

Abstract
Genome stability is essential for neural development and the prevention of neurological disease. Here we determined how DNA damage signaling from dysfunctional telomeres affects neurogenesis. We found that telomere uncapping by Pot1a inactivation resulted in an Atm-dependent loss of cerebellar interneurons and granule neuron precursors in the mouse nervous system. The activation of Atm by Pot1a loss occurred in an Atr-dependent manner, revealing an Atr to Atm signaling axis in the nervous system after telomere dysfunction. In contrast to telomere lesions, Brca2 inactivation in neural progenitors also led to ablation of cerebellar interneurons, but this did not require Atm. These data reveal that neural cell loss after DNA damage selectively engages Atm signaling, highlighting how specific DNA lesions can dictate neuropathology arising in human neurodegenerative syndromes.
Keywords: ATM, cerebellum, DNA damage, neural development, telomeres
Introduction
A hallmark example of human genome instability syndromes is ataxia telangiectasia (A-T), a neurodegenerative disease that results from inactivation of ataxia telangiectasia, mutated (ATM), a DNA damage signaling serine/threonine protein kinase and a member of the phosphatidylinositol kinase-related kinase family (Lavin, 2008; McKinnon, 2012; Shiloh and Ziv, 2013). An important function of ATM in the nervous system is the activation of DNA damage-induced apoptosis (McKinnon, 2012). An ATM-related kinase, ATR (ATM and Rad3-related) is also central to neural development and function. Hypomorphic mutations in the ATR gene result in Seckel syndrome, a disease characterized by microcephaly and mental retardation in humans (O'Driscoll et al., 2003). Both kinases can respond to DNA damage to activate cell cycle arrest, DNA repair, or apoptosis. A distinguishing feature of these kinases is the specific activating DNA lesion. Replication protein A (RPA)-coated single-stranded DNA, which can occur during replication stress, activates ATR, whereas DNA double-strand breaks (DSBs) activate ATM (Cimprich and Cortez, 2008; Lavin, 2008; Shiloh and Ziv, 2013).
The shelterin multiprotein complex, located at the telomere region of the chromosome, is critical to prevent chromosomal DNA ends being recognized as DNA damage. Six distinct proteins comprise this complex: POT1, TRF (TERF) 1 and 2, TPP1, TIN2, and RAP1 (for review, see de Lange, 2009, 2010). TRF1 and TRF2 (telomeric repeat binding factor 1 and 2) recognize telomeric sequences and bind as homodimers to double-stranded regions of the telomere, whereas POT1 (protection of telomeres 1) binds the 3′ single-stranded G-overhang; POT1 also binds to TPP1 (TIN2 and POT1 interacting protein 1) (Chan and Chang, 2010; de Lange, 2010). TIN2 (TERF1 interacting nuclear protein 2) provides a bridge between double-stranded and single-stranded portions of telomere by interacting with TRF1, TRF2, and TPP1 (de Lange, 2009).
DNA damage signaling resulting from telomeres with a compromised shelterin component is specific and involves the ATM or ATR kinases. It is generally accepted that disruption of POT1 activates ATR, whereas disruption of TERF2 activates ATM signaling (Denchi and de Lange, 2007; Guo et al., 2007; Sfeir and de Lange, 2012). This selective damage signaling results from the specific DNA substrates created by inactivation of these shelterin components. For example, loss of Pot1a can generate RPA-coated single-strand loops in telomeres, which is an activating lesion for ATR, but not ATM (Gong and de Lange, 2010; Flynn et al., 2011). TRF2 is responsible for forming T-loop structure that sequester the ends of a chromosome, thereby preventing its recognition as a DSB and the activation of ATM (Sfeir and de Lange, 2012; Doksani et al., 2013).
Although functional integration within the shelterin complex maintains telomere structure and prevents the activation of a DNA damage response, how this process operates during neurogenesis remains largely unknown. Here, we show that in the nervous system, rather than Pot1a deficiency exclusively activating Atr, perturbation in neural homeostasis involving cellular attrition after disruption of Pot1a depends upon Atm. Our physiologic data also highlight the importance of a novel crosstalk between these two key DNA damage response kinases after telomere perturbation resulting from disruption of a shelterin component.
Materials and Methods
Animals.
The Pot1a conditional allele has been previously described (Wu et al., 2006). To restrict inactivation of the Pot1a gene in the nervous system Nestin-Cre mice (B6.Cg-Tg(Nes-Cre)1Kln/J, JAX#003771) were used to generate Pot1aLoxP/LoxP;Nestin-cre (here after Pot1aNes-cre) animals. Nestin-cre was maintained maternally to minimize ectopic cre recombinase expression in organs outside of the nervous system. Pot1aNes-cre mice were also intercrossed with Atm−/− or p53−/− germline knock-outs (Lee et al., 2001, 2012b) or a conditional Atr allele (Lee et al., 2001, 2012b) to generate compound mutants to investigate the DNA damage signaling pathways. Brca2LoxP, Lig4LoxP, and Xrcc2LoxP mice were as previously described (Orii et al., 2006; Frappart et al., 2007; Shull et al., 2009). Genotypes of Pot1a animals were determined using PCR with the following primers; forward primer, 5′-CTCGAATTCCATCTCCTCCCAGTACTCTCTCAG and reverse primer, 5′-GGAACTGGTACGTATCAGTGTGTGTGG. Deletion of Pot1a after cre recombinase activity was done using forward primer, 5′-ACACGGATCCTGAGCCATAAACATGCCACACAAAGG and reverse primer, 5′-CTCGAATTCCATCTCCTCCCAGTACTCTCTCAG. PCR products were amplified for 30 cycles of 94°C for 45 s, 60°C for 45 s, and 72°C for 45 s. PCR conditions for genotyping Atm, Atr, p53, Brca2LoxP, Lig4LoxP, and Xrcc2LoxP have been described previously (Lee et al., 2001, 2012b; Orii et al., 2006; Frappart et al., 2007; Shull et al., 2009). The presence of a vaginal plug was indicated as embryonic day 0.5 (E0.5) and the day of birth as postnatal day 0 (P0). Experiments were performed on embryos and postnatal animals of either sex. All animals were housed in an Association for Assessment and Accreditation of Laboratory Animal Care accredited facility and were maintained in accordance with the National Institutes of Health' Guide for the care and use of laboratory animals. The institutional animal care and use committee at St Jude Children's Research Hospital approved all procedures for animal use.
Histology.
Most antibodies used for immunohistochemistry and immunocytochemistry have been described previously (Lee et al., 2009, 2012a). Additionally, anti-Rpa32T21 (rabbit, 1:200, Abcam) was also used. Histology procedures were performed as previously described (Lee et al., 2009, 2012a). Briefly, embryos and brains were collected at indicated time points after fixation with 4% buffered PFA. Cryosections (10 μm) for immunohistochemistry were cut using a HM500M cryostat (Microm). Localization of primary antibodies on tissue sections or cultured cells was done using either a VIP substrate kit (Vector Laboratories) with biotinylated secondary antibodies, or FITC/CY3-conjugated secondary antibodies (Jackson Immunologicals); 0.1% methyl green or DAPI/PI (Vector Laboratories) was used for counterstaining for VIP or FITC/CY3, respectively. All histology experiments were performed at least three times.
For cell cycle analysis experiments, brains from BrdU-injected animals (60 μg/g i.p. body weight) were isolated 2 h after transcardial perfusion with 4% PFA. Both ssDNA immunoreactivity and TUNEL assay using Apoptag (Millipore Bioscience Research Reagents) were applied to measure programmed cell death. All slides were examined and imaged using an Axio Imager A1 microscope (Zeiss), and images were captured and processed using Photoshop (Adobe).
Quantification analysis of cell proliferation and cell death using TUNEL, ssDNA, and BrdU incorporation was achieved by calculating positive immunosignals per unit area. Data were collected from at least 6 different sections per each group obtained from 3 different animals. Student's t tests were used for statistical analysis.
Western blot analysis.
Tissues or cells used for Western blot analysis were collected at indicated time points. Whole animals or cell lines were gamma-irradiated using a cesium irradiator. Routine Western blot procedures have been described previously (Lee et al., 2009, 2012a). Lysates from tissues or cells were prepared using a lysis buffer containing 50 mm Tris buffer, pH 7.5, 150 mm NaCl, 50 mm NAF, 0.2% NP-40, 1% Tween 20, 1 mm AEBSF (Roche), 1 mm dithiothreitol, protease inhibitor mixture (Complete Mini, Roche), and Phosphatase inhibitor mixture (PhosSTOP, Roche). Protein was quantified using Bio-Rad Protein Assay reagent. Samples were separated in NuPAGE Bis-Tris gels with MOPS buffer system and transferred onto the nitrocellulose or PVDF membrane according to the manufacturer's recommendation (Bio-Rad). Equal protein loading was verified by anti-actin (goat, 1:500, Santa Cruz Biotechnology) immunoreactivity and Ponceau staining. For Rpa32 immunoprecipitation experiments, anti-Rpa32 (rat, 1:100, Cell Signaling) was used together with protein A/G agarose beads (GE Healthcare), which were rinsed in RIPA buffer containing phosphate buffer, pH 7.6, 1% Triton X-100, 1% sodium deoxycholate, 0.1% SDS, and 2 mm EDTA after immunoprecipitation. Positive signals on the membranes were visualized by chemiluminescence method using ECL reagents (GE Healthcare).
The following antibodies were used for Western blot analysis: Rpa32 phosphothreonine21 (rabbit, 1:500, Abcam), Rpa32 phosphoserine33 (rabbit, 1:5000, Bethyl), Rpa32 phosphoserine4/8 (rabbit, 1:1000, Bethyl), Rpa32 (rabbit, 1:1000, Abcam; or rat, 1:1000, Cell Signaling), Chk2 (mouse, 1:1000, Millipore), Chk1 phosphoserine317 (rabbit, 1:1000, Bethyl), Chk1 (rabbit, 1:500, Cell Signaling), Kap1 phosphoserine824 (rabbit, 1:1000, Bethyl), Atm phosphoserine 1981 (clone 10H11.E12, a gift from Dr. Mike Kastan), Atm (mouse, 1:1000, Sigma), and actin (goat, 1:1000, Santa Cruz Biotechnology).
Astrocyte isolations.
Astrocytes were isolated from 3- to 4-d-old brains. Cerebral cortices were mechanically dissociated by passing through a glass pipette, and single-cell suspensions were washed with DMEM and Ham's nutrient mixture F-12 (1:1, Invitrogen-BRL), then cultured in media supplemented with 10% FBS, 1× glutamax, 100 U/ml penicillin, 100 μg/ml streptomycin, and 20 ng/ml epidermal growth factor (Sigma). Astrocytes were maintained in Primeria tissue culture flasks (Falcon) at 37°C in a 5% CO2 incubator and split every 4–5 d. For proliferation analysis, equal numbers of cells were plated in duplicate and counted every 2 d. Immunoreactivity was assessed in cells after permeabilization with 0.4% PBS buffered Triton X-100. Up to 500 cells per each cell line in triplicate were counted for quantification analysis. Student's t tests were used for statistical analysis.
Senescence-associated β-gal staining.
Cells were briefly fixed in 2% buffered PFA on ice and exposed to 1 mg/ml X-gal (5-bromo-4-chloro-3-indoyl β-d-galactoside, Promega) in 40 mm citric acid/sodium phosphate, pH 6.0, buffer, 5 mm potassium ferrocyanide, 5 mm potassium ferricyanide, 150 mm NaCl, and 2 mm MgCl2. In parallel, lysosomal β-galactosidase also was visualized in pH 4.0 citric acid/sodium phosphate buffer to detect a basal level of β-gal staining, which did not differ in astrocyte cell lines in different genetic backgrounds.
Cytogenetics analysis.
Astrocytes were subjected to spectral karyotyping and telomere FISH. Cells at metaphase were harvested 4 h after treatment of Colcemid (10 μg/ml). A commercially prepared spectral karyotyping probe was used from applied spectral imaging. The hybridization and detection procedures were performed according to the Applied Spectral Imaging Protocols. For telomere FISH, plasmid DNA containing a TTAGGG repeat sequence was labeled with digoxigenin-11-dUTP (Roche Molecular Biochemicals). After hybridization of labeled probes, positive signals were visualized with fluorescein-labeled antidigoxigenin antibody (Roche Molecular Biochemicals) and counterstained with DAPI. Images were captured with a Nikon Eclipse 80i microscope equipped with Applied Imaging Genus (version 3.2) using the probe measurement application.
Statistical analysis.
All statistical analyses were performed using Prism (version 4.0, Graphpad), and differences were considered significant when p < 0.05.
Results
Pot1a deficiency leads to cellular senescence in a p53-dependent manner
A single gene encodes human POT1, whereas the mouse contains two separate orthologs, Pot1a and Pot1b that share ∼75% homology to each other and to the human POT1 gene (He et al., 2006; Hockemeyer et al., 2006; Wu et al., 2006). Of the two, Pot1a is primarily responsible for preventing DNA damage activation at the telomere (Hockemeyer et al., 2006; Wu et al., 2006). We selectively induced telomere dysfunction by inactivation of Pot1a in neural progenitors using Nestin-cre to generate Pot1aNes-cre mice (Fig. 1). In contrast to early embryonic lethality seen in Pot1a germ-line-null animals (Hockemeyer et al., 2006), these conditional knock-out animals were born at normal Mendelian ratios with an overall body and brain size that was smaller than control counterparts; Pot1aNes-cre mice also had a shorter life span (Fig. 1). Coincident inactivation of p53, but not Atm, rescued overall brain size, implicating a role for the DNA damage pathway in the Pot1aNes-cre phenotype (Fig. 1).
Figure 1.

Pot1a is required for normal development of the nervous system and animal survival. A, Whereas brain size is reduced after inactivation of Pot1a in neural progenitors, overall development and histology is relatively intact. Coincident p53 loss partially rescues brain size in Pot1aNes-crep53−/− mice. Middle panels of sagittal brain section show relatively similar histology, as visualized with hematoxylin and eosin staining. B, A Kaplan–Meier plot shows that loss of Pot1a in the nervous system leads to premature death compared with controls. Survival is not rescued by coincident inactivation of either p53 or Atm.
Because Pot1aNes-cre mice were viable, we initially generated primary cortical astrocytes to determine the cellular consequences of Pot1a deficiency in vitro. We found that Pot1a−/− astrocytes accumulated DNA damage and underwent premature senescence, as determined using senescence-associated β-Gal staining (Fig. 2A). GFAP immunostaining in astrocytes also appeared enhanced after Pot1a loss, which was further exacerbated after additional inactivation of Atm. However, this was likely a feature of cell culture, as in vivo analysis of GFAP showed a mild gliosis after Pot1a loss that was not altered by Atm inactivation (Fig. 2B). Proliferation of Pot1a-null astrocytes was also markedly compromised but recued by p53 deficiency (but not Atm deficiency; Fig. 2C) similar to the situation in Pot1a-null MEFs (Hockemeyer et al., 2006; Wu et al., 2006). Pot1a-deficient astrocytes also accumulated γH2AX foci, a marker for DNA breaks, indicating that telomere dysfunction results in DNA damage accumulation (Fig. 2A,D). Finally, we examined genomic instability and found that Pot1a deficiency resulted in a spectrum of chromosomal defects, including chromosome breaks, translocation, and chromosome duplication (Fig. 2E). Telomere dysfunction was confirmed using telomere FISH, demonstrating that sites of chromosome fusions contained telomere signals (Fig. 2E). These data confirm that Pot1a inactivation in neural cells results in DNA damage leading to genome instability.
Figure 2.
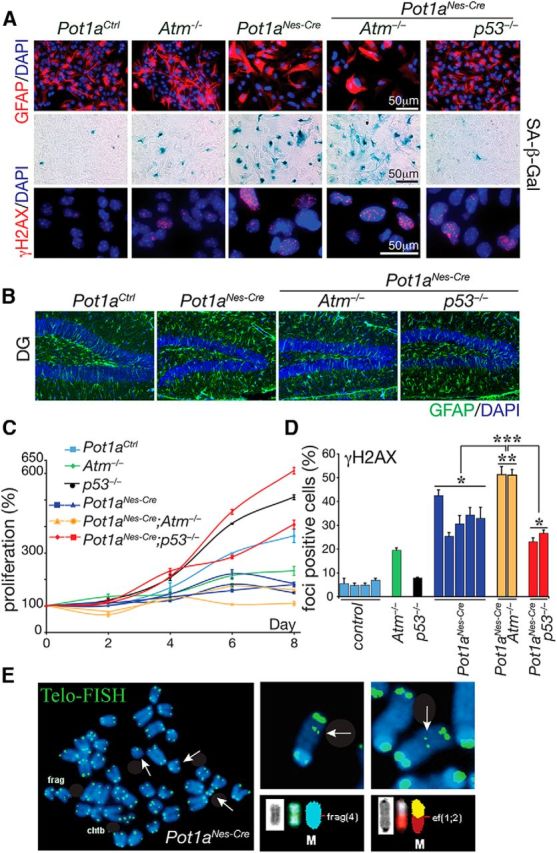
Pot1a-deficient astrocytes undergo premature senescence and genome instability. A, Astrocyte identity and morphology are shown using immunostaining for GFAP. Pot1a deficiency leads to premature senescence, as identified using SA β-gal staining. Pot1a−/− astrocyte cultures accumulate DNA damage as shown by γH2AX immunostaining. B, GFAP immunostaining in vivo after Pot1a loss shows mild gliosis in Pot1aNes-cre brain, which is not exacerbated after Atm or p53 loss. Region shown is the dentate gyrus (DG) of 2-month-old animals. C, Pot1a deficiency led to an impairment of replication that was relieved by coincident p53 deficiency. D, Quantification of γH2AX foci indicates that Pot1a−/− astrocytes accumulate substantial DNA damage, which is significantly exacerbated after Atm loss. ***p < 0.0001. **p < 0.001. *p < 0.05. E, Cytogenetic analysis of telomeres in low passage Pot1a−/− astrocytes indicates abnormalities, such as chromosomal fragments and fusions (white arrows). M, Marker chromosomes. Wild-type, Atm−/−, or p53−/− at similar passage number did not show elevated abnormalities. Frag, Fragmentation; Chtb, chromatid break; Ef, end-to-end fusion.
Atm inactivation rescues interneuron loss in the Pot1aNes-Cre cerebellum
We next examined the nervous system of Pot1aNes-cre mice to assess the physiologic consequence of Pot1a loss during neural development. Histological analysis revealed a mild effect throughout the brain after Pot1a inactivation, as Pot1aNes-cre brains showed a relatively normal histology (Fig. 1). However, despite this, we found a marked defect in the cerebellum of the Pot1a-deficient animals as they showed the loss of interneurons, including Stellate, Basket, and Golgi neurons and also a loss of granule neurons (Fig. 3A). This phenotype is reminiscent of loss of the base excision repair factor Xrcc1 throughout the nervous system, where p53-dependent cell-cycle arrest occurred in interneuron progenitors in the cerebellar white matter (WM), resulting in proliferation arrest and the loss of interneuron progenitor expansion after birth (Lee et al., 2009).
Figure 3.
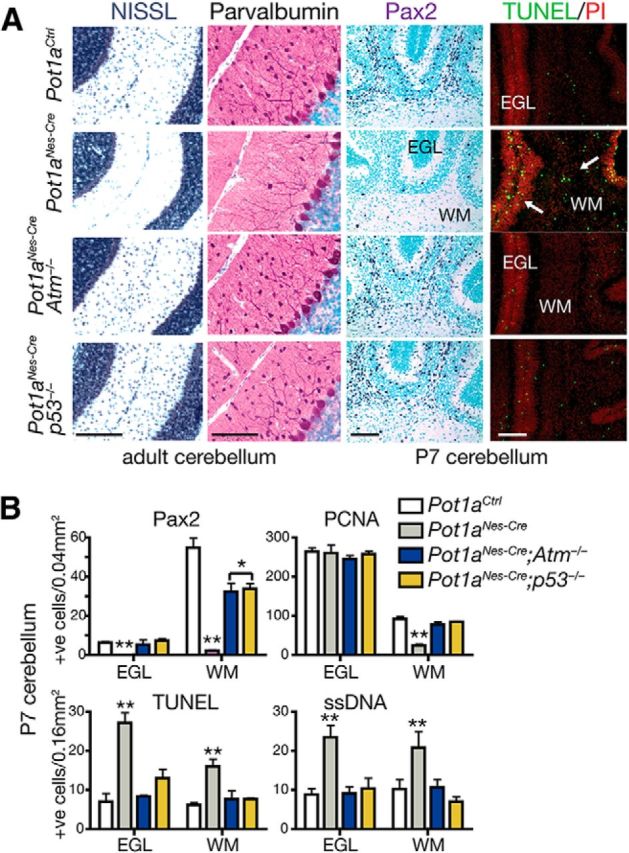
Cerebellar interneurons are lost after Pot1a deficiency in an Atm- and p53-dependent manner. A, Analysis of postnatal day 7 (P7) and adult Pot1aNes-cre cerebellum revealed a selective loss of cerebellar interneurons, which were identified using Nissl staining and immunostaining for Pax2 (a marker for interneuron precursors) and parvalbumin (a marker for mature interneurons and Purkinje cells). Pot1aNes-cre deficiency leads to increased apoptosis in the developing postnatal cerebellum, as shown using TUNEL staining. Coincident inactivation of either Atm or p53 resulted in substantial rescue of the interneurons. Scale bars, 300 μm. B, Quantification of interneuron numbers in the P7 cerebellar WM and EGL using Pax2 shows a marked reduction after Pot1a loss that is significantly rescued when either Atm or p53 is coincidently inactivated. Proliferating cell nuclear antigen (PCNA) immunostaining to identify dividing cells showed a reduction in proliferating cells in the cerebellar WM, the location of interneuron genesis. Apoptosis was determined using TUNEL and ssDNA assays and showed increased cell loss in the Pot1aNes-cre WM and EGL. **p < 0.0001. *p < 0.001.
Given the similar cerebellar phenotypes between Xrcc1Nes-cre and Pot1aNes-cre, we determined whether Pot1a deficiency also triggered cell cycle arrest in the WM progenitors. However, in contrast to Xrcc1Nes-cre, the Pot1aNes-cre cerebellum showed widespread apoptosis throughout the WM progenitors (Fig. 3B). We then assessed involvement of Atm and p53 signaling, as these two factors are central to the DNA damage response (DDR), by generating Pot1aNes-cre;Atm−/− and Pot1aNes-cre;p53−/− animals. Again, in contrast to Xrcc1Nes-cre mice where coincident inactivation of Atm did not rescue interneuron loss, both Pot1aNes-cre;p53−/− and Pot1aNes-cre;Atm−/− cerebellum contained a full complement of cerebellar interneurons (Fig. 3A). Consistent with DNA damage accumulation in response to Pot1a inactivation, we observed high levels of γH2AX immunostaining in interneuron and granule neuron progenitors and elsewhere throughout the brain (Fig. 4). γH2AX immunostaining was also observed in various brain regions of older animals, suggesting that loss of Pot1a results in chronic DNA damage signaling in mature neurons (Fig. 4).
Figure 4.
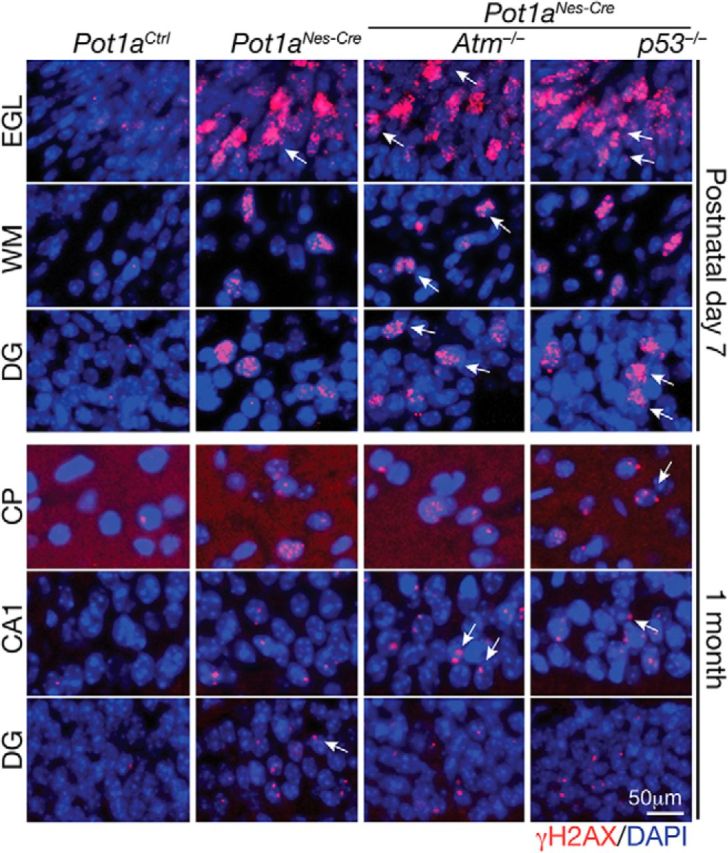
Loss of Pot1a results in chronic DNA damage. The inactivation of Pot1a in neural progenitors leads to an accumulation of DNA damage as shown by γH2AX immunostaining. DNA damage is found in different brain regions, including the developing cerebellum and cortex at postnatal day 7 and persists in older animals at 1 month. Arrows indicate representative γH2AX foci. EGL is of the cerebellum; WM is the cerebellar WM. DG, Dentate gyrus; CTX, cerebral cortex; CA1, cornu ammons region 1 of the hippocampus. Sections shown are from WT, Pota1aNes-cre, Pota1aNes-cre;Atm−/−, and Pota1aNes-cre;p53−/− mice.
The rescued Pot1aNes-cre;p53−/− and Pot1aNes-cre;Atm−/− interneurons were positive for several interneuron-specific markers, including parvalbumin, suggesting normal differentiation (Fig. 3A). In addition to interneurons, granule neuron progenitors of the external germinal layer of developing cerebellum also underwent apoptosis that was both Atm and p53-dependent (Fig. 3A,B). Purkinje cells appeared unaffected by Pot1a loss driven by Nestin-cre, possibly as they undergo differentiation by E14, before migration and synaptogenesis, and are therefore more resistant to apoptosis. The relatively mild effect of Pot1a loss in other brain regions likely relates to stochastic damage that does not impact overall development. We also analyzed glial cells using GFAP immunostaining and found areas reflective of gliosis throughout the older Pot1aNes-cre brains that indicated general perturbation in neural homeostasis (Fig. 2B and data not shown). Finally, in contrast to other animal models in which medulloblastoma arises after induced genomic instability on a p53-null background (Lee and McKinnon, 2002; Orii et al., 2006; Lee et al., 2009), Pot1aNes-cre;p53−/− animals did not develop neural tumors, at least through 6 months of age.
Brca2 is required for genesis of cerebellar interneurons
The Pot1aNes-cre phenotype was surprisingly selective and relatively mild, particularly in the context of germline Pot1a deletion, which results in very early embryonic lethality (Wu et al., 2006). To further understand the nature of the DNA damage signal resulting from Pot1a-induced telomere uncapping, we compared the Pot1a-deficient nervous systems to mice that have DNA DSB repair defects. To do this, we used mice in which homologous recombination was disabled by Brca2 or Xrcc2 inactivation, or nonhomologous end-joining was disrupted by DNA ligase IV (Lig4) loss (Orii et al., 2006; Frappart et al., 2007; Shull et al., 2009). Brca2Nes-cre, but not Xrcc2Nes-cre or Lig4Nes-cre, mice showed a similar loss of cerebellar interneurons to Pot1aNes-cre (Fig. 5A,D). However, dual inactivation of Xrcc2 and Lig4 reduced interneuron numbers (Fig. 5D), suggesting that the levels of DSBs determine cerebellar interneuron fate.
Figure 5.
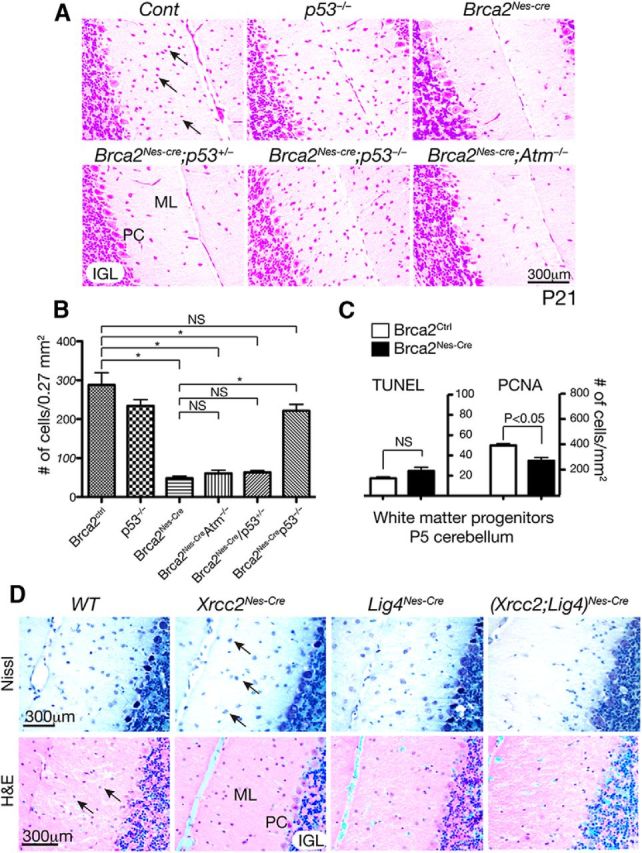
Cerebellar interneuron loss after Brca2 inactivation does not require Atm. A, The 21-day-old (p21) Brca2Nes-cre cerebellum shows a marked loss of interneurons. Nissl-stained cerebellum shows an absence of interneurons in the molecular layer; and whereas p53 inactivation (but not p53+/−) rescues this loss, coincident inactivation of Atm does not affect cell loss. B, Quantitative analysis indicates a significant rescue of Brca2Nes-cre interneurons by p53 loss but not by Atm loss. *p < 0.001. NS, Not significant. C, Proliferation was decreased in the Brca2Nes-cre cerebellar WM region containing interneuron progenitors (p < 0.05), as determined by counting proliferating cellular nuclear antigen (PCNA)-positive cells. In contrast, levels of apoptosis, determined by TUNEL staining, were not different (NS) between mutant and control tissue. D, Cerebellar interneurons are affected by dual inactivation of Xrcc2 and DNA ligase IV, whereas inactivation of the doubled break repair factors Xrcc2 or DNA ligase IV (Lig4) did not alter cerebellar interneuron numbers, dual inactivation markedly reduced the numbers of these cells. Top sections are stained with Nissl; bottom sections are stained with hematoxylin and eosin (H&E). ML, Molecular layer (cerebellar); PC, Purkinje cell layer; IGL, inner granule layer. Arrows indicate interneurons. Scale bar, 300 μm.
To determine whether Atm-dependent DNA damage signaling was important for cerebellar interneuron elimination after Brca2 inactivation as it is for Pot1a, we generated Brca2Nes-cre;Atm−/− mice. Given the central role played by p53 after DNA damage, we also generated Brca2Nes-cre;p53−/− mice. Whereas p53 loss rescued Brca2Nes-cre cerebellar interneurons, Atm inactivation failed to do this (Fig. 5A,B). We next determined whether DNA damage after Brca2 inactivation led to interneuron loss via apoptosis or cell cycle arrest. We found that, although apoptosis (TUNEL staining) was not increased, proliferation (proliferating cell nuclear antigen-positive cells) was significantly reduced in the Brca2Nes-cre cerebellar WM region (Fig. 5C). Thus, DNA damage signaling after DSBs is qualitatively different from telomere dysfunction, indicating that Atm selectively responds to different types of DNA damage in the developing nervous system.
Both Atr and Atm signal Pot1a deficiency during neurogenesis
Our finding that Atm signaling is critical for cerebellar neuron loss after Pot1a inactivation was unexpected since it has been previously shown that ATR, but not ATM, activated the DDR after POT1 loss (Denchi and de Lange, 2007; Guo et al., 2007), suggesting that the physiologic responses to POT1 deficiency shows important differences to an in vitro setting. To explore this apparent discrepancy, we examined whether Atr signaling was also relevant after Pot1a inactivation in the nervous system. Previously, we found that inactivation of Atr in neuroprogenitors via Nestin-cre resulted in a relatively complex neurological phenotype after embryonic day 16.5 (E16.5), and AtrNes-cre animals were born runted and only survived until a week of age (Lee et al., 2012b). We attempted to generate (Pot1a;Atr)Nes-cre animals, but we were unable to find any live births of the double mutants. Therefore, we analyzed (Pot1a;Atr)Nes-cre embryos and found that by E13.5 high levels of DNA damage, indicated by γH2AX immunostaining, occurred throughout the CNS (Fig. 6). Consistent with the eventual cerebellar defects (Fig. 3), DNA damage-associated apoptosis in Pot1aNes-cre embryos was observed in the E13.5 embryonic cerebellar ventricular zone, which is the site of genesis of interneuron progenitors (Fig. 6A). Additionally, apoptosis occurs in the rhombic lip, from where the external germinal layer (EGL) arises. Importantly, we found that apoptosis resulting from Pot1a loss was rescued by Atr deficiency during neurogenesis, similar to that found after Atm deficiency (Fig. 6).
Figure 6.

Atm and Atr are required for apoptosis after Pot1a loss. A, DNA damage identified by γH2AX foci (top) is apparent in the cerebellum as early as E13.5 after Pot1a loss; apoptosis (bottom; white arrows) is Atm- and Atr-dependent. VZ, Ventricular zone; RL, rhombic lip. B, Apoptosis in the E15.5 forebrain also occurs after Pot1a loss, and this apoptosis is abrogated by coincident inactivation of Atm or Atr. Proliferation defects in the Pot1aNes-cre forebrain after coincident loss of Atr were not fully rescued, which is likely associated with the complex neurological phenotype that eventually develops after Atr loss during neural development (Lee et al., 2001, 2012b). Analysis of proliferation using phospho-Ser10 of histone H3 or BrdU labeling indicates that Atm loss rescues the proliferation defects after Pot1a loss. **p < 0.0001. *p < 0.001.
We also observed increased apoptosis in regions of the Pot1aNes-Cre forebrain (Fig. 6B), which likely accounts for the mild reduction in cortical size (Fig. 1). Quantitative analysis after Pot1a inactivation at E15.5 showed that loss of either Atm or Atr reduced apoptosis as shown by the reduction of TUNEL-positive cells (Fig. 6B). Similarly, proliferation levels were recovered in in Pot1aNes-cre;Atm−/− tissue as judged by H3pSer10 and BrdU-positive cells. Because Atm loss does not rescue the brain size in Pot1aNes-cre mice (Fig. 1), DNA damage-induced apoptosis in the Pot1aNes-cre;Atm−/− brain is likely still sufficient to impact neural development.
However, in the case of (Pot1a;Atr)Nes-cre, proliferation was not recovered, most likely because at this developmental stage Atr loss itself begins to impact the developing forebrain (Lee et al., 2001, 2012b). Elevated apoptosis in the Pot1aNes-cre;Atm−/− and (Pot1a;Atr)Nes-cre E15.5 forebrain may also indicate some tissue-specific differences compared with the cerebellum. Therefore, these data indicate that, in addition to Atm, Atr is also activated by telomere dysfunction after Pot1a deficiency in the embryonic nervous system, and Atm-dependent apoptosis is a major physiologic outcome resulting from shelterin inactivation during neurogenesis.
Atm is activated via Atr in Pot1a-deficient embryonic brains
Given that inactivation of either Atm or Atr can block apoptosis after Pot1a loss (Fig. 6), we assessed DNA damage signaling hierarchy in embryonic tissue from various Pot1a-null genotypes. We initially examined phosphorylation on Atm serine 1987 (equivalent to serine 1981 in humans), as a marker for Atm activation (Bakkenist and Kastan, 2003). Atm serine 1987 phosphorylation occurred in Pot1aNes-cre neural tissue, but not in (Pot1a;Atr)Nes-cre-deficient tissue (Fig. 7A, red asterisk), implying that Atr is required for Atm phosphorylation. In agreement with this, Atm-dependent activation of Chk2 was also observed in Pot1aNes-cre but not in (Pot1a;Atr)Nes-cre embryonic brains. However, after ionizing radiation (IR)-induced DNA damage in (Pot1a;Atr)Nes-cre embryonic brain tissue, the Atm substrates Chk2 and Kap1, readily underwent modification by Atm as determined by reduced mobility of endogenous Chk2 and phosphorylation of Kap1 on Ser824 (Fig. 7A). Collectively, these data indicate that after Pot1a loss Atr subsequently activates Atm, which facilitates DNA damage-induced apoptosis.
Figure 7.

Atr is required to activate Atm-dependent DNA damage signaling after Pot1a loss. A, Western blot analysis of DNA damage signaling in isolated E13.5 embryonic brain shows that Atm (Ser1987) phosphorylation and Chk2 activation (red asterisk) occur after Pot1a loss in an Atr-dependent manner. Chk2 activation is shown as slower mobility of endogenous Chk2. IR was used to show that Pot1a-deficient tissue are competent for DNA damage signaling. Kap1(Ser824), Chk1(Ser317) phosphorylation and Chk2 activation (reduced mobility; red asterisk) are increased after radiation in an Atm-dependent manner. Phospho-serine specific antibodies are listed. Actin immunostaining shows relative protein loading. Numerical values reflect the ratio of endogenous Atm, Chk1, and Kap1 to phosphorylated levels. Western blots were done two independent times using pooled embryonic tissue. B, Phosphorylation of the RPA2 subunit in E13.5 embryonic brain shows endogenous phosphorylation at RPA2 at ser33 after Pot1a loss in an Atr-dependent manner (asterisks). Phosphorylation of other RPA2 serine and threonine residues is increased 2 h after 10 Gy IR. C, Immunocytochemistry with RPA2-Thr21 of astrocytes shows increased Atm-dependent phosphorylation of RPA2 after Pot1a loss, which is increased after IR (white arrows). Quantification of multiple astrocyte lines shows the relative RPA32/pT21 levels after Pot1a loss. ***p < 0.0001. **p < 0.001. *p < 0.05.
It has been suggested that single-stranded regions of telomeres that form after Pot1a loss are stabilized by RPA, followed by interaction with the Rad9-Rad1-Hus1 (9–1-1) complex and TopBP1 to activate ATR (Choi et al., 2010; Gong and de Lange, 2010; Flynn et al., 2011, 2012). RPA is a heterotrimeric protein complex composed of RPA1, RPA2, and RPA3, and RPA2 undergoes phosphorylation upon DNA damage (Wold, 1997; Vassin et al., 2009). To further examine Atr signaling in the Pot1aNes-cre nervous system, we measured phosphorylation of RPA2 at serine 4/8, threonine 21, and serine 33, which can be phosphorylated by ATM, DNA-PKCS, or ATR depending on the type of DNA damage (Oakley and Patrick, 2010). We found increased phosphorylation of RPA2 at serine 33 in Pot1aNes-cre and Pot1aNes-cre;Atm−/− embryonic brain compared with (Pot1a;Atr)Nes-cre tissue, indicating that Pot1a loss leads to specific Atr-dependent RPA phosphorylation (Fig. 7B). The reliance upon Atr to phosphorylate RPA2 serine 33 is consistent with reports of this residue as an ATR target (Anantha et al., 2008; Oakley and Patrick, 2010). Serine 33 phosphorylation of RPA2 by Atr during late S and G2 has been previously reported and may function to activate an S-phase checkpoint (Olson et al., 2006; Anantha et al., 2007; Vassin et al., 2009). Whereas RPA2 serine 33 was selectively phosphorylated after Pot1a inactivation, other RPA2 phosphorylation at serine 4/8 or threonine 21 was readily identified after IR, confirming RPA T21 being Atm-dependent and serine 4/8 and seine 33 requiring Atr after IR (Fig. 7B). Thus, Pot1a loss activates Atr signaling, but in a more selective manner than DNA strand breaks from IR. However, this signaling is distinct to the Atr to Atm axis above (Fig. 7A).
Given the increased DNA damage after Pot1a loss in astrocytes (Fig. 2), we also examined these cells for RPA2 phosphorylation. In contrast to embryonic brain, we found that Rpa2 phosphorylation at threonine 21 was readily detected in Pot1a-null astrocytes and these nuclear foci and were increased after radiation (Fig. 7C). These in vitro versus in vivo differences in RPA phosphorylation may reflect lower endogenous Rpa2(pThr21) levels rather than a difference between astrocytes and neurons because Pot1a deficiency effects neural progenitors that give rise to both lineages. In vitro neural cell cultures may show differences to tissue because of the added DNA damage from oxidative stress that occurs under tissue culture conditions (Parrinello et al., 2003). Radiation of E13.5 embryos shows a similar Atm dependency of Thr21 phosphorylation, although this is not affected by Pot1a status.
Discussion
Here we show a critical role for telomere end-protection by Pot1a during neurogenesis. In the setting of telomere dysfunction in the nervous system, our data show that apoptosis is a major outcome after Pot1a inactivation and that this involves both Atm and Atr. Notably, our data contrast previous studies demonstrating that telomere dysfunction due to POT1 inactivation triggers an ATR-dependent, but ATM-independent, signaling cascade to facilitate cell cycle arrest and senescence in vitro (Denchi and de Lange, 2007). However, Atm activation after Pot1a disruption has been previously shown in fibroblasts (Wu et al., 2006). In the nervous system, we have found that Atm activation, although involving Atr, is nonetheless centrally responsible for disruption of neurogenesis after Pot1a inactivation. Therefore, Pot1 loss contrasts the situation involving ATM and ATR signaling after DNA DSBs or replication-associated damage (see summary in Fig. 8).
Figure 8.

DNA damage signaling pathways after different genotoxic stress. A, ATM and ATR respond to different types of DNA damage, where DNA DSBs activate ATM via the MRE11-RAD50-NBS1 (MRN) sensor to phosphorylate Chk2 and p53 to activate a signaling cascade leading to apoptosis or cell cycle arrest. In contrast, ATR is activated by RPA-coated single-stranded DNA in a TOPBP1-dependent manner in response to replication stress. Activated ATR then activates p53 or Chk1 to facilitate cell cycle arrest or apoptosis. B, Proposed DNA damage signaling based on Pot1a inactivation in neural progenitors. In contrast to DNA strand breaks, telomere disruption in the form of inactivation of the shelterin component Pot1a generates a single-stranded lesion that activates ATR, which in turn activates ATM-dependent apoptosis. After Pot1a activation, ATR also phosphorylates other potential targets involved in DNA damage signaling, DNA replication, and cellular proliferation (dashed line). Inset, A functional unit of shelterin containing at least five distinct proteins. Many copies of shelterin coat the telomeric region of DNA.
Our data also illustrate that the physiologic signaling response to Pot1a inactivation reveals additional levels of complexity to the functional repertoire of ATM during neurogenesis. For instance, although Atm is required for cerebellar interneuron elimination after Pot1a loss, Atm status does not affect loss of these cells when the base excision repair factor, Xrcc1 is disrupted (Lee et al., 2009). TRF2, another shelterin factor, which suppresses ATM signaling (Karlseder et al., 1999; Guo et al., 2007; de Lange, 2009), has also been reported to be a key factor for neuronal survival during neurogenesis (Cheng et al., 2007; Zhang et al., 2007). These findings implicate a broad role for Atm during telomere maintenance in the nervous system.
Direct connections between Atm and Atr signaling pathways have been reported, although as distinct from our data, most of these other studies showed that ATM activated ATR. For example, in response to DNA DSBs, there is an ATM-dependent requirement for ATR signaling to the checkpoint kinase Chk1, whereby in the absence of ATM there is a failure to activate a Chk1-dependent cell cycle checkpoint after IR (Adams et al., 2006; Cuadrado et al., 2006; Jazayeri et al., 2006; Myers and Cortez, 2006). Additionally, ATM and ATR act cooperatively with Mre11 during the initiation and regulation of replication, and function to halt these processes when DNA damage is detected (Shechter et al., 2004; Adams et al., 2006; Trenz et al., 2006). Our data indicate a cooperative strategy for these two DNA damage-sensing kinases, whereby Pot1a loss promotes Atr activation, which in turn is necessary for Atm signaling. Initial activation of Atr occurs because Pot1a loss allows RPA binding at uncapped telomeres (Gong and de Lange, 2010; Flynn et al., 2012), and activated Atr may then directly phosphorylate Atm. Direct phosphorylation of ATM by ATR has been found in lymphoid cells after UV damage or replication fork stalling (Stiff et al., 2006). In this scenario, ATM phosphorylation by ATR did not involve ATM autophosphorylation, occurred in the presence of an ATM inhibitor and was not reliant upon the MRN complex (Stiff et al., 2006). Therefore, telomere dysfunction may be an additional setting in which ATR phosphorylation of ATM is an important signaling response to specific types of DNA damage.
In conclusion, our study identifies a requirement for Pot1a during neurogenesis and reveals a central role for Atm signal transduction after Pot1a inactivation, involving crosstalk between Atr and Atm. These two kinases cooperatively signal DNA damage after telomere dysfunction to efficiently eliminate neuroprogenitors with compromised telomeres, thereby maintaining genome integrity in the nervous system. Neural progenitors have a propensity to engage apoptosis as a means of eliminating genomically compromised cells because they are readily replaced during neurogenesis (McKinnon, 2009, 2013). The long lifespan of mature neurons likely mandates very stringent monitoring of genome integrity at stages where cell replacement is an option.
Footnotes
P.J.M. was supported by the National Institutes of Health Grants NS-37956 and CA-96832, the Cancer Center Support Grant P30 CA21765, and the American Lebanese and Syrian Associated Charities of St Jude Children's Research Hospital. E.J.B. was supported by the National Institutes of Health Grant AG-027376 and an American Recovery and Reinvestment Act supplement. S.C. was supported by the National Cancer Institute Grant CA-129037. Y.L. was supported by the Scientific Research Center (SRC) program 2011-0030833. We thank the Hartwell Center for biotechnological support, the cytogenetics core for chromosome analysis, the ARC for animal husbandry, and Jingfeng Zhao for technical support during this project.
The authors declare no competing financial interests.
References
- Adams KE, Medhurst AL, Dart DA, Lakin ND. Recruitment of ATR to sites of ionising radiation-induced DNA damage requires ATM and components of the MRN protein complex. Oncogene. 2006;25:3894–3904. doi: 10.1038/sj.onc.1209426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anantha RW, Vassin VM, Borowiec JA. Sequential and synergistic modification of human RPA stimulates chromosomal DNA repair. J Biol Chem. 2007;282:35910–35923. doi: 10.1074/jbc.M704645200. [DOI] [PubMed] [Google Scholar]
- Anantha RW, Sokolova E, Borowiec JA. RPA phosphorylation facilitates mitotic exit in response to mitotic DNA damage. Proc Natl Acad Sci U S A. 2008;105:12903–12908. doi: 10.1073/pnas.0803001105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bakkenist CJ, Kastan MB. DNA damage activates ATM through intermolecular autophosphorylation and dimer dissociation. Nature. 2003;421:499–506. doi: 10.1038/nature01368. [DOI] [PubMed] [Google Scholar]
- Chan SS, Chang S. Defending the end zone: studying the players involved in protecting chromosome ends. FEBS Lett. 2010;584:3773–3778. doi: 10.1016/j.febslet.2010.06.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheng A, Shin-ya K, Wan R, Tang SC, Miura T, Tang H, Khatri R, Gleichman M, Ouyang X, Liu D, Park HR, Chiang JY, Mattson MP. Telomere protection mechanisms change during neurogenesis and neuronal maturation: newly generated neurons are hypersensitive to telomere and DNA damage. J Neurosci. 2007;27:3722–3733. doi: 10.1523/JNEUROSCI.0590-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Choi JH, Lindsey-Boltz LA, Kemp M, Mason AC, Wold MS, Sancar A. Reconstitution of RPA-covered single-stranded DNA-activated ATR-Chk1 signaling. Proc Natl Acad Sci U S A. 2010;107:13660–13665. doi: 10.1073/pnas.1007856107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cimprich KA, Cortez D. ATR: an essential regulator of genome integrity. Nat Rev Mol Cell Biol. 2008;9:616–627. doi: 10.1038/nrm2450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cuadrado M, Martinez-Pastor B, Murga M, Toledo LI, Gutierrez-Martinez P, Lopez E, Fernandez-Capetillo O. ATM regulates ATR chromatin loading in response to DNA double-strand breaks. J Exp Med. 2006;203:297–303. doi: 10.1084/jem.20051923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Lange T. How telomeres solve the end-protection problem. Science. 2009;326:948–952. doi: 10.1126/science.1170633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Lange T. How shelterin solves the telomere end-protection problem. Cold Spring Harb Symp Quant Biol. 2010;75:167–177. doi: 10.1101/sqb.2010.75.017. [DOI] [PubMed] [Google Scholar]
- Denchi EL, de Lange T. Protection of telomeres through independent control of ATM and ATR by TRF2 and POT1. Nature. 2007;448:1068–1071. doi: 10.1038/nature06065. [DOI] [PubMed] [Google Scholar]
- Doksani Y, Wu JY, de Lange T, Zhuang X. Super-resolution fluorescence imaging of telomeres reveals TRF2-dependent T-loop formation. Cell. 2013;155:345–356. doi: 10.1016/j.cell.2013.09.048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Flynn RL, Centore RC, O'Sullivan RJ, Rai R, Tse A, Songyang Z, Chang S, Karlseder J, Zou L. TERRA and hnRNPA1 orchestrate an RPA-to-POT1 switch on telomeric single-stranded DNA. Nature. 2011;471:532–536. doi: 10.1038/nature09772. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Flynn RL, Chang S, Zou L. RPA and POT1: friends or foes at telomeres? Cell Cycle. 2012;11:652–657. doi: 10.4161/cc.11.4.19061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frappart PO, Lee Y, Lamont J, McKinnon PJ. BRCA2 is required for neurogenesis and suppression of medulloblastoma. EMBO J. 2007;26:2732–2742. doi: 10.1038/sj.emboj.7601703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gong Y, de Lange T. A Shld1-controlled POT1a provides support for repression of ATR signaling at telomeres through RPA exclusion. Mol Cell. 2010;40:377–387. doi: 10.1016/j.molcel.2010.10.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guo X, Deng Y, Lin Y, Cosme-Blanco W, Chan S, He H, Yuan G, Brown EJ, Chang S. Dysfunctional telomeres activate an ATM-ATR-dependent DNA damage response to suppress tumorigenesis. EMBO J. 2007;26:4709–4719. doi: 10.1038/sj.emboj.7601893. [DOI] [PMC free article] [PubMed] [Google Scholar]
- He H, Multani AS, Cosme-Blanco W, Tahara H, Ma J, Pathak S, Deng Y, Chang S. POT1b protects telomeres from end-to-end chromosomal fusions and aberrant homologous recombination. EMBO J. 2006;25:5180–5190. doi: 10.1038/sj.emboj.7601294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hockemeyer D, Daniels JP, Takai H, de Lange T. Recent expansion of the telomeric complex in rodents: two distinct POT1 proteins protect mouse telomeres. Cell. 2006;126:63–77. doi: 10.1016/j.cell.2006.04.044. [DOI] [PubMed] [Google Scholar]
- Jazayeri A, Falck J, Lukas C, Bartek J, Smith GC, Lukas J, Jackson SP. ATM- and cell cycle-dependent regulation of ATR in response to DNA double-strand breaks. Nat Cell Biol. 2006;8:37–45. doi: 10.1038/ncb1337. [DOI] [PubMed] [Google Scholar]
- Karlseder J, Broccoli D, Dai Y, Hardy S, de Lange T. p53- and ATM-dependent apoptosis induced by telomeres lacking TRF2. Science. 1999;283:1321–1325. doi: 10.1126/science.283.5406.1321. [DOI] [PubMed] [Google Scholar]
- Lavin MF. Ataxia-telangiectasia: from a rare disorder to a paradigm for cell signalling and cancer. Nat Rev Mol Cell Biol. 2008;9:759–769. doi: 10.1038/nrm2514. [DOI] [PubMed] [Google Scholar]
- Lee Y, McKinnon PJ. DNA ligase IV suppresses medulloblastoma formation. Cancer Res. 2002;62:6395–6399. [PubMed] [Google Scholar]
- Lee Y, Chong MJ, McKinnon PJ. Ataxia telangiectasia mutated-dependent apoptosis after genotoxic stress in the developing nervous system is determined by cellular differentiation status. J Neurosci. 2001;21:6687–6693. doi: 10.1523/JNEUROSCI.21-17-06687.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee Y, Katyal S, Li Y, El-Khamisy SF, Russell HR, Caldecott KW, McKinnon PJ. The genesis of cerebellar interneurons and the prevention of neural DNA damage require XRCC1. Nat Neurosci. 2009;12:973–980. doi: 10.1038/nn.2375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee Y, Katyal S, Downing SM, Zhao J, Russell HR, McKinnon PJ. Neurogenesis requires TopBP1 to prevent catastrophic replicative DNA damage in early progenitors. Nat Neurosci. 2012a;15:819–826. doi: 10.1038/nn.3097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee Y, Shull ER, Frappart PO, Katyal S, Enriquez-Rios V, Zhao J, Russell HR, Brown EJ, McKinnon PJ. ATR maintains select progenitors during nervous system development. EMBO J. 2012b;31:1177–1189. doi: 10.1038/emboj.2011.493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McKinnon PJ. DNA repair deficiency and neurological disease. Nat Rev Neurosci. 2009;10:100–112. doi: 10.1038/nrn2559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McKinnon PJ. ATM and the molecular pathogenesis of ataxia telangiectasia. Annu Rev Pathol. 2012;7:303–321. doi: 10.1146/annurev-pathol-011811-132509. [DOI] [PubMed] [Google Scholar]
- McKinnon PJ. Maintaining genome stability in the nervous system. Nat Neurosci. 2013;16:1523–1529. doi: 10.1038/nn.3537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Myers JS, Cortez D. Rapid activation of ATR by ionizing radiation requires ATM and Mre11. J Biol Chem. 2006;281:9346–9350. doi: 10.1074/jbc.M513265200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oakley GG, Patrick SM. Replication protein A: directing traffic at the intersection of replication and repair. Front Biosci. 2010;15:883–900. doi: 10.2741/3652. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O'Driscoll M, Ruiz-Perez VL, Woods CG, Jeggo PA, Goodship JA. A splicing mutation affecting expression of ataxia-telangiectasia and Rad3-related protein (ATR) results in Seckel syndrome. Nat Genet. 2003;33:497–501. doi: 10.1038/ng1129. [DOI] [PubMed] [Google Scholar]
- Olson E, Nievera CJ, Klimovich V, Fanning E, Wu X. RPA2 is a direct downstream target for ATR to regulate the S-phase checkpoint. J Biol Chem. 2006;281:39517–39533. doi: 10.1074/jbc.M605121200. [DOI] [PubMed] [Google Scholar]
- Orii KE, Lee Y, Kondo N, McKinnon PJ. Selective utilization of nonhomologous end-joining and homologous recombination DNA repair pathways during nervous system development. Proc Natl Acad Sci U S A. 2006;103:10017–10022. doi: 10.1073/pnas.0602436103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parrinello S, Samper E, Krtolica A, Goldstein J, Melov S, Campisi J. Oxygen sensitivity severely limits the replicative lifespan of murine fibroblasts. Nat Cell Biol. 2003;5:741–747. doi: 10.1038/ncb1024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sfeir A, de Lange T. Removal of shelterin reveals the telomere end-protection problem. Science. 2012;336:593–597. doi: 10.1126/science.1218498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shechter D, Costanzo V, Gautier J. ATR and ATM regulate the timing of DNA replication origin firing. Nat Cell Biol. 2004;6:648–655. doi: 10.1038/ncb1145. [DOI] [PubMed] [Google Scholar]
- Shiloh Y, Ziv Y. The ATM protein kinase: regulating the cellular response to genotoxic stress, and more. Nat Rev Mol Cell Biol. 2013;14:197–210. doi: 10.1038/nrm3546. [DOI] [PubMed] [Google Scholar]
- Shull ER, Lee Y, Nakane H, Stracker TH, Zhao J, Russell HR, Petrini JH, McKinnon PJ. Differential DNA damage signaling accounts for distinct neural apoptotic responses in ATLD and NBS. Genes Dev. 2009;23:171–180. doi: 10.1101/gad.1746609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stiff T, Walker SA, Cerosaletti K, Goodarzi AA, Petermann E, Concannon P, O'Driscoll M, Jeggo PA. ATR-dependent phosphorylation and activation of ATM in response to UV treatment or replication fork stalling. EMBO J. 2006;25:5775–5782. doi: 10.1038/sj.emboj.7601446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trenz K, Smith E, Smith S, Costanzo V. ATM and ATR promote Mre11 dependent restart of collapsed replication forks and prevent accumulation of DNA breaks. EMBO J. 2006;25:1764–1774. doi: 10.1038/sj.emboj.7601045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vassin VM, Anantha RW, Sokolova E, Kanner S, Borowiec JA. Human RPA phosphorylation by ATR stimulates DNA synthesis and prevents ssDNA accumulation during DNA-replication stress. J Cell Sci. 2009;122:4070–4080. doi: 10.1242/jcs.053702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wold MS. Replication protein A: a heterotrimeric, single-stranded DNA-binding protein required for eukaryotic DNA metabolism. Annu Rev Biochem. 1997;66:61–92. doi: 10.1146/annurev.biochem.66.1.61. [DOI] [PubMed] [Google Scholar]
- Wu L, Multani AS, He H, Cosme-Blanco W, Deng Y, Deng JM, Bachilo O, Pathak S, Tahara H, Bailey SM, Deng Y, Behringer RR, Chang S. Pot1 deficiency initiates DNA damage checkpoint activation and aberrant homologous recombination at telomeres. Cell. 2006;126:49–62. doi: 10.1016/j.cell.2006.05.037. [DOI] [PubMed] [Google Scholar]
- Zhang P, Dilley C, Mattson MP. DNA damage responses in neural cells: focus on the telomere. Neuroscience. 2007;145:1439–1448. doi: 10.1016/j.neuroscience.2006.11.052. [DOI] [PMC free article] [PubMed] [Google Scholar]