

Abstract
Herein we describe the development of a new class of antimicrobial and anti-infective peptidomimetics – cyclic lipo-α-AApeptides. They have potent and broad-spectrum antibacterial activity against a range of clinically relevant pathogens, including both multidrug-resistant Gram-positive and Gram-negative bacteria. Fluorescence microscopy suggests that cyclic lipo-α-AApeptides kill bacteria by disrupting bacterial membranes, possibly through a mechanism similar to that of cationic host defense peptides (HDPs). Furthermore, the cyclic lipo-α-AApeptide can mimic cationic host-defense peptides by antagonizing Toll-Like Receptor 4 (TLR4) signaling responses and suppressing pro-inflammatory cytokines such as tumor necrosis factor-α (TNF-α). Our results suggest that by mimicking host-defense peptides (HDPs), cyclic lipo-α-AApeptides may emerge to be a new class of antibiotic agents through direct bacteria killing, as well as novel anti-infective agents through immunomodulation.
Keywords: α-AApeptides, peptidomimetics, Toll-like receptors, TNF-α, host-defense peptides, antimicrobial activity, anti-infective activity
INTRODUCTION
Resistance to conventional antibiotics is a great threat to global public health.[1] As an alternative approach, host defense peptides (HDPs) have emerged as promising antibiotic agents. HDPs are present in virtually all life forms, acting as the first line of defense against microbial infection.[1–2] The current paradigm is that HDPs initially adopt globally amphipathic structures, which in turn disrupt negatively charged bacterial membranes.[1–2] Intriguingly, in addition to direct bacterial killing, some HDPs are reported to be able to modulate the innate immune response by antagonizing toll-like receptors (TLRs).[3] Immunomodulatory HDPs suppress production of harmful pro-inflammatory cytokines, thereby preventing complications from infection.[1] For instance, the human cathelicidin LL-37 can inhibit the induction of tumor necrosis factor-α (TNF-α) by Gram-positive and Gram-negative bacterial infections.[4]
Despite their promising potential, HDPs face obstacles for further development due to their susceptibility to protease degradation.[1] There is significant interest in the development of unnatural mimics of HDPs, including peptoids,[5] β-peptides,[6] other classes of oligomers and polymers.[7] Antimicrobial peptidomimetics function similarly to HDPs in their ability to assume globally amphipathic structures, embracing segregated hydrophobic and cationic patches that facilitate bacterial membrane disruption. We have previously reported a group of unnatural HDP mimics, α-AApeptides (N-acylated-N-aminoethyl peptides), as effective antimicrobial agents against various bacterial strains.[7h, 7i] Further lipidation have been shown to improve the antimicrobial activities of α-AApeptides and reduce antibiotic resistance.[7i] Herein, we report the development of modified α-AApeptides, through simultaneous lipidation and cyclization. These novel cyclic lipo-α-AApeptides demonstrate significantly improved potency and broad-spectrum activity against a range of clinically relevant Gram-positive and Gram-negative bacteria. Furthermore, the lead cyclic lipo-α-AApeptide is the first of its kind shown to mimic some HDPs by antagonizing TLR4-induced nuclear factor kappa-light-chain-enhancer of activated B cells (NF-κB) and nitric oxide (NO) signaling responses, as well as inhibiting production of pro-inflammatory cytokines such as tumor necrosis factor-α (TNF-α).[4] The linear analogue of the lead cyclic lipo-α-AApeptide shows weaker antimicrobial and inferior anti-inflammatory activity, indicating that both lipidation and cyclization are critical for the dual functions. As such, cyclic lipo-α-AApeptides may emerge as a new class of antibiotic agents with both direct bacterium killing and immunomodulation ability.
RESULTS AND DISCUSSIONS
Our current design is based on the finding that cyclization decreases sequence flexibility and stabilizes amphipathic structures, which leads to antimicrobial agents with improved potency.[7e, 7f, 7j] Clinically approved cyclic lipopeptides daptomycin[8] and polymyxin[9] are used to treat infections caused by Gram-positive and Gram-negative bacteria, respectively. However, the development of antimicrobial peptidomimetics with lipo-cyclic motifs is rare. We have previously shown that antimicrobial α-AApeptides can be designed in a straightforward manner by joining α-AApeptide building blocks together. These building blocks take up an amphipathic confirmations upon interaction with bacterial membranes.[7h, 7i] Thus, we hypothesize that cyclic α-AApeptides containing amphipathic α-AApeptide building blocks and a membrane-directing lipid tail will lead to improved antimicrobials.
Previous studies have shown that both the number of amphipathic building blocks and the length of lipid tails affect antimicrobial activity.[7h, 7i] This is because the specific interactions between bacterial membranes and peptide sequences are largely governed by overall hydrophobicity and cationic charges. In addition, the length of the lipid tails is believed to be important in penetrating bacterial membranes. [7h, 7i] Thus, a series of cyclic α-AApeptides containing different numbers of amphipathic α-AApeptide building blocks and different lengths of lipid tails were prepared following the synthetic route shown in Scheme 1.
Scheme 1.

Solid phase synthesis of cyclic lipo-α-AApeptides.
We designed amphipathic α-AApeptide building blocks that possess a cationic Lys side chain and hydrophobic lipid tails of varying lengths. Thus, different lipid tails can be conveniently introduced to cyclic α-AApeptides on the solid phase. C6-C16 alkyl tails are most commonly observed in lipopeptide antibiotics.[1] Therefore, we chose C6, C12 and C16 lipid tails to study their impact on the antimicrobial activity of cyclic-lipo-α-AApeptides. The synthesis was accomplished by first attaching an α-AApeptide building block to the solid support, followed by the addition of an allyl carboxylate ester-containing AApeptide building block, which enables cyclization directly on solid phase.[7j] After the α-AApeptide oligomers were assembled, the allyl protecting group was removed using Pd(PPh3)4. Following the removal of the Fmoc group, the sequence was cyclized in the presence of PyBop. Such an approach circumvents the cyclization of sequences in solution after cleavage from solid support, greatly simplifying the purification process.
We then tested these sequences against several clinically-relevant, multidrug-resistant bacteria, including both Gram-positive and Gram-negative strains (Table 1). Both the number of cationic charges and the extent of hydrophobicity have shown to be important for potent antimicrobial activity. Cyclic lipo-α-AApeptide 1, which contains three amphiphilic building blocks in the ring, as well as a C6 lipid tail, does not show any antimicrobial activity under tested conditions. However, with the same ring structure, replacement of the C6 lipid tail in 1 with a C12 lipid tail yields 2, which shows good activity against Gram-positive bacteria. Further increase of lipid tail length to C16 leads to 3, which has excellent potency against both Gram-positive and Gram-negative bacteria. The relationship between tail length and potency perhaps suggests that short lipid tails cannot penetrate bacterial membranes. This is especially relevant to Gram-negative bacteria, which have both outer and inner membranes. A similar correlation between lipid tail length and potency has also been observed in 5 and 6. Additionally, it seems that increasing ring size does not significantly affect antimicrobial activity, as seen with 3, 4, and 6. In fact, 3 possesses a smaller ring size and has the most potent antimicrobial activity against Gram-positive bacteria. Our results indicate that we may have reached an optimal ring size. This is also consistent to the findings that many natural cyclic peptide antimicrobial agents also contain 6–8 amino acid residues in their ring structures.[8–9] When compared to the previously developed linear α-AApeptide NB-119-2 and the HDP analogue Pexiganan, this new class of cyclic lipo-α-AApeptides are more potent.
Table 1.
The antimicrobial and hemolytic activities of cyclic lipo-α-AApeptides. The microbial organisms used are Klebsiella. pneumoniae (ATCC 13383), and Pseudomonas aeruginosa (ATCC 27853), Methicillin-resistant Staphylococcus epidermidis (RP62A), Vancomycin-resistant Enterococcus faecalis (ATCC 700802), Methicillin-resistant Staphylococcus aureus (ATCC 33591). The minimum inhibitory concentration (MIC) is the lowest concentration that completely inhibits growth after 20 h. HC50 is the concentration causing 50% hemolysis. NB-119-2 [7i] and Pexiganan [10] are listed for comparison.[7i, 10]
| Oligomers | n | Z | MIC, μg/mL (μM) | Hemolysis | ||||
|---|---|---|---|---|---|---|---|---|
| Gram-negative | Gram-positive | |||||||
| K. pneumoniae | P. aeruginosa | S. epidermidis | E. faecalis | S. aureus | HC50 μg/mL (μM) | |||
| 1 | 3 | C5H11 | >25 (17) | >25 (17) | >25 (17) | >25 (17) | >25 (17) | >250 (170) |
| 2 | 3 | C11H23 | >25 (16) | 20 (13) | 12 (8) | 12 (8) | 12 (8) | >250 (160) |
| 3 | 3 | C15H31 | 5 (3) | 10 (6) | 1 (0.6) | 1 (0.6) | 4 (2.4) | 150 (93) |
| 4 | 4 | C15H31 | 5 (3) | 10 (6) | 2 (1) | 2 (1) | 3 (2) | >250 (131) |
| 5 | 5 | C11H23 | >25 (12) | 10 (5) | >25 (12) | >25 (12) | >25 (12) | >250 (120) |
| 6 | 5 | C15H31 | 5 (2) | 10 (4) | 1 (0.4) | 5 (2) | 5 (2) | >250 (113) |
| NB-119-2 | - | - | 8 (5) | 12 (9) | 10 (6) | 4 (2) | 8 (5) | 250 (153) |
| Pexiganan | - | - | 8 (3) | 16 (6) | 8 (3) | 32 (12) | 16 (6) | 120 (45) |
The capability of cyclic lipo-α-AApeptides to mimic HDPs and disrupt bacterial membranes was assessed by fluorescence microscopy. MRSA ATCC 33591 was stained with the membrane-permeable dye 4′,6-diamidino-2-phenylindole (DAPI) and the non-membrane permeable dye propidium iodide (PI) in the presence or absence of cyclic lipo-α-AApeptide 3 (Figure 1). Upon treatment with 3, both DAPI and PI were able to permeate MRSA membranes, indicating perturbed integrity. The aggregation of bacteria after treatment may be due to the loss of membrane potential,[7h, 7k] consistent with the observation of membrane disruption.
Figure 1.

Fluorescence micrographs of MRSA treated with 5 μg/mL cyclic lipo-α-AApeptide 3 for 2 h. a1, control, no treatment, DAPI stained; a2, control, no treatment, PI stained; a3, control, no treatment, the merged view. b1, 3 treatment, DAPI stained; b2, 3 treatment, PI stained; b3, 3 treatment, the merged view.
In addition to potent antimicrobial activity, an interesting behavior of HDPs is that they may repress inflammation,[4] providing a new approach for the treatment of bacterium-induced inflammatory disorders. Our recent data has shown that certain HDP-mimicking γ-AApeptides also have the similar functions.[11] Therefore, as structural and functional mimics of HDPs, cyclic lipo-α-AApeptides are speculated to harness the innate immune system in a similar fashion. As such, we investigated the ability of 3 to modulate immune responses through TLR4 signaling. TLRs are evolutionarily conserved type I transmembrane proteins responsible for the inflammatory response to invasive pathogens. The native ligand for TLR4 is lipopolysaccharide (LPS), a component of Gram-negative bacterial cell walls.[4b] Through TLR4, LPS activates the NF-κB signaling pathway and downstream production of cytokines such as TNF-α. Currently, there is great interest in developing molecules to modulate TLR signaling as therapeutic candidates for immunity malfunctions. For an elegant example, Tew and co-workers have demonstrated that antimicrobial agents may exhibit immunomodulatory activities.[10a, 12]
To test the ability of cyclic lipo-α-AApeptides to modulate TLR4 signaling, we first tested the inhibitory effects of the lead compound, 3, in a nitric oxide (NO) production assay (Figure 2A).[13] NO produced in the downstream inflammatory signaling cascade for all TLRs plays an important role in immunological processes such as the generation of free radical bacterial toxins and regulation of phagocytosis.[14] RAW 264.7 murine monocyte macrophage cells were treated with varying concentrations of 3 in the presence of 20 ng/mL LPS. As shown in Figure 3, 3 is capable of inhibiting TLR4-induced NO production in a dose dependent fashion, with an IC50 value of 3.06 ± 0.21 μM. Importantly, 3 showed negligible cytotoxicity up to 100 μM (Figure 2B), confirming that its TLR4 suppression is not due to cytotoxicity.[15]
Figure 2.
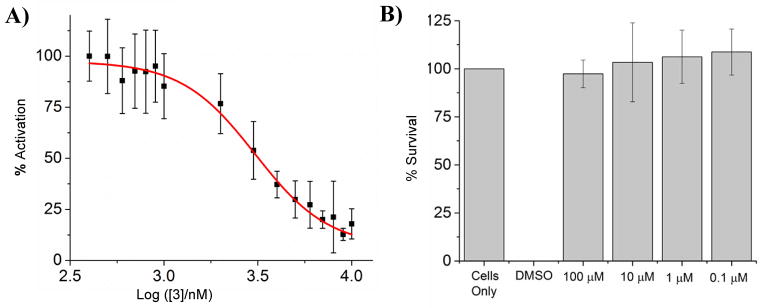
A) Reduced TLR4-induced NO production in the presence of 3. RAW 264.7 were treated with 20 ng/mL LPS and varying concentrations of 3. Data is normalized to 20 ng/mL LPS as 100% activation, and untreated cells as 0% activation. Our results indicate that 3 reduces NO production in a dose dependent fashion. B) 3 is non-toxic up to 100 μM as demonstrated by a Crystal Violet cell viability assay. Data is normalized with untreated cells as 100% survival, and 100% DMSO as 0% survival.
Figure 3.
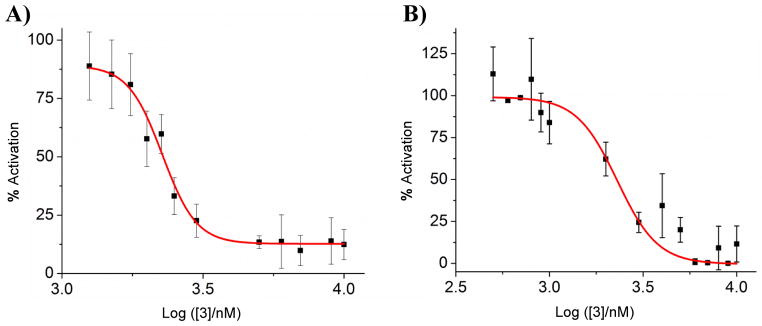
A) NF-κB activation is inhibited in HEK 293 cells by 3. Cells were plated in 96-well plate and treated with 20 ng/mL LPS and various concentrations of 3. Data is normalized to 20 ng/mL LPS as 100% activation, and untreated cells as 0% activation. B) Inhibition of TLR4-induced TNF-α production by 3. Mouse TNF (Mono/Mono) enzyme-linked immunosorbent assay (ELISA) in RAW 264.7 cells demonstrates that TNF-α production is decreased with treatment of 3. Data is normalized to 20 ng/mL LPS as 100% activation, and untreated cells as 0% activation.
To further confirm the effect of 3 on TLR signaling, a previously established secreted embryonic alkaline phosphatase (SEAP) assay was used to assess NF-κB activation in TLR4-overexpressing HEK 293 cells.[16] NF-κB transcription is directly correlated to TLR4 activation, as signaling results in NF-κB nuclear translocation.[17] Compound 3 inhibits NF-κB with an IC50 of 2.27 ± 0.08 μM (Figure 3A). Furthermore, the production of pro-inflammatory cytokine TNF-α is also inhibited with a comparable IC50 (Figure 3B), confirming the inflammation-suppressing effects of 3. As TNF-α dysregulation importantly contributes to medical conditions such as systemic lupus erythematosus and multiple cancers,[18] cyclic lipo-α-AApeptides may provide a group of novel candidates for the treatment of these diseases.
We also investigated if cyclization is critical for anti-inflammatory activity by comparing 3 with its linear analogue, 3-Linear. Interestingly, the ability of 3-Linear to decrease NO production is comparable to that of 3 (Figure S4b, supporting information) while its potency of inhibiting NF-κB and TNF-α is significantly less potent (Figure S5 and S6, supporting information). These results suggest that while linear and cyclic AApeptides do not behave identically, both possess anti-inflammatory activity. As such, we conclude that cyclization is not required to suppress inflammation. However, 3-Linear shows much decreased antimicrobial activity when compared to 3 (Figure S4a, supporting information), suggesting cyclization is more critical for the antimicrobial activity than anti-inflammatory activity.
It is known that non-peptidic antimicrobial agents suppress TLR signaling by binding specific TLR ligands (e.g. LPS).[10a, 12] By contrast, cyclic lipo-α-AApeptides appear to function through a different mechanism. In our experiment, both pre-treatment and co-treatment with 3 results in complete inhibition of NO production at 10 μM. Our data suggests that cyclic lipo-α-AApeptides reduce inflammatory responses using a novel mechanism, while simultaneously fighting bacterial infection.
CONCLUSIONS
In summary, we report a new class of antimicrobial and anti-inflammatory peptidomimetics, cyclic lipo-α-AApeptides that mimic the structures and functions of HDPs. These cyclic lipo-α-AApeptides can potently arrest the growth of multi-drug-resistant Gram-negative and Gram-positive bacteria. Furthermore, they can modulate immune responses by strongly antagonizing TLR4 signalling and suppressing LPS-induced production of pro-inflammatory cytokines. Here we report not only the design and synthesis of an unprecedented cyclic-lipo version of α-AApeptides, but also the first example of α-AApeptides that display both potent antimicrobial and anti-inflammatory activity. Cyclic-lipo-α-AApeptides may serve as a proof-of-concept for immunomodulatory antimicrobial peptidomimetics that do not bind to an extracellular ligand, but instead directly interact with components of the TLR signalling cascade. As the mechanistic nature of HDPs remains an outstanding problem to the field, these cyclic-lipo-α-AApeptides may help to understand an alternative mechanism for how HDPs function.
EXPERIMENTAL PROCEDURES
Solid-phase synthesis, purification and characterization of cyclic lipo-α-AApeptides
Standard Fmoc-chemistry protocol of solid phase synthesis was used to synthesize lipidated cyclic α-AApeptides on a Burell Wrist-Action shaker on Rink amide resin using peptide vessels (Figure S2). [7h–j, 19] Every coupling cycle consisted of Fmoc deprotection using 20% piperidine/DMF, 6 h coupling of 2 equiv. of building blocks in the presence of 4 equiv. of DIC (Diisopropylcarbodiimide) and DhbtOH (3–4-Dihydro-3-hydroxy-4-oxo-1-2-3-benzotriazine) in DMF. Firstly, the lipidated building block was attached to the solid phase support, followed by coupling with the building blocks using standard Fmoc-chemistry. After the desired sequence was assembled, the allyl group was removed by usage of Pd(PPh3)4/PhSiH3 (0.2 equiv./10 equiv.) in CH2Cl2 for 2 hours (repeated twice). The Fmoc group was then removed and the intramolecular cyclization was achieved by PyBop/HOBt/DIEA/DMF. Lastly, the resin was transferred into a 4 mL vial and the cyclic-lipo-α-AApeptide was cleaved from the solid support in 50:48:2(v/v) TFA/ CH2Cl2/Triisopropylsilane in 5 hours. The solvent was evaporated and the residues were analyzed and purified by Waters HPLC system on analytical (1 mL/min) and preparative (20 mL/min) modules, respectively. The purities of cyclic lipo-α-AApeptides (>95%) were determined by analytical HPLC.
Antimicrobial assay for MIC determination
The bacterial strains used for testing the efficacy of cyclic lipo-α-AApeptides were multidrug-resistant S. epidermidis (RP62A), Vancomycin-resistant E. faecalis (ATCC 700802), methicillin resistant S. aureus (ATCC 33591), K. pnuemoniae (ATCC 13383) and multidrug-resistant P. aeruginosa (ATCC 27853). The antimicrobial activities of the cyclic lipo-α-AApeptides developed were determined in by the serial dilution method. Bacterial cells were grown overnight at 37 °C in 5 mL medium after which a bacterial suspension of approximately 106 CFU/mL in Luria broth or trypticase soy was prepared ensuring that the bacterial cells were in the mid-logarithmic phase. Aliquots of bacterial suspension (50 μL) were added to 50 μL of medium containing the cyclic lipo-α-AApeptides for a total volume of 100 μL in each well. The cyclic lipo-α-AApeptides were dissolved in PBS buffer in two–fold serial dilutions. The concentration range used for peptides was 25 to 0.5 μg/mL. The 96-well plates were incubated at 37 °C for about 20 h, and the optical density (OD) at a wavelength of 600 nm. The lowest concentration at which complete inhibition of bacterial growth is observed is defined as the minimum inhibitory concentration (MIC). The experiments were repeated for three times and each time in duplicate.
Hemolysis assay
Freshly drawn human red blood cells (hRBC’s) were used for the assay. The blood sample was washed with PBS buffer several times and centrifuged at 700 g for 10 min until a clear supernatant was observed. The hRBC’s were re-suspended in 1 × PBS to get a 5% v/v suspension which was used to perform the assay. Two-fold serial dilutions of cyclic lipo-α-AApeptides were prepared in PBS buffer. Concentrations ranging from 250 μg/mL through 1.56 μg/mL were tested by adding the cyclic lipo-α-AApeptides solutions to sterile 96-well plates to make up to a total volume of 50 μL in each well. Then 50 μL of 5%v/v hRBC solution was added to make up a total volume of 100 μL in each well. The 0% hemolysis point and 100% hemolysis point were determined in 1 X PBS and 0.2% Triton-X-100 respectively. The 96 well plate was incubated at 37 °C for 1 h and centrifuged at 3500 rpm for 10 min. The supernatant (30 μL) was then diluted with 100 μL of 1 × PBS and hemoglobin was detected by measuring the optical density at 360nm by Biotek microtiter plate reader (Type: Synergy HT). % hemolysis = (Abs sample -Abs PBS)/(Abs Triton –Abs PBS) x 100. Peptide concentrations corresponding to 50% hemolysis were determined from the dose-response curves. The experiments were repeated for three times and each time in duplicate.
Fluorescence microscopy
A double staining method with DAPI (4′,6-Diamidino-2-phenylindole dihydrochloride, Sigma,>98%) and PI (Propidium iodide, Sigma) as fluorophores was used to visualize and differentiate the viable from the dead S. aureus (ATCC 33591) cells. DAPI being a double stranded DNA binding dye, stains all bacterial cells irrespective of their viability. Ethidium derivatives such as propidium iodide (PI) is capable of passing through only damaged cell membranes and intercalates with the nucleic acids of injured and dead cells to form a bright red fluorescent complex. The bacterial cells were grown until they reached mid-logarithmic phase and then cells (2 × 10 3) were incubated with the cyclic lipo-α-AApeptide 3 at 5 μg/mL for 4 h. Then the cells were pelleted by centrifugation at 3000 g for 15 min. The supernatant was decanted and the cells were washed with 1X PBS several times and then incubated with PI (5 μg/mL) in the dark for 15 min at 0°C. The excessive PI was removed by washing the cells several times with 1 × PBS several times. Lastly, the cells were incubated with DAPI (10 μg/mL in water) for 15 min in the dark at 0 °C. Then the excessive DAPI solution was removed and the cells were washed with 1 × PBS. The controls were performed following the exactly same procedure for bacteria in the absence of 3. The bacteria were examined by using the Zeiss Axio Imager Z1optical microscope (100 ×).
Fluorescent Detection of Nitric Oxide
Raw 264.7 (Mouse leukaemic monocyte macrophage cell line) cells were grown in RPMI 1640 medium with 10% fetal bovine serum (FBS), 1% Penicillin/streptomycin, and 1% L-glutamine. On day one, cells were plated in a 96-well plate at 75,000 cells/well in complete RPMI 1640 medium. Cells were grown overnight at 37 °C and 5% CO2 in a humidified incubator. On day 2, media was removed, and cells were placed in unsupplemented RPMI 1640 medium. LPS (20 ng/mL) and the appropriate concentration of AApeptides were added to a final volume of 200 μL. All stock solutions of AApeptides as 20 mM stocks were prepared in PBS, and then diluted to the desired concentration with PBS. There are PBS controls in each experiment. Plates were then incubated for 24 h after treatment. Following incubation, 100 μL of media was removed from each well and added to a flat black 96-well microfluor plate (Thermo Scientific, MA, USA). 10 μL 2,3-diaminonamthalene (0.05 mg/mL) in HCl (0.62 M) was added to the media and incubated for 20 min in the dark. The reaction was quenched with NaOH (3.0 M, 5 μL), and the plate was read on a Beckman Coulter DTX880 plate reader (Beckman Coulter, CA, USA). Data was collected with excitation at 360 nm and emission at 430 nm. Data was normalized with the ligand only control as 100% activation, and the untreated cells as 0% activation. Fold inhibition = [(Sample 430 nm – Untreated cells 430 nm)/(Ligand Control 430 nm – Untreated cells 430 nm)]. The IC50 values were calculated graphically using OriginPro v8.6 software.
Secreted Embryonic Alkaline Phosphatase (SEAP) Reporter of NF-κB Transcription
HEK293 (Human Embryonic Kidney 293) cells were grown in Dulbecco’s Modified Eagle Medium (DMEM) with 10% fetal bovine serum (FBS), 1% Penicillin/streptomycin, and 1% L-glutamine. HEK293 cells have been stably transfected with human TLR4, as well as the required accessory proteins MD-2 and CD14. Additionally, the cells possess an optimized alkaline phosphatase reporter gene under the control of a NF-κB inducible promoter. [20] On day one, cells were plated in a 96-well plate at 40,000 cells/well in complete DMEM medium. Cells were grown overnight at 37 °C and 5% CO2 in a humidified incubator. On day 2, media was removed, and cells were placed in Optimem + 0.5% FBS medium. 20 ng/mL LPS and the appropriate concentration of AApeptides were added to a final volume of 200 μL. All stock solutions of AApeptides were prepared as 20 mM stocks in PBS, and then diluted to the desired concentration with PBS. There were PBS controls in each experiment. Plates were then incubated for 24 h after treatment. Following incubation, the medium was assayed per the instructions of the Phospha-Light™ SEAP Reporter Gene Assay System (Applied Biosystems, NY, USA). The plate was read on a Beckman Coulter DTX880 plate reader (Beckman Coulter, CA, USA). Data was collected with luminescence at 430 nm. Data was normalized with the ligand only control as 100% activation, and the untreated cells as 0% activation. Fold inhibition = [(Sample 430 nm – Untreated cells 430 nm)/(Ligand Control 430 nm – Untreated cells 430 nm)]. The IC50 values were calculated graphically using OriginPro v8.6 software.
Enzyme-Linked Immunosorbent Assay (ELISA) Detection of TNF-α
Raw 264.7 (Mouse leukaemic monocyte macrophage cell line) cells were grown in RPMI 1640 medium with 10% fetal bovine serum (FBS), 1% Penicillin/streptomycin, and 1% L-glutamine. On day one, cells were plated in a 96-well plate at 75,000 cells/well in complete RPMI 1640 medium. Cells were grown overnight at 37 °C and 5% CO2 in a humidified incubator. On day 2, media was removed, and cells were placed in unsupplemented RPMI 1640 medium. LPS (20 ng/mL) and the appropriate concentration of AApeptides were added to a final volume of 200 μL. We prepared all stock solutions of AApeptides as 20 mM stocks in PBS, and then diluted to the desired concentration with PBS. There are PBS controls in each experiment. Plates were then incubated for 24 hours after treatment. Following incubation, samples were assayed for TNF-α per the method outlined in the BD Biosciences Mouse TNF (Mono/Mono) ELISA Set (BD Biosciences, CA, USA). The plate was read on a Beckman Coulter DTX880 plate reader (Beckman Coulter, CA, USA). Data was collected with absorbance at 450 nm. Data was normalized with the ligand only control as 100% activation, and the untreated cells as 0% activation. Fold inhibition = [(Sample 450 nm − Untreated cells 450 nm)/(Ligand Control 450 nm − Untreated cells 450 nm)]. The IC50 values were calculated graphically using OriginPro v8.6 software.
Crystal Violet Toxicity Assay
Cells which were treated with compound for nitric oxide experimentation were also tested for compound toxicity using crystal violet stain. Media was decanted, and cells were fixed for 20 min in paraformaldehyde (4%). After fixing, formaldehyde was removed and cells were incubated for one hour with crystal violet stain (0.05%). After incubation, cells were rinsed with deionized water to remove excess stain, and reconstituted in 100% methanol for 10 min. The plate was read on a Beckman Coulter DTX880 plate reader (Beckman Coulter, CA, USA). Data was collected with absorbance at 535 nm. Data was normalized with the untreated cells control as 100% survival, and 100% DMSO as 0% survival. Fold inhibition = [(Sample 535 nm − 100% DMSO 535 nm)/(Untreated cells 535 nm − 100% DMSO 535 nm)].
Supplementary Material
Acknowledgments
We thank financial support from USF start-up for (JC) and NIH GM101279 and GM103843 for (HY).
Contributor Information
Prof. Hang Yin, Email: hubert.yin@colorado.edu.
Prof. Jianfeng Cai, Email: jianfengcai@usf.edu.
References
- 1.Hancock RE, Sahl HG. Nat Biotechnol. 2006;24:1551–1557. doi: 10.1038/nbt1267. [DOI] [PubMed] [Google Scholar]
- 2.Marr AK, Gooderham WJ, Hancock RE. Curr Opin Pharmacol. 2006;6:468–472. doi: 10.1016/j.coph.2006.04.006. [DOI] [PubMed] [Google Scholar]
- 3.Mookherjee N, Brown KL, Bowdish DME, Doria S, Falsafi R, Hokamp K, Roche FM, Mu RX, Doho GH, Pistolic J, Powers JP, Bryan J, Brinkman FSL, Hancock REW. J Immunol. 2006;176:2455–2464. doi: 10.4049/jimmunol.176.4.2455. [DOI] [PubMed] [Google Scholar]
- 4.a) Bowdish DME, Davidson DJ, Lau YE, Lee K, Scott MG, Hancock REW. J Leukocyt Biol. 2005;77:451–459. doi: 10.1189/jlb.0704380. [DOI] [PubMed] [Google Scholar]; b) Scott MG, Gold MR, Hancock REW. Infect Immun. 1999;67:6445–6453. doi: 10.1128/iai.67.12.6445-6453.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.a) Olsen CA, Ziegler HL, Nielsen HM, Frimodt-Moller N, Jaroszewski JW, Franzyk H. ChemBioChem. 2010;11:1356–1360. doi: 10.1002/cbic.201000232. [DOI] [PubMed] [Google Scholar]; b) Chongsiriwatana NP, Patch JA, Czyzewski AM, Dohm MT, Ivankin A, Gidalevitz D, Zuckermann RN, Barron AE. Proc Natl Acad Sci U S A. 2008;105:2794–2799. doi: 10.1073/pnas.0708254105. [DOI] [PMC free article] [PubMed] [Google Scholar]; c) Chongsiriwatana NP, Miller TM, Wetzler M, Vakulenko S, Karlsson AJ, Palecek SP, Mobashery S, Barron AE. Antimicrob Agents Chemother. 2011;55:417–420. doi: 10.1128/AAC.01080-10. [DOI] [PMC free article] [PubMed] [Google Scholar]; d) Kapoor R, Wadman MW, Dohm MT, Czyzewski AM, Spormann AM, Barron AE. Antimicrob Agents Chemother. 2011;55:3054–3057. doi: 10.1128/AAC.01516-10. [DOI] [PMC free article] [PubMed] [Google Scholar]; e) Huang ML, Shin SB, Benson MA, Torres VJ, Kirshenbaum K. ChemMedChem. 2012;7:114–122. doi: 10.1002/cmdc.201100358. [DOI] [PubMed] [Google Scholar]; f) Kapoor R, Eimerman PR, Hardy JW, Cirillo JD, Contag CH, Barron AE. Antimicrob Agents Chemother. 2011;55:3058–3062. doi: 10.1128/AAC.01667-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.a) Karlsson AJ, Pomerantz WC, Weisblum B, Gellman SH, Palecek SP. J Am Chem Soc. 2006;128:12630–12631. doi: 10.1021/ja064630y. [DOI] [PubMed] [Google Scholar]; b) Karlsson AJ, Pomerantz WC, Neilsen KJ, Gellman SH, Palecek SP. ACS Chem Biol. 2009;4:567–579. doi: 10.1021/cb900093r. [DOI] [PubMed] [Google Scholar]; c) Karlsson AJ, Flessner RM, Gellman SH, Lynn DM, Palecek SP. Biomacromolecules. 2010;11:2321–2328. doi: 10.1021/bm100424s. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.a) Claudon P, Violette A, Lamour K, Decossas M, Fournel S, Heurtault B, Godet J, Mely Y, Jamart-Gregoire B, Averlant-Petit MC, Briand JP, Duportail G, Monteil H, Guichard G. Angew Chem Int Ed. 2010;49:333–336. doi: 10.1002/anie.200905591. [DOI] [PubMed] [Google Scholar]; b) Hua J, Scott RW, Diamond G. Mol Oral Microbiol. 2010;25:426–432. doi: 10.1111/j.2041-1014.2010.00591.x. [DOI] [PMC free article] [PubMed] [Google Scholar]; c) Hua J, Yamarthy R, Felsenstein S, Scott RW, Markowitz K, Diamond G. Mol Oral Microbiol. 2010;25:418–425. doi: 10.1111/j.2041-1014.2010.00590.x. [DOI] [PMC free article] [PubMed] [Google Scholar]; d) Choi S, Isaacs A, Clements D, Liu D, Kim H, Scott RW, Winkler JD, DeGrado WF. Proc Natl Acad Sci U S A. 2009;106:6968–6973. doi: 10.1073/pnas.0811818106. [DOI] [PMC free article] [PubMed] [Google Scholar]; e) Srinivas N, Jetter P, Ueberbacher BJ, Werneburg M, Zerbe K, Steinmann J, Van der Meijden B, Bernardini F, Lederer A, Dias RL, Misson PE, Henze H, Zumbrunn J, Gombert FO, Obrecht D, Hunziker P, Schauer S, Ziegler U, Kach A, Eberl L, Riedel K, DeMarco SJ, Robinson JA. Science (New York, NY. 2010;327:1010–1013. doi: 10.1126/science.1182749. [DOI] [PubMed] [Google Scholar]; f) Obrecht D, Robinson JA, Bernardini F, Bisang C, DeMarco SJ, Moehle K, Gombert FO. Curr Med Chem. 2009;16:42–65. doi: 10.2174/092986709787002844. [DOI] [PubMed] [Google Scholar]; g) Niu Y, Padhee S, Wu H, Bai G, Harrington L, Burda WN, Shaw LN, Cao C, Cai J. Chem Commun. 2011;47:12197–12199. doi: 10.1039/c1cc14476f. [DOI] [PMC free article] [PubMed] [Google Scholar]; h) Padhee S, Hu Y, Niu Y, Bai G, Wu H, Costanza F, West L, Harrington L, Shaw LN, Cao C, Cai J. Chem Commun. 2011;47:9729–9731. doi: 10.1039/c1cc13684d. [DOI] [PubMed] [Google Scholar]; i) Hu Y, Amin MN, Padhee S, Wang R, Qiao Q, Ge B, Li Y, Mathew A, Cao C, Cai J. ACS Med Chem Lett. 2012;3:683–686. doi: 10.1021/ml3001215. [DOI] [PMC free article] [PubMed] [Google Scholar]; j) Wu H, Niu Y, Padhee S, Wang RE, Li Y, Qiao Q, Ge B, Cao C, Cai J. Chem Sci. 2012;3:2570–2575. [Google Scholar]; k) Niu Y, Padhee S, Wu H, Bai G, Qiao Q, Hu Y, Harrington L, Burda WN, Shaw LN, Cao C, Cai J. J Med Chem. 2012;55:4003–4009. doi: 10.1021/jm300274p. [DOI] [PubMed] [Google Scholar]; l) Gibney KA, Sovadinova I, Lopez AI, Urban M, Ridgway Z, Caputo GA, Kuroda K. Macromol Biosci. 2012;12:1279–1289. doi: 10.1002/mabi.201200052. [DOI] [PMC free article] [PubMed] [Google Scholar]; m) Kuroda K, DeGrado WF. J Am Chem Soc. 2005;127:4128–4129. doi: 10.1021/ja044205+. [DOI] [PubMed] [Google Scholar]; n) Tew GN, Scott RW, Klein ML, Degrado WF. Acc Chem Res. 2010;43:30–39. doi: 10.1021/ar900036b. [DOI] [PMC free article] [PubMed] [Google Scholar]; o) Lienkamp K, Tew GN. Chem Eur J. 2009;15:11784–11800. doi: 10.1002/chem.200900049. [DOI] [PubMed] [Google Scholar]
- 8.Weis F, Beiras-Fernandez A, Schelling G. Curr Opin Investig Drugs. 2008;9:879–884. [PubMed] [Google Scholar]
- 9.Zavascki AP, Goldani LZ, Li J, Nation RL. J Antimicrob Chemother. 2007;60:1206–1215. doi: 10.1093/jac/dkm357. [DOI] [PubMed] [Google Scholar]
- 10.a) Thaker HD, Som A, Ayaz F, Lui DH, Pan WX, Scott RW, Anguita J, Tew GN. J Am Chem Soc. 2012;134:11088–11091. doi: 10.1021/ja303304j. [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Ge Y, MacDonald DL, Holroyd KJ, Thornsberry C, Wexler H, Zasloff M. Antimicrob Agents Chemother. 1999;43:782–788. doi: 10.1128/aac.43.4.782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Li Y, Smith C, Wu H, Padhee S, Manoj N, Cardiello J, Qiao Q, Cao C, Yin H, Cai J. ACS Chem Biol. 2013 doi: 10.1021/cb4006613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Som A, Navasa N, Percher A, Scott RW, Tew GN, Anguita J. Clin Vacc Immunol. 2012;19:1784–1791. doi: 10.1128/CVI.00291-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Cheng K, Wang XH, Yin H. J Am Chem Soc. 2011;133:3764–3767. doi: 10.1021/ja111312h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.MacMicking J, Xie QW, Nathan C. Annual Rev Immunol. 1997;15:323–350. doi: 10.1146/annurev.immunol.15.1.323. [DOI] [PubMed] [Google Scholar]
- 15.a) Vanparys P, Deknudt G, Sysmans M, Teuns G, Coussement W, Vancauteren H. Toxicol in Vitro. 1993;7:471–476. doi: 10.1016/0887-2333(93)90049-b. [DOI] [PubMed] [Google Scholar]; b) Raj L, Ide T, Gurkar AU, Foley M, Schenone M, Li XY, Tolliday NJ, Golub TR, Carr SA, Shamji AF, Stern AM, Mandinova A, Schreiber SL, Lee SW. Nature. 2011;475:231–234. doi: 10.1038/nature10167. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 16.Hutchinson MR, Zhang YN, Shridhar M, Evans JH, Buchanan MM, Zhao TX, Slivka PF, Coats BD, Rezvani N, Wieseler J, Hughes TS, Landgraf KE, Chan S, Fong S, Phipps S, Falke JJ, Leinwand LA, Maier SF, Yin H, Rice KC, Watkins LR. Brain Behavior Immun. 2010;24:83–95. doi: 10.1016/j.bbi.2009.08.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Zoubir M, Flament C, Gdoura A, Bahleda R, Litvinova E, Soumelis V, Conforti R, Viaud S, Soria JC, Kroemer G, Zitvogel L, Chaput N. Cell Cycle. 2011;10:118–126. doi: 10.4161/cc.10.1.14445. [DOI] [PubMed] [Google Scholar]
- 18.a) Locksley RM, Killeen N, Lenardo MJ. Cell. 2001;104:487–501. doi: 10.1016/s0092-8674(01)00237-9. [DOI] [PubMed] [Google Scholar]; b) Sabry A, Sheashaa H, El-husseini A, Mahmoud K, Eldahshan KF, George SK, Abdel-Khalek E, El-Shafey EM, Abo-Zenah H. Cytokine. 2006;35:148–153. doi: 10.1016/j.cyto.2006.07.023. [DOI] [PubMed] [Google Scholar]; c) Li J, Wang X, Zhang F, Yin H. Pharmacol Therapeu. ASAP; 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Hu Y, Li X, Sebti SM, Chen J, Cai J. Bioorg Med Chem Lett. 2011;21:1469–1471. doi: 10.1016/j.bmcl.2011.01.005. [DOI] [PubMed] [Google Scholar]
- 20.Hutchinson MR, Zhang YN, Brown K, Coats BD, Shridhar M, Sholar PW, Patel SJ, Crysdale NY, Harrison JA, Maier SF, Rice KC, Watkins LR. Eur J Neurosci. 2008;28:20–29. doi: 10.1111/j.1460-9568.2008.06321.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.