

Abstract
M13 bacteriophages are assembled via a covalent layer-by-layer process to form a highly nanoporous network capable of organizing nanoparticles and acting as a scaffold for templating metal-oxides. The morphological and optical properties of the film itself are presented as well as its ability to organize and disperse metal nanoparticles.
Keywords: M13 bacteriophage, layer-by-layer assembly, bio-template, nanoporous network, localized surface plasmon resonance
For several electrochemical and photovoltaic applications, nanoporous structures confer advantages over bulk, macro-, or micro-porous materials. As the pore size decreases, the available surface area increases for a constant volume of material. Such an active surface can be inherently catalytic or photoactive, modified to display functional groups, or loaded with active materials. Porous nanostructures have been employed to improve the device performance for batteries,[1] catalytic electrodes,[2] biosensors,[3] and photovoltaics.[4]
For instance, in bulk heterojunction solar cells, especially those relying on conjugated polymers for light absorption, a nanoporous bi-continuous morphology is essential to achieving high performance because of the limited lifetime of photogenerated excitons in conjugated polymers.[5] In addition, systems such as tandem solar cells require the stacking of different layers of materials for selective light absorption, which calls for a high degree of control over the spatial organization of nanomaterials inside the thin film.[6] [7] [8] For electroluminescent, photovoltaic, and photocatalytic devices, electron donor and electron acceptor materials must be in close contact and share a large interfacial area to improve charge transport and dissociation. Finally, for any form of electro- or photocatalysis, the pathways for charge transport to the electrodes must be as continuous and direct as possible. Therefore, a nanoporous network of nanowires can serve as a useful template for the assembly of many classes of active materials.
Typically, nanopore arrays are created with energy intensive specialized processes such as electron beam etching, nanoimprinting lithography,[4a] or atomic layer deposition.[9] The simple template approach is also commonly used, and generally consists of assembling biological scaffolds or block co-polymers into organized networks followed by nucleating materials, specifically or not, onto the resulting structure. However, most of these templating techniques generate mesopores[10] or micropores,[11] as opposed to nanopores.
It has previously been demonstrated that the M13 bacteriophage, which is ~ 880 nm in length and 6.6 nm in diameter, can be utilized as a template to generate singular nanowires through a sequential nucleation, growth, and annealing process.[12] The 2,700 copies of its pVIII coat protein can be engineered to display affinity for specific materials, making these phages excellent templates for nanoscale manipulation of metals[13], metal oxides[11], conjugation of organic compounds, and even binding of carbon nanostructures such as nanotubes and graphene.[14] We have found that it is possible to construct nanoporous networks via layer-by-layer (LbL) assembly of the high aspect ratio M13 bacteriophage and that these networks can be assembled with nanoscale control of their composition and maintenance of their binding and templating capabilities. With such functional networks, the performance of photovoltaic, optical and electrochemical devices relying on high aspect ratio structures for charge transport and nanoscale features could be greatly improved.
Layer-by-layer assembly provides a means to assemble nano-objects in a tightly controlled manner in terms of thickness, morphology, and composition at the nanoscale. Additionally, it has the key advantage of being a low-cost, aqueous solution-based technique.[15] LbL assembly leverages electrostatic interactions, hydrogen bonding, or covalent crosslinks to build films. The latter allows for the assembly of singular nano-objects, such as the M13 bacteriophage, onto a substrate. Conversely, techniques that randomly crosslink nanomaterials such as phage in the bulk state using conventional ‘one-pot’ chemistry often results in gels with high porosity but contain aggregated or bundled nanostructures and larger pores, which decreases the functional surface area available for electrochemical or photocatalytic reactions.[16]
In previous work, M13 bacteriophages have been adsorbed onto the surface of layer-by-layer assembled polymer films[17] and have been used as polyanions in combination with polymers to tune the morphology of LbL films.[11] Herein, we describe a novel method to assemble nanoporous LbL films composed solely of M13 bacteriophages. As illustrated in Scheme 1, these bacteriophages can be functionalized with nanoparticles or quantum dots, and then serve as vehicles for controllably incorporating them into the thin film. The resulting bacteriophage scaffolds can be further used to template a variety of semiconducting or metallic materials via sol-gel syntheses, thereby generating nanoporous networks of inorganic wires. This work focuses on presenting the properties of the bacteriophage nanotemplate, and demonstrating its ability to assemble porous nanocomposites by controllably introducing gold nanoparticles in the structure, and synthesizing crystalline titania nanoporous networks via biomineralization.
Scheme 1.
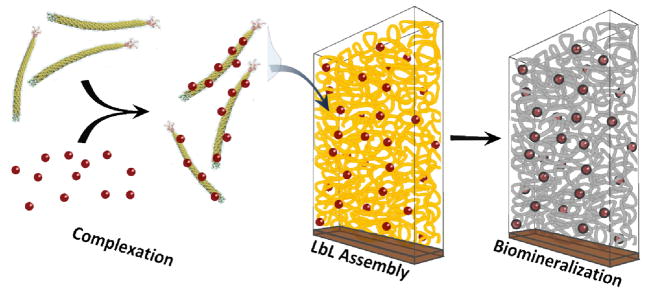
M13 bacteriophage complexation with nanoparticles, layer-by-layer assembly onto a substrate, and biomineralization.
The bacteriophages are assembled in an LbL fashion using 1-ethyl-3-(3-dimethylaminopropyl)carbodiimide (EDC) as a crosslinking agent in a novel and versatile covalent LbL process (see Figure 1A). This process relies on the crosslinking of carboxylic acid and amine groups, which are ubiquitously present on the surface of any M13 bacteriophage variant. Thus, the process is very versatile and allows for films to be built using bacteriophages with peptides that display many different functionalities. Covalent LbL assembly based on EDC chemistry has been employed by other groups to assemble films of proteins[18] or polymers[19]; the growth of these films was reported to be linear with the number of repeated cycles. A single protein with a negative surface charge at neutral pH, human serum albumin, could be layered in this fashion,[18] which indicates that repulsive electrostatic interactions do not override the reaction-diffusion driving force that transports the charged nanomaterials from the bulk solution to the surface, enabling the formation of covalent amide bonds.
Figure 1.

Bacteriophage film growth and morphology. A. Atomic force microscopy of a silicon substrate functionalized with a single layer of bacteriophages, and schematic representation of the covalent LbL process. B. Grow curve as a function of the number of EDC-phage bilayers, for three different assembly conditions: Diamond (1 bL = 36 min; 75 mM EDC, 5×1012 phage mL−1), Circle (1 bL = 14 min; 75 mM EDC, 1013 phage mL−1), Triangle (1 bL = 14 min; 150 mM EDC, 1013 phage mL−1). C. Growth curve as a function of the total assembly time for the same conditions. D. Surface morphology of the bacteriophage film after 16, 32, 48, 64 and 80 (EDC-phage) bilayers, characterized by AFM. E. Optical image of a 300 bL bacteriophage film constructed on silicon and cross-sectional TEM image of the film, illustrating the nanoporous morphology of the film.
The films can also be grown on a variety of substrates provided that the surfaces can be either silanized or rendered negatively charged via plasma treatment. We have successfully constructed bacteriophage films on glass, silicon wafers, plastics, and metal foils, and have shown that different surface functionalization methods result in the growth of films with the same morphology using covalent LbL assembly (see Figure S1).
The bacteriophage films grow in a linear manner, characteristic of LbL assembly, as shown in Figure 1B and C. Figure 1B shows that the thickness of the film can be predicted based on the number of EDC-phage bilayers (bL), regardless of the solution concentrations and dipping parameters. However, as shown in Figure 1C, the time-rate of growth of a film is dependent on the concentrations of EDC and the bacteriophage, as well as the dwell time during dips. As the film grows, it evolves from a flat bacteriophage-functionalized surface to a porous and rough film, as illustrated in Figure 1D. In the AFM images shown in this figure, the high aspect ratio features observed in the 16 bL image correspond to deposited bacteriophages. These individual features become less detectable as the thickness and roughness of the films increase. The surface roughness is generally proportional to 40% of the overall film thickness, and the porosity of a bacteriophage film was estimated to be 59% based on quartz crystal microbalance measurements (see Table S1 for details). As shown in Figure 1E, a film built on a silicon wafer appears uniform and translucent, yet a TEM cross-section demonstrates its nanoscale features. The diameter of the wire-like features in this TEM image is approximately equivalent to the diameter of a bacteriophage.
Sol-gel syntheses are employed to nucleate nanoparticles onto the phage scaffold and these nanoparticles are then sintered together to form a continuous mesh. Titania, a solution-processable photoactive wide-bandgap semiconductor, is an example of a useful material that can be nucleated on the bacteriophage template. To do so, the films are dipped vertically into an aqueous titanium tetrachloride (TiCl4) solution and hydrolysis is allowed to proceed at 80 °C for an hour. The films are then rinsed with deionized water, dried, and annealed at 450 °C in air. This annealing step has a dual role: it converts the amorphous titania to crystalline anatase nanoparticles (See Figure 2G and H for XRD and TEM characterization of the crystalline nanoparticles) and forms continuous structures along the virus template by interconnecting the titania nanoparticles, as well as removes all organic materials from the film via combustion (see Figure S2).
Figure 2.

Titanium dioxide nanoporous structures. A and B. Top surface SEM images of the phage film coated with titania and annealed at 450°C. In A, the reaction was stopped early (after 40 minutes) and in B the coating was allowed to proceed fully for an optimal reaction time of an hour. The color of the titania solution at each time point is show in the respective inset. Images of films coated with TiO2 for an hour are shown at two different magnifications. C. Pore size distribution based on 5 different top surface SEM images. The mean pore diameter is 8.1 nm, with a standard deviation of 6.5 nm for an hour reaction in TiCl4. D and E. TEM images of the cross-section of a titania network formed after reacting in TiCl4 for one hour reaction. The film was constructed on silicon and cross-sectioned with a focused ion beam, (D – STEM-Bright Field (BF) image, E – TEM image). F. STEM-HAADF (high angle annular dark field) image contrasting the titania nanowire mesh (brighter), from the silicon nitride support film that the phage film was constructed on (pale grey), and empty spaces (black). G and H. Crystallinity of titania-coated phage films after annealing at 450°C (G – TEM image showing lattice fringes of crystalline titania nanoparticles, H – X-Ray diffraction spectrum of resulting anatase titania). I. STEM-Bright Field image and elemental mapping of titanium for a titania nanowire mesh constructed on a silicon nitride support film.
The structure of a titania film templated from the bacteriophage network is depicted in Figure 2. The surface morphology reveals a nanowire-like interconnected mesh, with a wire diameter of 10 nm. Both on the surface and through the cross-section of the film, the vast majority of the pores have a diameter between 4 and 15 nm (see pore size distribution in Figure 2C), which is a critical size scale for many energy applications.
Precise control over the reaction time for TiO2 formation and the annealing step parameters are critical for obtaining a nanoporous film that maintains the structure of the bacteriophage template. The dilute TiCl4 solution is initially transparent, but its color changes as nanoparticles are formed. The solution turns light blue and then white as the nanometer-sized particles grow and scatter light. If the hydrolysis reaction is stopped prematurely (Figure 2A), not enough titania has nucleated on the virus to produce well-defined interconnected wires, as compared with films made with a longer reaction time (Figure 2B). However, if the reaction proceeds for too long, the film grows thicker on the template surfaces and the nanoporosity is lost, as nanoparticles merge and occlude the pores. The rate at which the temperature is elevated to 450 °C is also a key parameter for the successful preparation of the nanoporous titania. If the temperature is elevated slowly (over more than 30 min), then the bacteriophage scaffolds burn off before the titania nanoparticles are sintered together, resulting in a loss of structure, and the collapse of the film onto the substrate. A fast increase in temperature preserves the three-dimensional structure, but can also cause cracking of the film if the rate is too high (if increased to 450 °C over 10 min or less) (Figure S3).
Figure 2D and E show cross-sectional TEM images of a titania-coated bacteriophage film. The dark field image shown in Figure 2D uses contrast to reveal that the titania film is highly porous throughout its depth. In Figure 2E, the morphology of the cross-section can be visualized and a fine interconnected mesh structure is observed uniformly throughout the section.
TEM also allows for a clearer visualization of the bacteriophage-mediated organization of the titania nanoparticles. Figure 2F shows a STEM image of a thin film (10 EDC-phage bilayers) constructed, templated with titania, and annealed on a silicon nitride support film. The pores can clearly be visualized, as well as the sintered nanoparticles arranged into wire-like structures. An elemental mapping of titanium is also shown in Figure 2I.
Noble metal nanoparticles embedded in semiconducting networks produce plasmons, which can enhance the performance of light-absorbing and light-emitting devices. In the bacteriophage films, the distribution of nanoparticles varies significantly with the method of incorporation. Gold nanoparticles (AuNPs) can be added to the structure post-assembly, either by immersing the film into a nanoparticle solution, or by dropcasting this solution onto the film and allowing it to dry. When immersed in a NP solution, the majority of the particles remains on the surface of the film; whereas, capillary forces tend to pull the NPs deeper into the film when they are dropcast on the surface of the film (see XPS depth profiling results in Figure 3A, left panel). The dropcast method results in a more uniform NP distribution, although the concentration of NPs is still higher on the surface. For different M13 variants, different kinds of complexing behavior can be observed within the networks. For instance, the p8#9 variant expresses pVIII proteins with several serine amino acids, which through their hydroxyl groups, can coordinate, stabilize, and organize gold or silver nanoparticles.[13] Another variant, the E3 bacteriophage, expresses three glutamic acids on each of its pVIII proteins, and thus has a negative surface charge at neutral pH[11] due to its available carboxylic acid groups. Figure 3B shows a titania-coated p8#9 phage film onto which AuNPs were dropcasted before titania deposition. The functional groups on the p8#9 pVIII coat protein complex the NPs and uniformly distribute them on the surface. This phenomenon is not observed for all M13 bacteriophage variants. For example, more aggregation is observed when NPs are dropcast onto E3 bacteriophage films, which have a greater tendency to bind metals with less specificity (see Figure S4).
Figure 3.

Tight spatial distribution control of the bacteriophage-mediated incorporation of gold nanoparticles. A. XPS depth profiling analysis of the gold distribution as a function of the film depth, converted to nanoparticle to phage ratio for different film architectures. The top panel shows the distribution for nanoparticles infiltrated post-assembly, while the bottom panel shows films that were assembled with phage-nanoparticle complexes. B. SEM image of a phage film infiltrated with AuNPs, and coated with titania. The bright circles are the AuNPs dispersed within the nanowire mesh. C. Comparison of optical properties of AuNPs under different conditions; freely suspended in water, complexed to bacteriophages in solution, and incorporated into 59% porous dried bacteriophage films during LbL, or dropcast onto a film post-assembly. The theoretical prediction for each peak is shown with a dotted vertical line corresponding to the color of the experimental data. D. Shift in AuNPs absorption peak observed upon coating of the bacteriophage film with titania, before and after crystallization.
A significant advantage of the LbL assembly method is that nanoparticles can also be incorporated in a more controlled fashion. In the covalently-assembled bacteriophage films, we show that layers of viruses can be precisely assembled on top of each other and remain segregated. Because covalent bonds are linking these layers, the film does not disassemble upon changes in pH or salt concentration, as evidenced by its resistance to the highly acidic TiCl4 solution used for titania nucleation. In addition, these covalent crosslinks provide a means to tightly control the spatial organization of different templates and their resulting nanomaterials within the films.
As reported previously in our group, M13 bacteriophages can be used as a vehicle to carry nanoparticles.[20] To demonstrate control of this process during LbL assembly, two different film architectures were constructed with bacteriophages that were complexed with AuNPs in solution pre-assembly: (1) the AuNP-phage complexes were assembled in the layers at the bottom half of a multilayer film, while naked bacteriophages were assembled in the layers at top half, or (2) vice-versa. Through XPS depth profiling analysis, as shown in the right panel of Figure 3A, we show that the NPs can be localized near the bottom or top regions of the film, with control within a resolution of 50 nm when using 8 to 20 nm diameter AuNPs. When AuNPs are incorporated during the film assembly, their presence can be visualized macroscopically, as they give a uniform red color to the film (see optical images of films constructed on glass slides in Figure S5). When only bacteriophages are assembled, the resulting film is colorless. The precise control over localization of nanoparticles inside bacteriophage films would allow for the flexible design of functional and porous thin films. One could imagine tuning the distribution and concentration of NPs as a function of film thickness, and as a result, create on-demand localized plasmon resonances that affect only certain regions of the film. If, instead, the type of metallic NP is varied in different layers of the film, then the plasmon resonance wavelength could be defined in different sections of the film. Both of these scenarios find applications in designing photovoltaic devices that enhance the electric field and optimize light absorption in specific photoactive materials. Of course, all nanomaterials that M13 bacteriophages are programmed to bind could be incorporated in different layers of the film. For instance, the viruses could carry different nanoparticles into the LbL films in a hierarchical order, for instance quantum dots with different bandgaps for tandem solar cells or optical displays, particles with different dielectric constants for various optical nanostructures (e.g. antireflective coatings, gradient index materials, etc.), and metal nanoparticles for plasmonic-based biosensing or photocatalytic applications.
In order to further investigate the ability of bacteriophages to organize nanoparticles within the film, bacteriophage-AuNP complexes were assembled via LbL and the absorption spectrum of the film was experimentally collected for comparison against controls. Due to the phenomenon of localized surface plasmon resonance (LSPR), the location and width of the absorption peak for spherical AuNPs is very sensitive to two factors: (1) the dielectric function of the medium surrounding the nanoparticle and (2) the distance between neighboring nanoparticles in the system. By comparing the absorption measurements of AuNPs in different environments to theoretical predictions, inferences can be made about the degree of particle aggregation present in virus-based composites. Mie theory provides the extinction cross-secion, Cext, for a nanoparticle much smaller than the wavelength of incident light, which can be employed to predict the absorption spectra of isolated particles immersed in different media[21]:
| (1) |
where, a is the nanoparticle radius, ∊ = ∊1 + i∊2 is the complex dielectric function of the nanoparticle, and ∊m is the dielectric function of the medium surrounding it.
The dielectic function of a bacteriophage film with a porosity of 59% for wavelengths between 500 and 1700 nm was experimentally determined using ellipsometry (see Supporting Information). By fitting the ellipsometry results to the Bruggeman equation for an effective medium, estimates of the complex dielectric function for pure phage were obtained, which enabled the estimation of the effective dielectric constant observed by AuNPs when complexed onto the surfaces of bacteriophages suspended in aqueous solution (see Supporting Information). By inserting these dielectric functions into ∊m of Equation 1 and using the experimental dielectric function for gold as measured by Johnson and Christy for ∊,[22] the absorption peaks were predicted for 8 nm diameter AuNPs suspended in aqueous solution, complexed onto the bacteriophage in aqueous solution, or incorporated into a dried bacteriophage film.
It is well known that the formation of AuNP aggregates drastically changes the observed absorption spectra because higher order modes arise with resonance at lower energies, which collectively act to generate a broadened and red-shifted absorption peak.[23] In the simplest case of a AuNP dimer, the electromagnetic fields will couple and a additional orientation-dependent resonance peaks will arise at lower energies due to the longitudinal plasmon mode (see Figure S14 of Supporting Information). As shown in Table 1, the predicted peak positions are in excellent agreement with experimental observations and additional peaks or shoulders ares not observed in any of the absorption measurements. Together these two observations indicate that aggregates do not occur in significant enough abundance to be detected in the samples.
Table 1.
Predicted and measured absorption peak positions for 8 nm gold nanoparticles in different media
| Sample | Measured Absorption Peak Wavelength [nm] | Predicted Extinction Cross-Section Peak Wavelength [nm] |
|---|---|---|
| AuNPs in Water | 521.5 | 521.5 |
| AuNP-Phage Complexes in Water | 533.5 | 533.5 |
| AuNPs in 59% Porous Phage Film | 525.0 | 523.5 |
In contrast, when AuNPs are dropcast onto the bacteriophage film after it has been formed, they tend to fill the pores and form larger aggregates because the citrate capping agent loses its negative charge once the solvent is evaporated and an attractive van der Waals force causes neighboring nanoparticles to cluster. As shown in Figure 3C, the measured absorption peak of such films is significantly broadened and red-shifted from the LbL film peak, because, as opposed to isolated sub-wavelength AuNPs that only support the dipole resonance mode, AuNP clusters are able to support multiple higher-order resonance modes. The effect is the same as that observed when citrate-capped gold nanoparticles lose stability in the presence of salt and ethanol[24] or are aggregated via ligand exchange.[25]
By exploring the optical properties of the AuNP-bacteriophage films, two important observations have arisen: first, the effective index of bacteriophage-air films for wavelengths between 500 and 700 nm is close to that of water (nflim(λ = 500 nm) = 1.33+i0.10 to nflim(λ = 700 nm) = 1.45+i0.07 and nH2O= 1.33, see Figure S6). Therefore, if a plasmonic nanoparticle has a LSPR peak in this range when dispersed in water, the peak will be only slighly shifted when incorporated into the nanoporous bacteriophage film via LbL. Secondly, AuNPs must be incorporated into the bacteriophage films during the LbL process in order to remain well-dispersed and preserve the resonance peak location. AuNPs incorporated after the film has been built by LbL will result in aggregation with significantly red-shifted and broadened spectra.
When the AuNP-bacteriophage films created by LbL are templated with amorphous titania and then converted to anatase by annealing, we observe a red-shift in the measured absorbance spectra (Figure 3D). This shift is not a result of aggregation, but is due rather to the increase in the effective index of the porous medium upon the introduction of TiO2 into the system. Although the exact peak shift is dependent upon the degree of titania crystallinity and shell thickness, the finding is consistent with previously reported silver-titania[26] and gold-titania[27] nanoparticle composites. Quartz crystal microbalance was not used to measure the porosity of the anatase titania films because the substrate would not resist the required annealing temperatures. However, by using Bruggemann effective medium theory (see Supporting Information), the film porosity can be estimated by the position of the absorption peak. Based on the observed absorption peak at 567 nm, we estimated the final phage-based anatase titania film to be 53% porous.
In conclusion, we have shown that a covalent LbL assembled film composed only of bacteriophages generates a nanoporous structure that can serve as a scaffold for the nucleation of nanoparticles and nanowire meshes synthesized by a sol-gel method. Although this work focused on nucleating titania, the bacteriophage film can also serve as a scaffold for other materials. Because the bacteriophages have an affinity for gold and are assembled in an LbL fashion, AuNPs can be controllably loaded into specific regions of the film. This method is not limited to AuNPs but could be extended to other nanomaterials that can be directly bound to other M13 bacteriophage variants. For example, certrain variants can bind photoactive quantum dots or silver nanoparticles, which could facilitate the assembly of tandem or plasmon-enhanced solar devices. Furthermore, the nanoporosity of titania films described here and the nanoscale compositional control demonstrated herein are difficult to generate by other methods, making the bacteriophage nanotemplate promising for a variety of thin film devices where both nanoscale pores and high porosity are essential. Finally, the method described here to predict the optical properties of gold nanoparticles in nanoporous organic and inorganic films could be used to rationally design plasmonic-bacteriophage systems for a variety of optical applications.
Experimental
Experimental materials and methods can be found in Supporting Information.
Supplementary Material
Acknowledgments
This work was supported by Eni, S.p.A (Italy) through the MIT Energy Initiative Program. This work made use of the MIT MRSEC Shared Experimental Facilities supported by the National Science Foundation under award number DMR-0819762, and of the Institute for Soldiers Nanotechnology (ISN) Shared Facilities supported by the U.S. Army Research Office under contract W911NF-13-D-0001. Finally, the authors would like to thank the Koch Institute Swanson Biotechnology Center for the use of its core facilities. N.-M.D.C. gratefully acknowledges the Postgraduate Scholarship support from the Natural Sciences and Engineering Research Council of Canada (NSERC). N.-M.D.C, M.T.K. and P.-Y.C. acknowledge support from the MIT Energy Initiative Eni-MIT Energy Fellowship. ((Supporting Information is available online from Wiley InterScience or from the author)).
Footnotes
The authors wish to dedicate this paper to the memory of Officer Sean Collier, for his caring service to the MIT community and for his sacrifice.
Contributor Information
Noémie-Manuelle Dorval Courchesne, Department of Chemical Engineering, The David H. Koch Institute for Integrative Cancer Research, Massachusetts Institute of Technology, 77 Massachusetts Avenue, Cambridge, MA, 02139, USA.
Matthew T. Klug, Department of Mechanical Engineering, The David H. Koch Institute for Integrative Cancer Research, Massachusetts Institute of Technology, 77 Massachusetts Avenue, Cambridge, MA, 02139, USA
Po-Yen Chen, Department of Chemical Engineering, The David H. Koch Institute for Integrative Cancer Research, Massachusetts Institute of Technology, 77 Massachusetts Avenue, Cambridge, MA, 02139, USA.
Dr. Steven E. Kooi, The Institute for Soldier Nanotechnologies, Massachusetts Institute of Technology, 77 Massachusetts Avenue, Cambridge, MA, 02139, USA
Dr. Dong Soo Yun, The David H. Koch Institute for Integrative Cancer Research, Nanotechnology Materials Core, Massachusetts Institute of Technology, 77 Massachusetts Avenue, Cambridge, MA, 02139, USA
Dr. Nina Hong, J.A. Woollam Co., Inc., 645 M Street, STE 102, Lincoln, NE, 68508, USA
Prof. Nicholas X. Fang, Department of Mechanical Engineering, Massachusetts Institute of Technology, 77 Massachusetts Avenue, Cambridge, MA, 02139, USA
Prof. Angela M. Belcher, Email: Belcher@mit.edu, Department of Biological Engineering, Department of Materials Science and Engineering, The David H. Koch Institute for Integrative Cancer Research, Massachusetts Institute of Technology, 77 Massachusetts Avenue, Cambridge, MA, 02139, USA
Prof. Paula T. Hammond, Email: Hammond@mit.edu, Department of Chemical Engineering, The David H. Koch Institute for Integrative Cancer Research, Massachusetts Institute of Technology, 77 Massachusetts Avenue, Cambridge, MA, 02139, USA
References
- 1.Wang J, Du N, Song Z, Wu H, Zhang H, Yang D. RSC Advances. 2013;3:7543–7548. [Google Scholar]
- 2.a) Scavetta E, Mignani A, Tonelli D, Impellizzeri G, Romano L, Bongiorno C, Fraboni B, Grimaldi MG. Electrochemistry Communications. 2013;30:83–86. [Google Scholar]; b) Wittstock A, Wichmann A, Biener J, Baumer M. Faraday Discussions. 2011;152:87–98. doi: 10.1039/c1fd00022e. [DOI] [PubMed] [Google Scholar]
- 3.Cui HF, Zhang K, Zhang YF, Sun YL, Wang J, Zhang WD, Luong JHT. Biosensors and Bioelectronics. 2013;46:113–118. doi: 10.1016/j.bios.2013.02.029. [DOI] [PubMed] [Google Scholar]
- 4.a) Baek WH, Seo I, Yoon TS, Lee HH, Yun CM, Kim YS. Solar Energy Materials and Solar Cells. 2009;93:1587–1591. [Google Scholar]; b) Oosterhout SD, Wienk Martijn M, van Bavel Svetlana S, Thiedmann Ralf, Jan Anton Koster L, Gilot Jan, Loos Joachim, Schmidt Volker, Janssen Rene AJ. Nat Mater. 2009;8 doi: 10.1038/nmat2533. [DOI] [PubMed] [Google Scholar]
- 5.a) Nicholson PG, Castro FA. Nanotechnology. 2010;21 doi: 10.1088/0957-4484/21/49/492001. [DOI] [PubMed] [Google Scholar]; b) Wang H, Oey CC, Djurisic AB, Xie MH, Leung YH, Man KKY, Chan WK, Pandey A, Nunzi JM, Chui PC. Applied Physics Letters. 2005;87 [Google Scholar]
- 6.Yang J, You J, Chen C-C, Hsu W-C, Tan H-r, Zhang XW, Hong Z, Yang Y. ACS Nano. 2011;5:6210–6217. doi: 10.1021/nn202144b. [DOI] [PubMed] [Google Scholar]
- 7.Gan Q, Bartoli FJ, Kafafi ZH. Advanced Materials. 2013;25:2385–2396. doi: 10.1002/adma.201203323. [DOI] [PubMed] [Google Scholar]
- 8.Choi JJ, Wenger WN, Hoffman RS, Lim Y-F, Luria J, Jasieniak J, Marohn JA, Hanrath T. Advanced Materials. 2011;23:3144–3148. doi: 10.1002/adma.201100723. [DOI] [PubMed] [Google Scholar]
- 9.Knez M, Nielsch K, Niinistö L. Advanced Materials. 2007;19:3425–3438. [Google Scholar]
- 10.a) Wang Y, He C, Xing W, Li F, Tong L, Chen Z, Liao X, Steinhart M. Advanced Materials. 2010;22:2068–2072. doi: 10.1002/adma.200903655. [DOI] [PubMed] [Google Scholar]; b) Zakaria MB, Suzuki N, Torad NL, Matsuura M, Maekawa K, Tanabe H, Yamauchi Y. European Journal of Inorganic Chemistry. 2013;2013:2330–2335. [Google Scholar]
- 11.Chen PY, Ladewski R, Miller R, Dang X, Qi J, Liau F, Belcher AM, Hammond PT. Journal of Materials Chemistry A. 2013;1:2217–2224. [Google Scholar]
- 12.Mao C, Solis DJ, Reiss BD, Kottmann ST, Sweeney RY, Hayhurst A, Georgiou G, Iverson B, Belcher AM. Science. 2004;303:213–217. doi: 10.1126/science.1092740. [DOI] [PubMed] [Google Scholar]
- 13.a) Lee Y, Kim J, Yun DS, Nam YS, Shao-Horn Y, Belcher AM. Energy & Environmental Science. 2012;5:8328–8334. doi: 10.1039/C2EE21156D. [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Chiang CY, Mello CM, Gu J, Silva ECCM, Van Vliet KJ, Belcher AM. Advanced Materials. 2007;19:826–832. [Google Scholar]; c) Huang Y, Chiang CY, Lee SK, Gao Y, Hu EL, Yoreo JD, Belcher AM. Nano Letters. 2005;5:1429–1434. doi: 10.1021/nl050795d. [DOI] [PubMed] [Google Scholar]
- 14.Oh D, Dang X, Yi H, Allen MA, Xu K, Lee YJ, Belcher AM. Small. 2012;8:1006–1011. doi: 10.1002/smll.201102036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.a) Decher G. Science. 1997;277:1232–1237. [Google Scholar]; b) Hammond PT. Advanced Materials. 2004;16:1271–1293. [Google Scholar]
- 16.Chen P-Y, Dang X, Klug MT, Qi J, Dorval Courchesne N-M, Burpo FJ, Fang N, Hammond PT, Belcher AM. ACS Nano. 2013 doi: 10.1021/nn4014164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Yoo PJ, Nam KT, Belcher AM, Hammond PT. Nano Letters. 2008;8:1081–1089. doi: 10.1021/nl073079f. [DOI] [PubMed] [Google Scholar]
- 18.Tengvall P, Jansson E, Askendal A, Thomsen P, Gretzer C. Colloids and Surfaces B: Biointerfaces. 2003;28:261–272. [Google Scholar]
- 19.Serizawa T, Nanameki K, Yamamoto K, Akashi M. Macromolecules. 2002;35:2184–2189. [Google Scholar]
- 20.Flynn CE, Lee SW, Peelle BR, Belcher AM. Acta Materialia. 2003;51:5867–5880. [Google Scholar]
- 21.Hu M, Chen J, Li ZY, Au L, Hartland GV, Li X, Marquez M, Xia Y. Chemical Society Reviews. 2006;35:1084–1094. doi: 10.1039/b517615h. [DOI] [PubMed] [Google Scholar]
- 22.Johnson PB, Christy RW. Physical Review B. 1972;6:4370–4379. [Google Scholar]
- 23.Ghosh SK, Pal T. Chemical Reviews. 2007;107:4797–4862. doi: 10.1021/cr0680282. [DOI] [PubMed] [Google Scholar]
- 24.Han X, Goebl J, Lu Z, Yin Y. Langmuir. 2011;27:5282–5289. doi: 10.1021/la200459t. [DOI] [PubMed] [Google Scholar]
- 25.Basu S, Ghosh SK, Kundu S, Panigrahi S, Praharaj S, Pande S, Jana S, Pal T. Journal of colloid and interface science. 2007;313:724–734. doi: 10.1016/j.jcis.2007.04.069. [DOI] [PubMed] [Google Scholar]
- 26.Qi J, Dang X, Hammond PT, Belcher AM. ACS Nano. 2011;5:7108–7116. doi: 10.1021/nn201808g. [DOI] [PubMed] [Google Scholar]
- 27.Kalathil S, Khan M, Banerjee A, Lee J, Cho M. J Nanopart Res. 2012;14:1–9. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.