

Abstract
Human immunoglobulin heavy chain variable domains (VH) are promising scaffolds for antigen binding. However, VH is an unstable and aggregation-prone protein, hindering its use for therapeutic purposes. To evolve the VH domain, we performed in vivo protein solubility selection that linked antibiotic resistance to the protein folding quality control mechanism of the twin-arginine translocation pathway of E. coli. After screening a human germ-line VH library, 95% of the VH proteins obtained were identified as VH3 family members; one VH protein, MG2x1, stood out among separate clones expressing individual VH variants. With further screening of combinatorial framework mutation library of MG2x1, we found a consistent bias toward substitution with tryptophan at the position of 50 and 58 in VH. Comparison of the crystal structures of the VH variants revealed that those substitutions with bulky side chain amino acids filled the cavity in the VH interface between heavy and light chains of the Fab arrangement along with the increased number of hydrogen bonds, decreased solvation energy, and increased negative charge. Accordingly, the engineered VH acquires an increased level of thermodynamic stability, reversible folding, and soluble expression. The library built with the VH variant as a scaffold was qualified as most of VH clones selected randomly were expressed as soluble form in E. coli regardless length of the combinatorial CDR. Furthermore, a non-aggregation feature of the selected VH conferred a free of humoral response in mice, even when administered together with adjuvant. As a result, this selection provides an alternative directed evolution pathway for unstable proteins, which are distinct from conventional methods based on the phage display.
Introduction
The variable domain of heavy or light chain (VH or VL) of a human immunoglobulin G (IgG) molecule is the smallest part of the antibody that preserves the original binding activity. Although variable domains have short serum half-lives and lack effector function, their format flexibility by adopting immune cell engaging strategy or introducing a long-acting module can ameliorate these defects [1]–[3]. Furthermore, their ability to access occluded or hidden epitopes, superior bio-distribution, and cost-effective production make variable domains potentially useful in therapeutic applications for which full IgG molecules are not appropriate [4]–[6].
When not assembled with each other, instability problem of VH and VL of human IgG is a major concern for biotechnological applications since Ward et al. reported that such VH domains are relatively sticky resulting in tendency to aggregate [7]. This aggregation is primarily due to interactions between hydrophobic patches residing at the interface between VH and VL. Direct replacement of the interfacial hydrophobic residues of VH or VL with hydrophilic amino acids has been partially successful in improving protein stability. Three hydrophilic substitutions (G44E/L45R/W47G) improve the solubility of VH [8]–[10], but these changes also decrease expression yield and thermal stability due to the resultant deformations of the β-sheet structure [11]–[13].
In addition to rational mutation strategies, several groups have adopted combinatorial approaches to engineer human VH or VL. Jespers et al. screened a combinatorial CDR library bound to protein A for aggregation-resistant VH, using panning phage display under heat-denatured conditions [14]. They found that mutations in the CDRs of human VH can increase solubility and promote reversible folding. Without the use of heat denaturation in phage display, Barthelemy et al. isolated various mutant VH domains with an increased stability and solubility [15]. To eliminate the complicated step involving in vitro protein A panning, To et al. selected monomeric human VH domains directly from bacterial lawns by plaque size [16]. These variant techniques notwithstanding, most screenings of engineered VH domains have been conducted using phage display and protein A-binding activity.
On the other hand, in vivo genetic selection methods distinct from in vitro phage display have been applied in efforts to improve protein solubility [17], [18]. In one such in vivo method, the twin-arginine translocation (Tat) pathway was exploited as an in vivo protein fitness filter for fast folding and solubility of protein of interests including single chain Fv [19]–[21]. However, such approaches have not been attempted for VH or VL alone. In the current study, we applied this system to evolve human VH toward greater stability and characterized the structural hallmarks to greater stability and solubility.
Materials and Methods
Ethics Statement
All animal experiments were performed in accordance with the guidelines for the care and use of laboratory animals recommended by the Ministry of Food and Drug Safety of Republic of Korea. The experimental procedures were approved by the Mogam Animal Care and Use Committee. Currently, Mogam Animal Care and Use Committee changed the name as the Green Cross Central Research laboratory Animal Care and Use Committee, by which the animal experiment closing report was reviewed and approved.
Construction of the Tat-based genetic selection vector
The vector system for screening of stable VH domains was modified from the previous report [20], [22]. Briefly, TEM-1 β-lactamase (BLA) was ligated with the Tat signal sequence of trimethylamine N-oxide reductase (ssTorA) of E. coli in pET9a, yielding pET-TAPE (Figure 1A). Next, a fusion gene of ssTorA with the representative human immunoglobulin heavy chain variable domain VH family type 2 (VH2) was synthesized (GenScript, USA). VH2 was used as a template for PCR using a 5′ primer (Table S1, primer 1) including an NdeI restriction site and a 3′ primer (Table S1, primer 2) including a NotI site, a 6×His tag, and a BamHI site, to yield the NdeI-ssTorA-VH2-NotI-6×His-BamHI gene. This gene was inserted between the Ndel and BamHI sites in the multi-cloning site of pET9a to yield pET9a-ssTorA-VH2. The NotI-BLA-BamHI segment was generated by PCR (Table S1: primer 3 as sense, primer 4 as antisense) using BLA as a template. This gene was inserted between the Notl and BamHI sites of pET9a-ssTorA-VH2, yielding pET9a-ssTorA-VH2-BLA, which was named pET-TAPE. A synthetic or human germ-line VH library was constructed by replacing the VH2 gene in pET-TAPE.
Figure 1. Verification of the selection system, TAPE.
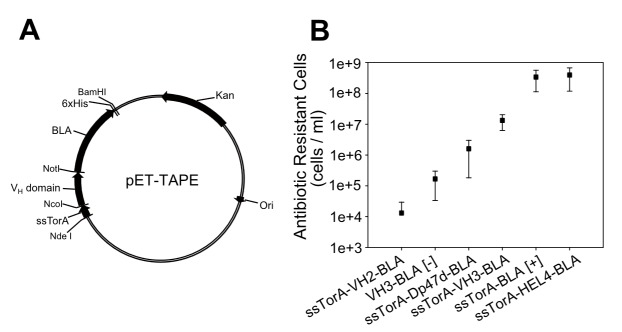
(A) Plasmid map of pET-TAPE. (B) Average number of ampicillin-resistant colonies from cultures harboring constructs for expression of a negative control (no Tat signal sequence, VH3-BLA [−]), positive control (Tat signal sequence and reporter gene only, ssTorA-BLA [+]), and published VH domains (HuCal VH2, HuCal VH3, Dp47d, and HEL4). Each construct was expressed in LB medium containing 50 µg/ml ampicillin. Cultures were induced by the addition of 1 mM isopropylβD-1-thiogalactopyranoside for 3 h after inoculation. After the induction, cultures were spread onto agar plates containing 50 µg/ml ampicillin for colony counting. Data points are means and standard deviation for three independent experiments.
Library design and construction
cDNA for the human VH library was obtained by reverse transcription of mRNAs from the liver, peripheral blood mononuclear cells, spleen, and thyroid (Clontech, Madison, WI, US) using various primers (Table S1: primers 5–12 as sense, and primers 13–15 as antisense). Each of cloned human VH gene family (VH1, VH3, and VH5) was inserted between the NdeI and BamHI sites of pET-TAPE, yielding a pET-TAPE-VH library with approximately 109 distinct clones. Mutations were introduced by PCR using MG2x1 as the template and primers that introduced mutations at the first fragment (Table S1: primers 16 and 17) and the second fragment (Table S1: primers 18 and 19). Next, MG2x1 variant genes were synthesized by overlapping PCR of the two gene fragments using primers 16 and 19 (Table S1). After digestion of the MG2x1 variants with NcoI and NotI, the inserts were cloned into pET-TAPE, yielding the frame-mutation VH library with approximately 108 distinct clones.
Setup for Tat-associated protein engineering (TAPE) system
Along with the construction of pET-TAPE, the protocol implementing a liquid culture and rescuing correct size of gene of interests was conducted to screen protein solubility in high-throughput manner. The antibiotic resistance of E. coli is correlated to the translocation of soluble VH-BLA fusion protein into the periplasm via the Tat pathway. The TAPE system differs from previously described systems [20] in that soluble proteins are enriched in consecutive rounds of liquid culture with increasing concentrations of antibiotic. E. coli T7 Express LysY/Iq was transformed with the pET-TAPE-VH library by electroporation. Transformants were cultured in SOC (20 g/l Bacto tryptone, 5 g/l Bacto yeast extract, 10 mM NaCl, 2.5 mM KCl, 10 mM MgCl2, 10 mM MgSO4, and 20 mM glucose) at 37°C for 1 h, and then inoculated and cultured in liquid LB media containing 50 µg/ml ampicillin. When OD (600 nm) reached 0.6, cells were collected by centrifugation and plasmid DNA was isolated. To prevent enrichment of false-positives in subsequent rounds of selection, isolated plasmids were restricted with NcoI and BamHI, and digests were subjected to gel electrophoresis to allow size selection of full-length VH-BLA genes. The size-selected VH-BLA genes were cloned between the NcoI and BamHI sites of pET-TAPE, and the resultant plasmids were transformed into E. coli. Subsequently, liquid culture was performed in repeated rounds with stepwise increases in the concentration of ampicillin up to 500 µg/ml. (Figure 2). After performing 3–5 consecutive cycles of liquid culture, clones were separated on an LB agar plate containing ampicillin and 50 µg/ml kanamycin.
Figure 2. Schematic procedure for screening protein solubility using TAPE (‘liquid screen’).
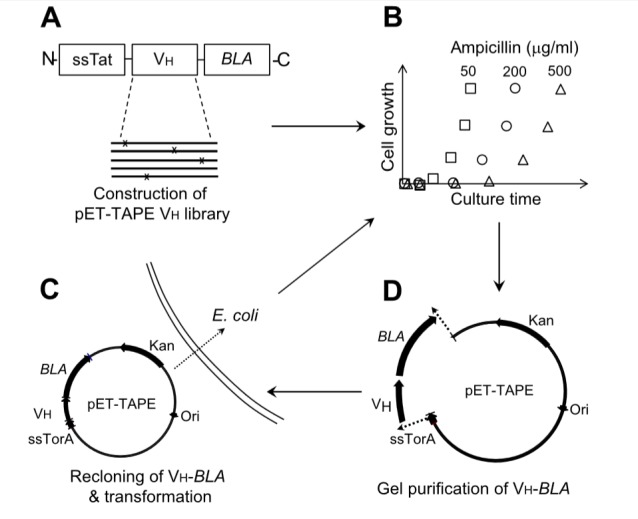
(A) Construction of the pET-TAPE VH library (either germ-line or mutated) and transformation of the library into E. coli. (B) Liquid culture of the library with stepwise increases in antibiotic concentration. (C) Collection of plasmids and purification of the intact VH-BLA coding region. (D) Re-cloning of the VH-BLA gene into pET-TAPE between the NcoI and BamHI sites, and transformation into E. coli. Steps (B), (C), and (D) were repeated four times for each ampicillin concentration (50, 100, 250, and 500 µg/ml).
Host strains and plasmids
E. coli T7 Express LysY/Iq (New England BioLabs, MA, USA) was used as the host for the expression of the VH domains and their fusion proteins. pET9a (New England BioLabs, MA, US) was used to construct the TAPE system, i.e., for expression of fusion proteins of various VH domains and BLA. pET22b (New England Biolabs, MA, USA) was used to express the VH domain alone. All other DNA manipulations were conducted according to common methods.
Fractionation of soluble and insoluble VH
To determine the degree of soluble expression, individual VH domains alone (i.e., without the BLA fusion) were expressed in E. coli. The soluble and insoluble fractions were separated after induction of VH expression, followed by SDS-PAGE. Soluble and insoluble proteins were fractionated in lysis buffer (B-PER Reagent, Thermo Scientific, USA). The pellet was washed with PBS, and then resuspended in solubilization buffer (pH 7.4, 50 mM NaH2PO4, 6 M urea, 0.5 M NaCl, and 4 mM DTT) to obtain the insoluble fraction. Each fraction was prepared from the same quantity of cells to allow band intensities to be compared after gels were stained with Coomassie blue.
Circular Dichroism
Purified VH domains were diluted to 0.2 mg/ml. The purity of VH domains used for CD measuremnt was demonstrated with SDS-PAGE (Figure S1). CD was measured using a spectropolarimeter (Jasco J-715 model, Jasco Inc, Easton, MD, US). Tm was defined as the temperature at which a 50% reduction in the soluble protein fraction was observed. The profile was recorded at a wavelength of 235 nm as the temperature gradually increased from 25 to 85°C at a rate of 1°C/min. All CD measurement were repeated 3 times for each VH domain. The p-value (paired t-test) between two VH domains was less than 0.005 for all possible pairs of the tested VH domains.
Recovery yield
The recovery yield was defined as the level of soluble VH after heat denaturation. After aggregates were removed by centrifugation, the concentration of soluble VH was determined according to the equation, c = A/ (E×b), where A is the absorbance at 280 nm, E is the molar extinction coefficient (M−1cm−1), b is the pathway length (cm), and c is the molar concentration (mol/l). The extinction coefficient was calculated using the amino acid composition, assuming that all pairs of cysteine residues were involved in disulfide bonds (web.expasy.org/protparam). Protein quality was confirmed by size-exclusion chromatography.
Humoral immune response of mice to the screened VH
BALB/c mice (six per group) were intravenously injected with 10 µg MG2x1, MG8-14, or VHH on 3 consecutive days. The injections were repeated at weeks 1, 4, and 8. Samples of immune sera were obtained every week, and mice were sacrificed at day 65. For intramuscular and subcutaneous injections, BALB/c mice (six per group) were injected with 1 or 10 µg of MG8-14 or VHH. VHH is identical to VHH #3E, which binds to tumor necrosis factor-α [23]. The injection was repeated every 2 weeks with a total of five injections. The mice were sacrificed 2 weeks after the final injection. Samples of immune sera were obtained every 2 weeks, 1 day before the next injection. To measure antibody titers, enzyme-linked immunosorbent assays were performed using 96-well plates coated with MG2x1, MG8-14, or VHH, and HRP-labeled goat anti-mouse antibody as a secondary antibody, followed by the addition of 3,3′,5,5′-tetramethylbenzidine and measurement of OD (490 nm).
Results
Verifying TAPE
To verify whether TAPE system can discriminate between proteins of different solubilities, we applied this system to various published VH domains whose soluble expression levels are well known. The VH domains were cloned into the pET-TAPE vector (Figure 1A), allowing them to be expressed in E. coli as fusions with BLA and the Tat signal sequence of ssTorA. The antibiotic resistance of strains carrying each construct was measured by counting the cell number in cultures containing 50 µg/ml ampicillin. Cells expressing BLA alone (ssTorA-BLA [+], positive control) exhibited the highest resistance, and cells expressing HEL4 were approximately as resistant as the positive control (Figure 1B) [22]. Cells expressing the other representative VH3 family genes, Dp47d and VH3 (HuCAL), exhibited resistances intermediate between those of the positive and negative controls [24], [25]. The resistance of cells expressing the antibiotic resistance gene with no Tat signal sequence (VH3-BLA [-], negative control) was lower than that of cells expressing any other construct, with the notable exception of the VH2 (HuCAL) construct (ssTorA-VH2-BLA). Cells expressing VH2 exhibited the lowest ampicillin resistance, even lower than that of the negative control. Since most of the VH2 was expressed exclusively as inclusion bodies (Figure 3A), the biostatic effect of VH2 aggregate formation in E. coli might have further slowed cell growth beyond the bactericidal effect of the antibiotic.
Figure 3. SDS-PAGE of soluble and insoluble fractions of E.x coli.
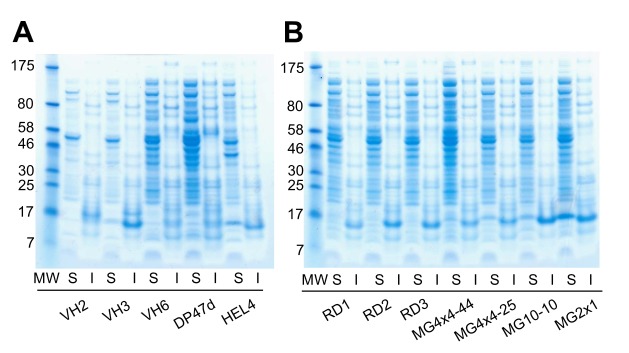
(A) Previously characterized VH domains (VH2, VH3, VH6, DP47d, and HEL4). (B) VH domains chosen randomly from the human VH germ-line library (RD1–3) or selected from the human germ-line library using TAPE (MG4x4-44, MG4x4-25, MG10-10, and MG2x1). Cultures expressing each VH domain were harvested after induction at 25°C for 3.5 h, and soluble (S) and insoluble (I) fractions were prepared. Lane ‘MW’ contains a protein size marker; the size of each marker is indicated (in kD) to the left of each panel. In both panels, the mobilities of VH domains correspond to the 15-kD protein size marker. Different parts from separating gels are grouped to align expression patterns for soluble and insoluble fraction of each VH domain.
Screening of human germ-line VH library via TAPE
In the Tat-associated screening system using ampicillin-containing agar plates, false-positive clones containing small VH peptide fragments were often enriched because such fragments are highly compatible with the Tat pathway. To overcome this problem, previous screens have included a step to exclude clones with excessively high antibiotic resistance (i.e., counter-selection) [20]. In this study, to perform VH solubility screening in a high-throughput manner, we enriched antibiotic-resistant clones in liquid cultures (‘liquid screen’) containing various concentrations of ampicillin (50–500 µg/ml) (Figure 2). Furthermore, to avoid enrichment of short VH gene fragments that might yield false-positive results, full-size VH-BLA fusion genes were recovered by gel purification. In contrast to the limitation of library size in the plate-based method, the liquid screen with a culture larger than 100 ml can cover library sizes greater than 109 because 1 ml overnight culture of E. coli in the LB with ampicillin contains normally about 109 cells. The size of the human germ-line VH library for TAPE was about 2.17×109.
After the third round of TAPE through selection of antibiotic resistance, 154 VH sequences were selected from the human germ-line VH library that had been constructed using primers specific for the VH1, VH3, and VH5 families. These 154 VH sequences were classified into 19 different VH family types. Of the 154 total VH hits, 146 (94.8%) were identified as members of the VH3 family; this frequency is significantly higher than the VH3 family frequency in the library prior to TAPE (101 VH3 family members out of 144 sequences: 70.1%). Among the VH3 family genes isolated from the germ-line VH library, the VH3–30 and VH3–23 genes were predominant. On the other hand, the frequencies of the VH1 and VH5 families decreased by 0.1-fold and 0.3-fold, respectively. Overall, as a result of TAPE, the VH3 family was enriched 1.4-fold (i.e., from 70.1% to 94.8%), whereas the other families became less abundant (Table 1).
Table 1. Isolated germ-line VH genes after the third round of TAPE.
| V gene name | % of VH after TAPEa | % of VH before TAPEb | Foldincreasec |
| VH3-7 | 6.5 | 4.3 | 1.5 |
| VH3-9 | 0.6 | 2.1 | 0.3 |
| VH3-15 | 7.1 | 3.9 | 1.8 |
| VH3-21 | 1.3 | 6.1 | 0.2 |
| VH3-23 | 14.3 | 16.0 | 0.9 |
| VH3-30 | 46.8 | 11.4 | 4.1 |
| VH3-33 | 1.3 | 2.9 | 0.4 |
| VH3-43 | 0.6 | 0.4 | 1.5 |
| VH3-48 | 7.1 | 3.9 | 1.8 |
| VH3-49 | 0.6 | 0.4 | 1.5 |
| VH3-53 | 0.6 | 1.0 | 0.6 |
| VH3-72 | 0.6 | 1.0 | 0.6 |
| VH3-74 | 7.1 | 2.5 | 2.8 |
| others | 0 | 13.7 | - |
| VH3 family | 94.8 | 70.1 | 1.4 |
| VH1-2 | 0.6 | 1.4 | 0.4 |
| VH1-8 | 0.6 | 0.0 | - |
| VH1-46 | 0.6 | 3.5 | 0.2 |
| VH1-69 | 0.6 | 4.7 | 0.1 |
| others | 0 | 9.7 | - |
| VH1 family | 2.6 | 19.4 | 0.1 |
| VH5-51 | 1.3 | 4.3 | 0.3 |
| VH5-a | 1.3 | 5.7 | 0.2 |
| others | 0 | 0.7 | - |
| VH5 family | 2.6 | 10.5 | 0.2 |
Proportion of each identified VH gene among the 154 sequences selected after TAPE.
Proportion of each identified VH gene among 144 sequences randomly selected from the library.
Ratio of ‘% of VH gene after TAPE’ to ‘% of VH gene before TAPE’.
To determine the degree of soluble expression of isolated individual VH domains lacking the BLA fusion, the soluble and insoluble fractions were separated after expression of the corresponding genes, and their expression patterns were compared with those of various VH domains published previously [24], [25]. VH domains randomly selected from the germ-line VH library were expressed predominantly as inclusion bodies (Figure 3B, RD1–3), whereas the soluble expression levels of VH domains selected by TAPE, e.g., MG4x4-44, MG4x4-25, MG10-10, and MG2x1, were significantly increased (Figure 3B). Moreover, the VH domains selected by TAPE exhibited a higher ratio of soluble to insoluble protein than the previously characterized VH domains described above, i.e., VH2 (HuCAL), VH3 (HuCAL), VH6 (HuCAL), VH3 (DP47d), and HEL4 (Figure 3A).
An artificial library comprising 25 individual VH domains, either selected from the germ-line library or previously characterized VH domains (HEL4, DP47d, HuCal VH3, and HuCal VH2) were subjected to TAPE. Only one clone, MG2x1, grew out at the third round of TAPE. This clone was used as the backbone for the frame-mutation library with selected mutation sites, described below.
Screening of the frame-mutation library of MG2x1 via TAPE
To confer additive solubility and stability to MG2x1, combinatorial mutations were introduced into seven specific sites of MG2x1 to generate the MG2x1 frame-mutation library. The number of distinct clones in the library was 1.4x108, which covers all the possible combinations of mutations with NNK degeneration codon (theoretically, 6.4×107 combinations). The selected mutation sites are distributed over the CDRH1 (S35), frame 2 (Q39, L45, and W47), and the CDRH2 (A50, Y58, and A60) with the kabat numbering system (Figure 4A, residues in red). These sites were selected by referring to the crystal structure of MG2x1 (PDB ID: 3ZHK) to identify amino acids that stretch their side chains outward from the surface. Also, all of these sites are located in the β-sheet structure away from the flexible loop of the CDRs. The frame-mutation library of MG2x1 was screened by TAPE, with the concentration of ampicillin increased (50, 100, 250, and 500 µg/ml) in successive rounds. After the final round of TAPE, 41 clones were randomly selected for sequencing of their VH domains. Changes at positions 50 and 58 (Kabat scheme) were biased toward tryptophan (W): alanine (A) at position 50 was replaced by W in 39% (16/41) of the clones, and tyrosine (Y) at position 58 was replaced by W in 58% (24/41) of the clones (Table 2). The other mutation sites were not particularly biased. Sequence alignment of the selected VH domains after TAPE revealed the biased amino acids at positions 50 and 58 (Figure 4B, dashed box). Based on the biased mutation frequencies at positions 50 and 58, we generated a MG8-14 mutant in which leucine (L) at position 50 was replaced with W (MG8-14 [L50W]) for further analyses of its physicochemical properties.
Figure 4. Rationale for designing of the combinatorial frame-mutation library.
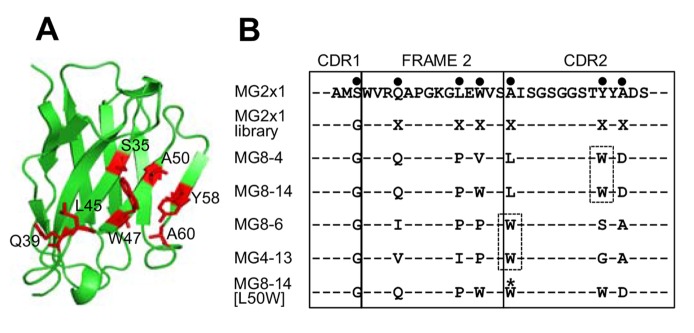
(A) Positions chosen for randomization based on the crystal structure of MG2x1. Residues are numbered according to the Kabat scheme for the VH sequence. (B) Representative sequences (MG8-4, MG8-14, MG4-13, and MG8-6) selected from the MG2x1 frame-mutation library by TAPE were aligned with the original MG2x1 sequence. Mutation sites in the sequence of MG2x1 are shown as bold dots. All mutations were introduced using degenerate codons (NNK), except that serine (S) 35 was replaced by glycine (G). X represents all amino acids. At positions 50 and 58, the mutations converged primarily onto tryptophan, indicated by dashed boxes.
Table 2. Frequency of mutations of the randomly chosen VH domains after screening of the combinatorial frame-mutation library of MG2x1 via TAPE.
| Mutations | Q39 | L45 | W47 | A50 | Y58 | A60 | Codon Degeneracy (%)a |
| F | 4 | 5 | 3 | 3 | 3.13 | ||
| W | 2 | 1 | 5 | 16 | 24 | 3.13 | |
| Y | 3 | 5 | 1 | 2 | 3 | 3.13 | |
| A | 2 | 2 | 3 | 1 | 7 | 6.25 | |
| C | 2 | 1 | 2 | 5 | 7 | 3.13 | |
| I | 4 | 4 | 4 | 3.13 | |||
| L | 3 | 7 | 3 | 6 | 3 | 1 | 9.38 |
| M | 3 | 1 | 6 | 2 | 1 | 3.13 | |
| P | 6 | 4 | 6.25 | ||||
| V | 5 | 5 | 6 | 3 | 1 | 1 | 6.25 |
| G | 1 | 1 | 6.25 | ||||
| N | 3 | 1 | 4 | 3.13 | |||
| Q | 3 | 3.13 | |||||
| S | 1 | 1 | 1 | 1 | 6 | 9.38 | |
| T | 3 | 4 | 2 | 1 | 3 | 6.25 | |
| H | 2 | 2 | 3.13 | ||||
| K | 2 | 1 | 3.13 | ||||
| R | 1 | 2 | 1 | 2 | 4 | 9.38 | |
| D | 4 | 3.13 | |||||
| E | 1 | 3.13 | |||||
| Total | 41 | 41 | 41 | 41 | 41 | 41 |
Possible substitution of amino acids into the frame-mutation library (%) according to the given codon degeneracy of primers used for mutagenesis of MG2x1 as shown in the Table S1.
Soluble expression level and thermodynamic stability are correlated in VH domains selected by TAPE
Among the hits obtained from the combinatorial frame-mutation library of MG2x1, 23 unique sequences were selected from the final round of TAPE. Most of the selected VH domains were expressed as soluble proteins. In particular, MG8-14, MG2-55, MG4-5, MG-4-13, MG8-4, and MG8-6 were expressed exclusively in their soluble forms (Figure 5). A previous study using the Tat pathway to express a protein fused to an antibiotic resistance marker showed that the ability to confer growth was correlated to both the solubility profile and the molecular weight of the protein [26]. The thermodynamic stabilities of the VH domains selected from the naïve human VH library by TAPE were higher than those of wild-type VH3 domains. The melting temperatures (Tm) of the selected germ-line VH domains were 55.6–65.2°C, whereas the Tm of the randomly chosen VH domains from the germ-line library were generally below 50°C, e.g., 46.5°C for VH3–15 (Figure 6A). Among the selected germ-line VH domains, MG2x1 had the highest Tm. Furthermore, the Tm of VH domains selected from the combinatorial frame-mutation library of MG2x1 (65.2–77.5°C) were significantly higher than that of the parental VH (MG2x1) (Figure 6B). The thermodynamic stabilities of the engineered VH domains identified in this study were generally higher than that of HEL4, which was selected from a combinatorial CDR3 library based on Dp47d by heat-resistant phage display selection [24].
Figure 5. Soluble expression level of screened VH domains.
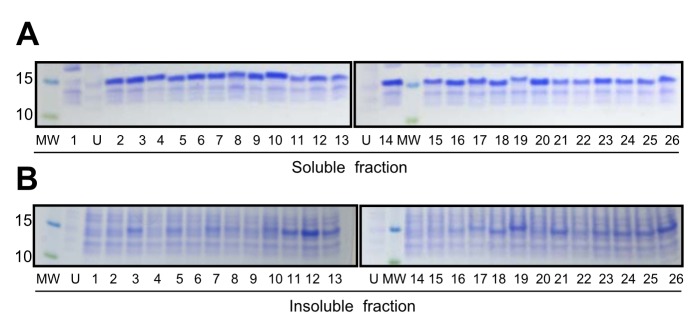
SDS-PAGE of soluble (A) and insoluble (B) fractions of E. coli expressing each VH domains selected from the frame-mutation library of MG2x1 were loaded as follows: 1, VHH (camel single-domain antibody); 2, HEL4; 3, MG2x1; 4, MG8-14; 5, MG2-47; 6, MG2-55; 7, MG2-57; 8, MG2-59; 9, MG4-2; 10, MG4-5; 11, MG4-6; 12, MG4-7; 13, MG4-12; 14, MG4-13; 15, MG4-17; 16, MG4-20; 17, MG4-28; 18, MG4-32; 19, MG4-33; 20, MG8-4; 21, MG8-5; 22, MG8-6; 23, MG8-8; 24, MG8-11; 25, MG8-12; 26, MG8-13. Lanes labeled ‘MW’ contained protein size markers (10 and 15 kD). U indicates fractions from a culture with no isopropylβD-1-thiogalactopyranoside.
Figure 6. Thermodynamic stability.
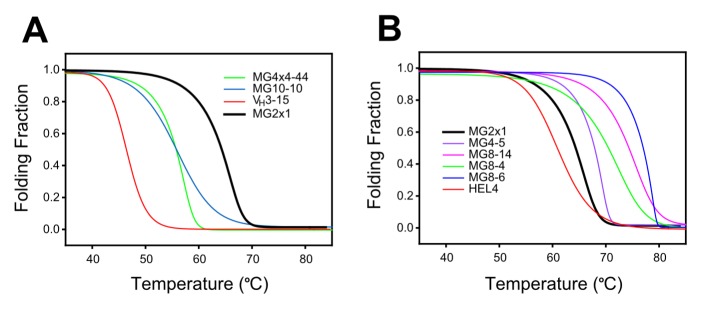
(A) Representative VH domains selected from the germ-line library. (B) Representative VH domains selected from the MG2x1 frame-mutation library. The black bold line indicates the profile of the parental VH, MG2x1, prior to mutation. Folding fraction was converted from the temperature-scouting CD profile at a fixed wavelength (230 nm).
Selected VH domains fold autonomously after denaturation
Proteins exist in thermodynamic equilibrium between their folded and unfolded states. Hence, unstable proteins are much more vulnerable to heat and pH disturbance because exposure of their hydrophobic core during occupancy of the unfolded state promotes aggregation. Many VH3 family domains are soluble and aggregation-resistant. However, once these proteins are denatured, they never refold into their native conformation. This was the case for all VH domains selected from the germ-line library in this study, including MG2x1. However, some of the VH domains selected from the frame-mutation library of MG2x1 by TAPE were folded reversibly after denaturation. Far-UV circular dichroism (CD) spectra suggested that MG8-14 could be reversibly folded after denaturation heating at 85°C (Figure 7C), whereas the parental VH domain, MG2x1, could not (Figure 7A). Furthermore, the modified MG8-14 [L50W] had a perfect renaturation profile (Figure 7D). MG8-6 had the highest Tm, but could not refold after denaturation (Figure 7B). The recovery yield for the selected VH after heat denaturation reached 95% (Table 3), in contrast to that of the parental sequence (MG2x1), which was below 5%.
Figure 7. Far-UV CD spectra for the detection of reversible folding.
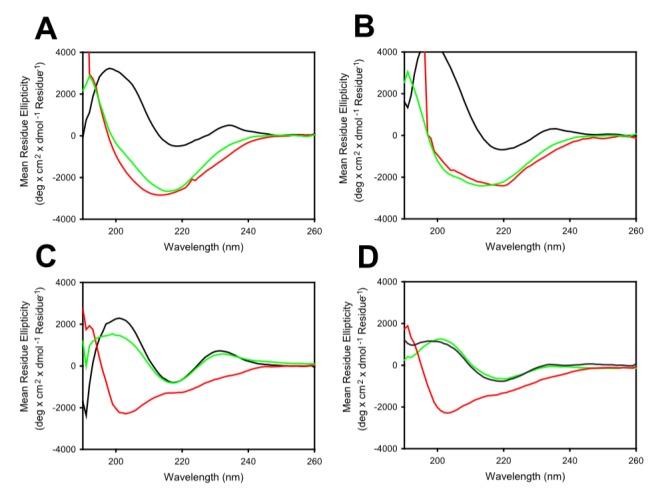
(A) MG2x1. (B) MG8-6. (C) MG8-14. (D) Modified MG8-14 [L50W]. Black lines indicate profiles for VH in native state at 25°C; red lines indicate the profile for VH denatured at 85°C; and green lines indicate profiles for VH renatured at 25°C.
Table 3. The recovery yields of selected VH after thermal stress. Data are means and standard deviation for three independent treatment of heat denaturation within the same sample.
| Initial A280 nm a | Final A280 nm b | Recovery yield (%)c | |
| MG2x1 (parent) | 0.964±0.011 | 0.041±0.006 | 3.843±0.662 |
| MG8-4 | 0.714±0.013 | 0.653±0.049 | 91.389±5.241 |
| MG8-14 | 0.726±0.010 | 0.643±0.062 | 88.458±7.370 |
| MG8-14 [L50W] | 0.722±0.011 | 0.695±0.022 | 96.257±2.305 |
| HEL4 | 0.964±0.011 | 0.878±0.032 | 91.097±2.348 |
Absorbance at 280 nm at 25°C.
Absorbance at 280 nm after heating (85°C) followed by cooling (25°C). Aggregates after heating were removed by centrifugation.
The recovery yield was defined as the fraction of soluble VH remaining after heating at the denaturation temperature (85°C).
Structural features underlying the superior biophysical properties of selected VH domains
Superimposition of crystal structures of the parental VH, MG2x1 (PDB ID: 3ZHK), and the modified VH domains MG8-4 (PDB ID: 3ZHD) and MG8-14 (PDB ID: 3ZHL) revealed that these proteins have the same overall topology: two β-sheets connected by a disulfide bond between C22 and C96, yielding a typical β-sandwich lectin fold structure (Figure 8A and Table S2). The random amino acid changes introduced in the combinatorial frame-mutation library of MG2x1 are positioned on the β-strand that forms the sandwich scaffolds; in particular, they are located on the side of the sandwich corresponding to the hydrophobic interface region between heavy and light chains in the typical Fab complex arrangement (Figure 8B). Mutations in MG8-4 and MG8-14 altered the conformation of the flexible CDRH3 loop, whereas the CDRH1 and CDRH2 loops remained in their original conformations (Figure 8C and Table S2).
Figure 8. Structures of MG2x1, MG8-4, and MG8-14.
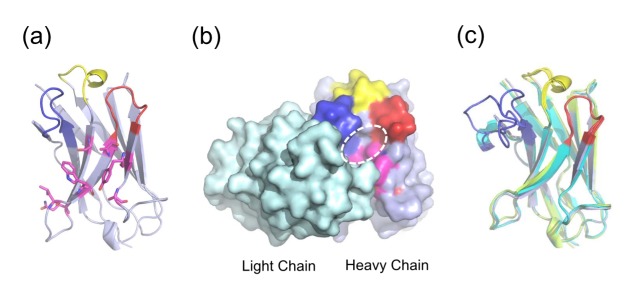
(A) Structure of MG2x1 with CDRH1 (yellow), CDRH2 (red), and CDRH3 (blue). Mutation sites for the MG2x1 mutation library are indicated as sticks (magenta). (B) Antibody light chain, in surface rendering (cyan), is shown to highlight the relative locations of CDR H1–H3 and the mutation sites in MG2x1 (magenta). The circle indicates the cavity area. (C) Superposition of three VH domains (MG2x1, MG8-4, and MG8-14), showing the variation in the loop in the CDRH3 region (blue).
Surface electrostatic calculations revealed that MG8-4 and MG8-14 exhibited increased partial negative charge next to the hydrophobic patch, possibly due to the introduction of a charged group such as aspartate (D) at position 60, whereas substantial positive charge was detected next to the exposed surface of the heavy chain in all three structures (MG2x1, MG8-4, and MG8-14) (Figure 9A and 9B). The solvation energies of MG8-4 and MG8-14 (−1166.8 kcal/mol and −1153.4 kcal/mol, respectively) were significantly lower than that of MG2x1 (−1047.5 kcal/mol), suggesting that the charged residues on the surface contribute to the solvation energy, and hence the solubility, of the protein. Analysis of surface features revealed an significantly increased number of hydrogen bonds between side chains of the residues of MG8-4 and MG8-14 (26 and 39, respectively), whereas only 19 hydrogen bonds were observed in MG2x1, indicating that the architecture of MG8-14 is more stable than that of MG2x1. In addition, the structures of MG8-4 and MG8-14 contained more charge-charge interactions (8 and 9, respectively) than the structure of MG2x1 (5) (Table 4).
Figure 9. Surface features of MG2x1, MG8-14, and modified MG8-14 [L50W].
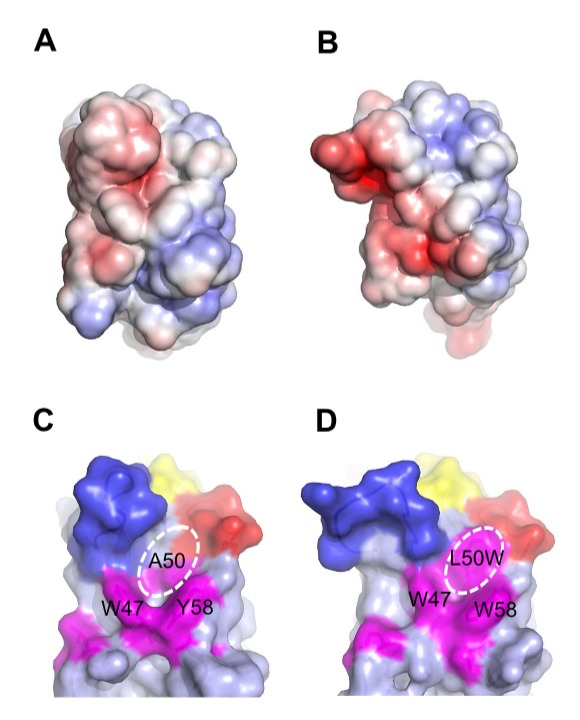
(A) Electrostatic charge distribution on the solvent-accessible surface of MG2x1 (red, −5 k; blue, +5 k). (B) Electrostatic charge distribution on the solvent-accessible surface of MG8-14. (C) Surface representation of MG2x1 showing the prominent cavity around residues 50 and 58. CDR regions are colored in yellow (CDRH1), red (CDRH2), and blue (CDRH3). Mutation sites are colored in magenta to highlight the cavity. (D) Surface representation of the structural model of the modified MG8-14 [L50W], in which L50 is replaced by W.
Table 4. Analyses of the structural features for MG2x1, MG8-4, and MG8-14.
| MG2x1 | MG8-4 | MG8-14 | MG8-14 [L50W] | |
| Solvent accessible surface (Å2) | 5958.6 | 6009.2 | 6393.5 | 6141.6 |
| Aromatic aromatic interaction | 4 | 4 | 4 | 4 |
| Main chain-sidechain hydrogen bonds | 48 | 49 | 48 | 48 |
| Side chain-side chain hydrogen bonds | 19 | 26 | 39 | 35 |
| Ionic interactions | 5 | 9 | 8 | 8 |
| Solvation energy (kcal/mol) | −1047.5 | −1166.8 | −1153.4 | −1155.9 |
MG2x1 contains a prominent pocket comprising residues W47, A50, and Y58, with a cavity area of 32 Å2 and a volume of 19.5 Å3, centered at residue A50 (Figure 8B and Figure 9C). Sequence analysis of VH domains selected by TAPE revealed that two positions in the framework, A50 and Y58, were consistently biased toward W. Residue A50 was also replaced by leucine (L) or W in representative selected VH domains such as MG8-4, MG8-14, MG8-6, and MG4-13, suggesting that replacement of this residue with a bulky side chain is related to the stability of the molecule. The structural model of the modified MG8-14 [L50W] suggests that the cavity is filled with a triad bulky side chains consisting of 50 W, W47, and W58 (Figure 9D). Accordingly, the modified MG8-14 [L50W] exhibited high thermodynamic stability as well as reversible folding after heat denaturation (Figure 7D).
Validation of the combinatorial CDRH synthetic library built on MG8-14 scaffold
To confirm the effects of CDR variation on the stability of VH scaffold, we examined the soluble expression level of VH domains containing CDRH3 regions of various lengths (7–13 amino acids), using a combinatorial CDRH synthetic library based on MG8-14. Eight or nine different sequences of each length were randomly selected and expressed in E. coli; 64 of 73 (88%) VH clones were expressed in soluble form. In addition, 11 different sequences from a rational mutation library (CDRH3 length fixed and seven positions of CDRH1, 2 and 3 of MG8-14 were randomized) were randomly tested; all of the test sequences were expressed in soluble form in the cytoplasm of E. coli under reducing conditions (Figure 10). Thus, aggregation was infrequently occurred regardless of CDR alteration in a combinatorial CDRH library that used the MG8-14 framework as a scaffold.
Figure 10. Validation of the combinatorial CDRH synthetic library built on MG8-14 scaffold.
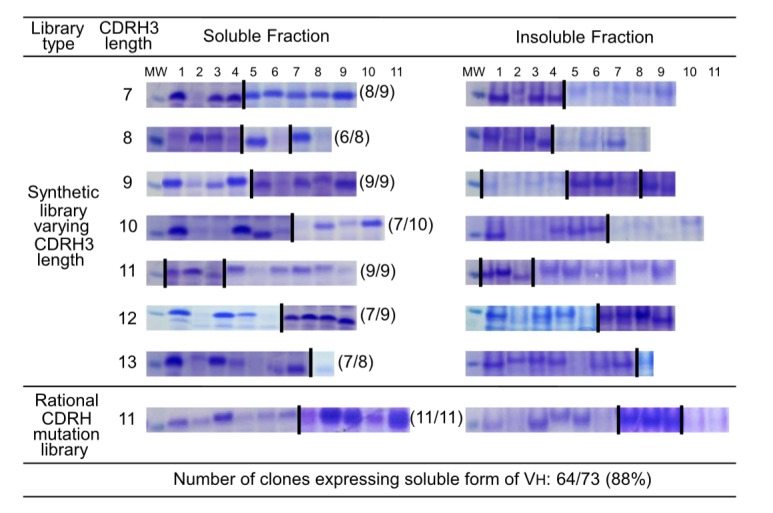
SDS-PAGE of soluble and insoluble fractions of E. coli expressing VH domains selected randomly from the combinatorial CDRH3 synthetic libraries. Coomassie-stained gels are aligned by lane numbers (columns) and amino acid lengths of CDRH3 (rows). Images depict the region of the gel corresponding to the size of VH. Some images were combined with separate gels for the purpose of alignment (indicating with a dividing bar between gels). ‘MW’ indicates the protein size marker corresponding to a molecular weight of 15 kD.
Humoral response to MG2x1 and MG8-14 in mouse
To test the humoral immune response of the selected VH domains, BALB/c mice were subjected to repeated immunization with selected VH domains, administered by various routes. Antibody against MG2x1 was undetectable after nine intravenous injections of 10 µg protein over 9 weeks (Figure 11A). Furthermore, there was no antibody-boosting response, even when injections included Freund′s Complete Adjuvant (CFA), in four of six mice at week 9 (Figure 11A). In the case of MG8-14, there was no detectable anti–MG8-14 antibody until week 6, although a mild antibody response was present in half of the tested mice at week 9 (Figure 11A). On the other hand, a camel single-domain antibody, VHH [23], was more immunogenic than MG2x1 and MG8-14, as shown by the high titer after the first injection (with CFA) at week 3 (Figure 11B). Intramuscular and subcutaneous injection of 1 µg MG8-14 resulted in no antibody response against MG8-14 throughout a 10-week course of immunization (Figure 11D), whereas VHH injection caused an increase in antibody titer starting at week 6 (Figure 11E). When mice were injected intramuscularly with 10 µg MG8-14, anti-MG8-14 antibody was elicited moderately at week 10 in only one of six mice. Subcutaneous injection of 10 µg MG8-14 elicited no antibody response until the fourth injection at week 6; moderate levels of anti-MG8-14 antibody were detectable after this time point (Figure 11D). Among mice subjected to intramuscular and subcutaneous injection of VHH, most animals exhibited an anti-VHH antibody response at week 4, immediately after the second injection (Figure E).
Figure 11. Humoral immune response of BALB/c mice.
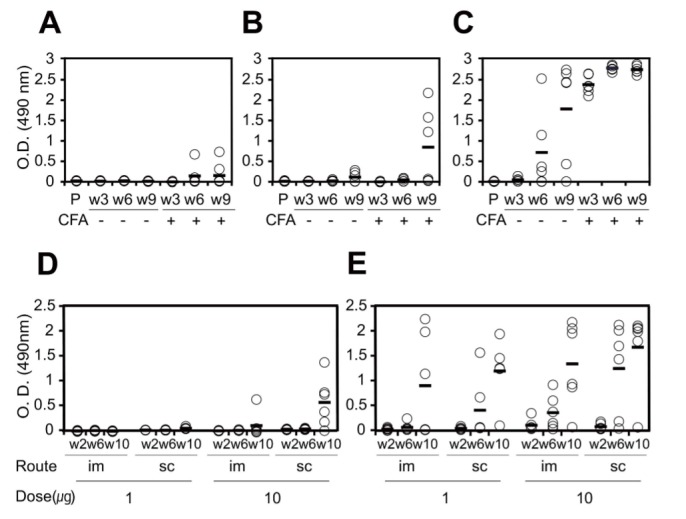
Six mice (per group) were received multiple intravenous injections of PBS (P), MG2x1 (A), MG2x1 plus CFA (A), MG8-14 (B), MG8-14 plus CFA (B), VHH (C), or VHH plus CFA (C) at week 3 (w3), week 6 (w6), and week 9 (w9). Six mice (per group) were received multiple injections of 1 µg MG8-14 intramuscularly (im, D), 1 µg MG8-14 subcutaneously (sc, D), 10 µg MG8-14 intramuscularly (im, D), 10 µg MG8-14 subcutaneously (sc, D), 1 µg VHH intramuscularly (im, E), 1 µg VHH subcutaneously (sc, E), 10 µg VHH intramuscularly (im, E), or 10 µg VHH subcutaneously (sc, E) at week 2 (w2), week 6 (w6), and week 10 (w10).
Discussion
The external diameter of the TatABC complex is around 160 Å, but its pore is relatively small [27]. Variations in complex size may result in variations in pore size, influencing the compatibility of each complex with differently sized Tat substrate proteins [28]. The capacity of the Tat system to export proteins via membrane-bound TatABC complexes varies among species of Gram-negative bacteria. For example, the A. tumefaciens TatABC complex is capable of exporting large (>80 kD) proteins [29], whereas in E. coli, the correlation between protein folding and export to the periplasm via the Tat pathway is poorer for proteins larger than 30 kDa than proteins of a lower molecular weight [26]. The molecular weight of the VH domain is around 14 kDa; therefore, this group of proteins was predicted to be compatible with the Tat pathway of E. coli. Consistent with this expectation, in this study, the export of VH in vivo corresponded well with properties related to protein stability in vitro. Accordingly, because the VH3 family is the most soluble of the seven VH families (VH1–7), the VH3 family was enriched via TAPE (Table 1) in a screen of a human germ-line library. This suggests that selection was driven by the function of the Tat pathway, which serves as a ‘molecular sieve’ in vivo as already discussed in many previous works [28], [30], [31].
We tried to compare ampicillin resistance of VH variants to the other variants by using visual measurement. For example, spot analyses of serial diluents of the culture containing ampicillin [22] was not sensitive to demonstrate the direct comparison of their resistance in this study (data not shown). To overcome this limitation, we performed a head-to-head competition of the ampicilline resistance among the 25 germ-line VH domains (the artificial library) with the third round of selection in liquid culture. This experiment resulted in MG2x1 as a sole survivor, a VH3 family member (VH3–23), which was used for the backbone of a frame-mutation library. This library was then subjected to another round of TAPE, with the goal of improving the physicochemical properties of this protein. Considering that MG2x1 is already relatively soluble and stable, one might expect only a marginal improvement from directed evolution via TAPE. However, subjecting the frame-mutation library to selection resulted in a significant improvement in folding-related properties.
Studies of the protein folding quality control mechanism of the E. coli Tat pathway have primarily focused on the tendency of proteins to be expressed in soluble form [19], [20]. However, the correlation between the selection via Tat-mediated protein folding and increases in the thermodynamic stabilities of proteins of interest has not been clearly demonstrated. In this study, we showed that both protein expression in soluble form and properties related to thermodynamic stability were clearly improved by Tat-associated screening. Foit et al. also demonstrated that antibiotic resistance bestowed by the tripartite fusion protein is correlated with stability in vivo and thermodynamic stability in vitro [32]. Although both methods use the same reporter gene, i.e., BLA, the protein folding occurs in a different environment, i.e., periplasm for the tripartite system and cytoplasm for TAPE. With the reduced condition of TAPE for protein folding, some of the evolved VH was capable of autonomous refolding over repeated cycles of heating and cooling. More reversible refolding and a higher recovery yield should increase resistance to mechanical or thermal stresses during the purification process, as well as improve long-term storage due to the low exposure rate of hydrophobic patches [33].
Christ et al. demonstrated that the frequency of aggregation-resistant domain was about 80% in the repertoire after heat-cooling selection and about 71% in the large aggregation-resistant repertoire generated by combinatorial ligation of CDR-encoding regions [34]. In this study, the frequency of aggregation-resistant VH domains in combinatorial CDRH3 repertoires with a fixed scaffold (MG8-14) screened by TAPE was 88%, regardless of the length of the CDRH3 region (Figure 10). With the exception of the CDRH3 region, the crystal structure of MG8-4 and MG8-14 superimposed closely with the parental VH, MG2x1, despite containing mutations in the frame region (Figure 8C). In addition, the atomic mobility of MG8-14 at residue L50 had the lowest observed B-factor (32), whereas the average B-factor was 43.3. These observations suggest that the core of this region is very rigid, but is still capable of accommodating various structures of CDRH3. As framework and CDR regions of the scaffold are conformational, a stability-functional tradeoffs are fully anticipated when the stability-enhancing mutation are introduced to the given functional protein, for example, scFv [20]. In contrast, we screened out the stable VH scaffold first and then generated the combinatorial CDRH synthetic library to give functionality later. As the affinity of VH domains we screened from the library against several antigens, including HER3, TNF-α, and albumin were all sub-nanomolar range, we can expect that the problems on a stability-functional tradeoffs would be a minimal when we screen the functional VH domains with this quality of the library (data not shown).
The modified MG8-14 [L50W] contains three W residues that fill a large cavity of MG2x1 near the VH/VL interface. Van der Waals interactions in this region would enhance stable architecture, allowing reversible folding of the antibody during the refolding process after denaturation. Within the cavity structure, high temperature leads to thermal destabilization as a result of water permeation [35], [36]. Therefore, water molecules in the hydrophobic cavity of MG2x1 may directly affect thermal resilience and promote structural perturbation. Taken together, these data demonstrate that surface properties are important factors in selection of single-domain antibodies with high solubility and thermodynamic stability.
VH domains that had been selected by heat-denatured phage display from a combinatorial CDR repertoire exhibited an enrichment of certain amino acids at several positions within the CDR regions, including glycine at position 35 and glutamate at position 32 [37]. Our differentiated in vivo selection strategy, using the Tat pathway in E. coli, resulted in a unique preference for tryptophan at positions 50 and 58, leading to the creation of a bulky ring structure. We believe that this preference helps VH to acquire a stable conformation, preventing structural perturbation during folding and refolding.
MG2x1 contains a negatively charged amino acid, aspartic acid (D) at position 61, which was previously identified as a determinant of protein aggregation and solubility [38]. In MG8-4 and MG8-14, which were selected from the MG2x1 frame-mutation library, D was incorporated consecutively at positions 60 and 61, significantly increasing the net negative charge. This preference for adjacent D residues has also been observed in other protein stability screens of combinatorial CDR repertoires. For example, positions 32 and 33 of VH and positions 52 and 53 of VL are determinants for aggregation resistance [37].
One important safety issue in protein therapeutics is related to immunogenicity. Many previous studies suggest that formation of sub-visible aggregates exerts a major influence on the humoral immune response [39], [40]. In this work, the antibody titer represents both the quantity and quality (affinity) of IgG that is specific to certain VH domain. Although we cannot discriminate which factor affects the titer more than the other does, it is obvious that the mouse immune system hardly responded to the selected VH domains even with CFA, compared to VHH as shown in Figure 11. This may be attributed to a favorable folding properties of the selected VH domains preventing aggregation, as we employed Tat-associated protein folding fitness filter.
Database access codes
The atomic coordinates and structure factors have been deposited in the Protein Data Bank. www.pdb.org (PDB ID: 3ZHL, 3ZHK and 3ZHD).
Supporting Information
SDS-PAGE of the purified VH domains used for the measurement of Far-UV CD spectra. (A) VH domains for testing thermodynamic stability were loaded as follows: 1, MG4x4-44; 2, MG10-10; 3, MG3-15; 4, MG2x1; 5, HEL4; 6, MG4-5; 7, MG8-14; 8, MG8-4; 9, MG8-6. (B) VH domains for testing reversible folding were loaded as follows: 1, MG2x1; 2, MG8-6; 3, MG8-14; 4, modified MG8-14 [L50W]. Lanes labeled ‘MW’ contained protein size markers.
(PDF)
Oligonucleotides used in this study.
(PDF)
Data collection and structure Solution parameters.
(PDF)
Funding Statement
The authors are grateful to Green Cross Corporation (http://www.greencross.com) for funding this work. The funder had no role in study design, data collection and analysis, decision to publish,or preparation of the manuscript.
References
- 1. Els Conrath K, Lauwereys M, Wyns L, Muyldermans S (2001) Camel single-domain antibodies as modular building units in bispecific and bivalent antibody constructs. J Biol Chem 276: 7346–7350. [DOI] [PubMed] [Google Scholar]
- 2. Bargou R, Leo E, Zugmaier G, Klinger M, Goebeler M, et al. (2008) Tumor regression in cancer patients by very low doses of a T cell-engaging antibody. Science 321: 974–977. [DOI] [PubMed] [Google Scholar]
- 3. Tijink BM, Laeremans T, Budde M, Stigter-van Walsum M, Dreier T, et al. (2008) Improved tumor targeting of anti-epidermal growth factor receptor Nanobodies through albumin binding: taking advantage of modular Nanobody technology. Mol. Cancer Ther 7: 2288–2297. [DOI] [PubMed] [Google Scholar]
- 4. Wesolowski J, Alzogaray V, Reyelt J, Unger M, Juarez K, et al. (2009) Single domain antibodies: promising experimental and therapeutic tools in infection and immunity. Med Microbiol Immunol 198: 157–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Enever C, Batuwangala T, Plummer C, Sepp A (2009) Next generation immunotherapeutics-honing the magic bullet. Curr Opin Biotechnol 20: 405–411. [DOI] [PubMed] [Google Scholar]
- 6. Hudson PJ, Souriau C (2003) Engineered antibodies. Nat Med 9: 129–134. [DOI] [PubMed] [Google Scholar]
- 7. Ward ES, Güssow D, Griffiths AD, Jones PT, Winter G (1989) Binding activities of a repertoire of single immunoglobulin variable domains secreted from Escherichia coli. Nature 341: 544–546. [DOI] [PubMed] [Google Scholar]
- 8. Dolk E, van Vliet C, Perez JM, Vriend G, Darbon H, et al. (2005) Induced refolding of a temperature denatured llama heavy-chain antibody fragment by its antigen. javascript:AL_get(this, ‘jour’, ‘Proteins.’). Proteins 59: 555–564. [DOI] [PubMed] [Google Scholar]
- 9. Ewert S, Cambillau C, Conrath K, Plückthun A (2002) Biophysical properties of camelid V(HH) domains compared to those of human V(H)3 domains. Biochemistry javascript:AL_get(this, ‘jour’, ‘Biochemistry.’). 41: 3628–3636. [DOI] [PubMed] [Google Scholar]
- 10. Kortt AA, Guthrie RE, Hinds MG, Power BE, Ivancic N, et al. (1995) Solution properties of Escherichia coli-expressed VH domain of anti-neuraminidase antibody NC41. J Protein Chem 14: 167–178. [DOI] [PubMed] [Google Scholar]
- 11. Davies J, Riechmann L (1994) ‘Camelising’ human antibody fragments: NMR studies on VH domains. FEBS Lett 339: 285–290. [DOI] [PubMed] [Google Scholar]
- 12. Aires da Silva F, Santa-Marta M, Freitas-Vieira A, Mascarenhas P, Barahona I, et al. (2004) Camelized rabbit-derived VH single-domain intrabodies against Vif strongly neutralize HIV-1 infectivity. J Mol Biol 340: 525–542. [DOI] [PubMed] [Google Scholar]
- 13. Riechmann L (1996) Rearrangement of the former VL interface in the solution structure of a camelised, single antibody VH domain. J Mol Biol 259: 957–969. [DOI] [PubMed] [Google Scholar]
- 14. Jespers L, Schon O, Famm K, Winter G (2004) Aggregation-resistant domain antibodies selected on phage by heat denaturation. Nat Biotechnol 22: 1161–1165. [DOI] [PubMed] [Google Scholar]
- 15. Barthelemy PA, Raab H, Appleton BA, Bond CJ, Wu P, et al. (2008) Comprehensive analysis of the factors contributing to the stability and solubility of autonomous human VH domains. J Biol Chem 283: 3639–3654. [DOI] [PubMed] [Google Scholar]
- 16. To R, Hirama T, Arbabi-Ghahroudi M, MacKenzie R, Wang P, et al. (2005) Isolation of monomeric human V(H)s by a phage selection. J Biol Chem 280: 41395–41403. [DOI] [PubMed] [Google Scholar]
- 17. Wigley WC, Stidham RD, Smith NM, Hunt JF, Thomas PJ (2001) Protein solubility and folding monitored in vivo by structural complementation of a genetic marker protein. Nat Biotechnol 19: 131–136. [DOI] [PubMed] [Google Scholar]
- 18. Cabantous S, Terwilliger TC, Waldo GS (2005) Protein tagging and detection with engineered self-assembling fragments of green fluorescent protein. Nat Biotechnol 23: 102–107. [DOI] [PubMed] [Google Scholar]
- 19. Karlsson AJ, Lim HK, Xu H, Rocco MA, Bratkowski MA, et al. (2012) Engineering antibody fitness and function using membrane-anchored display of correctly folded proteins. J Mol Biol 416: 94–107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Fisher AC, DeLisa MP (2009) Efficient isolation of soluble intracellular single-chain antibodies using the twin-arginine translocation machinery. J Mol Biol 385: 299–311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Ribnicky B, Van Blarcom T, Georgiou G (2007) A scFv antibody mutant isolated in a genetic screen for improved export via the twin arginine transporter pathway exhibits faster folding. J Mol Biol 369: 631–639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Fisher AC, Kim W, DeLisa MP (2006) Genetic selection for protein solubility enabled by the folding quality control feature of the twin-arginine translocation pathway. Protein Sci 15: 449–458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Silence K, Lauwereys M, De Haard H (2011) Single domain antibodies directed against tumor necrosis factor-alpha and uses therefor. EP2390268A1.
- 24. Jespers L, Schon O, James LC, Veprintsev D, Winter G (2004) Crystal structure of HEL4, a soluble, refoldable human V(H) single domain with a germ-line scaffold. J Mol Biol 337: 893–903. [DOI] [PubMed] [Google Scholar]
- 25. Rothe C, Urlinger S, Löhning C, Prassler J, Stark Y, et al. (2008) The human combinatorial antibody library HuCAL GOLD combines diversification of all six CDRs according to the natural immune system with a novel display method for efficient selection of high-affinity antibodies. J Mol Biol 376: 1182–1200. [DOI] [PubMed] [Google Scholar]
- 26. Lim HK, Mansell TJ, Linderman SW, Fisher AC, Dyson MR, et al. (2009) Mining mammalian genomes for folding competent proteins using Tat-dependent genetic selection in Escherichia coli. Protein Sci 18: 2537–2549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Gohlke U, Pullan L, McDevitt CA, Porcelli I, de Leeuw E, et al. (2005) The TatA component of the twin-arginine protein transport system forms channel complexes of variable diameter. Proc Natl Acad Sci USA 102: 10482–10486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Lee PA, Tullman-Ercek D, Georgiou G (2006) The bacterial twin-arginine translocation pathway. Annu Rev Microbiol 60: 373–395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Oates J, Mathers J, Mangels D, Kühlbrandt W, Robinson C, et al. (2003) Consensus structural features of purified bacterial TatABC complexes. J Mol Biol 330: 277–286. [DOI] [PubMed] [Google Scholar]
- 30. DeLisa MP, Tullman D, Georgiou G (2003) Folding quality control in the export of proteins by the bacterial twin-arginine translocation pathway. Proc Natl Acad Sci USA 100: 6115–6120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Berks BC, Palmer T, Sargent F (2005) Protein targeting by the bacterial twin-arginine translocation (Tat) pathway. Curr Opin Microbiol 8: 174–181. [DOI] [PubMed] [Google Scholar]
- 32. Foit L, Morgan GJ, Kern MJ, Steimer LR, von Hacht AA, et al. (2009) Optimizing protein stability in vivo. Mol Cell 36: 861–871. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Joubert MK, Luo Q, Nashed-Samuel Y, Wypych J, Narhi LO (2011) Classification and characterization of therapeutic antibody aggregates. J Biol Chem 286: 25118–25133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Christ D, Famm K, Winter G (2007) Repertoires of aggregation-resistant human antibody domains. Protein Eng Des Sel 20: 413–416. [DOI] [PubMed] [Google Scholar]
- 35. Jung TY, Li D, Park JT, Yoon SM, Tran PL, et al. (2012) Association of novel domain in active site of archaic hyperthermophilic maltogenic amylase from Staphylothermus marinus. J Biol Chem 287: 7979–7989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Bleicher L, Prates ET, Gomes TC, Silveira RL, Nascimento AS, et al. (2011) Molecular basis of the thermostability and thermophilicity of laminarinases: X-ray structure of the hyperthermostable laminarinase from Rhodothermus marinus and molecular dynamics simulations. J Phys Chem B 115: 7940–7949. [DOI] [PubMed] [Google Scholar]
- 37. Dudgeon K, Rouet R, Kokmeijer I, Schofield P, Stolp J, et al. (2012) General Strategy for the generation of human antibody variable domains with increased aggregation resistance. Proc Natl Acad Sci USA 109: 10879–10884. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Shaw BF, Schneider GF, Bilgiçer B, Kaufman GK, Neveu JM, et al. (2008) Lysine acetylation can generate highly charged enzymes with increased resistance toward irreversible inactivation. Protein Sci 17: 1446–1455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Schellekens H (2002) Bioequivalence and the immunogenicity of biopharmaceuticals. Nat Rev Drug Discov 1: 457–462. [DOI] [PubMed] [Google Scholar]
- 40. Hermeling S, Crommelin DJ, Schellekens H, Jiskoot W (2004) Structure immunogenicity relationships of therapeutic proteins. Pharm Res 21: 897–903. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
SDS-PAGE of the purified VH domains used for the measurement of Far-UV CD spectra. (A) VH domains for testing thermodynamic stability were loaded as follows: 1, MG4x4-44; 2, MG10-10; 3, MG3-15; 4, MG2x1; 5, HEL4; 6, MG4-5; 7, MG8-14; 8, MG8-4; 9, MG8-6. (B) VH domains for testing reversible folding were loaded as follows: 1, MG2x1; 2, MG8-6; 3, MG8-14; 4, modified MG8-14 [L50W]. Lanes labeled ‘MW’ contained protein size markers.
(PDF)
Oligonucleotides used in this study.
(PDF)
Data collection and structure Solution parameters.
(PDF)