

Abstract
Current methods of understanding microbiome composition and structure rely on accurately estimating the number of distinct species and their relative abundance. Most of these methods require an efficient PCR whose forward and reverse primers bind well to the same, large number of identifiable species, and produce amplicons that are unique. It is therefore not surprising that currently used universal primers designed many years ago are not as efficient and fail to bind to recently cataloged species. We propose an automated general method of designing PCR primer pairs that abide by primer design rules and uses current sequence database as input. Since the method is automated, primers can be designed for targeted microbial species or updated as species are added or deleted from the database. In silico experiments and laboratory experiments confirm the efficacy of the newly designed primers for metagenomics applications.
Introduction
DNA extraction and PCR amplification are essential steps in a number of different applications including forensics, sequencing, metagenomic analyses (i.e., study of community profiles) and comparison of ecosystems using their microbial profiles [1]. Design of primer pairs used in PCR depends on the target application and can be specific to a particular species, gene, ortholog group, taxon or community. In projects that aim to discover community composition and structure, primers are mainly required to (1) be universal in their ability to bind to a maximum number of different target species and (2) produce maximally distinguishable amplicons for the taxa of interest, which is the basis for the post PCR data analyses and species identification.
Launched in 2008, the Human Microbiome Project (HMP) [2] is the project du jour for studying microbial community composition and structure in different environmental niches within the human body. HMP primarily aims to understand the differences in the microbial composition between unhealthy and healthy states of five body sites: oral, skin, vaginal, gut and nasal/lung. HMP studies often utilize universal primers that amplify regions of the 16S rRNA, a popular target gene for studying taxonomic and evolutionary relationship between microbial organisms [3,4]. However, universal primers were developed over 20 years ago [5-7]. Since then many new bacterial and archaeal species have been discovered [8], so much so that a number of 16S rRNA databases have been created and updated many times. (RDP [9] has been updated 31 times in the last 5 years and has roughly quadrupled in size; Silva [10] with 16 full releases in 6 years, has grown six-fold with 449 new species in the last release.) Inevitably the universal primers have become too specific/biased, i.e., they fail to extract newly added species from the current database. Necessitated by particular projects, a number of taxon-specific primer pairs [11,12] have been developed and efforts have been made to improve complementarity of the majority of universal primers [13-15].
In this paper, primer design is cast as a constrained optimization problem, in which the pairs of primers must satisfy a range of criteria for being functional, while maximizing the distinguishability with respect to a given set of microbial organisms. Experimental results show the efficacy of the proposed method.
Background and motivation
In PCR, forward and reverse primers hybridize to specific locations on the target DNA sequence and the fragment between the primer binding sites is amplified. Efficient and effective PCR requires that the primers satisfy a set of widely accepted interdependent conditions (For example, see http://www.premierbiosoft.com/tech_notes/PCR_Primer_Design.html): (1) melting temperatu-re (Tm) range, (2) primer specificity, (3) GC clamp, (4) primer-primer interactions, (5) bounded degeneracy, and (6) amplicon length. When primers hybridize to multiple locations, the efficiency of PCR is reduced. PCR efficiency depends greatly on the strength of hydrogen bonds formed between the primer and the template. The distinction between a strong base (C or G) and a weak base (A or T) is based on the number of hydrogen bonds (3 and 2, respectively) that a base requires to bind to the reverse strand. Primers with greater GC content result in stronger anchoring and greater energy is needed to break the bonds. Given that the PCR elongation happens at the 3' end, it is important that 3' end anchors well and better (more strongly) than the 5' end. Still, it is recommended that the 3' end have at most two strong bases. PCR works efficiently only within the temperature range defined by the primers it uses. Thus, the temperature ranges defined by the forward and reverse primers must overlap. At higher temperatures primer-template binding is more specific and it is harder for primers to form secondary structures. However, the primer-template bonds that are formed are broken more easily, which reduces PCR efficiency. Hence the need for high GC content. At low temperatures, although the primer-template bonds are not easily broken, primers hybridize more easily. Furthermore, efficiency of PCR is greatly diminished by allowing forward or reverse primer to either bind to itself (selfDimer) or to each other (primerDimer). It is particularly important that the 3' ends of forward and reverse primers do not create primer dimers. This is implicitly taken care of by the rule of not having more than two strong bases at the 3' end. High primer degeneracy could also be another factor contributing to decreased primer efficiency. A degenerate primer is a mixture of non-degenerate primers. Hence, the increase in primer degeneracy is proportional to decrease in concentration of the individual primers.
Our studies show that most of the universal primers and their derivatives in current use do not abide by all of these primer design rules. In Table 1, the primers whose IDs start with U are the universal primers. Violations of the rules governing optimal primer design are underlined and in bold font. The column labeled nHits indicates the number of RDP database sequences that the primer is likely to anneal to. For PCR to produce enough amplicon for sequencing or for metagenomic analysis this number must be as high as possible. The two best universal primers in the list, U518R and U337F, do not have a compatible reverse primer. The first one is always paired with U8F to amplify the V13 region (e.g., HMPV13 primer pair). However, U8F has a very low number of hits from RDP. That is because the sequenced data in the RDP database often does not provide sequence data in that region. The U337F primer does not have compatible reverse primer, since its temperature range does not overlap with any of the universal reverse primers (U805R, U907R, U1492R).
Table 1.
| primerID | 5'→3' seq | deg | sDimer | Tm range | GC% range | length | nHits |
|---|---|---|---|---|---|---|---|
| U8F | AGAGTTTGATCCTGGCTCAG | 1 | 4 | 51.78→51.78 | 0.50→0.50 | 20 | 1553 |
| U336R | ACTGCTGCSYCCCGTAGGAGTCT | 4 | 4 | 60.62→62.40 | 0.61→0.65 | 22 | 7408 |
| U337F | GACTCCTACGGGAGGCWGCAG | 2 | 6 | 60.21→60.21 | 0.67→0.67 | 21 | 7689 |
| U518R | GTATTACCGCGGCTGCTGG | 1 | 6 | 55.41→55.41 | 0.63→0.63 | 18 | 8417 |
| U533F | GTGCCAGCMGCCGCGGTAA | 2 | 6 | 57.56→59.72 | 0.68→0.74 | 19 | 8929 |
| U785F | GGATTAGATACCCTGGTA | 1 | 4 | 45.77→45.77 | 0.44→0.44 | 18 | 8183 |
| U805R | GACTACCAGGGTATCTAATC | 1 | 4 | 49.73→49.73 | 0.45→0.45 | 20 | 8171 |
| U907R | CCGTCAATTCCTTTRAGTTT | 2 | 4 | 45.63→47.68 | 0.35→0.40 | 20 | 7296 |
| U928F | TAAAACTYAAAKGAATTGACGGG | 4 | 4 | 48.14→51.71 | 0.30→0.39 | 23 | 5090 |
| U1100F | YAACGAGCGCAACCC | 2 | 4 | 44.67→47.41 | 0.60→0.67 | 15 | 8980 |
| U1492R | GGTTACCTTGTTACGACTT | 1 | 3 | 46.77→46.77 | 0.42→0.42 | 19 | 449 |
| HMPV1F | GAGTTTGATCCTGGCTCAG | 1 | 4 | 51.09→51.09 | 0.53→0.53 | 20 | 1778 |
| HMPV3R | ATTACCGCGGCTGCTGG | 1 | 6 | 51.88→51.88 | 0.65→0.65 | 17 | 8428 |
| HMPV3F | CCTACGGGAGGCAGCAG | 1 | 3 | 54.29→54.29 | 0.71→0.71 | 17 | 8735 |
| HMV5R | CCCGTCAATTCMTTTRAGT | 4 | 4 | 44.62→48.93 | 0.37→0.47 | 19 | 8533 |
| HMPV6F | GYAACGAGCGCAACCC | 2 | 4 | 48.50→51.06 | 0.63→0.69 | 16 | 8507 |
| HMPV9R | AAGGAGGTGATCCAGCCGCA | 1 | 4 | 55.88→55.88 | 0.60→0.60 | 20 | 1193 |
| N337F | GGAGGCAGCAGTRRGGAAT | 4 | 2 | 51.09→55.41 | 0.53→0.63 | 19 | 8570 |
| N775R | CTACCRGGGTATCTAATCC | 2 | 4 | 48.93→51.09 | 0.47→0.53 | 19 | 8427 |
| K331F | CCTACGGGNGGCWGCAG | 8 | 6 | 54.29→56.70 | 0.71→0.76 | 17 | 8882 |
| K775R | GACTACHVGGGTATCTAATCC | 9 | 10 | 50.45→54.36 | 0.43→0.52 | 21 | 8868 |
| S899R | CCGTCAATTYMTTTRAGT | 8 | 4 | 38.93→45.77 | 0.28→0.44 | 18 | 8779 |
HMP commissioned a study that produced a set of recommended primer pairs, their experimental settings and post experimental data processing and analysis workflow [17]. The resulting primers, shown in Table 1, whose ID starts with HMP, are variants of universal primers, with degeneracy increased so as to increase the number of hits in the database, which in turn allows for more precise sequenced read classification and hence better estimation of sample diversity and richness, both of which are fundamental measures used in metagenomic studies.
The recent ubiquity and affordability of HMP-supported studies is due to the advent of Next Generation Sequencing (NGS) technologies that produce large volumes of sequenced reads cheaply. (Note that even though NGS makes it possible to cheaply sequence the entire genome instead of just the 16S region, it may not much help distinguishability of the microbes in the community because of the proliferation of highly conserved genes and the presence of horizontally transferred genes between members of the community.) NGS costs are affordable, but they are not low enough to allow for an experiment to be repeated a number of times. Thus, in silico studies have been conducted to assess the diversity of the amplicons using universal and HMP primers [4,18-20]. Although primers such as UV34, HMPV35 and UV6 are predicted to result in high diversity of the amplicons, they are flawed and can be improved.
We see two ways of improving metagenomic studies. First, we propose an automated, general method of designing new primers based on the current content of a target database (Table 2) that abide by primer design rules (Table 3). Since the method is automated, it will be easy to update the primers as new species are added to the database or a different target set is selected from the database. Second, we observe that different species are distinguishable by different variable regions. Given, the affordability of NGS sequencing, and the difficulty in designing primers that are both, universal and abide by primer design rules, we propose the use of multiple sets of primer pairs. These sets are automatically chosen so as to maximize the number of distinguishable species. In this paper we present a software package that achieves both objectives.
Table 2.
RDP Sequence set
| Number of Sequences | 9175 |
|---|---|
| Number of Species | 8372 |
| Number of Genera | 1779 |
| Number of Phyla | 29 |
| Avg Sequence Length | 1468 |
| Max Sequence Length | 1847 |
| Min Sequence Length | 1225 |
| Strain | Typed |
| Size | ≥ 1200 |
| Source | Isolates |
| Quality | Good |
Table 3.
Primer Design Rule Parameters
| Design Parameters | Values Used |
|---|---|
| allowable 3' ends | S[SW,WS][SW,WS,SS] |
| allowable 5' ends | WW, [WS,SW]SW |
| MINTEMP | 48 |
| MAXTEMP | 70 |
| MAXTEMPRANGE | 8 |
| MINCG% | 40 |
| MAXCG% | 60 |
| MINPRIMERLENGTH | 17 |
| MAXPRIMERLENGTH | 24 |
| MAXDEGENERACY | 64 |
| MAXSELFDIMER | 6 |
| MAXPRIMERDIMER | 6 |
| MAXRUNLENGTH (bp) | 4 |
| MINAMPLICONLENGTH | 150 |
| MAXAMPLICONLENGTH | 600 |
Algorithm
We start with some required notation. Let denote a set (database) of sequences over the alphabet Σ = {A, C, G, T, R, Y, S, W, K, M, B, D, H, V, N}, which corresponds to the letters used in the IUPAC code (http://www.bioinformatics.org/sms/iupac.html). Each letter in Σ corresponds to a set of bases from the DNA alphabet {A, C, G, T}. For example, the letter H corresponds to the set {A, C, T}. Each letter of has a degeneracy value, which equals the cardinality of its base set. We denote this by DEG(a), for . The degeneracy of a sequence S is defined as the product of the degeneracy values of each of its letters. More formally, , where S(j, l) is a contiguous subsequence of length l starting at position j in S. For example, let S1 = AAGGATCG, S2 = WGSANS, and S3 = AGGSTD, then DEG(S1) = 1, DEG(S2) = 32 and DEG(S3) = 6. A sequence is called non-degenerate if its degeneracy equals 1. Degeneracy of a sequence is therefore a measure of the number of different non-degenerate sequences that it represents (or matches).
Match. A letter, p, from a primer sequence is said to MATCH a letter, t, from a target sequence, if the set of bases corresponding to t is a subset of the set of the bases corresponding to p. Thus, for example, if the target base is R and the primer base is D then MATCH(D, R) = true, while match(R, D) = false. On the other hand, MATCH(G, H) = MATCH(H, G) = false. Extending this definition to sequences, a primer sequence P of length j is said to match a target sequence T, if there exists a location l, such that the letters of P match the corresponding letters of T (j, l). For example, for the sequences mentioned above, MATCH(S2, S3) = false, while MATCH(S2, S1) = true since S2 matches the subsequence of S1 of length 6 starting from location 2. Biologically, if a primer sequence matches a target sequence, then the primer sequence would hybridize to the target sequence at the location of the match.
Degenerate codes such as S = {C, G}, W = {A, T} and N = {A, C, G, T} mean different things in different contexts. A degenerate code N in the database (e.g. RDP) means that the value of that base is not precisely determined. Thus, an N suggests that the base is A or C or G or T and any choice of the base (other than N) for the primer at that position could fail to match. On the other hand, a degenerate code in the primer means that all possible bases are present in the primer at that position. Thus, an N suggests that the base is A and C and G and T, i.e., different copies of the primer with different bases in that position are used. Consequently, it would match any letter in the corresponding target location.
Common. Two letters from are said to be in COMMON if the sets of bases they represent have a non-empty intersection. Thus, for example, COMMON(R, D) = TRUE, while COMMON(G, H) = false. The definition can be extended to two sequences being in common as follows: Two sequences are said to be in COMMON if every letter of one of the sequences is in common with the corresponding letter from the other sequence.
Design parameters for optimal degenerate primers. As mentioned earlier, the PCR reaction requires a number of conditions to be satisfied by the primers in order to work well. The following parameters and their constraints have been compiled from various design manuals. (For example, see http://www.premierbiosoft.com/tech_notes/PCR_Primer_Design.html.) We will refer to them henceforth as the Optimal Primer Design Rules.
(1) SELFDIMER(P) is defined as the length of the longest contiguous COMMON subsequence between the primer P and its reverse complement. Good primer design requires that this quantity be minimized.
(2) PRIMERDIMER(F, R) is defined as the length of the longest contiguous COMMON subsequence between the forward (F ) and the reverse (R) primers. This quantity should be minimized.
(3) AMPLICONLENGTH(F, R) is the length of sequence that starts with F and finishes with R. The amplicon length is defined in the range of 150-600 bp. The lower limit prevents amplification of conserved regions. The upper limit is determined by the current read length that sequencing technologies support.
(4) RUNLENGTH(P ) is the maximum number of consecutive occurrences of the same base from the set {A, C, G, T}. Thus, for the U336R primer in Table 1, the red colored subsequence has RUNLENGTH(U336R) = 6.
(5) CG%(P ) is the percent of {C, G} bases in P. If DEG(P ) = 1 or if only S or W degenerate bases are present then there is no ambiguity in the computation of CG%. Since other degenerate bases cause ambiguity in the count, we end up with a range of values [MINCG%(P), MAXCG%(P)]. Good primer design requires that CG%(P) be between 50 and 65. In practice, this constraint may be slightly relaxed.
(6) While many methods exist in the literature, TM(P ) is calculated by the Basic Tm equation (see http://www.promega.com/techserv/tools/biomath/calc11.htm): Tm(P) = 64.9 + 41 * (CG%(P) - 16.4)/length(P). If DEG(P) = 1, or if only S or W degenerate bases are present then there is no ambiguity in the above calculation. As with CG%, if other degenerate bases are present in the primer sequence, then a range of values is possible, and is denoted by [MINTM(P), MAXTM(P)]. Good primer design requires that the entire range lies between 55° and 65°. It also requires that RANGETM(P) = MAXTM(P)-MINTM(P) not be more than 5°. Again, in practice, a slight deviation from these conditions is acceptable.
Instead of allowing an arbitrary sequence to be a candidate primer, we define some more terms that help us narrow down the search space for our candidate primers.
Viable Primer. A viable primer is a sequence that satisfies the optimal primer design rules from above.
Common Viable Primer for . A common viable primer for a multiset of sequences is a viable primer if it matches at least minSupport number of sequences in .
Derivative. A sequence P is derived from a sequence T if MATCH(P, T) = true. (For example, P1 = AWGCT is a derivative of T = ATGCT, but P2 = ATGGT is not.)
Amplicon from sequence S with Primer Pair (F, R). An amplicon A from sequence S is a contiguous subsequence of S defined by a viable primer pair (F, R). A must start with a subsequence that matches the reverse complement of the forward primer F and must end with a subsequence that matches the reverse primer R.
Reference Target Sequence T. A sequence T from a database of sequences is said to be a reference target sequence if the forward and reference primers are derived from T .
Problem statement
The primary task of a viable primer is to match as many sequences as possible from a target database. For primer pairs, it is important to maximize the number of sequences that match both primers. In applications such as metagenomics, where the amplicons are sequenced and then used to identify the organism from which it originated, a third requirement is that amplicons generated by the primers be uniquely distinguishable.
The Degenerate Primer Design Problem. For a given set of parameters , a set of sequences and a reference target sequence , find a viable primer pair (F, R) that matches T for which the number of distinguishable amplicons generated by its primers (F, R) for the sequences in is maximized.
In other words, given a database (e.g., RDP), a reference sequence T (e.g, E. coli), and values for the design parameters, , including MAXSELFDIMER, MAXPRIMERDIMER, MAXRUNLENGTH, MINCG%, MAXCG%, MINTM, and MAXTM, develop a set of viable forward and reverse degenerate primer pairs with the maximum number of unique amplicons.
Algorithm description
The reference sequence T ensures that the number of possible primer sequences is finite. The observation below prunes the search space further.
Observation 1 If primer P1 is not viable then a primer P2 derived from P1 is not viable either.
Also, given primers P1, P2, such that DEG(P1) ≤DEG (P2) and MATCH(P1, P2) = true, then (a) SELFDIMER(P1) ≤ SELFDIMER(P2), (b) TEMPRANGE(P1) ≤ TEMPRANGE(P2), (c) CG%(P1) ≤ CG%(P2), and (d) RUNLENGTH(P1) ≤ RUNLENGTH(P2).
Scanning for Candidate Forward and Reverse Primers. To make the search more efficient, we focus on primer sequences derived from the target sequence. Observation 1 is also used to prune away candidate primers that are not viable.
Algorithm SELECTPRIMERCANDIDATES shown here designs set of viable common degenerate forward primers that correspond to all viable primers P from reference sequence T and sequence database . A similar design is performed to generate the set of viable common reverse primers . First, for each viable primer from T function FIND-BESTVALIDMATCH generates a set of unique best valid matches in . Note that not all target sequences contribute to . Some target sequences might not have a valid match or unique best valid match for the relevant subsequence of T.
Unique best valid match between a primer and a sequence. A match between a viable primer P and a sequence Si is valid if there exists a contiguous subsequence in Si of the same length as P and that has at most log2(MAXDEGENERACY) mismatches with P. If there is only one valid match between P and Si that has the least number of mismatches then that is the unique best valid match.
Algorithm DEVELOPDEGPRIMER develops a set of viable common primers corresponding to input multiset . From the aligned set of hits , we create a frequency count matrix for each position. If the highest frequency in a position (column) in the alignment is smaller than minSupport, then that position in the primer would require to be degenerate. We first check that the number of positions that require degeneracy is no greater than log2(MAXDEGENERACY). If it is, then the bound on degeneracy would be violated and we exit without computing a common degenerate primer. Otherwise, we construct the initial degenerate primer, degPrimer, with each of its bases set to the base that occurs most frequently in that position. If the initial degenerate primer is not a viable primer we exit since its derivates need not be considered by Observation 1.
Starting from the initial primer, we remove all entries of that match the current primer. To increase the number of hits in , we use a greedy method to iteratively increase primer degeneracy, one base at a time, as follows. In each iteration, from the remaining sequences in , we reconstruct the frequency count matrix, storing all positions that do not yet match the current primer, ordered by the frequency count of the highest frequency. We choose to increase degeneracy of a base with the highest frequency count, while making sure that the new primer remains viable. Ties are broken in favor of the base for which the increased degeneracy causes the least increase in overall degeneracy of the sequence. When a new common viable primer is found, it is added to the candidate primer set .
Algorithm DESIGNPRIMERPAIR takes a set of viable forward primers and a set of viable reverse primers, groups each set into subregions, sorting each group by their number of hits in the database. To allow for better primer pairing with regard to distinguishabilty, each conserved region is further subdivided into subregions, defined by the primer start location. The sorted lists of forward and reverse primers are scanned to select the pair with maximum number of simultaneous hits in the database that abide by optimal design rules. Once we have primer pairs and their number of hits in the database, we focus on amplicon distinguishability. In the context of degenerate primer design, an amplicon is distinguishable if and only if it has a match to exactly one sequence in the database. In order to select sets of candidate primer pairs, we extend the notion of amplicon distinguishability to two or more primer pairs. A sequence is distinguishable if it produces a distinguishable amplicon with at least one of the primer pairs in the chosen set.
Function FINDDISTINGUISHTOTAL creates a taxon matrix to easily gather and compare results for each primer pair and across two (or more) primer pairs. Data can be generated for any combination of two primer pairs. It is possible to request only a number of best combination for a desired metric (distinguishable/total number of hits per genus/species).
Results
In silico design of degenerate primers
We run the algorithm on sequences from the 16S rRNA RDP database (downloaded on February 12, 2012). The set is described in Table 2. We used the E. coli sequence as the template sequence and the set of parameters used for primer design and primer pair design are described in Table 3. The algorithm described above found 208 (244, resp.) viable common forward (reverse, resp.) primers from 8 forward and (10 reverse, resp.) regions. We note that there is no viable common primer in the conserved regions between variable regions one and two. Note that our procedure was able to identify the widely used universal primers, U337F and U518R, which abide by the optimal design rules. The second primer in Table 4 in region FV3 is a more degenerate version of U337F, and the second primer under the region RV3 is a degenerate version of U518R.
Table 4.
Select Designed Primers
| regionID | 5'→3' sequence | deg | sDimer | TmRange | CG%Range | nHits | posEcoli |
|---|---|---|---|---|---|---|---|
| FV3 | ACWCCTRCGGGWGGCWG | 16 | 4 | 51.88→54.29 | 0.65→0.70 | 8867 | 345 |
| FV3 | ACWCCTRCGGGWGGCWGCAG | 16 | 6 | 57.93→59.98 | 0.65→0.70 | 8854 | 345 |
| FV4 | AGCAGCCGCGGTAANACG | 4 | 6 | 52.60→54.88 | 0.61→0.67 | 8305 | 528 |
| FV6 | WACSCGMRGAACCTTACC | 16 | 6 | 48.04→52.60 | 0.50→0.61 | 8045 | 975 |
| FV7 | ATGGYYGTCGTCARCTCG | 8 | 4 | 48.04→54.88 | 0.50→0.67 | 8821 | 1062 |
| FV7 | AGTCCNRYAACGAGCGCAACC | 16 | 4 | 54.36→60.21 | 0.52→0.67 | 8788 | 1103 |
| FV8 | AGGAAGGHGDGGAYGASGTC | 36 | 6 | 53.83→59.98 | 0.55→0.70 | 8810 | 1185 |
| RV3 | TTACCGCGGCTGCTGGCAC | 1 | 6 | 57.56→57.56 | 0.68→0.68 | 8926 | 505 |
| RV3 | TWTYACCGCGGCTGCTGG | 4 | 6 | 52.60→54.88 | 0.61→0.67 | 8543 | 508 |
| RV4 | ACCAGGGTATCTAAKCCTG | 2 | 4 | 48.93→51.09 | 0.47→0.53 | 8292 | 773 |
| RV6 | TYACRRCACGAGCTGWCG | 16 | 5 | 48.04→54.88 | 0.50→0.67 | 9026 | 1052 |
| RV7 | ACGTCRTCCHCWCCTTCC | 12 | 4 | 50.32→54.88 | 0.56→0.67 | 8114 | 1166 |
| RV8 | TGWGTACAAGRYCCGRGAACG | 16 | 6 | 52.40→58.26 | 0.48→0.62 | 8411 | 1367 |
Note that our algorithm found a forward primer better than U337F in the FV3 region with more hits and and lower SELFDIMER value. Similarly, a better reverse primer was found in the RV3 region with almost 400 more matches in RDP. Table 4 represents only a sample of the viable common primers found by the algorithm and represent the best primers in terms of the number of RDP hits. Table 4 shows a number of FV7 primers to illustrate that within a region two primers can have significantly different parameters, but still are valid options in the PRIMERPAIRDESIGN. In Table 5 we include only a selection of designed primer pairs, together with select primer pairs from HMP and Universal primer pairs for comparison. Comparing primer pairs spanning roughly the same regions, we note that primer pair F528-R1052 is better than HMP_V3-5 by most measures. Pair F345-R505 improves upon U337F-518R, and pair F345-R775/873 compares favorably to U337F-805R. Pairs F1064-R1367 or F1185-R1367 are by most measures better than HMPV69. Primer F1064 is a better substitute for U1100F primer, used as HMPV6F. The table also shows that different primer pairs can be chosen depending on what features are important to a user. For example, for region V8, primer pair F1185-R1367 would be chosen if phylum distinguishability is considered important, but not if genus or species distinguishability is considered important. Furthermore, the results also indicate that total number of hits per taxon and the corresponding distinguishability do not necessarily correlate (compare F528-R1052 and F797-R1052). The same can be said for the distinguishability between genus and species (compare F345-R922 and F528-R775).
Table 5.
Comparison of Primer Pairs
| Primer Pair Parameters | Total Number of Hits per Taxon | Number of Distinguishable Amplicons per Taxon | |||||||
|---|---|---|---|---|---|---|---|---|---|
| PrimerPairID | TmRange | Dimer | Strain | Species | Genus | Phylum | Species | Genus | Phylum |
| F345-R1052 | 6.83 | 4 | 8738 | 7961 | 1702 | 29 | 7110 | 1696 | 29 |
| F345-R505 | 5.69 | 4 | 8654 | 7887 | 1684 | 27 | 4726 | 1554 | 27 |
| F345-R775 | 5.18 | 4 | 7981 | 7342 | 1565 | 24 | 5943 | 1549 | 24 |
| F345-R873 | 6.47 | 5 | 7961 | 7264 | 1537 | 27 | 6031 | 1527 | 27 |
| F345-R922 | 6.83 | 5 | 7740 | 7074 | 1452 | 22 | 5893 | 1443 | 22 |
| F528-R1052 | 6.83 | 4 | 8212 | 7520 | 1647 | 27 | 6295 | 1639 | 27 |
| F797-R1052 | 6.83 | 3 | 8399 | 7720 | 1639 | 25 | 5396 | 1610 | 25 |
| F1064-R1367 | 6.48 | 4 | 8155 | 7439 | 1596 | 27 | 4926 | 1534 | 27 |
| F1185-R1367 | 7.58 | 4 | 8099 | 7375 | 1568 | 28 | 3776 | 1407 | 28 |
| HMPV35 | 9.67 | 5 | 8174 | 7452 | 1573 | 24 | 6225 | 1564 | 24 |
| HMPV13 | 0.79 | 3 | 1572 | 1198 | 498 | 18 | 1104 | 497 | 18 |
| HMPV69 | 7.38 | 3 | 1162 | 818 | 304 | 20 | 646 | 301 | 20 |
| U337F-805R | 10.48 | 3 | 7011 | 6479 | 1405 | 17 | 5218 | 1392 | 17 |
| U337F-518R | 4.81 | 6 | 7059 | 6512 | 1429 | 20 | 3886 | 1325 | 20 |
| NossaV34 | 6.47 | 4 | 8042 | 7413 | 1546 | 17 | 5997 | 1533 | 17 |
To see whether and how amplicon length is correlated taxon distinguishability we ran the degenerate primer design algorithm with no upper bound for amplicon length. One may hypothesize that an increase in the number of variable regions in the amplicons generated by a primer pair will lead to an increase in taxon distinguishability. The best result was obtained with the first primer pair in Table 5 (F345-R1052). It flanks V3-V6 regions. Hence increase in amplicon length does not necessarily increase taxon distinguishability. We postulate that conserved regions of different species are not uniformly conserved. This brings us to the final step: combination of different primer pairs that extract different taxons to increase taxon distinguishability. We include HMP and universal primers from Table 5 in the input data set. The results are presented in Table 6 below. In Table 6 the first two pairs of primer pairs are the best combination for distinguishing the most number of species and most number of genera, respectively, if amplicon length is not bounded.
Table 6.
Pairs of Primer Pairs with number of distinguishable species, genera and phyla.
| PrimerPair1 | PrimerPair2 | Species: Distinguish/Total | Genus: Distinguish/Total | Phylum |
|---|---|---|---|---|
| V36-F348-R1050 | V48-F532-R1367 | 7635/8284 | 1745/1763 | 29 |
| V36-F348-R1042 | V47-F528-R1164 | 7537/8256 | 1757/1758 | 29 |
| HMPV35 | V46-F528-R1052 | 7270/8217 | 1737/1742 | 28 |
| V34-F345-R775 | V46-F528-R1052 | 7210/8091 | 1727/1734 | 29 |
| V3-F348-R496 | V46-F528-R1052 | 7016/8205 | 1746/1754 | 28 |
| HMPV35 | V58-F797-R1367 | 7340/8083 | 1689/1692 | 28 |
| V3-F345-R505 | V78-F1064-R1367 | 6760/8228 | 1734/1743 | 29 |
| V3-F345-R505 | V57-F797-R1166 | 6266/8227 | 1712/1745 | 29 |
| V3-F348-R496 | V46-F528-R1052 | 7016/8205 | 1746/1754 | 28 |
If we allow amplicons of arbitrary lengths then designed primers can generate and distinguish all the Genera and can generate all the Species, while failing to distinguish 563. As the lengths of the sequenced reads are increased, the number of taxons not generated or distinguished by two primer pairs gets smaller. In particular, note that if we do not bound amplicon length, only 12 genera failed to be distinguished by a combination of V36-F348-R1050 and V47-F528-R1164, neither one of which is the best primer pair (V36-F345-R1052).
Laboratory experiments with designed primers
The best designed primers were tested in the laboratory. Needless to say, the performance of the primers will depend on the microbial composition of the samples on which these primers are applied. We tested these on DNA extracted from lung biolavage samples from the lungs of 8 (unidentified) individuals. The experiments were performed in the Mathee laboratory. PCR was performed on the extracted DNA and gel electrophoresis was used to isolate the amplicons of the appropriate length. The results were compared using BioAnalyzer (http://www.genomics.agilent.com/).
Figure 1 shows gel images for a sample from a single patient with 11 different primers - 3 HMP primers and 8 new primers from this paper. Under identical cnditions, the figure shows that 5 out of the 8 newly designed primers and 2 of the 3 HMP primers produced sufficient and comparable amplification. We then picked the best performing HMP primer and a MJ primer and tested them with 8 different patient samples. The read length distribution from the BioAnalyzer results in Figure 2 shows that the MJ primer produced more amplification than the HMP primer for most samples. More detailed analyses (not reported here) show that new genera (Alterococcus, Coxiella, Isosphaera, Leptolinea, Rubritalea, and Zavarzinella) and phyla (Armatimonadetes and Lentisphaerae) were detected by the new primers.
Figure 1.
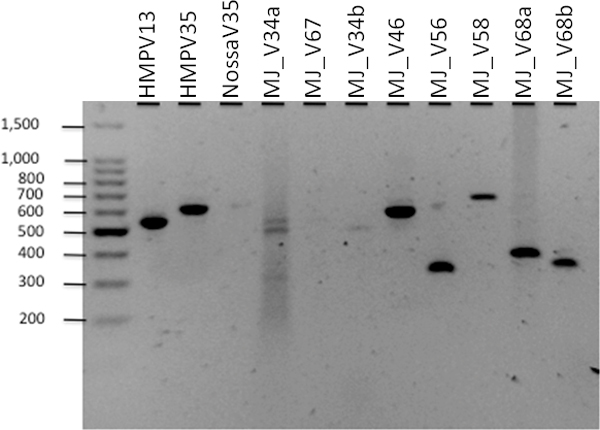
Comparing HMP and MJ primers: PCR amplification for one sample with 3 known HMP primer pairs and 8 newly designed primer pairs.
Figure 2.

Comparing HMP and MJ primers: BioAnalyzer read length distribution for 8 different samples. Top row is for MJV68 and bottom one is for HMPV35.
Discussion
The design paramater values shown in Table 3 are default values as suggested in the literature by PCR practitioners. However, the algorithm allows the user to change these values as they see fit. 16S rRNA variable regions are not uniformly variable across all species, i.e., for a group of species a particular variable region might be more conserved than others. Clearly, this differential variability of the regions affects their impact on the choice of primers and the ability of the primers to generate distinguishable amplicons.
Although intuitively it might seem that the greater the span of hypervariable regions of the amplicons from a primer pair, the greater would be its distinguishability, in reality it also depends on the specific taxa matched by the pair (F, R). A pair (F1, R1) with fewer hits than pair (F2, R2), and whose amplicon is contained in the amplicon for (F2, R2) is likely to distinguish more taxa. Different 16S rRNA regions have different degrees of variability. However, it has been noted that the set of taxa distinguished by two different regions may vary substantially, thus providing a basis for combining primer pairs. The primer pairs that result in the most number of distinguishable taxa is not necessarily the result of combining primer pairs with the best individual results. To summarize, in order to obtain as complete a picture of a microbiome as possible the number of uniquely identifiable extracted species must be maximized. In other words what is needed are compatible forward and reverse primers that bind well to the same, large number of identifiable species, and produce amplicons that are species specific. Taxon distinguishability depends on (1) how many hits a forward/reverse primer has (2) how many hits a primer pair has (3) how variable is the region a primer pair flanks (4) how many primer pairs distinguish different taxa and (5) how many additional taxa can be distinguished by combining two (or more) primer pairs.
In comclusion, we have developed an algorithm that takes as input a set of user-defined values for the primer design parameters and the sequence database, and outputs customized primer pairs that attempt to maximize their ability to produce distinguishable amplicons for the given database. In silico experiments prove that the newly designed primers are better than the primers currently used by practitioners. Laboratory experiments confirm the efficacy of the newly designed primers. This tool will positively impact the design of primers for the Human Microbiome Project and other related metagenomics projects.
Competing interests
The authors declare that they have no competing interests.
Authors' contributions
MJ performed all the computational work and analyses. MJ and GN wrote the paper. JS, ESH, and LS helped with testing the primers in the Mathee laboratory. All authors reviewed the paper.
Dedication
Sadly we announce the untimely passing of Melita Jaric and dedicate this paper to her memory.
Declaration
Publication of this work was supported by a grant (KM and GN) from the Florida Department of Health and support from Florida International University.
This article has been published as part of BMC Proceedings Volume 7 Supplement 7, 2013: Proceedings of the Great Lakes Bioinformatics Conference 2013. The full contents of the supplement are available online at http://www.biomedcentral.com/bmcproc/supplements/7/S7.
References
- Linhart C, Shamir R. The degenerate primer design problem. Bioinformatics. 2002;7(suppl 1):S172–S181. doi: 10.1093/bioinformatics/18.suppl_1.S172. [DOI] [PubMed] [Google Scholar]
- Methé B, Nelson K, Pop M, Creasy H, Giglio M, Huttenhower C, Gevers D, Petrosino J, Abubucker S, Badger J. et al. A framework for human microbiome research. Nature. 2012;7:215–221. doi: 10.1038/nature11209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fabrice A, Didier R. Exploring microbial diversity using 16S rRNA high-throughput methods. J Comput Sci Syst Biol. 2009;7:074–92. doi: 10.4172/jcsb.1000019. [DOI] [Google Scholar]
- Wang Y, Qian P. Conservative fragments in bacterial 16S rRNA genes and primer design for 16S ribosomal DNA amplicons in metagenomic studies. PLoS One. 2009;7(10):e7401. doi: 10.1371/journal.pone.0007401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lane D. 16S/23S rRNA sequencing. Nucleic acid techniques in bacterial systematics. 1991.
- Turner S, Pryer K, Miao V, Palmer J. Investigating Deep Phylogenetic Relationships among Cyanobacteria and Plastids by Small Subunit rRNA Sequence Analysis1. Journal of Eukaryotic Microbiology. 1999;7(4):327–338. doi: 10.1111/j.1550-7408.1999.tb04612.x. [DOI] [PubMed] [Google Scholar]
- Weisburg W, Barns S, Pelletier D, Lane D. 16S ribosomal DNA amplification for phylogenetic study. Journal of Bacteriology. 1991;7(2):697–703. doi: 10.1128/jb.173.2.697-703.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baker G, Smith J, Cowan D. Review and re-analysis of domain-specific 16S primers. Journal of Microbiological Methods. 2003;7(3):541–555. doi: 10.1016/j.mimet.2003.08.009. [DOI] [PubMed] [Google Scholar]
- Cole J, Wang Q, Cardenas E, Fish J, Chai B, Farris R, Kulam-Syed-Mohideen A, McGarrell D, Marsh T, Garrity G. et al. The Ribosomal Database Project: improved alignments and new tools for rRNA analysis. Nucleic Acids Research. 2009;7(suppl 1):D141–D145. doi: 10.1093/nar/gkn879. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pruesse E, Quast C, Knittel K, Fuchs B, Ludwig W, Peplies J, Glöckner F. SILVA: a comprehensive online resource for quality checked and aligned ribosomal RNA sequence data compatible with ARB. Nucleic Acids Research. 2007;7(21):7188–7196. doi: 10.1093/nar/gkm864. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hodkinson B, Lutzoni F. A microbiotic survey of lichen-associated bacteria reveals a new lineage from the Rhizobiales. Symbiosis. 2009;7(3):163–180. doi: 10.1007/s13199-009-0049-3. [DOI] [Google Scholar]
- Wei X, Kuhn D, Narasimhan G. Proceedings of the IEEE Conference on Computational Systems Bioinformatics, 2003. IEEE; 2003. Degenerate primer design via clustering; pp. 75–83. [PubMed] [Google Scholar]
- Doud M, Zeng E, Schneper L, Narasimhan G, Mathee K. Approaches to analyse dynamic microbial communities such as those seen in cystic fibrosis lung. Human Genomics. 2009;7(3):246–256. doi: 10.1186/1479-7364-3-3-246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klindworth A, Pruesse E, Schweer T, Peplies J, Quast C, Horn M, Glöckner F. Evaluation of general 16S ribosomal RNA gene PCR primers for classical and next-generation sequencing-based diversity studies. Nucleic Acids Research. 2012. [DOI] [PMC free article] [PubMed]
- Sim K, Cox M, Wopereis H, Martin R, Knol J, Li M, Cookson W, Moffatt M, Kroll J. Improved detection of bifidobacteria with optimised 16S rRNA-gene based pyrosequencing. PLoS One. 2012;7(3):e32543. doi: 10.1371/journal.pone.0032543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Universal primers for 16S ribosomal RNA. http://en.wikipedia.org/wiki/16S_ribosomal_RNA
- Schloss P. 16S rRNA Gene Sequencing Curation Pipeline. http://www.hmpdacc.org/doc/16SrRNA_GeneCuration_SOP.pdf
- Schloss P, Gevers D, Westcott S. Reducing the effects of PCR amplification and sequencing artifacts on 16S rRNA-based studies. PLoS One. 2011;7(12):e27310. doi: 10.1371/journal.pone.0027310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chakravorty S, Helb D, Burday M, Connell N, Alland D. A detailed analysis of 16S ribosomal RNA gene segments for the diagnosis of pathogenic bacteria. Journal of microbiological methods. 2007;7(2):330–339. doi: 10.1016/j.mimet.2007.02.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nossa C, Oberdorf W, Yang L, Aas J, Paster B, DeSantis T, Brodie E, Malamud D, Poles M, Pei Z. Design of 16S rRNA gene primers for 454 pyrosequencing of the human foregut microbiome. World journal of gastroenterology: WJG. 2010;7(33):4135. doi: 10.3748/wjg.v16.i33.4135. [DOI] [PMC free article] [PubMed] [Google Scholar]