

Abstract
The inclusion of phytase in monogastric animal feed has the benefit of hydrolyzing indigestible plant phytate (myo-inositol 1,2,3,4,5,6-hexakis dihydrogen phosphate) to provide poultry and swine with dietary phosphorus. An ideal phytase supplement should have a high temperature tolerance, allowing it to survive the feed pelleting process, a high specific activity at low pHs, and adequate gastric performance. For this study, the performance of a bacterial phytase was optimized by the use of gene site saturation mutagenesis technology. Beginning with the appA gene from Escherichia coli, a library of clones incorporating all 19 possible amino acid changes and 32 possible codon variations in 431 residues of the sequence was generated and screened for mutants exhibiting improved thermal tolerance. Fourteen single site variants were discovered that retained as much as 10 times the residual activity of the wild-type enzyme after a heated incubation regimen. The addition of eight individual mutations into a single construct (Phy9X) resulted in a protein of maximal fitness, i.e., a highly active phytase with no loss of activity after heating at 62°C for 1 h and 27% of its initial activity after 10 min at 85°C, which was a significant improvement over the appA parental phytase. Phy9X also showed a 3.5-fold enhancement in gastric stability.
Phytate is the major form of phosphorus storage in the cereal grains and legumes used in commercial animal feeds (19). Unfortunately, phytate is not hydrolyzed in the monogastric gut and phytate-associated phosphate remains unabsorbed, requiring the exogenous addition of phosphate to avoid phosphorus deficiency. In addition to increasing basic food costs, the phosphate in excreted phytate is available to soil microbial consortia, by which it is broken down to create phosphate runoff and serious environmental pollution (16, 17). This phenomenon has led to hypereutrophication of streams, rivers, and lakes (11, 12). In addition, phytate is antinutritive; it chelates important dietary minerals such as zinc, copper, iron, magnesium, tin, and calcium (5, 7, 14, 15, 17) and has been implicated in the precipitation of metal-binding enzymes and proteins (25). The inclusion of a phytase in animal feed can reduce these problems and has been shown for 10 years to be a safe alternative to the addition of exogenous phosphate (27).
The inclusion of phytases in animal feed is increasingly mandated as environmental and federal laws and penalties designed to regulate the addition of phosphate in animal feed are enacted. Because traditional enzymes cannot survive feed-pelleting temperatures, phytase use incurs an additional expense due to the need to apply it in liquid form after pelleting. A more economical approach would be the addition of a thermally stable phytase to feed mixtures followed by a simultaneous pelleting of enzyme and feed.
Phytase catalyzes the partial or complete hydrolytic removal of orthophosphate from phytate. The complete hydrolysis of phytate (see structure below) results in the production of one molecule of inositol and six molecules of inorganic phosphate.

Phytases can be found throughout nature and occur in plants, some animal tissues, and microbes (10). Phytases are also found in enteric bacteria, which account for 0.1% of the total bacteria present (20) and residing in the animal gut. However, although many gut microorganisms have the ability to produce phytase, little activity is available (2) because many microbes do not secrete the enzyme and because the pH of the intestine is above the enzyme optimum.
The appA gene of Escherichia coli encodes a phytase (EC 3.1.3.26) with a rate optimum at pH 4.5 and a high specific hydrolytic activity towards phytate (1,700 U/mg) that significantly surpasses the activities of other reported phytases (6, 29). The enzyme rapidly removes four of the six orthophosphate groups of phytate (9). Because its specific activity and specificity are high and its pH profile is favorable for gastric activity, the appA gene product was chosen as a candidate for increasing thermal tolerance to promote the survival of the enzyme during pelleting.
In order to create an optimized phytase gene, we employed gene site saturation mutagenesis (GSSM) technology to identify mutations that increase enzymatic performance. The technology, which generates a library of all possible single-site mutations in an enzyme, can target individual aspects of a phenotype in association with an appropriate high-throughput screening assay. In this case, a dual goal of increased thermal tolerance and a maintenance of high turnover was established. Combining GSSM technology and an assay that challenged all of the GSSM constructs with a heat step revealed mutations that increased thermal tolerance. Combining these mutations led to a phytase, Phy9X, with an optimal phenotype for economic use as an animal feed supplement. In addition to an increase in thermal tolerance, a concomitant increase in gastric stability was observed. Here we report the mutagenesis efforts that resulted in the creation of Phy9X. The field data demonstrating the efficacy of Phy9X added prior to pelleting for both poultry and swine feeding applications will be presented separately (unpublished data).
MATERIALS AND METHODS
Materials.
Dodecasodium phytate from rice (P-3168), ammonium metavanadate (A-1183), and ammonium molybdate (A-7302) were obtained from Sigma. All other reagents were obtained from Fisher unless stated otherwise.
Evolution by use of GSSM technology.
The appA gene from E. coli strain MG1655 was cloned into the expression vector pQE60 (Invitrogen), which included a C-terminal six-histidine affinity tag. The gene product was 432 amino acids long. GSSM technology was employed as previously described (3, 8, 13, 23), using a 32-fold degenerate mutagenic oligonucleotide for each position (NNK, where N is all four nucleotides and K is G or T). Statistically, this level of degeneracy will cover all 20 amino acids. Quality control consisted of sequencing of a 96-well plate of clones (threefold oversampling) and resulted in codons being found for 18 amino acids. Quality control with other GSSM libraries for which large samples were sequenced indicated that there is a 90% confidence that 150 clones (fivefold oversampling) from a pool will contain all 20 amino acids. The mutagenized appA library was screened with >10-fold oversampling to ensure complete coverage at each position (see below for details). All positions, excluding the N terminus, were mutagenized, resulting in 431 pools of variants.
High-throughput screening.
Enzymes that are used for animal feed supplements need to withstand the pelleting process, which is typically done at temperatures of approximately 80°C for short periods of time (<5 min). The enzyme must then function under the conditions that are prevalent in the stomachs of monogastric animals (pH < 4.5, 37°C). As in all cases of directed enzyme mutagenesis, it is imperative that the conditions of the screen adequately mimic the final application process. Therefore, we designed our high-throughput screening protocols to test for a low pH activity following a heat challenge. Subsequent assays were then used to ensure that no other property of the enzyme was negatively affected. Since our primary screen was for the retention of structural integrity, the model substrate 4-methyl-umbelliferylphosphate (4-MUP) was used in the high-throughput screen. Phosphatase activity was monitored by an increase in fluorescence over time due to the release of methyl-umbelliferone. We determined by experimentation that the wild-type enzyme expressed in E. coli and grown in a 384-well plate retained between 5 and 10% activity after heating for 60 min at 62°C; thus, these conditions were used when the GSSM library was screened.
Each GSSM pool was used to transform E. coli and was grown on a Luria-Bertani (LB)-solid agar plate (with 50 μg of ampicillin/ml). The resulting colonies were resuspended in a small volume of LB medium and dyed with propidium iodide. Fluorescence-activated cell sorting was used to place individual cells from a pool into separate wells of a 384-well plate; therefore, each well contained a single clone from the mixed population. One 384-well plate was used per pool and every plate contained a column of parental controls. Growth in the presence of 1 mM isopropyl-β-d-thiogalactopyranoside (IPTG) was allowed to proceed for 2 days at 30°C in a humidified incubator. After growth, the master plates were equally divided into two daughter plates as well as replicated by pin tooling onto LB-solid agar plates (with 50 μg of ampicillin/ml). One of the daughter plates was covered with adhesive foil, heated to 62°C for 60 min, and cooled to room temperature for 30 min. The second daughter plate acted as a control and remained at room temperature. After 30 min, 20 μl of a solution containing 500 mM sodium acetate (NaOAc), pH 4.5, and 5 mM 4-MUP was added to the wells of both daughter plates. The reaction was allowed to proceed for 30 min at 25°C and then quenched by the addition of 30 μl of 1 M Tris base (pH 10.0). The phosphatase activity was determined by the increase in the relative fluorescence (excitation wavelength, 360 nm; emission wavelength, 465 nm) (Molecular Devices Spectramax Gemini) due to the hydrolysis of 4-MUP to 4-methyl umbelliferone. Residual activity was defined by the relative fluorescence of the heated plate compared to that of the unheated control plate. Clones exceeding 20% residual activity were chosen and reassayed under the same conditions for confirmation. Those clones passing the secondary assay were regrown, and their plasmids were isolated and sequenced.
Combinatorial screening.
After the identification of all single point mutations that increased the thermal resistance of the phytase, a new screen was designed to discriminate between the most stable of the combinatorial products. The temperature screening conditions were identical to those for the high-throughput screen, with the exception that as the thermal tolerance of the enzyme combinatorial product increased, the temperature of the incubation was increased. When the top candidate from any round of addition reached or exceeded an 80% retention of activity (compared to the unheated sample), the incubation temperature was increased 4°C. Over the nine rounds of combination, the temperature was increased three times, to a maximum of 74°C.
Enzymatic activity assays.
Phytase-specific activity was measured with a colorimetric assay (4) to detect the amount of free phosphate liberated from dodecasodium phytate in 100 mM NaOAc, pH 4.5. An aliquot of enzyme (50 μl) was added to 950 μl of assay mix (5 mM phytate, 100 mM NaOAc, pH 4.5) in a microcentrifuge tube in a constant-temperature heat block. Aliquots (50 μl) were withdrawn at 2-min intervals and quenched by being added to 50 μl of a color-stop mix in a 96-well plate. The absorbance was determined at 415 nm with a Molecular Devices Spectramax Plus microplate reader.
The appA-encoded and Phy9X phytases were tested for the ability to remove orthophosphate (the number of phosphates removed per phytate was measured). Enzyme assays were performed at 37°C in a solution containing 0.16 mM phytic acid and 100 mM NaOAc, pH 4.5. Samples were withdrawn at selected time points and the reactions were stopped by the addition of an equal volume of 5% trichloroacetic acid. The amount of released inorganic phosphate was photometrically measured at 700 nm by monitoring the production of phosphomolybdate with an equal volume of developer (4 volumes of 1.5% ammonium molybdate solution in 5.5% sulfuric acid and 1 volume of 2.7% ferrous sulfate solution). The molar amount of phosphate released per mole of substrate (phytic acid) was calculated.
Enzyme purification.
The wild-type appA-encoded phytase and selected variants were purified by nickel-chelating Sepharose chromatography. Induced overnight cultures were centrifuged (8,000 × g, 15 min, 4°C) and resuspended in a solution containing 20 mM Tris (pH 7.9), 500 mM NaCl, and 5 mM imidazole. Cells were disrupted by sonication, and the insoluble cell debris was removed by centrifugation (20,000 × g, 15 min). The cell extract was then passed over a Ni-chelating Fast Flow column (Amersham-Pharmacia) equilibrated with binding buffer (20 mM Tris [pH 7.9], 5 mM imidazole, 500 mM NaCl), and the column was reequilibrated with 3 volumes of binding buffer. The bound protein was eluted with an imidazole gradient, and fractions with activity were pooled, dialyzed against 20 mM Tris, pH 7.5, and concentrated in an Amicon stirred-cell diafiltration unit.
Differential scanning calorimetry (DSC).
A Perkin-Elmer Pyris 1 calorimeter was used to determine the melting temperatures (Tm) of wild-type and mutant phytases. Protein samples were concentrated to 30 to 45 mg/ml in 50 mM 2-(N-morpholino)ethanesulfonic acid (MES) buffer, pH 7.0, sealed in stainless steel cells, equilibrated to 40°C, and then scanned from 40 to 90°C at a rate of 10°C/min. For assessments of reversibility, proteins were scanned from 90 to 40°C.
Thermal tolerance and thermal stability assays.
Thermal tolerance was determined by measuring the residual activity of an enzyme at 37°C following incubation at temperatures exceeding the Tm of the parental enzyme. The samples were incubated for 5 min at different temperatures to monitor temperature tolerance and were incubated for various amounts of time at one temperature to monitor thermal stability over time. After being incubated at an elevated temperature, the samples were cooled on ice and assayed against 5 mM phytate at 37°C in 100 mM NaOAc buffer, pH 4.5. Enzymes were incubated in the presence and absence of phytate. Thermal stabilities were determined by measuring the initial rates of the hydrolysis of phytate at elevated temperatures. Aliquots (950 μl) of assay buffer (5 mM phytate, 100 mM NaOAc, pH 4.5) were equilibrated at a temperature for 3 min, and then phytase was added and assayed for 10 min, with samples taken every 2 min.
Gastric stability assay.
The half-lives of the E. coli K-12 appA-encoded and Phy9X phytases were determined by incubating the enzymes in simulated gastric fluid (SGF) (26) containing 2 mg of NaCl/ml, 84 mM HCl, and 3.2 mg of pepsin/ml (pH 1.2) and then measuring the residual phytase activities. In vitro digestibility assays were performed by adding phytase to the SGF solution (1:4 [vol/vol]) and incubating it at 37°C. Aliquots of the digestion reaction mixture were removed at intervals and assayed for residual phytase activity. The degradation of the protein was also monitored in SGF containing no pepsin and in 100 mM acetate buffer, pH 5.5. The percent residual activity (based on initial rates) was plotted versus time. An exponential curve with the equation y = Ae−kt, where A is the absorbance, k is the first-order rate constant describing decay, and t is time, was fitted to the data, and the half-lives of the proteins were determined with the calculation t1/2 = ln 2/k. The half-life was defined as the time (in minutes) at which 50% of the activity was lost relative to the activity at the initiation of the digestion reaction (time zero).
RESULTS
GSSM technology was used to systematically mutate the parental appA gene and to produce 431 pools of variants. Each pool of daughter genes contained a collection of variants encoding all 20 possible amino acid side chains at the corresponding sequence position. Clones were sorted into an addressed microtiter format and a high-throughput screen was conducted in which the clones were challenged at an elevated temperature followed by activity determination at 37°C. Those clones retaining more residual activity than the parental controls were isolated, and their genes were sequenced to identify single residue replacements that conferred increased thermal tolerance. Fourteen single-site mutations were identified which retained more activity than the wild-type control activity following the heat challenge. Of the 14 up-regulated mutants, only one site included more than one residue replacement that led to increased thermal tolerance.
To efficiently take advantage of the potential additivity or residue synergies conferred by combining mutations, we used a sequential, recursive addition protocol to create the fittest ultimate construct. The recursive path tested all possible 2-, 3-, 4-, and so on side-chain combinations, and at each step of the mutational addition process, defined the best template for the next addition. This ensured that the mutant addition followed the shortest possible path to the ultimate daughter construct.
Among the single-site mutants, one with a Q84W substitution, the result of a three-nucleotide codon change, was significantly more thermally tolerant than the other mutants. Beginning with the Q84W mutant as a template, each of the 13 remaining single-site mutations was individually added to create a library of double mutants. Each double mutant was then assayed for thermal tolerance by the temperature challenge screening protocol. The fittest double mutant was then used as a template and the remaining 12 single mutations were added to create a pool of triple mutants. This addition screening procedure was continued until no additional tolerance was gained by combining single mutations. The maximal improvement in thermal tolerance was observed upon the addition of eight mutations into a single construct, Phy9X. Figure 1 illustrates the path taken to increase thermal tolerance during the recursive procedure and shows the fittest mutational combination at each step.
FIG. 1.

Comparison of the fitness of the most thermally tolerant phytase variant after each round of mutagenesis. Bars indicate the temperatures at which 50% residual activity remained after 60 min at the elevated temperature. The genotype of the clone is indicated below the bar.
DSC was used to determine the Tm of the purified Phy9X daughter construct relative to that of the similarly prepared wild-type appA-encoded enzyme (Fig. 2). DSC scans were run in the forward (40 to 90°C) and reverse directions. The reverse scans indicated that the melting of both the appA-encoded and Phy9X phytases was essentially irreversible (not shown). The Tm for Phy9X was elevated 12°C over that of the parental E. coli phytase (75.7°C versus 63.7°C).
FIG. 2.

DSC graph showing phytase Tms. Symbols: squares, K-12 appA-encoded phytase (parent), with a Tm of 63.7°C; triangles, Phy9X phytase, with a Tm of 75.7°C.
Figure 3 shows the inactivation kinetics of the appA-encoded and Phy9X phytases in SGF. The gastric stability, expressed as the activity half-life (t1/2), of the parental enzyme (t1/2 = 2.4 min) was increased 3.5-fold by the eight point mutations incorporated into Phy9X (t1/2 = 8.7 min). These kinetics are complex, but the data can be fit to a simple, single, exponential model resulting in the determination of the activity half-life.
FIG. 3.

Decay of phytase activity in SGF. Symbols: squares, K-12 appA-encoded phytase, with a t1/2 of 2.7 ± 0.2 min; triangles, Phy9X, with a t1/2 of 8.4 ± 1.1 min.
Because we selectively altered an aspect of the phytase phenotype, we were interested to determine whether the specificity, turnover, and effect of pH on activity were altered in the daughter enzyme. The appA-encoded enzyme has been shown to rapidly remove four phosphates from phytate and then to slowly remove the fifth phosphate, leaving monophosphate inositol (9, 28). The kinetics of orthophosphate removal by Phy9X were compared to those for the appA-encoded enzyme, and the enzymes were shown to be identical in rate (specific activity of 1,700 U/mg) and for the removal of four phosphate groups (data not shown). The pH activity profiles for the appA-encoded phytase and the Phy9X construct are essentially superimposable (data not shown). Both enzymes displayed a maximal rate at pH 4.5, with phytate hydrolysis activity extending below pH 2.5. In addition, the specific activity of the Phy9X mutant was similar to that of wild-type phytase despite its improvement in thermal tolerance and its resistance to proteolytic cleavage.
The goal of this work was to increase the performance of a highly active bacterial phytase to enable the inclusion of the enzyme in animal feed prior to pelleting and to ensure the survival of phytase activity postpelleting. The objective was achieved, as the new phytase (i) selectively increased the thermal tolerance of the parental enzyme, (ii) retained the wild-type pH and rate profiles and specific activity, (iii) maintained the orthophosphate hydrolysis stoichiometry, and (iv) increased the t1/2 for gastric stability.
DISCUSSION
For this study, we sought to evolve a bacterial phytase that is able to withstand the high-temperature feed pelleting process yet retain a high level of specific phosphate-releasing activity at the low pH of a monogastric gut environment. An ideal process-compatible enzyme would have the ability to withstand a brief challenge at pelleting temperature, have a high specific activity at physiological temperatures, and be sufficiently stable in the low-pH, proteolytic environment of the stomach to make ample phosphate available to the animal before the enzyme is degraded.
In a scheme (equation 1) describing the temperature-denatured behavior of proteins as they progress from a folded state to an unfolded state and then to an insoluble state, k1 and k−1 represent the equilibrium between the folded and unfolded states and k2 represents a combination of events which prevent a protein from refolding after denaturation. Among these events are the essentially irreversible chemical modification of side chains and the aggregation and precipitation of misfolded (or denatured) forms of theprotein.
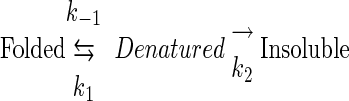 |
(1) |
An enhanced resistance to high temperatures could be obtained by engineering either a protein with an increased melting temperature (Tm) or an enzyme that refolds reversibly after partial or complete thermal denaturation. As a case in point, a previous application of GSSM technology targeted the thermostability of a dehalogenase enzyme to be used in an industrial by-product recovery process (8). Screening of a GSSM library constructed from a Rhodococcus dhlA gene identified eight mutations that enhanced thermostability. In this example, these mutations endowed the enzyme with the ability to reversibly renature. Optimization allowed the enzyme to function under ambient process temperatures of about 65°C. Explanations for the observed increase in thermal tolerance of the Phy9X enzyme over that of the parental enzyme include a stabilization of the hydrophobic core of the protein and an increasing surface polarity of the protein (24). In contrast to what was found for dehalogenase (8), no residues were identified by GSSM evolution for this protein that allowed the protein to refold and to regain its native structure and activity. Modeling of the point mutations (Fig. 4) on the crystal structure of wild-type E. coli AppA showed that the residue changes that increased thermal tolerance occurred throughout the enzyme sequence and that they were not necessarily predictable based on a sequence or structure analysis. All mutations were at or near the enzyme surface. Of the eight mutations in Phy9X, only one (Y277D) was the result of a single base change in a codon; three were double base changes and the remaining four were three-base changes (all positions in the codon were changed). These mutations would not have been captured by a typical mutagenesis protocol such as error-prone PCR, which accesses, at random, one base change per codon.
FIG. 4.

Model of Phy9X based on crystal structure of E. coli K-12 AppA. The model shows the locations and identities of the Phy9X mutations.
It has been noted that the stabilizing effects of point mutations may be cumulative (1, 8, 18, 21, 22, 28). Adding the single point mutations identified through GSSM evolution created a phytase that retained the desired activity properties while increasing the thermal tolerance and gastric stability of the enzyme. The most stable point mutation (Q84W) identified through high-throughput screening was then used as the foundation for all subsequent site-directed mutagenesis steps. The combination of the substitution that imparted the second highest thermal resistance of any single mutation with the Q84W mutant revealed a second possible outcome of combining point mutations. When these two stabilizing mutations were combined into a single construct, the stability of the double mutant was lower than that of either single mutation by itself. Though unexpected, this was not surprising, since each of the single mutations was found in the wild-type structural environment and since each mutation modifies this environment. One implication of this discovery is that simply combining all of the newly discovered stabilizing mutations into a single construct will not necessarily result in an enzyme with optimal thermal tolerance or stability. In order to circumvent problems associated with combining mutually exclusive residues into a single structure, we added each point mutation sequentially and recursively to the most stable sequence from the previous round of site-directed mutagenesis. As the mutations accumulated, the temperature of the heat challenge was raised to improve discrimination among the increasingly thermally tolerant new constructs (Fig. 1). We continued this iterative process until the addition of more point mutations yielded no discernible increase in thermal tolerance or stability.
The final criterion for an animal feed enzyme is resistance to gastric degradation by proteases and peptidases found in the digestive tracts of monogastric animals. These digestive enzymes require the availability of recognition sites on the surface of the substrate protein. Solvent-exposed loops and random coils represent ideal locations for the presence of proteolytic sites. The increase in gastric stability observed for Phy9X over that of the wild-type parent could be explained by a tighter packing of secondary structural elements and a less dynamic flexibility of the loops and random coils.
These data substantiate the power of directed mutagenesis and high-throughput screening for the enhancement of selected enzyme properties. GSSM technology is particularly useful, providing comprehensive codon variation to chart an optimal mutational path to selectively and rapidly target aspects of an enzyme's phenotype.
Acknowledgments
We thank Xuqiu Tan, John Macomber, Jonathan Eads, Mike Lafferty, Amit Vasavada, and Mark Wall for discussions, modeling, and the implementation of techniques described in this paper.
REFERENCES
- 1.Akasaka, A., M. Haruki, M. Oobtake, and S. Kanaya. 1995. High resistance of Escherichia coli ribonuclease H1 variant with quintuple thermostabilizing mutations to thermal denaturation, acid denaturation and proteolytic cleavage. Biochemistry 34:8115-8122. [DOI] [PubMed] [Google Scholar]
- 2.Cromwell, G. L., and R. D. Coffey. 1991. P—a key essential nutrient, yet a possible major pollutant. Its central role in animal nutrition, p. 133. In T. P. Lyons (ed.), Bio/technology in the feed industry. Proceedings of the Alltech 7th Annual Symposium. Alltech Technical Publications, Nicholasville, Ky.
- 3.DeSantis, G., K. Wong, B. Farwell, K. Chatman, Z. Zhu, G. Tomlinson, H. Huang, X. Tan, L. Bibbs, P. Chen, K. Kretz, and M. Burk. 2003. Creation of a productive, highly enantioselective nitrilase through gene site saturation mutagenesis (GSSM). J. Am. Chem. Soc. 125:11476-11477. [DOI] [PubMed] [Google Scholar]
- 4.Engelen, A. J., F. C. van der Heeft, P. H. Randsdorp, and E. L. Smit. 1994. Simple and rapid determination of phytase activity. J. AOAC Int. 77:760-764. [PubMed] [Google Scholar]
- 5.Erdman, J. W., Jr., and A. Poneros-Schneier. 1989. Phytic acid interactions with divalent cations in foods and in the gastrointestinal tract. Adv. Exp. Med. Biol. 249:161-171. [DOI] [PubMed] [Google Scholar]
- 6.Golovan, S. P., G. Wang, J. Zhang, and C. W. Forsberg. 2000. Characterization and overproduction of the Escherichia coli appA encoded bifunctional enzyme that exhibits both phytase and acid phosphatase activities. Can. J. Microbiol. 46:59-71. [DOI] [PubMed] [Google Scholar]
- 7.Graf, E. 1983. Calcium binding to phytic acid. J. Agric. Food Chem. 31:851-855. [Google Scholar]
- 8.Gray, K. A., T. H. Richardson, K. Kretz, J. M. Short, J. P. F. Bartnek, R. Knowles, L. Kan, P. E. Swanson, and D. E. Robertson. 2001. Rapid evolution of reversible denaturation and elevated melting temperature in a microbial haloalkane dehalogenase. Adv. Synth. Catalysis 343:607-617. [Google Scholar]
- 9.Greiner, R., N. Carlsson, and M. L. Alminger. 2000. Stereospecificity of myo-inositol hexakisphosphate dephosphorylation by a phytate-degrading enzyme of Escherichia coli. J. Biotechnol. 84:53-62. [DOI] [PubMed] [Google Scholar]
- 10.Greiner, R., U. Konietzny, and K. D. Jany. 1993. Purification and characterization of two phytases from Escherichia coli. Arch. Biochem. Biophys. 303:107-113. [DOI] [PubMed] [Google Scholar]
- 11.Jongbloed, A. W., Z. Mroz, and P. A. Kemme. 1992. The effect of supplementary Aspergillus niger phytase in diets for pigs on concentration and apparent digestibility of dry matter, total phosphorus, and phytic acid in different sections of the alimentary tract. J. Anim. Sci. 70:1159-1168. [DOI] [PubMed] [Google Scholar]
- 12.Kornegay, E. T. 2001. Digestion of phosphorous and other nutrients: the role of phytases and factors influencing their activity, p. 237-271. In M. R. Bedford and G. G. Partridge (ed.), Enzymes in farm animal nutrition. CABI Publishing, Oxon, United Kingdom.
- 13.Kretz, K. A., T. H. Richardson, K. A. Gray, D. Robertson, X. Tan, and J. M. Short. Methods Enzymol., in press. [DOI] [PubMed]
- 14.Lee, D. Y., J. Schroeder III, and D. T. Gordon. 1988. Enhancement of Cu bioavailability in the rat by phytic acid. J. Nutr. 118:712-717. [DOI] [PubMed] [Google Scholar]
- 15.Lei, X., P. K. Ku, E. R. Miller, D. E. Ullrey, and M. T. Yokoyama. 1993. Supplemental microbial phytase improves bioavailability of dietary zinc to weanling pigs. J. Nutr. 123:1117-1123. [DOI] [PubMed] [Google Scholar]
- 16.Nasi, M. 1990. Microbial phytase supplementation for improving the bioavailability of plant phosphorus in the diet of growing pigs. J. Agric. Sci. Finland 62:435-442. [Google Scholar]
- 17.Nayani, N. R., and P. Markakis. 1983. Effects of inositol phosphates on mineral utilization. Fed. Proc. 45:819-826. [Google Scholar]
- 18.Pantoliano, M. W., M. Whitlow, J. F. Wood, S. W. Dodd, K. D. Hardman, M. L. Rollence, and P. N. Bryan. 1989. Large increases in general stability for subtilisin BPN′ through incremental changes in the free energy of unfolding. Biochemistry 28:7205-7213. [DOI] [PubMed] [Google Scholar]
- 19.Reddy, N. R., S. K. Sathe, and D. K. Salunkhe. 1982. Phytates in legumes and cereals. Adv. Food Res. 28:1-92. [DOI] [PubMed] [Google Scholar]
- 20.Rosebury, T. 1962. Microorganisms indigenous to man. McGraw-Hill, New York, N.Y.
- 21.Serrano, L., A. G. Day, and A. R. Fersht. 1993. Step-wise mutation of barnase to binase. A procedure for engineering increased stability of proteins and an experimental analysis of the evolution of protein stability. J. Mol. Biol. 233:305-312. [DOI] [PubMed] [Google Scholar]
- 22.Shih, P. O., and J. F. Kirsch. 1995. Design and structural analysis of an engineered thermostable chicken lysozyme. Protein Sci. 4:1063-1072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Short, J. M. January 2001. Saturation mutagenesis in directed evolution. U.S. patent 6,171,820.
- 24.Szilagyi, A., and P. Zavodszky. 2000. Structural differences between mesophilic, moderately thermophilic and extremely thermophilic protein subunits: results of a comprehensive survey. Struct. Fold Des. 8:493-504. [DOI] [PubMed] [Google Scholar]
- 25.Urbano, G., P. Aranda, E. Gomez-Villalva, S. Frejnagel, J. M. Porres, J. Frias, C. Vidal-Valverde, and M. Lopez-Jurado. 2003. Nutritional evaluation of pea (Pisum sativum L.) protein diets after mild hydrothermal treatment and with and without added phytase. J. Agric. Food Chem. 51:2415-2420. [DOI] [PubMed] [Google Scholar]
- 26.U.S. Pharmacopeia. 2000. Simulated gastric fluid, TS. National Formulary no. 24/19, 2235. National Publishing, Philadelphia, Pa.
- 27.van Dijck, P. W. M. 1999. Chymosin and phytase made by genetic engineering. J. Biotechnol. 67:77-80. [Google Scholar]
- 28.von der Osten, C., S. Branner, S. Hastrup, L. Hedegaard, M. D. Rasmussen, H. Bisgard-Frantzen, S. Carlsen, and J. M. Mikkelsen. 1993. Protein engineering of subtilisins to improve stability in detergent formulations. Bio/Technology 28:55-68. [DOI] [PubMed] [Google Scholar]
- 29.Wyss, M., R. Brugger, A. Kronenberger, R. Remy, R. Fimbel, G. Oesterhelt, M. Lehmann, and A. P. van Loon. 1999. Biochemical characterization of fungal phytases (myo-inositol hexakisphosphate phosphohydrolases): catalytic properties. Appl. Environ. Microbiol. 65:367-373. [DOI] [PMC free article] [PubMed] [Google Scholar]