate digit number and identity. Nature. 2002;418:979–83. doi: 10.1038/nature01033. [DOI] [PubMed] [Google Scholar]
- Liu A, Wang B, Niswander LA. Mouse intraflagellar transport proteins regulate both the activator and repressor functions of Gli transcription factors. Development. 2005;132:3103–11. doi: 10.1242/dev.01894. [DOI] [PubMed] [Google Scholar]
- Long F, Zhang XM, Karp S, Yang Y, McMahon AP. Genetic manipulation of hedgehog signaling in the endochondral skeleton reveals a direct role in the regulation of chondrocyte proliferation. Development. 2001;128:5099–108. doi: 10.1242/dev.128.24.5099. [DOI] [PubMed] [Google Scholar]
- Louie CM, Gleeson JG. Genetic basis of Joubert syndrome and related disorders of cerebellar development. Hum Mol Genet. 2005;14(Spec No. 2):R235–42. doi: 10.1093/hmg/ddi264. [DOI] [PubMed] [Google Scholar]
- Malatesta P, Hack MA, Hartfuss E, Kettenmann H, Klinkert W, Kirchhoff F, Gotz M. Neuronal or glial progeny: regional differences in radial glia fate. Neuron. 2003;37:751–64. doi: 10.1016/s0896-6273(03)00116-8. [DOI] [PubMed] [Google Scholar]
- Marino S, Vooijs M, van Der Gulden H, Jonkers J, Berns A. Induction of medulloblastomas in p53-null mutant mice by somatic inactivation of Rb in the external granular layer cells of the cerebellum. Genes Dev. 2000;14:994–1004. [PMC free article] [PubMed] [Google Scholar]
- Marszalek JR, Ruiz-Lozano P, Roberts E, Chien KR, Goldstein LS. Situs inversus and embryonic ciliary morphogenesis defects in mouse mutants lacking the KIF3A subunit of kinesin-II. Proc Natl Acad Sci U S A. 1999;96:5043–8. doi: 10.1073/pnas.96.9.5043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marshall WF, Nonaka S. Cilia: tuning in to the cell’s antenna. Curr Biol. 2006;16(15):R604–14. doi: 10.1016/j.cub.2006.07.012. [DOI] [PubMed] [Google Scholar]
- May SR, Ashique AM, Karlen M, Wang B, Shen Y, Zarbalis K, Reiter J, Ericson J, Peterson AS. Loss of the retrograde motor for IFT disrupts localization of Smo to cilia and prevents the expression of both activator and repressor functions of Gli. Dev Biol. 2005;287:378–89. doi: 10.1016/j.ydbio.2005.08.050. [DOI] [PubMed] [Google Scholar]
- Morris RL, Scholey JM. Heterotrimeric kinesin-II is required for the assembly of motile 9+2 ciliary axonemes on sea urchin embryos. J Cell Biol. 1997;138:1009–22. doi: 10.1083/jcb.138.5.1009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Muresan V, Lyass A, Schnapp BJ. The kinesin motor KIF3A is a component of the presynaptic ribbon in vertebrate photoreceptors. J Neurosci. 1999;19(3):1027–37. doi: 10.1523/JNEUROSCI.19-03-01027.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nonaka S, Tanaka Y, Okada Y, Takeda S, Harada A, Kanai Y, Kido M, Hirokawa N. Randomization of left-right asymmetry due to loss of nodal cilia generating leftward flow of extraembryonic fluid in mice lacking KIF3B motor protein. Cell. 1998;95:829–37. doi: 10.1016/s0092-8674(00)81705-5. [DOI] [PubMed] [Google Scholar]
- Novak A, Guo C, Yang W, Nagy A, Lobe CG. Z/EG, a double reporter mouse line that expresses enhanced green fluorescent protein upon Cre-mediated excision. Genesis. 2000;28:147–55. [PubMed] [Google Scholar]
- Oliver TG, Read TA, Kessler JD, Mehmeti A, Wells JF, Huynh TT, Lin SM, Wechsler-Reya RJ. Loss of patched and disruption of granule cell development in a pre-neoplastic stage of medulloblastoma. Development. 2005;132:2425–2439. doi: 10.1242/dev.01793. [DOI] [PubMed] [Google Scholar]
- Osumi N, Hirota A, Ohuchi H, Nakafuku M, Iimura T, Kuratani S, Fujiwara M, Noji S, Eto K. Pax-6 is involved in the specification of hindbrain motor neuron subtype. Development. 1997;124:2961–72. doi: 10.1242/dev.124.15.2961. [DOI] [PubMed] [Google Scholar]
- Otto EA, Schermer B, Obara T, O’Toole JF, Hiller KS, Mueller AM, Ruf RG, Hoefele J, Beekmann F, Landau D, Foreman JW, Goodship JA, Strachan T, Kispert A, Wolf MT, Gagnadoux MF, Nivet H, Antignac C, Walz G, Drummond IA, Benzing T, Hildebrandt F. Mutations in INVS encoding inversin cause nephronophthisis type 2, linking renal cystic disease to the function of primary cilia and left-right axis determination. Nat Genet. 2003;34:413–20. doi: 10.1038/ng1217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Riddle RD, Johnson RL, Laufer E, Tabin C. Sonic hedgehog mediates the polarizing activity of the ZPA. Cell. 1993;75:1401–16. doi: 10.1016/0092-8674(93)90626-2. [DOI] [PubMed] [Google Scholar]
- Rohatgi R, Milenkovic L, Scott MP. Patched1 regulates Hedgehog signaling at the primary cilium. Science. 2007;317:372–376. doi: 10.1126/science.1139740. [DOI] [PubMed] [Google Scholar]
- Romer JT, Kimura H, Magdaleno S, Sasai K, Fuller C, Baines H, Connelly M, Stewart CF, Gould S, Rubin LL, Curran T. Suppression of the Shh pathway using a small molecule inhibitor eliminates medulloblastoma in Ptc1(+/-)p53(-/-) mice. Cancer Cell. 2004;6:229–240. doi: 10.1016/j.ccr.2004.08.019. [DOI] [PubMed] [Google Scholar]
- Rosenbaum JL, Witman GB. Intraflagellar transport. Nat Rev Mol Cell Biol. 2002;3:813–25. doi: 10.1038/nrm952. [DOI] [PubMed] [Google Scholar]
- Sanchez P, Ruiz i Altaba A. In vivo inhibition of endogenous brain tumors through systemic interference of Hedgehog signaling in mice. Mech Dev. 2005;122:223–30. doi: 10.1016/j.mod.2004.10.002. [DOI] [PubMed] [Google Scholar]
- Sarpal R, Todi SV, Sivan-Loukianova E, Shirolikar S, Subramanian N, Raff EC, Erickson JW, Ray K, Eberl DF. Drosophila KAP interacts with the kinesin II motor subunit KLP64D to assemble chordotonal sensory cilia, but not sperm tails. Curr Biol. 2003;13:1687–96. doi: 10.1016/j.cub.2003.09.025. [DOI] [PubMed] [Google Scholar]
- Sayer JA, Otto EA, O’Toole JF, Nurnberg G, Kennedy MA, Becker C, Hennies HC, Helou J, Attanasio M, Fausett BV, Utsch B, Khanna H, Liu Y, Drummond I, Kawakami I, Kusakabe T, Tsuda M, Ma L, Lee H, Larson RG, Allen SJ, Wilkinson CJ, Nigg EA, Shou C, Lillo C, Williams DS, Hoppe B, Kemper MJ, Neuhaus T, Parisi MA, Glass IA, Petry M, Kispert A, Gloy J, Ganner A, Walz G, Zhu X, Goldman D, Nurnberg P, Swaroop A, Leroux MR, Hildebrandt F. The centrosomal protein nephrocystin-6 is mutated in Joubert syndrome and activates transcription factor ATF4. Nat Genet. 2006;38:674–81. doi: 10.1038/ng1786. [DOI] [PubMed] [Google Scholar]
- Singla V, Reiter JF. The primary cilium as the cell’s antenna: signaling at a sensory organelle. Science. 2006;313:629–33. doi: 10.1126/science.1124534. [DOI] [PubMed] [Google Scholar]
- Snow JJ, Ou G, Gunnarson AL, Walker MR, Zhou HM, Brust-Mascher I, Scholey JM. Two anterograde intraflagellar transport motors cooperate to build sensory cilia on C. elegans neurons. Nat Cell Biol. 2004;6:1109–13. doi: 10.1038/ncb1186. [DOI] [PubMed] [Google Scholar]
- Soriano P. Generalized lacZ expression with the ROSA26 Cre reporter strain. Nat Genet. 1999;21:70–1. doi: 10.1038/5007. [DOI] [PubMed] [Google Scholar]
- Sotelo C. Cellular and genetic regulation of the development of the cerebellar system. Prog Neurobiol. 2004;72:295–339. doi: 10.1016/j.pneurobio.2004.03.004. [DOI] [PubMed] [Google Scholar]
- Spassky N, Goujet-Zalc C, Parmantier E, Olivier C, Martinez S, Ivanova A, Ikenaka K, Macklin W, Cerruti I, Zalc B, Thomas JL. Multiple restricted origin of oligodendrocytes. J Neurosci. 1998;18:8331–43. doi: 10.1523/JNEUROSCI.18-20-08331.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takeda S, Yonekawa Y, Tanaka Y, Okada Y, Nonaka S, Hirokawa N. Left-right asymmetry and kinesin superfamily protein KIF3A: new insights in determination of laterality and mesoderm induction by kif3A-/- mice analysis. J Cell Biol. 1999;145:825–36. doi: 10.1083/jcb.145.4.825. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Teng J, Rai T, Tanaka Y, Takei Y, Nakata T, Hirasawa M, Kulkarni AB, Hirokawa N. The KIF3 motor transports N-cadherin and organizes the developing neuroepithelium. Nat Cell Biol. 2005;5:474–482. doi: 10.1038/ncb1249. [DOI] [PubMed] [Google Scholar]
- Valente EM, Silhavy JL, Brancati F, Barrano G, Krishnaswami SR, Castori M, Lancaster MA, Boltshauser E, Boccone L, Al-Gazali L, Fazzi E, Signorini S, Louie CM, Bellacchio E, Bertini E, Dallapiccola B, Gleeson JG. Mutations in CEP290, which encodes a centrosomal protein, cause pleiotropic forms of Joubert syndrome. Nat Genet. 2006;38:623–5. doi: 10.1038/ng1805. [DOI] [PubMed] [Google Scholar]
- Vierkotten J, Dildrop R, Peters T, Wang B, Ruther U. Ftm is a novel basal body protein of cilia involved in Shh signalling. Development. 2007;134:2569–77. doi: 10.1242/dev.003715. [DOI] [PubMed] [Google Scholar]
- Wallace VA. Purkinje-cell-derived Sonic hedgehog regulates granule neuron precursor cell proliferation in the developing mouse cerebellum. Curr Biol. 1999;9:445–8. doi: 10.1016/s0960-9822(99)80195-x. [DOI] [PubMed] [Google Scholar]
- Wechsler-Reya RJ, Scott MP. Control of neuronal precursor proliferation in the cerebellum by Sonic Hedgehog. Neuron. 1999;22:103–14. doi: 10.1016/s0896-6273(00)80682-0. [DOI] [PubMed] [Google Scholar]
- Whitfield JF. The neuronal primary cilium--an extrasynaptic signaling device. Cell Signal. 2004;16:763–7. doi: 10.1016/j.cellsig.2003.12.002. [DOI] [PubMed] [Google Scholar]
- Yacubova E, Komuro H. Cellular and molecular mechanisms of cerebellar granule cell migration. Cell Biochem Biophys. 2003;37:213–34. doi: 10.1385/cbb:37:3:213. [DOI] [PubMed] [Google Scholar]
- Yue Q, Groszer M, Gil JS, Berk AJ, Messing A, Wu H, Liu X. PTEN deletion in Bergmann glia leads to premature differentiation and affects laminar organization. Development. 2005;132:3281–91. doi: 10.1242/dev.01891. [DOI] [PubMed] [Google Scholar]
- Zhuo L, Theis M, Alvarez-Maya I, Brenner M, Willecke K, Messing A. hGFAP-cre transgenic mice for manipulation of glial and neuronal function in vivo. Genesis. 2001;31:85–94. doi: 10.1002/gene.10008. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Low magnifications of confocal projections of double immunostaining with an anti-acetylated α-tubulin antibody (red) and an anti-γ-tubulin antibody (green) on sagittal sections of wild type (A, C) and conditional Kif3a mutant (B, D) at E18.5. White square in A, B shows the portion of the EGL that is magnified in C, D. White square in C, D shows which portion of the EGL is magnified in Fig. 2. Scale bar : 310 μm (A, B), 17 μm (C, D).
Serial sections of mutant GCPs at P0, which spans the entire thickness of a basal body, show the lack of primary cilia. The basal body (arrowhead) lacking the primary cilium is anchored to the cell membrane. A centriole (asterisk) is attached to the basal body. Scale bar: 0.2 μm.
GFAP immunostaining on cerebella sagittal cryosections from P5 wild type (A) and conditional Kif3a mutant (B) mice. Although glial fibers appear slightly disorganized in mutants compared to wild type, these cells are GFAP+ and their processes extend toward the pial surface. Scale bar: 20 μm.
Tunel staining of a control (A) and a conditional Kif3a mutant (B) on sagittal sections of cerebellum at P0. Scale bar: 200 μm.
Immunofluorescence of cerebellar sagittal sections from E18.5 wild type (A, C) and conditional Kif3a mutant (B, D) mice with anti-N-cadherin and anti-β-catenin antibodies. Scale bar: 20 μm.