

Abstract
Stable isotope fractionation was studied during the degradation of m-xylene, o-xylene, m-cresol, and p-cresol with two pure cultures of sulfate-reducing bacteria. Degradation of all four compounds is initiated by a fumarate addition reaction by a glycyl radical enzyme, analogous to the well-studied benzylsuccinate synthase reaction in toluene degradation. The extent of stable carbon isotope fractionation caused by these radical-type reactions was between enrichment factors (ɛ) of −1.5 and −3.9‰, which is in the same order of magnitude as data provided before for anaerobic toluene degradation. Based on our results, an analysis of isotope fractionation should be applicable for the evaluation of in situ bioremediation of all contaminants degraded by glycyl radical enzyme mechanisms that are smaller than 14 carbon atoms. In order to compare carbon isotope fractionations upon the degradation of various substrates whose numbers of carbon atoms differ, intrinsic ɛ (ɛintrinsic) were calculated. A comparison of ɛintrinsic at the single carbon atoms of the molecule where the benzylsuccinate synthase reaction took place with compound-specific ɛ elucidated that both varied on average to the same extent. Despite variations during the degradation of different substrates, the range of ɛ found for glycyl radical reactions was reasonably narrow to propose that rough estimates of biodegradation in situ might be given by using an average ɛ if no fractionation factor is available for single compounds.
Many biochemical reactions are known to cause the fractionation of stable isotopes. Molecules consisting of lighter isotopes are utilized preferentially, and consequently heavier molecules are enriched in the residual substrate pool. Well-known examples are autotrophic CO2 fixation by plants (22) and bacterial methanogenesis (14). Within the last few years, several studies investigated stable isotope fractionation during bacterial degradation of contaminants such as toluene (17), benzene (15), and chlorinated hydrocarbons (12) that were associated with a significant enrichment of 13C in the residual substrate fraction. These findings opened the door to assessing contaminant degradation at polluted sites qualitatively, and under certain conditions even quantitatively, and of outlining the future development of the contamination and its potential impact on the environment and the drinking water supply. This concept has been applied successfully in several field experiments so far (12, 16, 24, 25).
Basic features of microbial stable isotope fractionation have been examined for anaerobic and aerobic bacterial cultures with toluene as a model substrate (18). It was demonstrated that the first enzyme reaction of toluene degradation is the rate-limiting step and that this reaction is also the key process leading to isotope fractionation. Isotope effects due to the transport of toluene to and into the cells were negligible. Furthermore, it was shown that various aerobic and anaerobic degradation reactions led to characteristic degrees of fractionation (19). However, it is not possible to deduce the type of the underlying reaction mechanism from the extent of fractionation, because fractionation factors (α) can vary significantly between identical reactions. Studies dealing with the stable isotope fractionation of particular reaction mechanisms have provided information about the span of isotope fractionation effects caused by these mechanisms. Based on theoretical considerations and calculations, it was proposed that the rate-limiting step in the benzylsuccinate synthase reaction is the addition of fumarate to the benzyl radical (10).
Stable isotope fractionation during degradation reactions initiated by glycyl radical enzymes is of particular interest because this reaction mechanism seems to be predominant in the anaerobic degradation of contaminants such as aromatic hydrocarbons, alkyl phenols, and alkanes. Regardless of the electron acceptor employed, anaerobic bacterial toluene degradation proceeds via benzylsuccinate formation in all cases investigated so far (8, 26). The enzyme mechanism was first described for anaerobic toluene degradation by denitrifying bacteria, where the enzyme benzylsuccinate synthase catalyzes the addition of a fumarate molecule to the methyl group to form benzylsuccinate (3). Fumarate addition and subsequent activation and β oxidation convert the former methyl group to a carbonylic function that acts as an entry port for single electrons in the subsequent ring reduction (4). The glycyl radical formed in benzylsuccinate synthase and related enzymes derives from 5′-deoxyadenosyl that is a cleavage product of S-adenosylmethionine (7). The enzyme reaction withdraws one hydrogen atom from a highly conserved cysteine residue of the enzyme. This thiyl radical then takes one hydrogen atom from the methyl group of the aromatic substrate, which later is transferred from the cysteine residue to the Cβ of the succinyl side chain of the produced benzylsuccinate (7, 13) (Fig. 1).
FIG. 1.

Reaction mechanism proposed for benzylsuccinate synthase modified according to the work of Frey (7) for m-xylene, o-xylene, m-cresol, and p-cresol, with R1, R2, and R3 equal to H, CH3, and OH, respectively. E, polypeptide chain of the enzyme.
We investigated stable isotope fractionation in degradation pathways initiated by a glycyl radical mechanism with two bacterial strains. Desulfobacterium cetonicum, a toluene-degrading sulfate reducer, was shown to degrade m-cresol and p-cresol via fumarate addition (20, 21). Enzyme activities in cell extracts showed that the cresols are probably not converted by the toluene-degrading benzylsuccinate synthase, but whether the two different cresol isomers are attacked by the same enzyme remains undetermined.
Degradation of m-xylene and o-xylene by the sulfate-reducing strain OX39 also proceeds via fumarate addition, which was confirmed by the identification of methylbenzylsuccinate derivatives in culture supernatants (B. Morasch, unpublished data). Induction experiments with strain OX39 and with m-xylene, o-xylene, and toluene showed that every substrate needed a specific enzyme for degradation (B. Morasch, unpublished data).
The objective of the present study was to systematically investigate stable carbon isotope fractionation during radical enzyme reactions of aromatic compounds which are prominent groundwater contaminants. The study should elucidate whether this type of reaction produces consistent isotope fractionation during degradation. For better comparison of isotope fractionations of the various substrate molecules, data for the intrinsic isotope fractionation at the molecular site of the reactions are provided.
MATERIALS AND METHODS
Cultivation of bacteria.
The sulfate-reducing strain OX39 was isolated from soil from a site contaminated with BTEX (benzene, toluene, ethylbenzene, and xylene) and polycyclic aromatic hydrocarbons near Stuttgart, Germany (B. Morasch, unpublished data). D. cetonicum (DSM 7267) was taken from the lab culture collection.
Bacteria were cultivated at 30°C in bicarbonate-buffered freshwater (strain OX39) or brackish (D. cetonicum) mineral medium, pH 7.4, with sulfate (10 mM) as the electron acceptor. The medium was prepared under an atmosphere of N2-CO2 (80:20) and reduced with Na2S (1 mM) (27). A sterile, anoxic FeCl2 solution was added to the medium of strain OX39 to a final concentration of 3 mM.
Strains were grown in 120-ml serum bottles half filled with mineral medium and tightly sealed with Viton rubber stoppers (Maag Technic, Dübendorf, Switzerland). Cultures for isotope fractionation experiments were inoculated with 6 ml of precultures. m-Xylene and o-xylene were injected directly into the culture bottles through the rubber stoppers with microsyringes. m-Cresol and p-cresol were added from aqueous stock solutions (100 mM). Metabolic activity was monitored by observing sulfide production (6).
Hydrocarbon analysis.
Xylene concentrations were determined by high-performance liquid chromatography (Bischoff Chromatography, Leonberg, Germany) with a C18 reversed-phase column (Prontosil, 200 by 3 mm, 3-μm film thickness; Bischoff) at 30°C and by UV detection at 210 nm with a mix of acetonitrile (Chromasol V super gradient grade; Fluka, Buchs, Switzerland) and demineralized water (70:30 [vol/vol]) as eluents. Cresol concentrations were determined by using a mix of acetonitrile and ammonium phosphate buffer (100 mM, pH 3.5) (50:50 [vol/vol]) as the eluent. Culture samples were diluted 1:5 with ethanol (99.9% gradient grade) and centrifuged (20,000 × g, 5 min) to remove precipitates before analysis.
Isotope analysis.
13C/12C stable carbon isotope ratios were determined by isotope ratio-monitoring gas chromatography-mass spectrometry. The system consisted of a gas chromatograph (HP-5890; Hewlett-Packard Co., San Diego, Calif.) connected via a combustion unit (gas chromatograph-combustion interface; Finnigan, Bremen, Germany) with an isotope mass spectrometer (MAT 252; Finnigan). Samples were measured as described previously (18).
Calculations.
The 13C/12C isotope ratios of the substrate were calculated as relative δ13C values (per mille) according to equation 1 below, where Rsample is the 13C/12C isotope ratio of the sample and Rstd is the isotope ratio of the international Pee Dee Belemnite standard (11). Kinetic isotope α (αC) were calculated by using equation 2, which is derived from the Rayleigh equation for closed systems (11, 23). Ct/C0 is the fraction of the substrate remaining in the sample at time t. If ln(Rt/R0) is plotted over ln(Ct/C0), for the time intervals (t), the slope of the linear regression curve gives the αC as (αC − 1). Enrichment factors (ɛ) are a convenient expression of stable isotope fractionation that can be retrieved directly from αC using equation 3 (5).
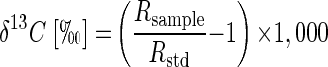 |
(1) |
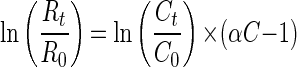 |
(2) |
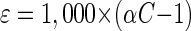 |
(3) |
Derivation of αintrinsic.
The general definition of the isotope fractionation factor α is
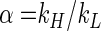 |
(4) |
where kH and kL are the reaction rate constants of the heavier and lighter isotopomeres, respectively. The intrinsic isotope fractionation factor α (αintrinsic) is defined as
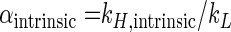 |
(5) |
with kH,intrinsic being the reaction rate constant at the position of the molecule where the chemical reaction takes place. For our model compound toluene, the heavier atom may be located at the methyl group (C-7) or at any position of the aromatic ring. Therefore, the overall reaction rate constant (kH) is the sum of the individual reaction rate constants (kH,i) for the location of the heavy atom at any position (i) of the labeled compound divided by the number of atoms (n) of the molecule, as shown in general equation 6.
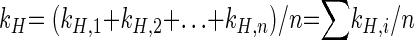 |
(6) |
In the specific case of the carbon stable isotope fractionation of toluene [kH = (kH,1 + kH,2 +… + kH,7)/7], the chemical reaction (fumarate addition) takes place at the methyl group of toluene (the C-7 atom). Therefore, only the reaction rate kH,7 (when the 13C atom is located at the methyl group) is subject to a kinetic isotope effect (kH,intrinsic). For all other reaction rate constants, kH,i, the 13C atom is located elsewhere in the molecule. With the assumption that secondary isotope effects are negligible compared to primary isotope effects, the kH,i rate constants for 12C atoms at the methyl group and 13C atoms at nonreactive positions of the molecule are assumed to be identical to the reaction rate constant kL for nonlabeled molecules. In addition, we assume that the probability of there being two 13C carbon atoms per toluene molecule is statistically unlikely due to the low natural abundance of 13C. With the exception of kH,intrinsic, the kH,i reaction rate constants are therefore equal to the reaction rate constant kL for the 12C isotopomeres. Equation 6 can therefore be written as
 |
(7) |
Equation 7 inserted into equation 4 gives
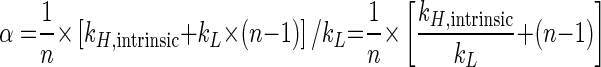 |
(8) |
Equation 5 inserted into equation 8 results in
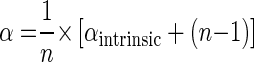 |
(9) |
The exponent of the Rayleigh equation 2 can therefore be written as
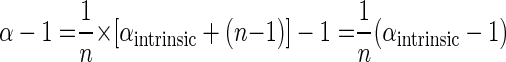 |
(10) |
or, according to equation 3, as
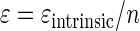 |
(11) |
RESULTS AND DISCUSSION
Stable 13C/12C isotope fractionation was determined during the growth of D. cetonicum with m-cresol and p-cresol that had initial δ13C signatures of −29.32‰ ± 0.36‰ and −27.50‰ ± 0.36‰, respectively. Anaerobic m-cresol degradation caused a strong increase in δ13C in the residual substrate fraction, which is shown in a representative experiment where degradation of 68% of the initial 1,030 μM led to an isotope shift from −29.04‰ ± 0.47‰ to −24.53‰ ± 0.41‰ (Fig. 2A). The average isotope ɛ of −3.9‰ ± 0.5‰ for anaerobic m-cresol degradation was determined from four replicates (Fig. 3). Anaerobic degradation of p-cresol produced a shift in δ13C from −27.11‰ ± 0.17‰ to −23.44‰ ± 0.25‰ during the degradation of 89% of the initial 1,052 μM (data not shown). The smaller increase in δ13C per amount of substrate degraded resulted in an average ɛ of −1.6‰ ± 0.1‰ calculated from three replicates (Fig. 3). For comparison, the isotope fractionation produced during toluene degradation by D. cetonicum resulted in an ɛ of −2.2‰ ± 0.4‰ (18).
FIG. 2.

13C enrichment in the residual substrate fraction during the anaerobic degradation of m-cresol (▴) by D. cetonicum (A) and o-xylene (•) by the sulfate-reducing strain OX39 (B). Changes in sulfide concentration (Δ and ○) and δ13C (⧫) were monitored over time. The carbon isotope composition is presented as an average of six individual measurements, with error bars indicating the standard deviations. The diagram shows the results of one representative experiment out of four and of three replicates for m-cresol and o-xylene. d, days.
FIG. 3.

Stable carbon isotope fractionation during the degradation of m-xylene (—) and o-xylene (---) by strain OX39 and of m-cresol (·-·-) and p-cresol (···) by D. cetonicum. Regression lines of the 13C/12C isotope data over the respective concentrations are plotted according to equation 2, with r2 values of 0.968, 0.893, 0.942, and 0.946 for m-xylene, o-xylene, m-cresol, and p-cresol, respectively.
Stable carbon isotope fractionation was determined during the degradation of m-xylene and o-xylene by strain OX39 in three independent growth experiments per substrate. The initial δ13C carbon signatures of the substrates were −27.66‰ ± 0.44‰ for m-xylene and −28.19‰ ± 0.06‰ for o-xylene. During the degradation of 82% of the initial m-xylene (203 μM), the 13C isotope became enriched to a level of −24.81‰ ± 0.16‰ in one of three growth experiments (data not shown). Three replicate m-xylene degradation experiments yielded an average ɛ of −1.8‰ ± 0.2‰ (Fig. 3). Degradation of an initial o-xylene concentration of 257 μM by strain OX39 resulted in a δ13C shift from −28.28‰ ± 0.13‰ to −24.72‰ ± 0.19‰ when 85% of the substrate was used (Fig. 2B). The average ɛ of three replicates was −1.5‰ ± 0.1‰ (Fig. 3) and thus similar to the factor found for the degradation of the m-xylene isomer. In previous degradation experiments using a sediment column filled with contaminated aquifer material under sulfate-reducing conditions, carbon isotope fractionation by an ɛ of −1.1‰ for o-xylene, which was lower than the ɛ reported here, was determined (24). Furthermore, carbon isotope fractionation resulting in an ɛ of −0.9‰ ± 0.1‰ was found for the degradation of 2-methylnaphthalene by a sulfate-reducing enrichment culture (9) which also initiates degradation via fumarate addition (2).
By far the most information on isotope fractionation by glycyl radical reactions is available concerning toluene degradation under various redox conditions. In a comparative study, carbon isotope α determined for bacterial pure cultures using Fe(III), NO3−, or SO42− as electron acceptors were found to be between an ɛ of −1.8‰ and an ɛ of −1.7‰ (17). These results were in the same order as those we obtained for p-cresol, m-xylene, and o-xylene. Another study reported isotope fractionation upon toluene degradation yielding ɛ of −0.8‰ and −0.5‰ by sulfate-reducing and methanogenic enrichment cultures, respectively, which were significantly lower than those measured in our experiments (1).
To summarize the data available on isotope fractionation during degradation reactions employing glycyl radical mechanisms, the lowest fractionation (ɛ = −0.5‰) was found for toluene degradation under methanogenic conditions, and the strongest fractionation (ɛ = −3.9‰) was found for m-cresol degradation by D. cetonicum. Most ɛ obtained were about −1.8‰. It should be emphasized that isotope fractionation in the radical reactions of the benzylsuccinate synthase-type enzymes involves just one methyl carbon atom at the reactive site. However, determination of ɛ by compound-specific isotope analysis gives overall δ13C values of the entire molecule. This discrepancy can be overcome by calculating the fractionation factor site-specific isotopes αintrinsic, which refers to the atom of the target molecule where the enzymatic reaction takes place (equation 9).
The use of this equation allows the normalization of isotope fractionation of aromatic compounds with different numbers of carbon atoms in order to study specific reactions in detail. However, the comparison of intrinsic enrichment factors (ɛintrinsic) upon reactions of the benzylsuccinate synthase type in Table 1 shows the same extent of variation of the average of the intrinsic enrichment factors (ɛintrinsic = −12.3 ± 5.8‰) as the average of the overall enrichment factors (ɛ = −1.67 ± 0.86‰). The standard deviations are 51 and 47%, respectively (Table 1). Obviously, the specific properties of every single enzyme catalyzing the same type of reaction but taking other substrates influence the degree of isotope fractionation. Thus, the use of the ɛintrinsic does not lead to additional information.
TABLE 1.
Comparison of compound-specific carbon isotope enrichment factor ɛ to the ɛintrinsic calculated for the carbon atoms at the molecular site of the reaction of the respective substratea
| Bacterial strain or culture | Substrate | ɛ | ɛintrinsic | Reference |
|---|---|---|---|---|
| Thauera aromatica | Toluene | −1.7 | −11.9 | 17 |
| Geobacter metallireducens | Toluene | −1.8 | −12.6 | 17 |
| Sulfate-reducing strain TRM1 | Toluene | −1.7 | −11.9 | 17 |
| Desulfobacterium cetonicum | Toluene | −2.2 | −15.4 | 18 |
| Sulfate-reducing enrichment culture | Toluene | −0.8 | −5.6 | 1 |
| Methanogenic enrichment culture | Toluene | −0.5 | −3.5 | 1 |
| Sulfate-reducing strain OX39 | m-Xylene | −1.8 | −14.4 | This study |
| Sulfate-reducing strain OX39 | o-Xylene | −1.5 | −12.0 | This study |
| Desulfobacterium cetonicum | m-Cresol | −3.9 | −27.3 | This study |
| Desulfobacterium cetonicum | p-Cresol | −1.6 | −11.2 | This study |
| Sulfate-reducing enrichment culture | 2-Methylnaphthalene | −0.9 | −9.9 | 9 |
| Avg | −1.67 ± 0.86 | −12.3 ± 5.8 |
Average 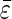 and
and 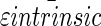 ± standard deviations were calculated from the sum of all data presented in the table.
± standard deviations were calculated from the sum of all data presented in the table.
Based on the observed isotope enrichment factor ɛ, we estimated the maximal molecular mass of the substrate that would still allow us to measure isotope fractionation accurately in the field. As shown above, the ɛintrinsic of enzyme reactions of the benzylsuccinate type are more or less equal. However, the larger the molecular mass of the substrate, the less overall isotope fractionation can be measured, because the isotope effect is diluted with an increasing number of carbon atoms. Taking an average inaccuracy of isotope analysis of ±0.5‰ at the detection limits, the absolute isotope shift needed to reliably detect isotope fractionation would be about 2‰ if about 90% of the substrate was degraded. This value correlates to an overall ɛ of −0.895‰. If we take the average intrinsic ɛ (ɛintrinsic) from Table 1, the presumptions would be fulfilled for molecules such as toluene with seven carbon atoms (ɛoverall = −1.76‰) or xylene with eight carbon atoms (ɛoverall = −1.54‰). The calculated borderline would be at molecules of 13 carbon atoms, with ɛ of −0.949‰. Beyond 14 carbon atoms, the overall ɛ (−0.881‰) is probably too low to allow significant isotope shifts upon degradation to be analyzed. 2-Methylnaphthalene with 11 carbon atoms (Table 1) is close to the analytical limit, but isotope fractionation can be measured under ideal conditions. However, the variations of the intrinsic isotope effects reported above emphasize the insecurity of exact measurements in the field, and the limitation for field applications will probably be at 11 to 12 carbon atoms.
In situ, the enrichment of heavier isotopes along a groundwater flow can be taken as qualitative evidence for biodegradation. In addition, the extent of bacterial degradation can be quantified by combining the isotope signatures determined in the field with the laboratory-derived α and using the Rayleigh equation (equation 2) (16, 24). The ɛ for m-cresol, p-cresol, m-xylene, and o-xylene from this study were used together with assumed changes in the isotope signatures between δ13C levels of −30‰ and −20‰ to calculate the portion of substrate remaining compared to the initial concentration (Fig. 4). The percentage of biodegradation is defined according to equation 12 (24):
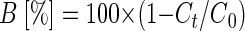 |
(12) |
To give an impression of how accurate an average α would be, we used an ɛ of −1.6‰ ± 0.86‰ from Table 1 to calculate the extent of biodegradation based on assumed isotope shifts. At small isotope shifts of 1% in the field, the extent of biodegradation would be 45.5% ± 16.15% (Fig. 4). The error becomes smaller as the extent of biodegradation increases; e.g., an isotope shift of 5‰ results in an average biodegradation of 95.1% ± 6.87% (Fig. 4). Thus, it might be possible to apply an average ɛ to calculate the biodegradation of methylated compounds by glycyl radical enzymes in contaminated aquifers if the shifts in δ13C are sufficiently high.
FIG. 4.

Calculation of the percentage of bacterial biodegradation based on the assumed amount of δ13C (per mille) isotope shifts. Curves with symbols are based on the isotope enrichment factors determined here for D. cetonicum degrading m-cresol (▴) and p-cresol (▾) and for strain OX39 degrading m-xylene (○) and o-xylene (□). The error range of biodegradation calculated for the average glycyl radical type enrichment factor is marked in grey.
Acknowledgments
We are grateful to Stefan Haderlein for continuous support and to Christian Griebler for assistance with the calculation of error propagations.
This work was financially supported by the Deutsche Forschungsgemeinschaft (grant Schi 180/7) and by the Bundesministerium für Bildung und Forschung (grant 02WT0022).
REFERENCES
- 1.Ahad, J. M. E., B. Sherwood Lollar, E. A. Edwards, G. F. Slater, and B. E. Sleep. 2000. Carbon isotope fractionation during anaerobic biodegradation of toluene: implications for intrinsic bioremediation. Environ. Sci. Technol. 34:892-896. [Google Scholar]
- 2.Annweiler, E., A. Materna, M. Safinowski, A. Kappler, H. H. Richnow, W. Michaelis, and R. U. Meckenstock. 2000. Anaerobic degradation of 2-methylnaphthalene by a sulfate-reducing enrichment culture. Appl. Environ. Microbiol. 66:5329-5333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Biegert, T., G. Fuchs, and J. Heider. 1996. Evidence that anaerobic oxidation of toluene in the denitrifying bacterium Thauera aromatica is initiated by formation of benzylsuccinate from toluene and fumarate. Eur. J. Biochem. 238:661-668. [DOI] [PubMed] [Google Scholar]
- 4.Boll, M., G. Fuchs, and J. Heider. 2002. Anaerobic oxidation of aromatic compounds and hydrocarbons. Curr. Opin. Chem. Biol. 6:604-611. [DOI] [PubMed] [Google Scholar]
- 5.Clark, I., and P. Fritz. 1997. Environmental isotopes in hydrogeology. Lewis Publishers, Boca Raton, Fla.
- 6.Cline, J. D. 1969. Spectrophotometric determination of hydrogen sulfide in natural waters. Limnol. Oceanogr. 14:454-458. [Google Scholar]
- 7.Frey, P. A. 2001. Radical mechanisms of enzymatic catalysis. Annu. Rev. Biochem. 70:121-148. [DOI] [PubMed] [Google Scholar]
- 8.Gibson, J., and C. S. Harwood. 2002. Metabolic diversity in aromatic compound utilization by anaerobic microbes. Annu. Rev. Microbiol. 56:345-369. [DOI] [PubMed] [Google Scholar]
- 9.Griebler, C., M. Safinowski, A. Vieth, H. H. Richnow, and R. U. Meckenstock. 2004. Combined application of stable carbon isotope analysis and specific metabolites determination for assessing in situ degradation of aromatic hydrocarbons in a tar oil-contaminated aquifer. Environ. Sci. Technol. 38:617-631. [DOI] [PubMed] [Google Scholar]
- 10.Himo, F. 2002. Catalytic mechanism of benzylsuccinate synthase, a theoretical study. J. Phys. Chem. B 106:7688-7692. [Google Scholar]
- 11.Hoefs, J. 1997. Stable isotope geochemistry, 4th ed. Springer Verlag, Berlin, Germany.
- 12.Hunkeler, D., R. Aravena, and B. J. Butler. 1999. Monitoring microbial dechlorination of tetrachloroethene (PCE) in groundwater using compound-specific stable carbon isotope ratios: microcosm and field studies. Environ. Sci. Technol. 33:2733-2738. [Google Scholar]
- 13.Krieger, C. J., W. Roseboom, S. P. J. Albracht, and A. M. Spormann. 2001. A stable organic free radical in anaerobic benzylsuccinate synthase of Azoarcus sp. strain T. J. Biol. Chem. 276:12924-12972. [DOI] [PubMed] [Google Scholar]
- 14.Krzycki, J. A., W. R. Kenealy, M. J. DeNiro, and J. G. Zeikus. 1987. Stable carbon isotope fractionation by Methanosarcina barkeri during methanogenesis from acetate, methanol, or carbon dioxide-hydrogen. Appl. Environ. Microbiol. 53:2597-2599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Mancini, S. A., A. C. Ulrich, G. Lacrampe-Couloume, B. Sleep, E. A. Edwards, and B. Sherwood Lollar. 2003. Carbon and hydrogen isotopic fractionation during anaerobic biodegradation of benzene. Appl. Environ. Microbiol. 69:191-198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Meckenstock, R. U., B. Morasch, M. Kästner, A. Vieth, and H. H. Richnow. 2002. Assessment of bacterial degradation of aromatic hydrocarbons in the environment by analysis of stable carbon isotope fractionation. Water Air Soil Pollut. Focus 2:141-152. [Google Scholar]
- 17.Meckenstock, R. U., B. Morasch, R. Warthmann, B. Schink, E. Annweiler, W. Michaelis, and H. H. Richnow. 1999. 13C/12C isotope fractionation of aromatic hydrocarbons during microbial degradation. Environ. Microbiol. 1:409-414. [DOI] [PubMed] [Google Scholar]
- 18.Morasch, B., H. H. Richnow, B. Schink, and R. U. Meckenstock. 2001. Stable hydrogen and carbon isotope fractionation during microbial toluene degradation: mechanistic and environmental aspects. Appl. Environ. Microbiol. 67:4842-4849. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Morasch, B., H. H. Richnow, B. Schink, A. Vieth, and R. U. Meckenstock. 2002. Carbon and hydrogen stable isotope fractionation during aerobic bacterial degradation of aromatic hydrocarbons. Appl. Environ. Microbiol. 68:5191-5194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Müller, J. A., A. Galushko, A. Kappler, and B. Schink. 1999. Anaerobic degradation of m-cresol by Desulfobacterium cetonicum is initiated by formation of 3-hydroxybenzylsuccinate. Arch. Microbiol. 172:287-294. [DOI] [PubMed] [Google Scholar]
- 21.Müller, J. A., A. S. Galushko, A. Kappler, and B. Schink. 2001. Initiation of anaerobic degradation of p-cresol by formation of 4-hydroxybenzylsuccinate in Desulfobacterium cetonicum. J. Bacteriol. 183:752-757. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.O'Leary, M. H. 1984. Carbon isotope fractionation in plants. Phytochemistry 20:553-567. [Google Scholar]
- 23.Rayleigh, J. W. S. 1896. Theoretical considerations respecting the separation of gases by diffusion and similar processes. Philos. Mag. 42:493-498. [Google Scholar]
- 24.Richnow, H. H., E. Annweiler, W. Michaelis, and R. U. Meckenstock. 2003. Microbial in situ degradation of aromatic hydrocarbons in a contaminated aquifer monitored by carbon isotope fractionation. J. Contam. Hydrol. 65:101-120. [DOI] [PubMed] [Google Scholar]
- 25.Sherwood Lollar, B., G. F. Slater, B. Sleep, M. Witt, G. M. Klecka, M. Harkness, and J. Spivack. 2001. Stable carbon isotope evidence for intrinsic bioremediation of tetrachloroethene and trichloroethene at area 6, Dover Air Force Base. Environ. Sci. Technol. 35:261-269. [DOI] [PubMed] [Google Scholar]
- 26.Spormann, A. M., and F. Widdel. 2000. Metabolism of alkylbenzenes, alkanes, and other hydrocarbons in anaerobic bacteria. Biodegradation 11:85-105. [DOI] [PubMed] [Google Scholar]
- 27.Widdel, F., and F. Bak. 1992. Gram-negative mesophilic sulfate-reducing bacteria, p. 3352-3378. In A. Balows, H. G. Trüper, M. Dworkin, W. Harder, and K.-H. Schleifer (ed.), The prokaryotes, 2nd ed., vol. 4. Springer Verlag, New York, N.Y.