

Abstract
Investigations of cardiomyopathy mutations in Ca2+ regulatory proteins troponin and tropomyosin provide crucial information about cardiac disease mechanisms, and also provide insights into functional domains in the affected polypeptides. Hypertrophic cardiomyopathy-associated mutations TnI R145G, located within the inhibitory peptide (Ip) of human cardiac troponin I (hcTnI), and TnT R278C, located immediately C-terminal to the IT arm in human cardiac troponin T (hcTnT), share some remarkable features: structurally, biochemically, and pathologically. Using bioinformatics, we find compelling evidence that TnI and TnT, and more specifically the affected regions of hcTnI and hcTnT, may be related not just structurally but also evolutionarily. To test for functional interactions of these mutations on Ca2+-regulation, we generated and characterized Tn complexes containing either mutation alone, or both mutations simultaneously. The most important results from in vitro motility assays (varying [Ca2+], temperature or HMM density) show that the TnT mutant “rescued” some deleterious effects of the TnI mutant at high Ca2+, but exacerbated the loss of function, i.e., switching off the actomyosin interaction, at low Ca2+. Taken together, our experimental results suggest that the C-terminus of cTnT aids Ca2+-regulatory function of cTnI Ip within the troponin complex.
Keywords: thin filament, motility assay, temperature, evolution
Introduction
Familial hypertrophic cardiomyopathy (FHC) is the leading cause of non-traumatic death in young athletes and accounts for 42% of diagnosed childhood cardiomyopathies [1]. FHC may be associated with any one of more than 1000 mutations in genes that encode sarcomere proteins [2]. While relatively few of those mutations are in the three distinct polypeptides of human cardiac troponin (hcTn), some of the most clinically severe mutations affect hcTn’s I and T subunits [3]. It can be challenging to characterize specific cardiomyopathy mutations in patients due to low prevalence, variable penetrance, age of onset, symptoms and severity. FHC’s genetic and phenotypic heterogeneity underscore the complexity in molecular pathways of cardiac contraction and dysfunction [4,5,6,7,8]. Clearly more information is needed on molecular mechanisms that underlie genetic cardiomyopathies to develop and optimize therapeutic strategies.
In addition to elucidating disease mechanisms, investigations of cardiomyopathy mutations often provide crucial insights into the normal molecular function of affected proteins. Interestingly, the structural domains of hcTn that have not yet been determined are often the very regions where FHC mutations are clustered, which may reflect the functional importance of flexible and/or disordered regions. The absence of this structural information, however, has not prevented investigation of physiological consequences of individual FHC mutations including hcTnI R145G [9] and hcTnT R278C [10,11], which are the focus of this investigation.
The hcTnI R145G mutation is particularly severe in some mouse models where the hearts are hypercontractile and exhibit diastolic dysfunction [12]. This is expected because R145 is a critical residue within TnI’s inhibitory peptide (Ip) region, as clearly—and presciently— demonstrated with the corresponding residue in skeletal TnI [13]. TnI R145G has been extensively investigated in vitro using a variety of experimental approaches. Reconstituted hcTn complex containing TnI R145G did not fully inhibit ATPase activity in the absence of Ca2+ for actin-Tm-Tn-myosin [14,15] or with Tn exchanged into myofibrils [16]. Ca2+-sensitivity of ATPase activity increased in solution [14,16] and also with isometric, skinned preparations from transgenic mice [17]. These effects are consistent with observations that the mutation weakens TnI affinity for actin-Tm [14], and also weakens affinity of the C-terminal domain of TnC for Ip in the presence of Ca2+ [18] even though mutant TnI does not directly alter TnC affinity for Ca2+ in the binary TnI-TnC complex [16]. Maximal Ca2+-activation of ATPase activity was significantly decreased [15,16] or was unaffected [14,17] with the mutation. Ca2+-sensitivity of isometric force development was increased [12,17], or decreased [19], in permeabilized preparations from transgenic mice with the TnI R145G mutation; was increased in permeabilized myocytes transfected with mutant TnI [20]; and was increased [15,16] or unchanged [19] following exchange of Tn complex containing TnI R145G into permeabilized preparations. At low Ca2+, the mutation did not fully inhibit force generation [15,16,17,19], and force relaxation was significantly prolonged by the mutant [19]. Maximum isometric force was [15,17] or was not [16,20] significantly reduced, although this effect depends on the proportion of mutant and WT TnI in the myofilaments [12]; in addition, shortening velocity was not affected [12]. Ca2+ transients were prolonged, along with force prolongation in electrically stimulated papillary muscles from TnI R145G transgenic mice [17], consistent with predictions [6]. Although there is some disparity in reported effects of TnI R145G, taken together, the results described above are most consistent with a disease mechanism in which the mutantion permits actomyosin interactions at Ca2+ levels where the myocardium would normally be relaxed, thus prolonging pressure generation which leads to diastolic dysfunction [6,12,14,15,16,17,19].
A second mutation, TnT R278C, could be in close proximity to TnI 145 just beyond the end of the α-helical coiled-coil IT arm of Tn because, even though neither residue is resolved in high resolution structures of Tn’s core domain [21,22], both are only a few amino acids C-terminal to the IT arm in the primary sequences of cTnI and cTnT (Fig. 1). The phenotypic defects in mouse models of the TnT R278C mutation appear to be somewhat less severe than some of the other identified TnT FHC mutations [23,24,25]. Functional assays have provided some insight into the effects of mutation at this site. TnT R278C increased Ca2+ sensitivity of ATPase activity in rabbit cardiac myofibrils [26], but not for skinned preparations from transgenic mouse hearts [24]. TnT R278C also increased Ca2+ sensitivity and reduced cooperativity of isometric force generation by skinned preparations from rabbit [27] or porcine [28] heart, but not from transgenic mice [23,24]. Maximum Ca2+-activated force was depressed by the mutation [24,27,28], while maximum ATPase activity was unaffected in the sarcomere [24,26] or was increased in solution studies [28]. Although transgenic TnT R278C mice neither exhibited extensive ventricular fibrosis nor developed significant hypertrophy [24], they did exhibit significant impairment due to diastolic function [25]. The latter change in cardiac function is consistent with in vitro experiments showing that solution ATPase activity and skinned fiber force are not fully inhibited in low Ca2+ conditions [28], and also suggests that there may be underlying disease mechanisms in common with the more severe TnI R145G mutation, which could derive from their presumptive structural relationship (Fig. 1).
Figure 1. Proximity of FHC mutation sites cTnI R145 and cTnT R278 in the troponin complex.
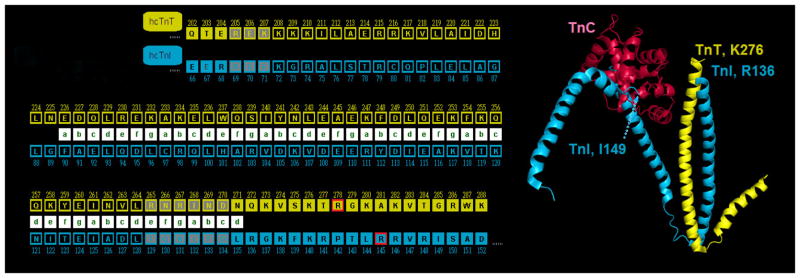
(Left) partial primary sequences of human cTnT (yellow) and cTnI (cyan) aligned according to structure of the α-helical coiled-coil IT arm [21]; heptad repeat is indicated by green letters in white boxes. Arg residues affected by FHC mutations examined in this study, cTnI R145G and cTnT R278C, are just C-terminal to the end of the α-helical coiled-coil IT arm, and are highlighted in boxes outlined with red. Note that the C-terminus of cTnT is the last residue shown, K288. Amino acids with gray/black background depict residues aligned between hcTnI and hcTnT (as yielded by BLAST); gray when aligned, but not part of the IT-arm and black when aligned and part of the IT-arm. (Right) Ca2+-saturated cTn52K (chains D = TnC = red, E = TnT = yellow, and F = TnI = cyan from PDB structure 1J1E) by Takeda et al. [21]; Ca2+ ions not shown. Note that the TnI and TnT constructs used by Takeda et al. [21] were not full length; also note that some portions of the three constructs, including residues cTnI R145 and cTnT R278, were not resolved in the x-ray crystal structures. α-helical coiled-coil of the IT-arm is oriented vertically near center of image, with C-terminal ends of the resolved TnI and TnT peptides (R136 and K276, respectively) at top; note that FHC-related residues cTnI R145 and cTnT R278 are just C-terminal to the portions of TnI and TnT resolved (also note that the next residue in the primary sequence of TnI that is resolved is Ile 149, which is visible in association with TnC). Rendered using Polyview-3D [78].
Interestingly, both the Ip region of hcTnI and hcTnT’s C-terminus have some unusual characteristics in common which, given their common location near the C-terminal end of the IT-arm (Fig. 1), might be more than coincidence. It has been suggested from molecular [29] and structural [30] evidence that TnI and TnT might have diverged from a common evolutionary ancestor. Both regions are “hot spots” for mutations that, individually, cause FHC [3,31,32]. Both regions have a high density of basic amino acids, and both hcTnI R145G and cTnT R278C mutations replace a positively charged arginine with an uncharged residue. Both mutations are located in regions of structural ambiguity [21,22], disorder that is likely related to both structural flexibility and functional roles of these regions. hcTnI R145 is situated in the inhibitory region, which is believed to move away from actin along with the TnI switch domain which can bind in the hydrophobic cleft of TnC’s N-lobe when Ca2+ binds at site II of TnC [33,34,35]. Binding of the TnI switch domain is thought to induce a conformational change in Ip from a β-turn/coil motif to an extended conformation [36,37,38,39]. Intra-domain distances within the Ip region, however, are more consistent with an α-helical structure [40]. Despite being located in different subunits of cTn, FHC mutations hcTnI R145G and hcTnT R278C are both responsible for disease-causing alteration of the interface between the cTn core domain and the rest of the thin filament.
Analysis of Tn’s structure (Fig. 1) along with disparate results from functional studies indicates that valuable structural and functional information about these unresolved regions of the hcTn complex, and the role of each individual mutation in cardiac dysfunction, could be obtained by considering relationships between the two targeted regions. Here, we present our use of bioinformatics to examine protein sequence alignments, located spatially near these unresolved regions, that support an evolutionary relationship between the two Tn subunits; these new findings add support to prior analyses [29]. In addition, we have constructed a recombinant double mutant containing both cTnT R278C and cTnI R145G (“DM”) as a molecular tool. Recombinant DM was expressed with the specific goal of gaining functional insights into these structurally unresolved portions of hcTn. Cardiomyopathy patients have not been documented with both mutations simultaneously, and natural occurrence of the double mutation would be unlikely. Our rationale for constructing DM differed from other studies that utilized double mutant constructs to determine the therapeutic potential that one cardiomyopathy mutation might have to “rescue” the physiological effects of another, more severe mutation [41,42,43]. We compared the following recombinant hcTn protein complexes with conventional and modified in vitro motility analyses: mutation-free (WT), FHC mutant hcTnT R278C, FHC mutant hcTnT R145G, and DM. Using in vitro motility assays, we studied the effects of varying Ca2+ concentration, motor density, and temperature on thin filament sliding speed of each of these genetically engineered protein complexes. Our hypothesis was that if both mutation positions are structurally proximal to each other within the hcTn complex, functional effects of both mutations would accumulate; changes in sliding speed would thus be more significant than that seen with either mutation alone. Our analyses provide strong support for the idea that there exist both structural and evolutionary relationships between hcTnI and hcTnT, and lead to a new model on the structure of unresolved hcTn regions affected by FHC mutations hcTnI R145G and hcTnT R278C.
Materials and methods
Bioinformatics
The evolutionary relatedness between protein sequences TnI and TnT were explored utilizing the Basic Local Alignment Search Tool (BLAST) (NCBI).
Preparation of actin, myosin, heavy meromyosin, and recombinant proteins troponin and tropomyosin
All procedures and protocols involving vertebrate animals were approved by Florida State University’s Institutional Animal Care and Use Committee. Adult male New Zealand White rabbits were handled in accordance with the relevant National Institutes of Health/National Research Council Guide for the Care and Use of Laboratory Animals. Briefly, rabbits were injected with intramuscular 3mg acepromazine, 10 mg kg−1 xylazine and 50 mg kg−1 ketamine. Following verification of appropriate surgical depth of anesthesia, rabbits were exsanguinated via laceration of the carotid artery. The animals were then skinned, eviscerated and chilled on ice. Back and leg muscles were removed for acetone powder, or back muscles only for myosin preparation [44,45,46]. Rabbit skeletal myosin was prepared as previously described [44,45,46,47] and was subjected to mild chymotryptic digestion to obtain rabbit skeletal heavy meromyosin (HMM) [48]. ATP-insensitive ‘dead heads’ were removed from HMM preparations by ultracentrifugation in the presence of MgATP and F-actin on the day of use [46,48]. Actin was extracted from acetone powder as described [8,44,45,49]. F-actin was labeled with RhPh for fluorescence microscopy as previously described [50].
Recombinant human α-Tm was expressed in Escherichia coli as a homodimeric fusion protein with maltose binding protein (MBP); α-Tm was purified following removal of the MBP affinity tag via thrombin cleavage as previously described [46,50,51,52,53]. After removal of the MBP tag, each of the two polypeptides in recombinant α-Tm has two extra, N-terminal amino acids (GS-); GS- is a conservative alternative to the AS-dipeptide in bacterially expressed Tm that substitutes functionally for acetylation of native Tm’s N-terminus in eukaryotic cells [54,55].
Purified Tn from human cardiac muscle (cTn) was obtained from Research Diagnostics (Flanders, NJ), or coexpressed recombinantly (rhcTn) in E. coli as a fusion protein with glutathione S-transferase (GST); the ternary rhcTn complex was purified following removal of the GST affinity tag via cleavage with TEV protease [46,50,52,56]. Human cardiac mutations of rhcTn were introduced via site-directed mutagenesis to the bacterial coexpression plasmid; changes were verified by DNA sequencing. Single mutants hcTnT R278C and hcTnI R145G were prepared as described [50], and DM hcTnT R278C- hcTnI R145G, where each ternary complex of Tn contains both mutations, was specifically generated for this study. rhcTn mutant protein preparations were assessed by Coomassie stained Tricine-SDS PAGE (Fig. 2) [57].
Figure 2. SDS-PAGE analysis of representative protein preparations for rhcTn WT and mutants used in this study.
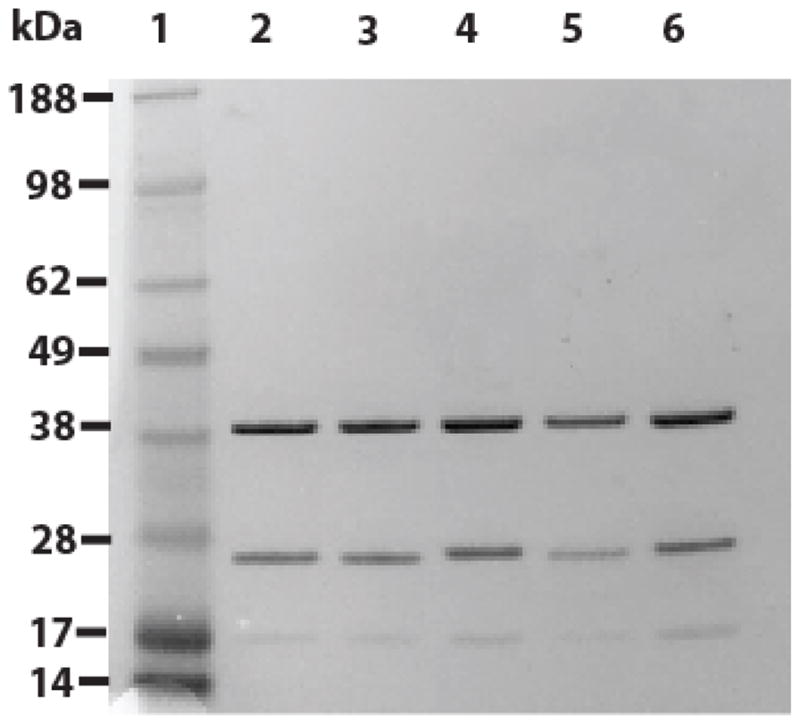
A 4–12% polyacrylamide gradient (Invitrogen) gel was stained with Simple Blue (Invitrogen). Lane 1: MW marker (Invitrogen See Blue Plus 2) with MWs indicated on left. Lane 2: rhcTn WT. Lane 3: rhcTn mutant TnI K206Q (expressed, but not used for this study). Lane 4: rhcTn mutant TnI R145G. Lane 5: rhcTn mutant TnT R278C. Lane 6: rhcTn mutant TnI R145G·TnT R278C.
In Vitro Motility Assays
The speed (s) of RhPh F-actin sliding over HMM-coated surfaces was measured to assess actomyosin kinetics with reconstituted thin filaments and their regulation by Ca2+. All aspects of the motility experiments with unregulated F-actin, such as flow cell assembly, solution preparation, assay procedures, and data collection were conducted as described [44,46,48,50,58,59]. In Ca2+- and temperature-dependence assays, [HMM] applied to flow cells was 250 μg mL−1 to achieve a high motor density of HMM (ρ) on the nitrocellulose-coated motility surface. Ca2+- and ρ-dependence assays were conducted at 30°C. Control assays with unregulated F-actin were conducted in actin buffer (AB: 25 mM KCl, 25 mM imidazole, 4 mM MgCl2, 1 mM EGTA, 1 mM DTT, and pH 7.4) [49] with 2 mM ATP and 0.3% MC, or at pCa 5 without cTn or Tm. Motility buffer composition for assays with regulated thin filaments was calculated as described previously [46,50,52,56,60,61]. Solutions contained 2 mM MgATP, 1 mM Mg2+, 10 mM EGTA, sufficient Ca(CH3COO)2 to achieve the desired pCa (pCa 9–4, for Ca2+−dependence experiments) (pCa = −log10([Ca2+]), where [Ca2+] is in molar), 50 mM K+, 15 mM Na+, 20 mM MOPS, pH 7.00 at 30°C, 0.5% MC, and cTn and Tm (see below). Temperature- and ρ-dependence experiments were conducted at pCa 5. Γ/2 was adjusted to 0.085 M with TrisOH and acetic acid. To minimize fluorophore photobleaching and photo-oxidative damage to the proteins, 3 mg mL−1 glucose, 100 μg mL−1 glucose oxidase, 18 μg mL−1 catalase, and 40 mM DTT were added to motility buffer.
To measure maximum Ca2+-activated s for regulated thin filaments (pCa 5) as a function of HMM density (ρ) on the flow cell surface, ρ was varied by applying different concentrations of HMM in the initial sequence of solutions added to each flow cell. In separate experiments, ρ of ATPase-competent HMM on the flow cell surface was estimated from K-EDTA ATPase assays, assuming that the enzymatic activity of surface-adhered HMM was the same as that measured in solution [53,58,62].
To measure the temperature-dependence of maximum Ca2+-activated thin filament sliding speed (pCa 5), motility data were collected while continuously varying temperature by employing modified flow cells containing microfabricated Au heater and thermometer elements as previously described [50].
Regulated thin filaments were reconstituted in the flow cell as described [46,50,60,61]. The minimum concentrations of WT Tn and Tm added to motility buffer to obtain “well regulated” filaments at 30°C were determined by titrations on each experimental day by applying the following criteria: filament sliding was inhibited at pCa 9, while motility was fast and uniform at pCa 5 and standard temperature of 30°C [45,46,50,56,60,61].
Fluorescence microscopy, data acquisition and data analysis
RhPh-labeled F-actin motility was observed by fluorescence microscopy and data were collected as described [46,59]. Motility speed was analyzed using MetaMorph software (Universal Imaging) as described [63]. Stacks of frames (one stack for each second of temperature transient data, or 10–12 stacks for each constant temperature experiment from one flow cell) were created from digitized movies as described [50].
Nonlinear regression analysis was performed to fit pCa dependence of s, weighted by the fraction of filaments moving uniformly, to a modified 4-parameter version of Sir A. V. Hill’s equation for cooperative binding [64]:
| Eq. 1 |
where regression parameter s0 represents the speed at low [Ca2+] conditions and sc represents the increase in sliding speed above s0 due to the addition of saturating [Ca2+]. Note that s0 + sc is equivalent to smax measured at high [Ca2+]. pCa50 is equal to the pCa at the midpoint of the relationship (i.e., for s = s0 + sc/2), and nH describes the steepness of the relationship around pCa50 and typically reflects cooperativity of the Ca2+ activation process.
An alternate version of the Hill equation was used to describe the ρ-dependence of sliding speed [62], expressed as η(ρ) × Sm:
| Eq. 2 |
where ρ50 characterizes the HMM density required to achieve (η(∞) x sm)/2, z is the Hill exponent that characterizes the apparent cooperativity of this process, and sm represents maximum sliding speed at η(∞). sm from this analysis is equivalent to smax defined above.
Statistical analyses
Averages and error estimates were calculated with Microsoft Excel 2000. Averages are given as unweighted mean ± SD. Linear regression analyses were performed using Microsoft Excel 2000 or MatLab (Version 7.10); nonlinear regression analyses were performed using MatLab (Version 7.10) or SigmaPlot (Version 11.2.0.5). Where expressed, regression errors are standard errors for the regression parameter estimates.
Results
Sequence alignments of cTnI and cTnT
We explored the possibility that there may exist gene homology between cTnI and cTnT in evolutionary history by performing BLAST searches amongst all species. The searches were designed to identify any species that might have conserved Tn sequences to such a degree that homology between TnT (as the query sequence) and hcTnI, or TnI (as the query sequence) and hcTnT, would yield a very low BLAST “Expect-value” (E-value)—the measurement used to denote significance of a BLAST match [65]. Based on our surprising results, we have hypothesized that hcTnI and hcTnT are essentially two separate end products from a gene duplication that took place during the Cambrian explosion.
Using hcTnI as the query protein sequence, the first, non-TnI sequence with the greatest similarity was TnT from Lycosa singoriensis (lsTnT), with an E-value of 2 × 10−9; conversely, using hcTnT as the query protein sequence, first, non-TnT sequence with the greatest similarity was TnI from Schistosoma japonicum (sjTnI), with an E-value of 6 × 10−11 (Fig. 3). If TnI and TnT are indeed the products of gene duplication of a common ancestor, then the parsimonious explanation for our results is that the sequences of both lsTnT and sjTnI still resemble the presumptive ancestral sequence. This hypothesis can be easily examined statistically (although not tested experimentally) because if true, then both the lsTnT and sjTnI protein sequences, despite being from two different genes and two different species, should have strong similarity. Alignment between lsTnT and sjTnI yielded a highly significant E-value of 1 × 10−14. Similar results were obtained with additional TnT and TnI sequences from invertebrates obtained by BLAST (data not shown). Alignment of all four sequences (lsTnT, sjTnI, hcTnI and hcTnT) reveals strong homology among the portions corresponding to the IT-arm region (Fig. 3), which is spatially located near unresolved regions including both FHC mutations hcTnI R145G and hcTnT R278C in the structure of cTn’s core domain (Fig. 1) [21].
Figure 3. BLAST alignment of TnI and TnT centers on, and corresponds with the α-helical coiled-coil IT arm and suggests a common evolutionary origin.

The best match between the hcTnI sequence (in blue) and any TnT sequence was identified using BLAST as TnT from Lycosa singoriensis (lsTnT; red sequence). Paired alignment of those sequences (blue and red; consensus sequence in black in between) results in an E-value of 2×10−09. Similarly, the best match determined between hcTnT (yellow sequence) and any TnI was TnI from Schistosoma japonicum (sjTnI, green sequence). Paired alignment of those two sequences (green and yellow, with consensus sequence in between) results in an E-value of 6×10−11. Paired alignment of lsTnT (red) and sjTnI (green), shown in a black box with the resulting consensus sequence in black, results in an E-value of 1×10−14.
Effects of FHC hcTn mutations on Ca2+ sensitivity of thin filament sliding at 30°C
To examine the effects of single mutants cTnT R278C and cTnI R145G, and DM cTnT R278C-cTnI R145G on Ca2+ sensitivity of thin filament sliding, experiments were conducted across a range of [Ca2+]. Fig. 4 illustrates a highly significant reduction in maximum filament sliding speed at saturating Ca2+ (smax) when WT hcTnI (smax = 8.33 ± 0.35 μm/s) was replaced with hcTnI R145G (smax = 6.34 ± 0.39 μm/s; p=0.012), but not with hcTnTR28C (smax = 8.21 ± 0.34 μm/s). This inhibition of smax by cTnI R145G was apparently “rescued” by cTnT R278C in DM (smax = 8.21 ± 0.18 μm/s). However, “rescue” was not observed in our analysis of filament sliding at sub-maximum Ca2+ (Fig. 5, Table 1).
Figure 4. Inhibition of maximum Ca2+-activated thin filament sliding speed (smax) by cTnI R145G is rescued double mutant (DM) cTnI R145G-cTnT R278C.

smax was measured at 30°C and pCa5 for thin filaments reconstituted with cTn WT, cTnI R145G, cTnT R278C, or DM. Each bar represents the mean (± SE) of 9 flow cells; 30 s recordings of six independent fields were analyzed for each flow cell. Note that the vertical (speed) axis does not begin at 0.
Figure 5. FHC mutations increase Ca2+ -sensitivity of thin filament sliding.

Ca2+-dependence of thin filament sliding was determined at 30°C using motility assays (Materials and methods). Each point represents the mean (± SE) of 3 flow cells; 30 s recordings of six independent fields were analyzed for each flow cell. Data were fit to the Hill Eq (Eq. 1) by nonlinear least squares regression, and then both data and the regression were normalized to s0 + sc for each troponin used: cTn WT (dark solid line), cTnI R145G (dashed line), cTnT R278C (light solid line), or DM (dash-dot line). Inset: Percent of immobile filaments as a function of Ca2+, which illustrates the additive effect of the mutations at low Ca2+.
Table 1.
Nonlinear least squares regression parameter estimates for speed-pCa relations in Fig. 5.
| Cardiac Troponin | ||||
|---|---|---|---|---|
| Parameter | WT | TnI R145G | TnT R278C | TnI R145G + TnT R278C |
| R2 | 0.908 | 0.761 | 0.878 | 0.895 |
| sc (normalized) | 0.90 ± 0.08 | 0.80 ± 0.15 | 0.90 ± 0.10 | 0.69 ± 0.07 |
| s0 (normalized) | 0.10 ± 0.04 | 0.20 ± 0.08 | 0.10 ± 0.05 | 0.31 ± 0.04 |
| pCa50 | 5.76 ± 0.04 | 6.02 ± 0.14 | 5.88 ± 0.06 | 6.01 ± 0.05 |
| nH | 4.3 ± 1.3 | 1.6 ± 0.7 | 2.8 ± 0.9 | 3.1 ± 1.0 |
Regression parameter estimates (± SE) for nonlinear least squares fits to Eq 1 were obtained as described (Materials and methods) for motility data shown in Fig. 5 using regulated thin filaments reconstituted with αTm and cTn WT, cTn-TnI R145G, cTn-TnT R278C, or double mutant cTn-TnI R145G-TnT 278C. The four fit parameters were sc, s0, pCa50 and nH; sc and s0 were normalized to their sum (sc + s0).
The pCa50, Hill coefficient (nH), sliding speed at low Ca2+ (s0), and maximum Ca2+-activated component of sliding speed (sc) were obtained for each hcTn by nonlinear least squares regression of the data on Eq. 1 (Fig. 5, Table 1). Although there was no difference in normalized so between WT and hcTnT R278C, so increased with hcTnI R145G as compared to WT; so was increased even further with DM (Fig. 5; Table 1). Parameter estimates of pCa50 were significantly higher for each hcTn mutant compared to WT (Table 1), indicating increased Ca2+ sensitivity by individual FHC mutants. Ca2+ sensitivity of the DM was increased to approximately the same extent as that of single mutant hcTnI R145G. The Hill coefficient (nH) was noticeably reduced for hcTnI R145G, hcTnT R278C, and also for the DM. In this case, nH for the DM was essentially the same as with single mutant hcTnT R278C. Each mutation on its own was less competent than WT at inhibition of actomyosin interaction at pCa 9 as indicated by the percentage of immobile filaments on the flow cell (WT = 87%, hcTnT R278C = 66%, and hcTnI R145G = 58%), and the DM exhibited a more dramatically decreased ability to render filaments immobile (DM = 17%) (inset, Fig. 5). Under low Ca2+ conditions, the effect of DM on so appears to be much more pronounced than either mutation alone.
Effect of FHC hcTn mutations on the HMM density-dependence of thin filament sliding at 30°C and pCa 5
We determined the effects of single mutants hcTnI R145G and hcTnT R278C, and the DM on filament sliding speed at different motor densities (ρ) at 30ºC and pCa 5 to (Fig. 6). To describe the ρ-dependence of the sliding speed with each hcTn, data were fit to Eq. 2 using nonlinear least squares regression. Comparable to results at high HMM density and saturating Ca2+ (smax and (sc + s0) in Fig. 4 and Table 1, respectively), maximum sliding speed (sm) was decreased notably when WT was replaced with TnI R145G at all ρ examined (WT = 8.1 ± 0.5 μm/s, hcTnI R145G = 6.0 ±0.6 μm/s) at the highest achievable motor densities. Interestingly, in comparison to WT, hcTnT R278C exhibited similar sm to WT at all ρ examined. Consistent with the observations described above, the loss of sm by hcTnI R145G was “rescued” by DM, with sm values at maximum ρ essentially the same as that of WT (DM = 8.3 ± 0.8 μm/s).
Figure 6. Effect of FHC hcTn mutations on the HMM density-dependence of thin filament sliding at 30°C and pCa 5.

HMM motor density-dependence of thin filament sliding was determined at 30°C and pCa 5 (Materials and methods). Each point represents the mean (± SE) of 3 flow cells; 30 s recordings of six independent fields were analyzed for each flow cell run for each troponin. Data were fit to Eq. 2 for thin filaments reconstituted with cTn WT (dark solid line), cTnI R145G (dashed line), cTnT R278C (light solid line), or DM (dash-dot line).
ρ50, which characterizes the HMM density required to achieve sm/2, was not noticeably different from WT for either single mutant or the DM. Hill exponent (z), which characterizes the apparent cooperativity of this process, was not significantly affected when WT hcTn was substituted with any of the hcTn mutants.
Effects of FHC mutations on the temperature dependence of thin filament sliding at pCa 5
To compare the temperature dependence of actomyosin kinetics amongst all three of our FHC mutants, we monitored the sliding speed response upon cyclic heating of reconstituted thin filaments at pCa 5 and saturating HMM density. Speed s was measured over a wide range of temperatures between 20°C and 55°C (Fig. 7) using a microfabricated thermoelectric controller [50,53,59]. As expected for a biochemically driven process, sliding speed increased as temperature was increased over the range examined. Speed s for the DM was similar to that for TnT R278C at physiological temperature and above, as expected (Figs. 4, 6; Table 1), but at lower temperatures, s for DM was intermediate between that of the single mutants (Fig. 7). We have previously demonstrated the existence of a transition temperature (Tt), around physiological temperature, that not only marks a shift in temperature sensitivity of filament sliding, but also likely reveals a change in the rate limiting step of the ATPase cycle [50]. The Arrhenius representation of all sliding speeds with temperature was, therefore, fit with two linear regression lines (Fig. 7). Tt of DM (39.7°C) was intermediate to that determined for the single mutants (hcTnI R145G = 38.4°C and hcTnT R278C = 41.0°C) (Fig. 7).
Figure 7. Effect of FHC hcTn mutations on the temperature-dependence of thin filament sliding.

Arrhenius plots of temperature dependence of thin filament sliding at pCa 5 for thin filaments reconstituted with DM (open symbols and dot-dash line); regressions for cTnI R145G (dashed line) and cTnT R278C (light solid line) were replotted from Brunet et al. [50]. Data were obtained during the rising phase of continuous temperature variation (Materials and methods), and each point represents the average speed during 1 s. Transition temperatures (Tt; solid symbols) are suggestive of variations in rate limiting factors between the low and high temperature regimes.
Discussion
Protein sequence analyses suggest a common evolutionary origin for TnI and TnT
The heptad repeats in the hcTnI and hcTnT sequences are the hallmark of a coiled-coil structure [66] which forms the IT arm in Tn’s core domain [21,22]. The sequences corresponding to the IT arm were found to lie within a region of local similarity between hcTnI and hcTnT, with a predicted alignment that coincided with the actual structural alignment of heptad repeats in the IT arm’s coiled-coil (Figs. 1, 3).
We used additional sequence alignments to derive more information on possible homology of hcTnI and hcTnT. BLAST searches utilizing either hcTnI or hcTnT as the query did not yield the partner subunit as a viable result in either case, even if algorithm parameters were adapted to minimize thresholds. Despite the lack of direct homology between hcTnI and hcTnT, there are suggestions that the genes coding for both sequences share a common ancestor [29,30]. If an ancestral, homodimeric protein did exist, the putative gene duplication giving rise to two separate Tn subunits must have occurred more than 500 million years ago, based on the presence of TnI and TnT sequences in various invertebrates as divergent as the model organism Drosophila melanogaster and the oyster Pinctada fucata (not shown). By using BLAST sequence alignments with hcTnI or hcTnT as queries to identify homology with the partner subunit, across sequences of all known species available in the protein database, we anticipated detection of sequences that resemble a putative common ancestor. Our approach yielded a number of invertebrate sequences, and we focused on lsTnT and sjTnI (Fig. 3); the efficiency of this approach was proven by showing that those independently obtained results are highly homologous to each other (E-value 1 × 10−14). The most likely explanation for the high homology between those two sequences from different genes and species is that they resemble the original “common ancestor” sequence before gene duplication took place. Based on our results, our conclusion is that these two subunits within the hcTn complex that transduce a Ca2+ signal into cardiac contraction actually evolved after duplication of a single, common ancestor gene. Since it is completely plausible that regions of the sequence prone to (FHC) mutation would have copied along with the rest of the gene sequence, we can now provide a possible explanation for the close proximity of hcTnI R145G and hcTnT R278C FHC mutation “hot spots.”
FHC mutants TnIR145G and TnTR278C were tested alone and together in motility assays
To investigate how these FHC mutations in hcTn might confer conformational changes in protein structure that contribute to inherited cardiomyopathies observed today, we engineered recombinant hcTn complexes containing either mutant hcTnI R145G or hcTnT R278C, and a “double mutant” carrying both mutations simultaneously. Although rare, it is not impossible that a single individual is affected by multiple FHC gene mutations [67]. Our goal, however, was not to investigate the clinical significance of this double mutation because the likelihood of the two FHC mutations occurring simultaneously in a single individual is, admittedly, small. Rather the goal was to derive functional information about these regions of the hcTn complex that have not yet been resolved in high resolution structural studies. We compared data obtained for WT, each of the individual mutants, and DM using in vitro motility assays at varying concentrations of Ca2+, densities of HMM, and temperature.
Inhibition of smax by TnIR145G is “rescued” by TnTR278C, but only at temperatures above Tt
In previously described permeabilzed muscle preparations from TnI R145G Tg mice [12], there was no change in Vmax (maximum velocity of sarcomere shortening) at saturating Ca2+. The results of our unloaded filament sliding assays differ. We found a significant reduction in smax with the cTnI R145G single mutant; however, when rhcTn contained both the hcTnI R145C and hcTnT R278C mutant, the loss in smax due to hcTnI R145G was effectively “rescued” (Fig. 4). At all ρ examined, the loss of smax by hcTnI R145G single mutant appeared to be “rescued” when hcTn contained both the hcTnI R145C and hcTnT R278C mutations (Fig. 6).
In addition, with all three mutant constructs, sliding speeds increased with increasing temperature, exhibiting a “break point” in the slope of each line (Fig. 7). We [50], and others [68,69], have previously shown that there are two distinct thermal regimes discernible in Arrhenius analyses of cross-bridge cycling, with the change in the rate limiting step occurring at around body temperature. It appears that the effect of the DM is different above Tt than below Tt—this, perhaps, implies different effects on the rate limiting steps for filament sliding. With the DM, we find that Tt of the rate limiting step is mid-way between that of each single mutant.
Sliding at low Ca2+ and changes in pCa50 with TnI R145G indicate diastolic dysfunction: so is even less inhibited in combination with TnT R278C
Most interestingly, the increase in so with the hcTnI R145G single mutant was not effectively restored to normal levels when the complex contained both mutations even though hcTnT R278C did not affect so on its own—rather, the DM exhibited an even greater increase in so (Fig. 5). The DM exhibits an even more significant decrease in regulation competency, with a noticeably decreased ability to render filaments immobile (inset, Fig. 5). Ca2+-sensitivity, as demonstrated by changes in pCa50, was increased for both single mutants and DM (Fig. 5, Table 1). Cooperative activation by Ca2+ (nH in Eq. 1) of filament sliding was altered by the mutations (Fig. 5, Table 1) although there was little or no effect on cross-bridge dependent cooperativity (z in Eq. 2) at saturating Ca2+ (Fig. 6). Thus, at diastolic Ca2+ levels, the single mutants are only partially competent to regulate actomyosin interactions (Fig. 5).
These findings are in agreement with some [12,17,26], but not all [19,24], earlier studies using different muscle preparations. The increase in Ca2+-sensitivity of filament sliding with hcTnT R278C (Fig. 5) contrasts with its lack of effect on isometric force generation in the intact sarcomere in preparations from Tg mice [24]. Such a disjunction between unloaded versus isometric conditions has previously been noted for some mutants such as cTnI K206Q [60]. The protein composition of the sarcomere lattice may also influence Ca2+-sensitivity as studies of cTnT R278C increased Ca2+-sensitivity of myofibrillar ATPase [26] and isometric force [27] in rabbit preparations, and isometric force in porcine preparations [28], but not in Tg mouse sarcomeres [24]. Taken together with most prior reports, the observed changes in pCa50, so and/or nH suggest mechanistic explanations for these two mutations’ roles in diastolic dysfunction [12,25].
Function and structure analyses suggest TnT C-terminus aids TnI inhibitory peptide
We have developed a working model (Fig. 8) to structurally explain the effects outlined in this investigation, based on the combination of results from our bioinformatics analyses and in vitro motility experiments. The presumptive evolutionary origins of TnI and TnT lead to the possibility that the function of TnT’s C-terminus could be evolutionarily and structurally related to the function of the Ip of TnI. In addition, there are plausible explanations for our results within the context of what is currently known about Ca2+-mediated dynamics of filament sliding. Although our studies do not provide direct evidence for specific structural interactions, the following scenario (Fig. 8) is developed to show plausible, and we believe likely, interactions of thin filament proteins to explain our functional data. Takeda et al. [21] speculated that hcTnT R278 might be located within a Ca2+-sensitive interface between TnT and Tm, although experimental evidence indicates that removal of TnT’s C-terminus does not affect TnT-Tm binding [70] and that the two Tm-binding regions of TnT are located elsewhere [71]. The C-terminus of cTnT is involved in some form of interaction with other components of the thin filament as removal of the last fourteen amino acids—including R278—negatively affects Ca2+ regulation of actomyosin function [72,73] and leads to FHC [74]. An alternative hypothesis is that TnT (residues 272–288) normally interacts with actin in the Ca2+ free state. This region of TnT is composed of positively charged residues that would naturally be attracted to the net negative charge along the actin filament. In the Ca2+-free state, TnI Ip binds actin; it seems possible that the C-terminus of TnT might also bind actin in diastolic conditions. In the presence of Ca2+, the Ip of TnI moves away from actin along with TnI’s switch peptide. Affinity chromatography [75] and NMR [18] studies have previously indicated that Ip binds TnC in the Ca2+-bound state; thus, it could logically follow—from our results suggesting a parallel between TnI’s Ip and the C-terminus of TnT (Figs. 1 and 3)—that the C-terminus of TnT could also interact with TnC in the presence of Ca2+. The TnI R145G mutation has been shown to weaken the interaction of Ip with TnC [18,75]; by inference, this proposed interaction of TnT and TnC might also be weakened with the R278C mutation.
Figure 8. Model of the structural and functional relationships between TnI R145 and TnT R278.

WT (left panels) and DM (right panels) troponin on actin-Tm in the absence (top panels) or presence (bottom panels) of Ca2+. TnC N-lobe (purple), TnI (blue), TnT (yellow), Tm (orange), and F-actin (grey). Myosin-binding sites on actin are represented by holes along the linearized actin filament. In the absence of Ca2+ (top left), the Ca2+-insensitive WT TnT-actin interface and the Ca2+-sensitive WT TnI-actin interface prevent Tm from traversing into the actin groove, which prevents activation of filament sliding. Destabilization of those interfaces by Ca2+ (bottom left) or mutations (top right) enables maximum or partial filament sliding, respectively. In the presence of Ca2+, TnI switch peptide binds TnC N-lobe (bottom panels); the WT TnI Ip, and possibly also the WT TnT C-terminus, may also interact with TnC C-lobe (not shown). The weakened TnT-actin bond with DM (bottom right) permits maximum filament sliding upon Ca2+-induced decoupling of actin and TnI.
We have suggested that the single mutations hcTnI R145G and hcTnT R278C result in a disorganization of α-helical structure normally present in WT Tn (Fig. 1). Our model predicts that the Ca2+ mediated dynamics which turn filament sliding off and on (Fig. 8, left panels) require two critical interactions: the first—a weak and Ca2+ insensitive interaction—is between actin and TnT (residues 272–288), the second—strong and Ca2+ sensitive—is between actin and TnI (residues 137–148). Weakening of either interaction alone will have a negative effect on the ability of the Tn complex to inhibit sliding in the absence of Ca2+. When both interactions are simultaneously weakened, the loss of contractile function is cumulative, and thus more pronounced (Fig. 8, top right; interpretation based on low Ca2+ results in Fig. 5, and Fig. 5 inset). In the presence of Ca2+, normally the weak Tn-actin bond is negligible, because it breaks easily once filament sliding commences via Ca2+-induced decoupling of TnI from actin (Fig. 8, bottom left). When decoupling between TnI and actin is hampered with the TnI R145G mutation, the weak Tn-actin interaction doesn’t break and thus becomes more relevant. We have demonstrated the relevance of this Tn-actin interaction by utilizing DM, because the effects of the hcTnI R145G mutation can only be somewhat “rescued” (at high Ca2+) by also weakening the Tn-actin interaction with the addition of hcTnTR278C to the construct (Fig. 8, bottom right; interpretation based on results in Figs. 5–7), which under certain conditions leads to normal sliding at saturating Ca2+ despite the presence of both mutations (Fig. 4).
Conclusions
We have utilized a “double mutant” construct, hcTnI R145G-hcTnT R278C, to gain structural insight into unresolved portions of the human cardiac troponin complex. Specifically, these mutations are located in the regions of hcTnI and hcTnT just C-terminal to the IT arm that is resolved in the crystal structure of Tn’s core domain [21]. Based on our bioinformatics analyses, TnI and TnT may have evolved from a common ancestral gene, with the strongest homology in the region of the IT arm. Based on our in vitro motility studies, we present a model of these two regions that suggests a functional interdependence between the inhibitory peptide region of TnI and the C-terminus of TnT, and corresponding structures for both domains based on intradomain distances within the inhibitory region of hcTnI obtained by Brown et al. [40]. Of novel interest in this study is the ability of our DM, under certain conditions, to “rescue” some aspects of the hcTnI R145G FHC mutation. This suggests overall that identification and use of therapeutic target sites on one troponin subunit could have direct and positive effects on malfunctions usually caused by specific mutations on another subunit. Nonetheless, our results at low Ca2+, where there is probably more relevance to diastolic dysfunction in hypertrophic cardiomyopathies, do not support the possibility that this particular double mutation might be therapeutic. Full understanding of the complete structure of the cardiac troponin complex, and how that structure affects interactions of all three subunits in the context of the thin filament [76] and other cellular locations [5,77], may lead to effective identification of therapeutic targets to alleviate the pathological symptoms of hypertrophic cardiomyopathies.
Highlights.
FHC mutants TnIR145G and TnTR278C were tested alone and together in motility assays
Inhibition of smax, but not left-shift of pCa50, by TnIR145G is rescued by TnTR278C
Sliding at low Ca2+ (diastolic dysfunction) with TnIR145G is worsened by TnTR278C
Protein sequence analyses suggest a common evolutionary origin for TnI and TnT
Function and structure analyses suggest TnT C-terminus aids TnI inhibitory peptide
Acknowledgments
We thank: Dr. Peng Xiong and Dr. Stephan von Molnár in FSU’s Department of Physics for assistance with constructing microthermal heaters to continuously measure temperature-dependence of filament sliding speed; Dr. David L. Swofford for valuable suggestions and guidance in bioinformatics; Dr. Nekeisha S. Sweeney for assistance with some motility assays; and Steve Miller in the DNA Sequencing Facility and Rani Dhanarajan in the Molecular Cloning Laboratory of FSU Department of Biological Science’s Core Facilities for assistance in generating and validating clones and mutations.
Funding
This work was supported by NIH/NHLBI HL63974 (PBC), American Heart Association FL/PR Affiliate Predoctoral Fellowship 0315097B (NMB), and NSF NIRT Grant ECS-0210332 (PX).
Funding sources did not have any role in: study design; collection, analysis and interpretation of data; writing or decision to submit this article for publication.
Abbreviations
- AB
actin buffer
- cTn
native troponin complex from human cardiac muscle
- cTnI
cardiac troponin I
- cTnT
cardiac troponin T
- DTT
dithiothreitol
- DM
hcTn double mutant (hcTnI R145G plus hcTnT R278C)
- EDTA
ethylenediaminetetraacetic acid
- EGTA
ethylene glycol tetraacetic acid
- F-actin
filamentous actin
- FHC
familial hypertrophic cardiomyopathy
- GST
glutathione S-transferase (affinity tag)
- hcTnI
human cardiac troponin I
- hcTnT
human cardiac troponin T
- hcTn
human cardiac troponin complex
- HMM
heavy meromyosin
- Ip
TnI inhibitory peptide
- lsTnT
troponin T from Lycosa singoriensis
- MBP
maltose binding protein (affinity tag)
- MC
methylcellulose
- MOPS
3-(N-morpholino)propanesulfonic acid
- ρ
density of ATPase-competent HMM on a flow cell surface
- rhcTn
recombinantly expressed human cardiac troponin complex
- RhPh
rhodamine phalloidin
- sjTnI
troponin I from Schistosoma japonicum
- TEV protease
tobacco etch virus protease
- Tm
tropomyosin
- Tn
troponin complex
- TnC
troponin C
- TnI
troponin I
- TnI Ip
troponin I’s inhibitory peptide region
- TnT
troponin T
- WT
wild type
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errorsmaybe discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Lipshultz SE, Sleeper LA, Towbin JA, Lowe AM, Orav EJ, Cox GF, Lurie PR, McCoy KL, McDonald MA, Messere JE, Colan SD. N Engl J Med. 2003;348:1647–55. doi: 10.1056/NEJMoa021715. [DOI] [PubMed] [Google Scholar]
- 2.Seidman CE, Seidman JG. Circ Res. 2011;108:743–50. doi: 10.1161/CIRCRESAHA.110.223834. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Gomes AV, Potter JD. Ann N Y Acad Sci. 2004;1015:214–24. doi: 10.1196/annals.1302.018. [DOI] [PubMed] [Google Scholar]
- 4.Ahmad F, Seidman JG, Seidman CE. Annu Rev Genomics Hum Genet. 2005;6:185–216. doi: 10.1146/annurev.genom.6.080604.162132. [DOI] [PubMed] [Google Scholar]
- 5.Chase PB, Szczypinski MP, Soto EP. J Muscle Res Cell Motil. 2013;34:275–284. doi: 10.1007/s10974-013-9356-7. [DOI] [PubMed] [Google Scholar]
- 6.Kataoka A, Hemmer C, Chase PB. J Biomech. 2007;40:2044–52. doi: 10.1016/j.jbiomech.2006.09.026. [DOI] [PubMed] [Google Scholar]
- 7.Parmacek MS, Solaro RJ. Prog Cardiovasc Dis. 2004;47:159–76. doi: 10.1016/j.pcad.2004.07.003. [DOI] [PubMed] [Google Scholar]
- 8.Schoffstall B, Kataoka A, Clark A, Chase PB. J Pharmacol Exp Ther. 2005;312:12–8. doi: 10.1124/jpet.104.073445. [DOI] [PubMed] [Google Scholar]
- 9.Kimura A, Harada H, Park JE, Nishi H, Satoh M, Takahashi M, Hiroi S, Sasaoka T, Ohbuchi N, Nakamura T, Koyanagi T, Hwang TH, Choo JA, Chung KS, Hasegawa A, Nagai R, Okazaki O, Nakamura H, Matsuzaki M, Sakamoto T, Toshima H, Koga Y, Imaizumi T, Sasazuki T. Nat Genet. 1997;16:379–82. doi: 10.1038/ng0897-379. [DOI] [PubMed] [Google Scholar]
- 10.Van Driest SL, Ellsworth EG, Ommen SR, Tajik AJ, Gersh BJ, Ackerman MJ. Circulation. 2003;108:445–51. doi: 10.1161/01.CIR.0000080896.52003.DF. [DOI] [PubMed] [Google Scholar]
- 11.Watkins H, McKenna WJ, Thierfelder L, Suk HJ, Anan R, O’Donoghue A, Spirito P, Matsumori A, Moravec CS, Seidman JG, Seidman CE. N Engl J Med. 1995;332:1058–64. doi: 10.1056/NEJM199504203321603. [DOI] [PubMed] [Google Scholar]
- 12.James J, Zhang Y, Osinska H, Sanbe A, Klevitsky R, Hewett TE, Robbins J. Circ Res. 2000;87:805–11. doi: 10.1161/01.res.87.9.805. [DOI] [PubMed] [Google Scholar]
- 13.Van Eyk JE, Hodges RS. J Biol Chem. 1988;263:1726–32. [PubMed] [Google Scholar]
- 14.Elliott K, Watkins H, Redwood CS. J Biol Chem. 2000;275:22069–74. doi: 10.1074/jbc.M002502200. [DOI] [PubMed] [Google Scholar]
- 15.Lang R, Gomes AV, Zhao J, Housmans PR, Miller T, Potter JD. J Biol Chem. 2002;277:11670–8. doi: 10.1074/jbc.M108912200. [DOI] [PubMed] [Google Scholar]
- 16.Takahashi-Yanaga F, Morimoto S, Harada K, Minakami R, Shiraishi F, Ohta M, Lu QW, Sasaguri T, Ohtsuki I. J Mol Cell Cardiol. 2001;33:2095–107. doi: 10.1006/jmcc.2001.1473. [DOI] [PubMed] [Google Scholar]
- 17.Wen Y, Pinto JR, Gomes AV, Xu Y, Wang Y, Potter JD, Kerrick WGL. J Biol Chem. 2008;283:20484–94. doi: 10.1074/jbc.M801661200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Lindhout DA, Li MX, Schieve D, Sykes BD. Biochemistry. 2002;41:7267–74. doi: 10.1021/bi020100c. [DOI] [PubMed] [Google Scholar]
- 19.Kruger M, Zittrich S, Redwood C, Blaudeck N, James J, Robbins J, Pfitzer G, Stehle R. J Physiol. 2005;564:347–57. doi: 10.1113/jphysiol.2004.079095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Westfall MV, Borton AR, Albayya FP, Metzger JM. Circ Res. 2002;91:525–31. doi: 10.1161/01.res.0000034710.46739.c0. [DOI] [PubMed] [Google Scholar]
- 21.Takeda S, Yamashita A, Maeda K, Maéda Y. Nature. 2003;424:35–41. doi: 10.1038/nature01780. [DOI] [PubMed] [Google Scholar]
- 22.Vinogradova MV, Stone DB, Malanina GG, Karatzaferi C, Cooke R, Mendelson RA, Fletterick RJ. Proc Natl Acad Sci U S A. 2005;102:5038–43. doi: 10.1073/pnas.0408882102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Baudenbacher F, Schober T, Pinto JR, Sidorov VY, Hilliard F, Solaro RJ, Potter JD, Knollmann BC. J Clin Invest. 2008;118:3893–903. doi: 10.1172/JCI36642. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Hernandez OM, Szczesna-Cordary D, Knollmann BC, Miller T, Bell M, Zhao J, Sirenko SG, Diaz Z, Guzman G, Xu Y, Wang Y, Kerrick WGL, Potter JD. J Biol Chem. 2005;280:37183–94. doi: 10.1074/jbc.M508114200. [DOI] [PubMed] [Google Scholar]
- 25.Sirenko SG, Potter JD, Knollmann BC. J Physiol. 2006;575:201–13. doi: 10.1113/jphysiol.2006.107557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Yanaga F, Morimoto S, Ohtsuki I. J Biol Chem. 1999;274:8806–12. doi: 10.1074/jbc.274.13.8806. [DOI] [PubMed] [Google Scholar]
- 27.Morimoto S, Nakaura H, Yanaga F, Ohtsuki I. Biochem Biophys Res Commun. 1999;261:79–82. doi: 10.1006/bbrc.1999.1000. [DOI] [PubMed] [Google Scholar]
- 28.Szczesna D, Zhang R, Zhao J, Jones M, Guzman G, Potter JD. J Biol Chem. 2000;275:624–30. doi: 10.1074/jbc.275.1.624. [DOI] [PubMed] [Google Scholar]
- 29.Huang QQ, Jin JP. J Mol Evol. 1999;49:780–8. doi: 10.1007/pl00006600. [DOI] [PubMed] [Google Scholar]
- 30.Chong SM, Jin JP. J Mol Evol. 2009;68:448–60. doi: 10.1007/s00239-009-9202-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Tardiff JC. Circ Res. 2011;108:765–82. doi: 10.1161/CIRCRESAHA.110.224170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Willott RH, Gomes AV, Chang AN, Parvatiyar MS, Pinto JR, Potter JD. J Mol Cell Cardiol. 2010;48:882–92. doi: 10.1016/j.yjmcc.2009.10.031. [DOI] [PubMed] [Google Scholar]
- 33.Sia SK, Li MX, Spyracopoulos L, Gagné SM, Liu W, Putkey JA, Sykes BD. J Biol Chem. 1997;272:18216–21. doi: 10.1074/jbc.272.29.18216. [DOI] [PubMed] [Google Scholar]
- 34.Spyracopoulos L, Li MX, Sia SK, Gagné SM, Chandra M, Solaro RJ, Sykes BD. Biochemistry. 1997;36:12138–46. doi: 10.1021/bi971223d. [DOI] [PubMed] [Google Scholar]
- 35.Vinogradova MV, Stone DB, Malanina GG, Karatzaferi C, Cooke R, Mendelson RA, Fletterick RJ. Proc Natl Acad Sci USA. 2005;102:5038–43. doi: 10.1073/pnas.0408882102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Dong WJ, An J, Xing J, Cheung HC. Arch Biochem Biophys. 2006;456:135–42. doi: 10.1016/j.abb.2006.08.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Dong WJ, Xing J, Robinson JM, Cheung HC. J Mol Biol. 2001;314:51–61. doi: 10.1006/jmbi.2001.5118. [DOI] [PubMed] [Google Scholar]
- 38.Dong WJ, Xing J, Villain M, Hellinger M, Robinson JM, Chandra M, Solaro RJ, Umeda PK, Cheung HC. J Biol Chem. 1999;274:31382–90. doi: 10.1074/jbc.274.44.31382. [DOI] [PubMed] [Google Scholar]
- 39.Tung CS, Wall ME, Gallagher SC, Trewhella J. Protein Sci. 2000;9:1312–26. doi: 10.1110/ps.9.7.1312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Brown LJ, Sale KL, Hills R, Rouviere C, Song L, Zhang X, Fajer PG. Proc Natl Acad Sci U S A. 2002;99:12765–70. doi: 10.1073/pnas.202477399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Davis J, Metzger JM. PLoS One. 2010;5:e9140. doi: 10.1371/journal.pone.0009140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Li Y, Charles PY, Nan C, Pinto JR, Wang Y, Liang J, Wu G, Tian J, Feng HZ, Potter JD, Jin JP, Huang X. J Mol Cell Cardiol. 2010;49:402–11. doi: 10.1016/j.yjmcc.2010.04.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Wei B, Gao J, Huang XP, Jin JP. J Biol Chem. 2010;285:27806–16. doi: 10.1074/jbc.M110.137844. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Chase PB, Chen Y, Kulin KL, Daniel TL. Am J Physiol Cell Physiol. 2000;278:C1088–98. doi: 10.1152/ajpcell.2000.278.6.C1088. [DOI] [PubMed] [Google Scholar]
- 45.Gordon AM, LaMadrid MA, Chen Y, Luo Z, Chase PB. Biophys J. 1997;72:1295–307. doi: 10.1016/S0006-3495(97)78776-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Schoffstall B, Clark A, Chase PB. Biophys J. 2006;91:2216–26. doi: 10.1529/biophysj.105.079061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Margossian SS, Lowey S. Methods Enzymol. 1982;85(Pt B):55–71. doi: 10.1016/0076-6879(82)85009-x. [DOI] [PubMed] [Google Scholar]
- 48.Kron SJ, Toyoshima YY, Uyeda TQ, Spudich JA. Methods Enzymol. 1991;196:399–416. doi: 10.1016/0076-6879(91)96035-p. [DOI] [PubMed] [Google Scholar]
- 49.Pardee JD, Spudich JA. Methods Enzymol. 1982;85(Pt B):164–81. doi: 10.1016/0076-6879(82)85020-9. [DOI] [PubMed] [Google Scholar]
- 50.Brunet NM, Mihajlović G, Aledealat K, Wang F, Xiong P, von Molnár S, Chase PB. J Biomed Biotechnol. 2012;2012:657523. doi: 10.1155/2012/657523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Loong CK, Zhou HX, Chase PB. PLoS One. 2012;7:e39676. doi: 10.1371/journal.pone.0039676. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Schoffstall B, LaBarbera VA, Brunet NM, Gavino BJ, Herring L, Heshmati S, Kraft BH, Inchausti V, Meyer NL, Moonoo D, Takeda AK, Chase PB. DNA Cell Biol. 2011;30:653–9. doi: 10.1089/dna.2010.1163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Wang F, Brunet NM, Grubich JR, Bienkiewicz EA, Asbury TM, Compton LA, Mihajlović G, Miller VF, Chase PB. J Biomed Biotechnol. 2011;2011:435271. doi: 10.1155/2011/435271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Heald RW, Hitchcock-DeGregori SE. J Biol Chem. 1988;263:5254–9. [PubMed] [Google Scholar]
- 55.Monteiro PB, Lataro RC, Ferro JA, Reinach FC. J Biol Chem. 1994;269:10461–6. [PubMed] [Google Scholar]
- 56.Loong CK, Takeda AK, Badr MA, Rogers JS, Chase PB. Cell Mol Bioeng. 2013;6:183–198. doi: 10.1007/s12195-013-0269-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Schägger H, von Jagow G. Anal Biochem. 1987;166:368–79. doi: 10.1016/0003-2697(87)90587-2. [DOI] [PubMed] [Google Scholar]
- 58.Gordon AM, Chen Y, Liang B, LaMadrid M, Luo Z, Chase PB. Adv Exp Med Biol. 1998;453:187–96. doi: 10.1007/978-1-4684-6039-1_22. [DOI] [PubMed] [Google Scholar]
- 59.Mihajlović G, Brunet NM, Trbović J, Xiong P, von Molnár S, Chase PB. Appl Phys Lett. 2004;85:1060–1062. [Google Scholar]
- 60.Köhler J, Chen Y, Brenner B, Gordon AM, Kraft T, Martyn DA, Regnier M, Rivera AJ, Wang CK, Chase PB. Physiol Genomics. 2003;14:117–28. doi: 10.1152/physiolgenomics.00101.2002. [DOI] [PubMed] [Google Scholar]
- 61.Liang B, Chen Y, Wang CK, Luo Z, Regnier M, Gordon AM, Chase PB. Biophys J. 2003;85:1775–86. doi: 10.1016/S0006-3495(03)74607-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Uyeda TQ, Kron SJ, Spudich JA. J Mol Biol. 1990;214:699–710. doi: 10.1016/0022-2836(90)90287-V. [DOI] [PubMed] [Google Scholar]
- 63.Grove TJ, McFadden LA, Chase PB, Moerland TS. J Muscle Res Cell Motil. 2005;26:191–7. doi: 10.1007/s10974-005-9010-0. [DOI] [PubMed] [Google Scholar]
- 64.Hill AV. J Physiol. 1910;40:iv–vii. doi: 10.1113/jphysiol.1910.sp001377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Altschul SF, Madden TL, Schaffer AA, Zhang J, Zhang Z, Miller W, Lipman DJ. Nucleic Acids Res. 1997;25:3389–402. doi: 10.1093/nar/25.17.3389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Stefancsik R, Jha PK, Sarkar S. Proc Natl Acad Sci U S A. 1998;95:957–62. doi: 10.1073/pnas.95.3.957. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Tsoutsman T, Bagnall RD, Semsarian C. Clin Exp Pharmacol Physiol. 2008;35:1349–57. doi: 10.1111/j.1440-1681.2008.05037.x. [DOI] [PubMed] [Google Scholar]
- 68.Homsher E, Nili M, Chen IY, Tobacman LS. Biophys J. 2003;85:1046–52. doi: 10.1016/S0006-3495(03)74543-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Moerland TS, Sidell BD. J Exp Zool. 1986;238:287–95. doi: 10.1002/jez.1402380303. [DOI] [PubMed] [Google Scholar]
- 70.Jha PK, Leavis PC, Sarkar S. Biochemistry. 1996;35:16573–80. doi: 10.1021/bi9622433. [DOI] [PubMed] [Google Scholar]
- 71.Jin JP, Chong SM. Arch Biochem Biophys. 2010;500:144–50. doi: 10.1016/j.abb.2010.06.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Franklin AJ, Baxley T, Kobayashi T, Chalovich JM. Biophys J. 2012;102:2536–44. doi: 10.1016/j.bpj.2012.04.037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Gafurov B, Fredricksen S, Cai A, Brenner B, Chase PB, Chalovich JM. Biochemistry. 2004;43:15276–15285. doi: 10.1021/bi048646h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Thierfelder L, Watkins H, MacRae C, Lamas R, McKenna W, Vosberg HP, Seidman JG, Seidman CE. Cell. 1994;77:701–12. doi: 10.1016/0092-8674(94)90054-x. [DOI] [PubMed] [Google Scholar]
- 75.Van Eyk JE, Hodges RS. J Biol Chem. 1988;263:1726–1732. [PubMed] [Google Scholar]
- 76.Gordon AM, Homsher E, Regnier M. Physiol Rev. 2000;80:853–924. doi: 10.1152/physrev.2000.80.2.853. [DOI] [PubMed] [Google Scholar]
- 77.Asumda FZ, Chase PB. Differentiation. 2012;83:106–115. doi: 10.1016/j.diff.2011.10.002. [DOI] [PubMed] [Google Scholar]
- 78.Porollo A, Meller J. BMC Bioinformatics. 2007;8:316. doi: 10.1186/1471-2105-8-316. [DOI] [PMC free article] [PubMed] [Google Scholar]