

Abstract
Research on myelination has focused on identifying molecules capable of inducing oligodendrocyte (OL) differentiation in an effort to develop strategies that promote functional myelin regeneration in demyelinating disorders. Here, we show that transforming growth factor β (TGFβ) signaling is crucial for allowing oligodendrocyte progenitor (OP) cell cycle withdrawal, and therefore, for oligodendrogenesis and postnatal CNS myelination. Enhanced oligodendrogenesis and subcortical white matter (SCWM) myelination was detected after TGFβ gain of function, while TGFβ receptor II (TGFβ-RII) deletion in OPs prevents their development into mature myelinating OLs, leading to SCWM hypomyelination in mice. TGFβ signaling modulates OP cell cycle withdrawal and differentiation through the transcriptional modulation of c-myc and p21 gene expression, mediated by the interaction of SMAD3/4 with Sp1 and FoxO1 transcription factors. Our study is the first to demonstrate an autonomous and crucial role of TGFβ signaling in OL development and CNS myelination, and may provide new avenues in the treatment of demyelinating diseases.
Keywords: myelination, oligodendrocyte, oligodendrocyte progenitor, proliferation, SMAD, TGFβ
Introduction
The transforming growth factor β (TGFβ) signaling pathway is a promising therapeutic target for the treatment of multiple sclerosis (MS) and other autoimmune diseases because of its capacity to modulate immune cell functions (Li and Flavell, 2008; Li et al., 2006; Mirshafiey and Mohsenzadegan, 2009). Previous studies have reported that systemic administration of TGFβ1 in mice attenuates experimental autoimmune encephalomyelitis (EAE), a mouse model of human MS (Kuruvilla et al., 1991; Racke et al., 1991; Johns and Sriram, 1993), suggesting a protective role for TGFβ in EAE. However, the specific roles and cellular targets of TGFβ in the CNS other than those in the immune system, and whether this signaling pathway directly controls oligodendrocyte (OL) development and myelination during physiological or pathophysiological conditions remains unclear.
TGFβ signaling plays a key role in a number of biological processes, including development, body patterning, carcinogenesis, and immune responses (Massagué et al., 2000). Activation of TGFβ signaling exerts a potent anti-proliferative action on multiple cell types, such as tumoral (Siegel and Massagué, 2003), immune (Li et al., 2008), epithelial, astroglial (Vergeli et al., 1995), and neural progenitor cells (García-Campmany and Martí, 2007; Misumi et al., 2008), limiting cell proliferation and inducing differentiation through a program of anti-mitotic gene responses (Siegel and Massagué, 2003). Cell cycle arrest in response to SMAD-mediated TGFβ signaling activation occurs at the “restriction point” in late G1 phase by transcriptional activation of cyclin-dependent kinase inhibitors (p15Ink4b, p21Cip1, and p27kip1; García-Campmany and Martí, 2007; Misumi et al., 2008) and repression of c-myc and inhibitors of differentiation 1 and 2 (Id1, Id2; Massagué and Gomis, 2006). Thus, loss of TGFβ signaling prevents cell differentiation and TGFβ signaling activation or overexpression induces cell cycle exit and differentiation in several cell types.
Indeed, in the context of oligodendrogenesis and myelination, in vitro studies have shown that O-2A progenitor cells express TGFβ1 and that TGFβ signaling activation exerts an anti-mitogenic effect countering PDGFRα signaling, in turn promoting cell cycle arrest (McKinnon et al., 1993). Moreover, Activin-A, a member of the TGFβ superfamily, has been proposed as one of the cytokines secreted by microglial cells that plays a role in OL regeneration and remyelination (Miron et al., 2013). These studies suggest that TGFβ signaling may be an important cue for OL development, myelination, and remyelination. However, an autonomous role for TGFβ signaling in OL development and CNS myelination and the molecular mechanisms meditated by this pathway are still unknown.
Here, by using loss-of-function and gain-of-function approaches, and in vivo and in vitro analysis, we provide the first evidence that TGFβ signaling exerts an essential and autonomous role in OL development during the critical periods of CNS myelination. We show that TGFβ signaling, by modulating c-myc and p21 expression through the cooperation of SMAD3/4 with FoxO1 and Sp1, exerts essential functions in the control of OP cell cycle exit and OL differentiation. Understanding the role of TGFβ signaling in oligodendrogenesis and CNS myelination will also aid in the design of strategies that promote myelin repair.
Materials and Methods
Animals.
All animal procedures were performed according to the Institutional Animal Care and Use Committee of DLAR, SUNY Stony Brook School of Medicine, and the National Institutes of Health (NIH) Guide for the Care and Use of Laboratory Animals. The generation and characterization of the CNP–EGFP (RRID:MGI_MGI:3850679; Yuan et al., 2002), NG2-dsRED (RRID:IMSR_JAX:008241; Zhu et al., 2008), NG2-Cre (RRID:IMSR_JAX:008533; Rivers et al., 2008), and PDGFRα creERT2 (RRID:IMSR_JAX:018280; Rivers et al., 2008) mouse lines were previously described. The TGFβ-RIIfl/fl (01XNs–B6129-S6-Tgfbr2tm1HLm; NCIMR:01XN5; Chytil et al., 2002) mouse line was provided by NCI Mouse Repository. The recombination efficiency in the PDGFRα-Cretm:TGFβ-RIIfl/fl:YFP and the corresponding w/w mice was calculated by injecting tamoxifen (20 mg/kg) into P6 male and female pups for 3 consecutive days (24 h apart), and mice were killed 24 h after the last tamoxifen injection. We performed immunofluorescence double labeling on brain sections with anti-YFP and anti-NG2 antibodies. The percentage of YFP+NG2+/total NG2+ cells was used to determine the recombination efficiency. Our data revealed that ∼85 ± 4% of the NG2 population was YFP+ in the subcortical white matter (SCWM). With this tamoxifen dosage schedule, we obtained the highest recombination efficiency with no lethality. TGFβ signaling pharmacological activation or inhibition was performed by intraperitoneal administration of TGFβ1 (100 ng/kg; BioLegend) or SB-431542 (5 mg/kg; Cayman), respectively, twice a day for 1 day or 3 consecutive days. BrdU administration in vivo was performed as described previously (Aguirre et al., 2007, 2010). Cell proliferation was assessed by injecting BrdU at 100 mg/kg into male mice at 2 h before the end of the experiment. When TGFβ1 was used to analyze cell cycle exit experiments (Ki67/BrdU ratios), BrdU was injected first, and 3 h later, male mice received TGFβ1 or vehicle administration (100 ng/kg, twice a day for 2 d) and tissue was analyzed 48 h later (Palazuelos et al., 2012).
Immunohistochemistry.
For characterization of OL lineage cells brain tissue was processed as previously described (Aguirre et al., 2007). In brief, 30-μm-thick brain sections were blocked with 5% goat serum and then incubated with the indicated primary antibodies (overnight at 4°C). The following day, sections were washed and fluorescent secondary antibodies were used to reveal the antigens in question in situ. The primary antibodies used are as follows: anti-p21 (AB_396415) and anti-p27 (AB_397636; BD Biosciences); anti-GST-π (AB_591792; MBL), anti-NG2 (AB_91789; Millipore), anti-BrdU (AB_609568; Accurate), anti-MBP (AB_510039), and anti-CNP (AB_510037; Covance); anti-MOG (AB_2282105; Epitomics), anti-TGFβ-RII (AB_1660532; Novus Biologicals), anti-Cdk2 (AB_631216), anti-PDGFRα (AB_631064), anti-c-Myc (AB_627268), anti-SMAD3 (AB_2255105), and anti-SMAD4 (AB_2255136;Santa Cruz Biotechnology); anti-Ki67 (AB_442102, Novocastra), anti-CC1 (AB_213434; Calbiochem), anti-YFP (AB_1196615), anti-pSMAD2/3 (AB_2193178), anti-pSMAD3 (AB_2193207), and anti-FoxO1 (AB_10831514; Cell Signaling Technology); anti-GFAP (AB_880201), anti-Vimentin (AB_778826), and anti-Sp1 (AB_1566756; Abcam).
The appropriate mouse, rat, and rabbit highly cross-adsorbed Alexa Fluor 488, Alexa Fluor 547, and Alexa Fluor 647 secondary antibodies (Invitrogen) were used.
Immunocytochemistry.
For immunocytochemistry, cells were plated (10,000 cells) onto poly-l-lysine-coated (Sigma; 50 mg/ml in sterile water) glass coverslips to test both proliferation and cell differentiation. At the conclusion of the respective experiments, cells were fixed with 4% PFA and then incubated with 20% goat serum for 10 min at room temperature. The coverslips were then processed with the indicated primary antibodies followed by secondary antibody incubation.
Confocal microscopy.
A confocal laser-scanning microscope TCS-SP5 (Leica DMI6000 B instrument) was used for image localization of FITC (488 nm laser line excitation; 522/35 emission filter), Cy3 (570 nm excitation; 605/32 emission filter), and Cy5 (647 excitation; 680/32 emission filter). Optical sections (z = 0.5 μm) of confocal epifluorescence images were sequentially acquired using a 63× objective (NA 1.40), with LAS AF software. NIH ImageJ (RRID:nif-0000-30467) software was then used to merge images. Merged images were processed in Photoshop Cs4 software with minimal manipulation of contrast. At least four different brains for each strain and each experimental condition were analyzed and counted. Cell counting was performed blindly, and tissue sections were matched across samples. For SCWM analysis, a minimum of six correlative slices from a 1-in-10 series located between +1 and −1 mm from bregma were analyzed. All cell quantification data were obtained by cell counting using ImageJ, and data are presented as the mean cell number per cubic millimeter (×1000). Statistical analysis was performed by an unpaired t test.
Electron microscopy.
Fifteen-day-old NG2-cre::TGFb-RII w/w and fl/fl mice were processed for electron microscopy analysis as previously described (Aguirre et al., 2007; Relucio et al., 2012). Mice were perfused intracardially with 2% PFA/2.5% glutaraldehyde in 0.1 m PBS followed by brain fixation overnight. SCWM tissue was sectioned along the sagittal plane on a Leica VT-1000 Vibratome at 50 μm. Free-floating sections were placed in 2% osmium tetroxide in 0.1 m phosphate and ethyl alcohols and vacuum infiltrated in Durcupan ACM embedding agent (Electron Microscopy Sciences). Ultrathin sections (70–80 nm) were obtained using a Reichert–Jung 701704 Ultracut E ultramicrotome and counterstained with uranyl acetate and lead citrate. Samples were analyzed with a Tecnai Spirit Bio-Twin G2 transmission electron microscope (FEI) coupled to an AMT XR-60 CCD Digital Camera System (Advanced Microscopy Techniques). Images were analyzed using Adobe Photoshop and ImageJ (NIH).
SCWM dissection.
SCWM tissue was microdissected from 200-μm-thick coronal sections of P4–P30 mouse brains. The SCWM was dissected out with fine forceps to avoid tissue contamination from surrounding areas. SCWM tissue was processed for RNA and protein extraction, but also for FACS sorting and cell cultures.
Western blots and immunoprecipitation.
SCWM tissue from wild-type and transgenic mice was microdissected from 200-μm-thick coronal sections, and SCWM tissue was then processed for whole-protein extraction using RIPA lysis buffer (Santa Cruz Biotechnology) with inhibitors–PMSF in DMSO, protease inhibitors, and sodium orthovanadate as recommended by the manufacturer. Protein samples (15 μg) were separated on 10% acrylamide (Bio-Rad) gels and transferred to polyvinylidene difluoride (PVDF) membranes (Millipore). Primary antibodies were used for detection of the indicated proteins on PVDF membranes. Primary antibodies were used in combination with secondary horseradish peroxidase-conjugate antibodies to detect the protein in question using an enhanced chemiluminescence substrate mixture (ECL Plus; GE Healthcare; Santa Cruz Biotechnology; 1:5000). Protein levels in question were quantified in arbitrary units (a.u.) after actin normalization. For immunoprecipitation experiments, SCWM tissue from wild-type and transgenic mice was processed with RIPA buffer to obtain protein extracts (Aguirre et al., 2007). Aliquots of 200 μg protein extract with antibodies against SMAD3 or SMAD4 (Santa Cruz Biotechnology; 2 μl) in the presence of 10 μl of protein A Agarose (Santa Cruz Biotechnology) were incubated overnight at 4°C. SMAD3/4 immunocomplexes were enriched by 12k rpm centrifugation for 3 min at 4°C and washed twice with cold RIPA buffer. SMAD complexes were resolved on a 10% acrylamide gel and detected as above using the appropriate antibodies.
FACS sorting.
FACS purification of CNP-EGFP+ cells was performed as previously described (Aguirre et al., 2010). Cell suspensions were analyzed for light forward and side scatter using a FACStar plus instrument (Becton Dickinson). Purified CNP-EGFP+ cells were used for cell culture analysis as described above. For FACS sorting of TGFβ-RIIfl/fl OP cells, wt and fl/fl mice received tamoxifen administration at P6 for 2 consecutive days, and 24 h later SCWM was dissected out and processed as above (Aguirre et al., 2010). Purified PDGFRα-Cre::YFP+:NG2+ cells were used for cell culture or RT-PCR analysis.
Cell cultures.
OP cell cultures were obtained from FACS-purified CNP-EGFP+ cells. Cells were dissociated and seeded (20,000 cells/well) on poly-l-lysine-coated coverslips in 24-well plates. Cells were cultured at different time points as indicated for each experiment. OPs were maintained in Neurobasal medium with N2, B27 (Supplements; Invitrogen), and 2% FBS. For proliferation conditions the growth factors PDGF and bFGF2 were added daily to cultures for 4 d in vitro (DIV), and for differentiation conditions, growth factors were removed and T3 was added for 3 DIV. For TGFβ receptor pharmacological activation studies, TGFβ1 was added to the medium at 5 ng/ml, and cells were fixed at the indicated time points using 4% PFA. In vitro cell proliferation assays were performed by adding BrdU at 200 ng/ml for 2 h before the end of the experiment. After culture, coverslips were processed for immunocytochemistry.
For gene promoter activity assays, CNP-EGFP+ FACS-sorted cells were transiently transfected with 0.5 μg of pGL-p21UTR and pRL c-myc3′UTR to analyze the promoter activity of p21 and c-myc, respectively, using the Renilla luciferase system (Aguirre et al., 2010). Transcriptional promoter-driven luciferase activity was performed by using the Dual-Luciferase Reporter Promega assay system and Renilla-derived luciferase activity as an internal transfection control in a TD-20/20 luminometer (Turner Biosystems; Aguirre et al., 2010). For forced p21 overexpression (gain of function), CNP-EGFP+ FACS-sorted cells were transiently transfected with 0.5 μg of pCMV-mp21CIP1(Addgene) and cells were processed for cell proliferation and differentiation analysis at 24 and 72 h, respectively.
Real-time qPCR.
SCWM tissue or FACS-purified cells were processed with TRIzol (Invitrogen) to obtain total RNA. Total RNA (1 μg) from each sample was reverse transcribed using the SuperScript First-Strand cDNA Synthesis kit (Invitrogen). Mouse gene-specific primers were obtained from Integrated DNA Technologies. Semiquantitative PCR amplifications were performed using the following conditions: 93°C for 1 min, two rounds (30 s at 59°C, 1 min at 72°C, and 30 s at 93°C), two rounds (30 s at 57°C, 1 min at 72°C, and 30 s at 93°C), and 25 cycles (30 s at 55°C, 1 min at 72°C, and 30 s at 93°C). Real-time qPCR was performed with the SYBR Green system (Invitrogen). Amplifications were run in a LightCycler 480 Real-time PCR system (Roche), and the values obtained were adjusted using 18S and β-actin RNA levels as references. The sequences of the primers used are as follows: TGF-β1-F:GTGCCCGAACCCCCATTGCT; TGF-β1-R:CGTTTGGGGCTGATCCCGTTGAT;TGF-β2-F:GTTGGGAACGCGTTGCATTT;TGF-β2-R:GTTGGGAACGCGTTGCATTT;TGF-β3-F:GGCCAGTTCATTGTGCCCGCC;TGF-β3-R:CGGTGATGACCCACGTCCCC;TGFβ-RI-F:CAGCTCCTCATCGTGTTGG;TGFβ-RI-R CAGAGGTGGCAGAAACACTG;TGFβ-RII-F:TCGGATGTGGAAATGGAAGCCCA;TGFβ-RII-R:TGGGGTCGTGGCAAACCGTC;TGFβ-RIII-F:AACTTGCCTCGCAAGCGGTG;TGFβ-RIII-F:GGTCCGGATGGCGCAGTTGA;Alk-1-F:GCTATGGGAGATCGCCCGGC;Alk-1-R:AGCCCGGAGAGGACCGGATCT;Alk-2-F:GGAGCAGTCAGTACCTCTCTGCTGG;Alk-2-R:AGACCCTGGATCCATTCATCCCTGT;p15-F:AATAACTTCCTACGCATTTTCTGC;p15-R:CCCTTGGCTTCAAGGTGAG;p21-F:TCCACAGCGATATCCAGACA;p21-R:GGACATCACCAGGATTGGAC;p27-F:GACAATCAGGCTGGGTTAGC,p27-R:TTCTGTTGGCCCTTTTGTTT;CNP-F:AAGGCCTTGCCATACGATCT;CNP-R:CGCTGGGGCAGAAGAATAC,MBP-F:AGCCCTCTGCCCTCTCAT,MBP-R:GGTAGTTCTCGTGTGTGAGTCCT;MOG-F:CTTCTTCAGAGACCACTCTTACCA;MOG-R:GTTGACCCAATAGAAGGGATCTT;c-myc-F:AGGAATTGATATGTGCCTTTGAG;c-myc-R:CAGGATTCCAAGGGCTTTCT;Olig1-F:GTTTCCTGGCATGCTTCC;Olig1-R:GCCCCTCTGGGTAGACACT;Olig2-F:AGACCGAGCCAACACCAG;Olig2-R:AAGCTCTCGAATGATCCTTCTTT;βActin-FCTCTTCCAGCCTTCCTTCCT;βActin-R:TGTTGGCGTACAGGTCTTTG.
Chromatin immunoprecipitation.
CNP-EGFP FACS-sorted cells were cultured under proliferating conditions and were treated with TGFβ1 or vehicle and 4 h later samples were processed as recommended by the manufacturer (EZ-ChIP; Millipore). Immunoprecipitation was performed with anti-SMAD3, anti-SMAD4 (Santa Cruz Biotechnology), anti-Sp1, and anti-FoxO1 (Cell Signaling Technology) antibodies or a nonspecific rabbit IgG as control. The presence of DNA bound to each transcription factor (TF) from the indicated regions of the two promoters analyzed was determined by qPCR analysis as above. All qPCR amplifying conditions were validated by semiquantitative PCR analysis. The enrichment for each gene was calculated by normalizing the TF/input ratio with the TF/input ratio of a DNA region that does not bind each TF. We analyzed the DNA sequence of the promoter regions of p21 and c-myc genes by using Genomatix software (RRID:nif-0000-10236) to determine the putative binding sites of each TF (SMAD3, SMAD4, FoxO1, and Sp1) in each promoter. Primers were designed by using Primer3 software (RRID:nlx_156833). The sequences of the primers used are as follows: c-myc-1F:TCCAGGGTACATGGCGTATT;c-myc-1R:GCTCCGGGGTGTAAACAGTA;c-myc-2F:TCCCTAAGTGGCTCTCCAAG;c-myc-2R:AAGGCTGGGGAAAACAGAAT;c-myc-3F:GCCTCTTGTGAAAACCGACT;c-myc-3R:CACACACACTCCAGCACCTC;p21-1F:TCCACAGCGATATCCAGACA;p21-1R:GGACACACCTGTGACTCTGG;p21-2F:CCCGAAACCCAGGATTTTAT;p21-2R;CCACACCTGGGCTATTCTCT;p21-3F:TGGGCTTGTTTTGTTTTTGA;p21-3R:CCTATTCCGATGGAGACCAA;p21-4F:GTGTGGTCCCAGTCAGGTCT;p21-4R:TCCACAGAAGGAAAGGATGC;p21-5F:GAAGAAAACCCCCTTCTGCT;p21-5R;ACATTCTCCGTTCCCACCTT;p21-6F:ACAGCTTCTCCAAAGCAGGA;p21-6R:CAGGAGACAGAAACAGGAGGA;p21-7F:AAGTGCAAGGCCAAGAATGT;p21-7:TTTGGGAGAAAACAGCTTGAA;p21-8F:AAGTGCAAGGCCAAGAATGT;p21-8R:GCTACCCAGGAAGGAAGAGG,p21-9F:GGTCTGGCTACCACCTTGTG;p21-9R:CATGCACACACCACACACAT;p21-10F:ACGTCTGTCTGTGGGTCCTT;p21-10R:TTCAGGTCACAAACCAGCAC.
Statistics.
Results shown represent the mean ± SEM, and the number of experiments is indicated in every case. Statistical analysis was performed by one-way or two-way ANOVA as appropriate. A post hoc analysis was made by the Student–Newman–Keuls test.
Results
Temporal expression of TGFβ signaling pathway elements in OL lineage cells
To test whether TGFβ signaling plays a role in OL development and CNS myelination, we first characterized the expression of TGFβ signaling components in the developing SCWM. Both real-time PCR and Western blot (WB) analysis of samples obtained from the SCWM at P8–P60 revealed a temporal regulation of TGFβ receptors RI, RII, and Alk2, and ligands TGFβ1, TGFβ2, and TGFβ3 expression during postnatal SCWM development. Interestingly, the peak of expression of TGFβ pathway elements in the SCWM coincides with the developmental period when OP differentiation and the initiation of myelination are most active (P15 and P30; Fig. 1A–D). Furthermore, increased activation levels of the canonical downstream target pSMAD2/3 were also observed during the critical period of CNS myelination. To further characterize the expression of TGFβ signaling elements in the context of oligodendrogenesis and myelination, we performed immunohistochemistry analysis by using tissue samples from transgenic mice that fluorescently label OL lineage cells (CNP-EGFP and NG2-dsRed mice). At P15, the vast majority of OPs (CNP-EGFPlow and NG2-dsRed+high cells) and OL lineage cells undergoing differentiation (pre-OL; CNP-EGFPhigh, CC1+) also displayed high levels of TGFβ-RII (Fig. 1E,F). However, pSMAD2/3 showed a stronger nuclear localization in cells undergoing OL differentiation (Fig. 1H,I). Both TGFβ-RII and pSMAD2/3 were downregulated after completion of the main process of SCWM myelination (Fig. 1G,J).
Figure 1.

TGFβ signaling pathway elements are expressed in OL lineage cells. A, B, Real-time PCR analysis of TGFβ signaling receptors (A) and ligands (B) from SCWM extracted at P8, P15, and P30. Results show that the expression of TGFβ-RI and II; Alk2; and TGFβ1, 2, and 3 in the SCWM peaks at P15–P30. Note that TGFβ-RIII and Alk1 were not found to be temporally regulated. Results are expressed in arbitrary units after actin normalization. C, D, Analysis of TGFβ-RII, pSMAD2/3, and MBP expression levels by WB during postnatal myelination (P8–P60), performed with SCWM protein extracts. E–J, Immunohistochemistry analysis demonstrates that TGF-βRII and its downstream targets pSMAD2/3 are expressed at the onset of myelination. At P15, TGFβ-RII (E, F) and pSMAD2/3 (H, I) are present in both, OPs (NG2dsRed+ cells and CNP-EGFPlow CC1neg, E, F1, H, and I1) and pre-OLs (CNP-EGFPhigh CC1+, F2 and I2). G, J, At P60 both TGFβ-RII and pSMAD2/3 were downregulated after the main process of SCWM myelination. K–O, CNP-EGFPlow OPs were FACS sorted and analyzed by RT-PCR or cultured in vitro. K, A typical FACS plot diagram of CNP-EGFP+ cells isolated at P5 from SCWM tissue of the CNP-EGFP mouse. L, M, Real-time PCR analysis of mRNA extracted from CNP-EGFPlow (OPs) and CNP-EGFPhigh (pre-OL) FACS-sorted cells. N, O, Immunofluorescence of TGFβ-RII (N) and pSMAD2/3 (O) in CNP-EGFPlow FACS-sorted cells cultured under differentiating conditions. Note that we found higher levels of TGFβ-RII and pSMAD2/3 in cells undergoing OL differentiation compared with OPs. Results are expressed in arbitrary units after actin normalization. Data are shown as mean ± SEM; n = 4 brains for each time point. Scale bars: E–J, 5 μm; N, O, 3 μm. *p < 0.05; **p < 0.01, ***p < 0.001.
In an extension of our characterization, we further analyzed the expression of TGFβ signaling pathway components in a purified population of OL lineage cells in vitro. To this end, we isolated CNP-EGFP+ cells from the SCWM of CNP-EGFP mice at P5 using FACS sorting (Fig. 1K). Real-time PCR analysis confirmed that both OPs (CNP-EGFP+lowNG2+ cells) and premyelinating OLs (CNP-EGFP+highNG2neg cells) express TGFβ-RII. However, a significant upregulation was observed in premyelinating OLs as compared with OPs (Fig. 1L,M). Consistent with our data above, pSMAD2/3 immunoreactivity was observed in both OPs and pre-OLs, but increased activation levels were found in cells undergoing differentiation (Fig. 1N,O). Altogether, our data demonstrate that TGFβ signaling is activated during the transition from OP to differentiating myelinating OL, suggesting a role for this signaling pathway during OL development and CNS myelination.
TGFβ signaling gain of function accelerates OL development and CNS myelination
To determine the functionality of TGFβ receptors in OL lineage cells and whether TGFβ signaling is involved in oligodendrogenesis and CNS myelination, we first performed a gain-of-function analysis in vitro. FACS-sorted CNP-EGFP+low OPs (Fig. 2A) were cultured with either TGFβ1 or vehicle (Veh) for 48 h and then cells were characterized by immunocytochemistry analysis. TGFβ1 treatment in OPs induced OL differentiation, as approximately a fourfold greater number of mature OLs (MBP+ and CNP+ cells) was observed in these conditions compared with controls (Fig. 2B,C). To confirm the effect of TGFβ1 on OPs in the context of oligodendrogenesis and CNS myelination in vivo, we systemically administered TGFβ1 or vehicle to P5 CNP-EGFP+ mice for 3 consecutive days and then characterized the development of the SCWM at 24 h after the last administration (P8; Fig. 2D). Strikingly, pharmacological activation of TGFβ signaling accelerated SCWM development and myelination, as our immunohistochemistry analysis showed higher expression levels of several myelin-related proteins, including MBP and CNP, in the SCWM of TGFβ1-treated mice (Fig. 2E–H). The enhanced myelination following TGFβ1 injection was observed throughout the entire SCWM including the corpus callosum, cingulum, and external capsule (Fig. 2E–H). This observation was further confirmed and quantified by WB (Fig. 2I) and RT-PCR (Fig. 2J) analysis, which revealed up to fivefold upregulation of the myelin proteins MBP and MOG in the SCWM of TGFβ1-treated mice.
Figure 2.
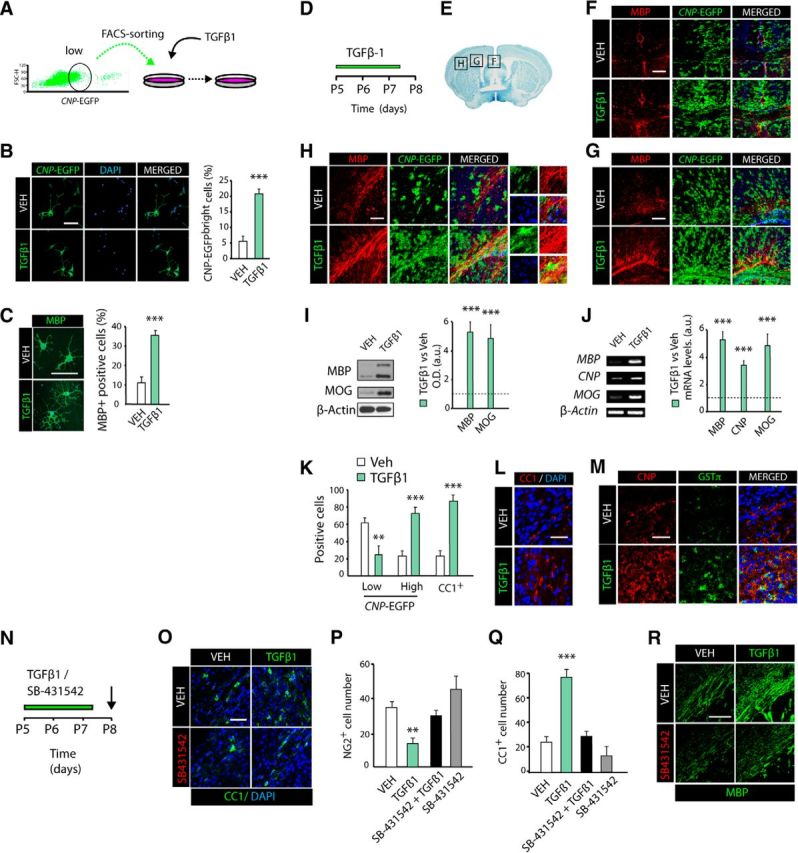
TGFβ signaling gain of function accelerates OL development and SCWM myelination. A–C, FACS-sorted OPs (CNP-EGFPlow) were cultured in the presence of TGFβ1 or Vehicle (VEH, control) for 48 h. Pharmacological activation of TGFβ signaling induces OL differentiation, as a higher number of mature OLs (CNP-EGFPhigh; B) and MBP+(C) cells were observed by immunofluorescence analysis compared with vehicle-treated cells. D, Time line representing the paradigm used for TGFβ1 or vehicle administration in vivo. P5 CNP-EGFP pups received vehicle or TGFβ1 intraperitoneal injections daily (100 ng/kg twice a day) for 3 d. E, Illustration of the SCWM brain regions used for analysis. F–H, Representative confocal images at P8 obtained from different regions of the SCWM, corpus callosum (F), cingulum (G), and external capsule (H) with anti-MBP antibodies. WB (I) and RT-PCR (J) analysis of SCWM extracts at P8 for myelin proteins further confirmed the potent effect of TGFβ1 treatment on oligodendrogenesis and myelination. K–M, TGFβ1 systemic administration accelerates OL differentiation. Representative confocal images and quantification of immature (NG2+, CNP-EGFPlow; K) OP markers, and for mature OL cell markers, including CC1+ (K, L), CNP-EGFPhigh (F–H, M), and GSTΠ (M) at P8 after 3 d of TGFβ1 treatment. N, Time line representing the paradigm used for TGFβ1, SB-431542 (TGFβ-RII antagonist), or vehicle treatment in vivo. P5 CNP-EGFP pups received vehicle, TGFβ1 (100 ng/kg), SB-431542 (5 mg/kg), or a combination of TGFβ1 and SB-431542 intraperitoneal injections twice a day for 3 d. O–R, Immunofluorescence analysis and quantification with anti-NG2, anti-CC1, and anti-MBP antibodies revealed that TGFβ1 failed to induce OL differentiation when SB-421542 was administrated previously. Histograms express results in arbitrary units after actin normalization. n = 5–6 mice of each group. Cells numbers are expressed as number of positive cells/mm3. Scale bars: B, C, 5 μm; F–H, M, R, 100 μm; L, O, 10 μm. **p < 0.01, ***p < 0.001.
Our data demonstrating higher expression levels of myelin-related proteins following TGFβ1 administration could come as a result of either increased differentiation rate of OPs and/or increased cell survival. To further define the cellular effects mediated by the TGFβ pathway in these two contexts, we characterized OL lineage cells in situ in the SCWM. We observed a threefold higher density of myelinating OLs (GSTπ+, CNP+high, and CC1+ cells) and approximately a twofold reduction in OPs (CNP-EGFP+low NG2+ cells) after TGFβ1 administration in vivo (Fig. 2K–M). Additionally, we did not observe differences in the total numbers of activated caspase-3+ or TUNEL+ cells among OL lineage cells between control and TGFβ1-treated mice (data not shown). These data indicate that TGFβ signaling, not only in vitro but also in vivo, promotes OL differentiation leading to enhanced SCWM development and myelination.
To further validate the selectivity of TGFβ1 administration in vivo on OL development and SCWM myelination, we administered a TGFβ-RII antagonist (SB-4311542) alone or in combination with TGFβ1 into P5 CNP-EGFP mice for 3 consecutive days (Fig. 2N). TGFβ-RII antagonist prevented the effect of TGFβ1 treatment on OL differentiation and SCWM myelination, as after systemic administration of SB-4311542, the number of OLs was unchanged compared with vehicle-treated mice (Fig. 2O–R). Together, these results confirm the selectivity of TGFβ1 administration in vivo and further demonstrate an essential role for TGFβ signaling in oligodendrogenesis and CNS myelination.
TGFβ signaling regulates OP cell cycle exit
Cellular responses to TGFβ signaling differ between cell types and physiological conditions, although a central function of the TGFβ pathway in neural cells is the inhibition of cell cycle progression (Vergeli et al., 1995; García-Campmany and Martí, 2007; Misumi et al., 2008). To determine whether the accelerated OL development and SCWM myelination by TGFβ1 treatment is due to OP cell cycle regulation, we first performed OP culture experiments and analyzed cell cycle dynamics after TGFβ1 stimulation (Fig. 3A). OPs were stimulated with TGFβ1 or vehicle for 16 h, and BrdU was added to the culture 2 h before the end of the experiment. TGFβ1 stimulation reduced the proliferation profile of OPs (BrdU+CNP-EGFPlow), as a twofold reduction in this population was observed in TGFβ1-treated cells (Fig. 3B,C), suggesting that TGFβ signaling regulates cell cycle exit in OPs.
Figure 3.

TGFβ signaling activation induces OP cell cycle withdraw and differentiation. A, FACS-sorted OPs (CNP-EGFPlow) were cultured in the presence of TGFβ1 or Vehicle (VEH, control) for 16 h. B, C, Immunofluorescence analysis with anti-BrdU antibodies and quantification of proliferating OPs (CNP-EGFP+ BrdU+/CNP-EGFP+ cells) indicates that pharmacological activation of TGFβ signaling induces cell cycle exit in OPs, as determined by the reduced number of OPs (CNP-EGFP+ cells) in S phase in TGFβ1-treated cells. D–F, Systemic administration of TGFβ1 in vivo induces OP cell cycle withdrawal. D, Time line showing the TGFβ1 treatment paradigm and time point selected for the analysis (black arrow). E, F, Immunofluorescence analysis with anti-BrdU and anti-NG2 antibodies after 16 h of TGFβ1 administration, and quantification of BrdU+NG2+CNP-EGFP+/ CNP-EGFP+ cells. F, TGFβ1 induces cell cycle exit, as indicated by the reduced number of OPs (NG2+CNP-EGFP+ cells) in S phase. G–I, TGFβ1 treatment induces cell cycle withdrawal in OPs during postnatal myelination of the SCWM. G, Time line representing the paradigm used to analyze cell cycle exit in OPs. H, I, Representative confocal images and quantification of double immunofluorescence with anti-Ki67 and anti-BrdU antibodies used to calculate cell cycle exit in OPs. Data are shown as mean ± SEM; n = 5 brains for each time point. Scale bars: B, 5 μm; E, 20 μm; H, 10 μm. **p < 0.01, ***p < 0.001.
To confirm the above results in vivo, we analyzed OP cell proliferation after TGFβ1 administration in the developing SCWM. To this end, TGFβ1 was administered systemically into P5 CNP-EGFP mice followed by BrdU injection 16 h later (Fig. 3D). Our data showed a significant reduction in the number of proliferating OP cells (CNP-EGFP+NG2+BrdU+ cells) compared with controls (Fig. 3E,F). Furthermore, cell cycle exit experiments confirmed the above findings, as a significant reduction in Ki67+BrdU+/BrdU+ OPs was found in the SCWM after 48 h of TGFβ1 administration (Fig. 3G–I). Our data here indicate that during postnatal development, TGFβ signaling gain of function induces OP cell cycle exit, and consequently, accelerates OL development and CNS myelination.
TGFβ signaling modulates c-myc and p21 expression in OPs to promote cell cycle exit and differentiation
The anti-proliferative actions of TGFβ signaling occur at the restriction point in late G1 phase by transcriptional activation of cyclin-dependent kinase inhibitors (p15Ink4b, p21Cip1, and p27kip1; Massagué and Gomis, 2006; García-Campmany and Martí, 2007; Misumi et al., 2008) and repression of c-myc and the inhibitors of differentiation Id1 and Id2 (Massagué et al., 2005). The transcriptional modulation of p21WAF1/Cip1 (p21 or cdkn1a) and c-myc by TGFβ signaling involves SMAD2/3 activation. SMAD3 together with SMAD4 functionally cooperates with Sp1 and FoxO1 transcription factors (TF) to activate the p21 promoter and repress the c-myc promoter (Siegel and Massagué, 2003; Massagué and Gomis, 2006). This transcriptional activation requires SMAD-mediated c-myc promoter repression and cytoplasmic transport of the c-myc protein, ultimately leading to p21 promoter de-repression (Siegel and Massagué, 2003).
To gain insight into the molecular mechanism by which TGFβ signaling modulates OP cell cycle exit and OL development, we analyzed the expression of the anti-mitotic program mentioned above in OP cell cultures. OPs were obtained and cultured in the presence of TGFβ1 or vehicle, and 8 h later, mRNA levels of members of the anti-mitotic program and oligodendrogenesis (Olig1 and Olig2; Rowitch et al., 2002; Fig. 4A) were measured. OP cell cultures stimulated with TGFβ1 showed increased mRNA levels of the cyclin-dependent kinase inhibitors p15, p21, and p27, and reduced c-myc mRNA levels (Fig. 4B). We also found increased mRNA levels of Olig1 and Olig2, as well as of myelin-related proteins, including Mbp, Cnp, and Mog in TGFβ1-treated cultures (Fig. 4B). To further confirm the transcriptional modulation of these genes by TGFβ, we analyzed the promoter activity of c-myc and p21 by using transfection experiments in OPs with luciferase constructs. Consistent with our data at the mRNA level, TGFβ1 repressed c-myc promoter luciferase activity, and increased p21 promoter activity (Fig. 4C).Together, our data reveal that the TGFβ signaling pathway controls cell cycle exit in OPs by the transcriptional modulation of p21 and c-myc genes.
Figure 4.

TGFβ signaling modulates c-myc and p21 expression in OP cells through the cooperation of SMAD3/SMAD4 with FoxO1 and Sp1. A, FACS-sorted OPs (CNP-EGFPlow) were cultured in the presence of TGFβ1 or Vehicle (VEH, control). B, Real-time PCR analysis for the indicated genes from cultured OPs (CNP-EGFPlow) stimulated for 8 h with TGFβ1. C, Luciferase reporter assay activity for the indicated genes was performed in CNP-EGFPlow FACS-sorted cells. D–F, Immunofluorescence analysis of OP cultures after 3 h of TGFβ1 treatment shows that TGFβ signaling activation in OPs induces high levels and nuclear localization of pSMAD2/3 (D, F), SMAD4, FoxO1 (E), and Sp1 (F). G, H, Coimmunoprecipitation from OP cell cultures treated with TGFβ1 or vehicle for the indicated antibodies. I, J, ChIP analysis after 4 h of TGFβ1 stimulation of OP cultures. I, Diagram representing the putative binding sites of all TFs analyzed to the promoter region of p21 and c-myc genes. Immunoprecipitation was performed with anti-SMAD3, anti-SMAD4, anti-Sp1, and anti-FoxO1 antibodies or a nonspecific rabbit IgG as a control. J, qPCR analysis revealed increased binding of these four TFs to the promoter region of c-myc and p21, in TGFβ1-stimulated cells. Results are expressed as arbitrary units after normalization. Data are shown as mean ± SEM; n = 5 brains for each time point. *p < 0.05; **p < 0.01; ***p < 0.001. Scale bars: 3 μm.
We next investigated the involvement of SMAD2/3, SMAD4, FoxO1, and Sp1 TFs in the transcriptional regulation of p21 and c-myc expression by TGFβ signaling. Immunofluorescence analysis in OP cell cultures stimulated with TGFβ1 demonstrated that TGFβ pathway activation induced a strong activation and nuclear localization of pSMAD2/3 and SMAD4 in OPs (Fig. 4D–F). Furthermore, TGFβ1 stimulation in OP cell cultures also induced the nuclear translocation of FoxO1 and Sp1 (Fig. 4E,F). To further define the interaction between these TFs during oligodendrogenesis, we performed immunoprecipitation experiments using protein extracts obtained from OP cultures stimulated with TGFβ1. Our data demonstrated that TGFβ1 induced the interaction of SMAD3/SMAD4/FoxO1 and SMAD3/SMAD4/Sp1, as higher levels of these complexes were observed in TGFβ1 culture conditions (Fig. 4G,H). To confirm the involvement of SMAD3/4, FoxO1, and Sp1 in the transcriptional modulation of p21 and c-myc in OPs, we performed chromatin immunoprecipitation (ChIP) analysis of protein extracts from OP cultures after 4 h of TGFβ1 or vehicle treatment. Interestingly, TGFβ1 promoted the recruitment of SMAD3, SMAD4, Sp1, and FoxO1 to multiple sites of the c-myc and p21 promoters in OP cultures (Fig. 4I,J). Our results indicate that TGFβ signaling promotes OP cell cycle exit and OL differentiation through the transcriptional modulation of c-myc and p21, which involves the cooperation between nuclear SMAD3/4 with the TFs Sp1 and FoxO1.
To confirm that the molecular mechanism observed in vitro also applies in vivo, we characterized the expression of TGFβ anti-mitotic program components in OL lineage cells after TGFβ1 administration (Fig. 5A). At 4 h after TGFβ1 administration, we observed increased levels and nuclear localization of pSMAD2/3 and SMAD4 in OPs and cells undergoing OL differentiation (Fig. 5B,C, insets 1 and 2), but not in mature OLs (Fig. 5B,C, inset 3). Immunofluorescence and IP analysis confirmed the induced interaction of SMAD3 and SMAD4 with Sp1 and FoxO1 in OPs after TGFβ1 administration in vivo (Fig. 5D–F). Furthermore, RT-PCR analysis from FACS-sorted CNP-EGFP+ cells obtained from the SCWM after 8 h of TGFβ1 administration also showed higher mRNA levels of p15, p21, p27, Olig1, and Olig2 and reduced mRNA levels of c-myc (Fig. 5G,H). WB analysis confirmed that TGFβ signaling activation promotes cell cycle withdrawal, as TGFβ1 treatment reduced Cdk2 and PDGFRα protein levels and increased p21 and p27 protein levels in the SCWM (Fig. 5I,J). Consistently, immunofluorescence analysis in the SCWM confirmed that TGFβ1 administration induced OP cell cycle exit, as TGFβ1 administration upregulated p21 and p27 expression and downregulated c-myc expression in OPs (CNP-EGFPlowNG2+ cells; Fig. 5K–M). Moreover, TGFβ1 administration induced c-myc cytoplasmic localization together with increased FoxO1 nuclear localization in OPs (CNP-EGFPlow; Fig. 5D,K). These results confirm that the molecular mechanisms by which TGFβ signaling modulates OP cell cycle exit and OL differentiation observed in vitro also occur in vivo.
Figure 5.

TGFβ signaling modulates c-myc and p21 expression in OP cells during SCWM. A, G, I, Time line representing the paradigm used for TGFβ1 or vehicle treatment in vivo. B, Immunofluorescence analysis in the SCWM was performed after 4 h of TGFβ1 administration. Activation of TGFβ signaling induces nuclear localization of pSMAD2/3 (B, C), SMAD4 (C, D), and FoxO1 (D) in OPs. E, F, Coimmunoprecipitation analysis of SCWM extracts from TGFβ1 or vehicle (VEH)-treated mice, at 4 h after treatment. TGFβ signaling activation increased the interaction of SMAD3 and SMAD4 with FoxO1 and Sp1 in OPs in the SCWM. SCWM immune complexes were incubated with antibodies against SMAD4 (E) or FoxO1 (F). As a loading control, coimmunoprecipitation samples were blotted with the same antibodies used to perform the immunoprecipitation. G, H, P5 CNP-EGFP pups received a single administration of Veh or TGFβ1 (100 ng/kg) and 8 h later, CNP-EGFPlow (OPs) were FACS sorted to isolate RNA and perform RT-PCR analysis for genes involved in the TGFβ-mediated anti-mitotic program and OL differentiation. I, J, Proteins involved in the TGFβ-mediated anti-mitotic program and OL differentiation were analyzed by WB analysis from total extracts of SCWM after 16 h of treatment. K–M, P5 CNP-EGFP mice received TGFβ1 or vehicle, and immunohistochemistry analysis was performed in the SCWM after 16 h of treatment. Representative confocal images of proteins involved in the cell cycle in OPs (CNP-EGFP+NG2+ cells), including c-myc (K), p21 (L), and p27 (M) after 16 h of TGFβ1 treatment. Histograms express results in a.u. after normalization. n = 5 brains for each time point. **p < 0.01. Scale bars: B, 50 μm; C, D, K–M, 10 μm.
TGFβ loss of function in OPs leads to defects in SCWM myelination
Our results above suggest that TGFβ1 treatment may play a direct role in promoting OL development and myelination in the CNS. To assess whether TGFβ signaling indeed regulates OL development and myelination in a cell-autonomous manner, we performed a lineage-specific loss-of-function analysis by generating a new PDGFRα-Cretm: TGFβ-RIIfl/fl:YFP mouse line (fl/fl mice). We focused on deleting the TGFβ-RII in OPs, as TGFβ ligand binding allocates the type II receptor to phosphorylate the type I receptor. This phosphorylation turns on receptor kinase activity and subsequently leads to the activation of SMAD proteins (SMAD2/3), which allows for their association with the common-mediator SMAD (SMAD4). The resulting SMAD complex comprised of two R-SMADs and a single SMAD4 (such as a SMAD2-SMAD3-SMAD4 complex) is then translocated into the nucleus, which allows for transcriptional regulation of target genes (Siegel and Massagué, 2003). In this transgenic mouse, we are able to genetically deplete the TGFβ-RII specifically in OPs by tamoxifen (TAM) administration, thus, allowing us to demonstrate the cell-autonomous effect of this pathway in OL development during SCWM myelination. To this end, mice received TAM injections for 3 d at P6, and were analyzed at 1 and 3 weeks after the last TAM injection (Fig. 6A). Immunohistochemistry analysis at 1 week after last TAM injection revealed that TGFβ-RII genetic deletion in OPs prevented their differentiation. A decrease in the percentage of recombined mature OLs (YFP+CC1+ cells) and an increase in OPs (YFP+NG2+ cells) were observed in the SCWM of fl/fl mice (Fig. 6B–E). Our data using fl/fl mice suggest that activation of TGFβ signaling is crucial in promoting OP cell differentiation. Consistent with this hypothesis, we observed increased numbers of proliferating OPs (NG2+BrdU+) and a reduced density of mature OLs (CC1+) in the SCWM of fl/fl mice compared with wt littermates (Fig. 6D,E). Furthermore, a morphological analysis of the recombined YFP+ cells showed that fl/fl cells displayed a typical morphology of OPs (cells lacking myelin fibers) compared with control wt recombined cells, which displayed a morphology typical of mature myelinating OL (myelin fibers oriented parallel to axons; Fig. 6F). WB and immunofluorescence analysis at 1 and 3 weeks after TAM injection indicated that the total levels of MBP, MOG, and CNP proteins were significantly reduced in fl/fl SCWM (Fig. 6G–I). Altogether, our data using fl/fl mice strongly indicate that TGFβ signaling is crucial for OL development and SCWM myelination. To investigate whether hypomyelination in the SCWM following TGFβ-RII deletion in fl/fl mice is mediated by the deregulation of p21, we performed immunohistochemistry and WB analysis. Consistently, our analysis in the SCWM of fl/fl mice revealed increased levels of Cdk2 and PDGFRα, and reduced levels of p21 and p27 in NG2+ OPs (Fig. 6J–M), suggesting that the observed deficits in fl/fl OP cell cycle exit is due to the deregulation of p21 expression.
Figure 6.

TGFβ signaling loss of function in OPs leads to defects in oligodendrogenesis and SCWM myelination. A, Time line depicting the developmental stage at which mice received TAM injections and the time points of analysis (arrows). PDGFRα-Cretm:TGF-βRIIw/w/YFP (w/w) and PDGFRα-Cretm:TGF-βRIIfl/fl/YFP (fl/fl) mice were injected with TAM (3 times, 24 h apart) starting at P6, and tissue was processed at different time points after the last tamoxifen injection (TAM). B, C, Immunofluorescence analysis with anti-NG2 and anti-CC1 antibodies was used to characterize OL lineage cells (YFP+) in the SCWM at 1 week after TAM. C, Histogram depicting the quantification of OPs (YFP+NG2+) and OLs (YFP+CC1+) at 1 week after TAM. Immunohistochemistry analysis was used to quantify the number of proliferating OPs (BrdU+NG2+cells; D) and total OLs (CC1+ cells; E) at 1 week after TAM injection. F, Representative confocal images in the SCWM with anti-MBP and anti-YFP antibodies at 1 week after TAM injection in w/w and fl/fl mice. Note that in w/w mice, YFP+ cells display a mature OL morphology and colocalize with MBP, but in fl/fl mice, YFP+ cells display an immature OL morphology and are negative for MBP. G, Representative confocal images in the SCWM with anti-MBP and anti-CNP antibodies at 3 weeks after TAM injection. H, I, The SCWM of w/w and fl/fl mice was dissected out at 1 (P15) and 3 weeks (P30) after TAM injection and WB analysis was performed to analyze total expression levels of myelin proteins. Immunofluorescence (J, K) and WB (L, M) analysis with antibodies against p21 (J) and p27 (K) proteins show that TGFβ-RIIfl/fl OPs display lower levels of p21 and p27 in the developing SCWM. Histograms express results in arbitrary units after actin normalization. N, O, Immunofluorescence analysis with anti-MBP antibodies of the SCWM of NG2-Cre:TGF-βRIIfl/fl:YFP (fl/fl) and WT littermate mice at P15. Note that in fl/fl mice, the overall levels of MBP are reduced relative to levels in WT mice, and YFP+ cells have a nonmyelinating morphology. P, At P15 an increased number of proliferating NG2-Cre:YFP+ cells was observed in fl/fl mice compared with WT mice. Nuclei are immunostained with DAPI. Q, Representative electron microscopy images from the NG2-Cre:TGF-βRIIfl/fl and wt mice at P15 in the SCWM. Electron microscopy studies of the NG2-Cre:TGF-βRIIfl/fl:YFP at P15 confirmed the myelination defects in the developing SCWM. Data are shown as mean ± SEM; n = 5 brains for each time point.**p < 0.01. Scale bars: B, D, F, J, K, O, P, 10 μm; N, G, 50 μm; Q, 500 nm.
These results were further confirmed using another mouse line that uses a different Cre driver to target the same population of OPs (NG2-Cre:TGFβ-RIIfl/fl:YFP). As in the PDGFRα-Cretm: TGFβ-RIIfl/fl:YFP mouse line, we observed a higher number of proliferating OPs and notably increased areas of hypomyelination in the developing SCWM of NG2-Cre:TGFβ-RIIfl/fl mice compared with wt mice (Fig. 6N–Q). The defects in postnatal myelination observed in this mouse line were further confirmed by using electron microscopy analysis. Our electron microscopy analysis of the SCWM at P15 evidenced that while compacted myelinated axons were observed in wt mice, NG2-cre::TGFβ-RIIfl/fl mice displayed severe deficits in axonal myelination at this developmental stage (Fig. 6Q). This unbiased evidence further confirms the essential function of TGFβ signaling in OP cell cycle exit, OL development, and CNS myelination.
TGFβ signaling promotes oligodendrogenesis and SCWM myelination in a cell-autonomous manner
Finally, to unambiguously confirm the cell-autonomous role of TGFβ signaling in directly supporting OL development and CNS myelination, and to exclude the participation of other cell types (microglia and astrocytes) in this phenotype, we performed experiments using TGFβ1 stimulation in fl/fl and wt mice. To this end, we injected TAM to P5 fl/fl and wt mice for 2 d, and 3 h after the last TAM injection, mice received TGFβ1 or vehicle, and tissue was then analyzed at 1 and 3 d later (Fig. 7A). Using this approach, we confirmed that TGFβ1 treatment induced OL differentiation and CNS myelination only in control littermate mice (Fig. 7B–E). In fl/fl SCWM, the number of OPs (NG2+ cells) and mature OLs (CC1+) were similar in vehicle and TGFβ1-treated mice (Fig. 7B–D). Remarkably, TGFβ1 treatment had no effect on SCWM development and myelination in fl/fl mice (Fig. 7E). In line with these results, TGFβ1 administration into fl/fl mice also failed to modulate pSMAD2/3 and p21 protein levels in OPs of the SCWM at 1 d after TGFβ1 administration (Fig. 7F,G), confirming the cell-autonomous action of TGFβ1 administration on the modulation of OP cell cycle exit and OL differentiation. Additionally, we did not observe differences in the total numbers of activated caspase-3+ or TUNEL+ cells among OL lineage cells between fl/fl and wt littermates at any time point analyzed (data not shown), indicating that the differences observed in SCWM myelination during development in fl/fl and wt mice were not as a consequence of OL lineage cell death. Moreover, we did not observe differences in the number of GFAP+ Vimentin+ astrocytes or Iba-1+ microglial cells at this time point in any of the groups analyzed (Fig. 7H; data not shown), further confirming the cell-autonomous action of TGFβ signaling in OL development and CNS myelination and excluding the participation of other glial cells in the cellular outcome mediated by this pathway.
Figure 7.

TGFβ signaling induces OP cell cycle exit and differentiation in a cell-autonomous manner. A, TAM was injected into w/w and fl/fl mice (2 times, 24 h apart), followed by vehicle (Veh) or TGFβ1 (100 ng/kg twice a day) treatment for 1 d (F, G) or 3 d (B–E, H). B–E, Immunofluorescence analysis was used to characterize the number of OPs (NG2+; B, C) and OLs (CC1+; B, D) and myelin-related protein levels (MBP; E) in the SCWM of w/w and fl/fl mice treated either with TGFβ1 or vehicle. F, G, Immunofluorescence analysis in the SCWM of w/w and fl/fl mice was performed to characterize components of the TGFβ pathway including pSMAD2/3 (F) and p21 (G) in OPs. H, Representative confocal images of immunofluorescence analysis of astroglial activation (GFAP and vimentin) in PDGFRα-Cretm:TGF-βRIIfl/fl/YFP (fl/fl) mice and wt littermates at P9, treated with either Veh or TGFβ1 (100 ng/kg, twice a day) at P6 for 3 consecutive days. Data show that genetic deletion of TGFβ-RII in OP cells does not affect astroglial activation. I, J, YFP+ cells were FACS purified from the SCWM of wt and fl/fl mice 1 d after the last TAM injection to isolate RNA to perform RT-PCR analysis. J, RT-PCR analysis shows that fl/fl OPs expressed lower mRNA levels of p21 and p27 and higher mRNA levels of c-myc compared with wt OPs. K, L, OPs from wt and fl/fl SCWM were FACS sorted and cultured under proliferating conditions and 24 h later cells were transfected with plasmid vectors that overexpress p21, or empty control. Cells were processed for cell proliferation analysis at 24 h after transfection. Histograms show results after actin normalization. Data are shown as mean ± SEM; n = 5 brains for each time point and 3 independent experiments. Scale bars: E, 50 μm; B, F–H, K, 20 μm. *p < 0.05; **p < 0.01; ***p < 0.001.
To confirm that the observed deficits in fl/fl OP differentiation are due to the deregulation of p21 expression, we performed in vitro experiments using fl/fl OP cultures. NG2+YFP+ OPs were FACS sorted from the SCWM of wt and fl/fl mice. First, we analyzed basal mRNA levels of p21, p27, and c-myc in fl/fl mice by real-time RT-PCR analysis. Lower p21 and p27 and higher c-myc mRNA levels were observed in fl/fl OPs compared with wt OPs (Fig. 7J). Furthermore, fl/fl OPs maintained in culture conditions that support differentiation failed to withdraw from the cell cycle (data not shown). Finally, we performed rescue experiments by forced expression of p21 in vitro using plasmid vectors (Fig. 7I,K). Immunofluorescence analysis showed that forced expression of p21 in fl/fl OPs restored cell cycle exit and OL differentiation (Fig. 7K,L), confirming the critical role of p21 as a key mediator in the anti-proliferative action of TGFβ signaling in OPs and the modulation of OL development and CNS myelination.
Discussion
To date, a large number of molecules have been implicated in the temporal regulation of OP cell cycle exit and OL differentiation and maturation. Positive and negative intrinsic factors and extracellular signals are tightly coordinated to regulate the proper timing of OP expansion and the switch from proliferation into a differentiating program. Here, we have demonstrated that TGFβ-RII genetic deletion in OPs causes hypomyelination in the developing SCWM, while TGFβ signaling gain of function promotes OP cell cycle exit and accelerates oligodendrogenesis and myelination.
Mechanistically, TGFβ signaling in OPs modulates the canonical downstream TGFβ-R effectors SMAD2/3/4. Upon TGFβ-R activation, the heteromeric SMAD2/3/4 complex establishes a nuclear localization, and cooperates with FoxO1 and Sp1 to modulate the transcription of c-myc and p21 (Fig. 8). These targets genes of TGFβ signaling not only belong to the anti-mitotic program described in other cells types (Massagué et al., 2000; Siegel and Massagué, 2003; Massagué et al., 2005), but also are essential players in the modulation of OP cell cycle exit and OL differentiation (Huang et al., 2013). Upon binding to their regulatory sequences, SMAD3/4, FoxO1, and Sp1 cooperate to inhibit c-myc transcription and activate p21 transcription, allowing for OP cell cycle withdrawal and progression toward development into myelinating OLs. It is important to note that we cannot exclude the possibility that TGFβ signaling can also modulate other TFs that positively (Nkx2.2; Qi et al., 2001; Zfp488, Sox10; Stolt et al., 2002; ZFP191; Howng et al., 2010; MRF, Sip1; Weng et al., 2012) or negatively (Sox5 and Sox6 or Hes5; Rowitch et al., 2002) influence OL development during CNS myelination.
Figure 8.

Proposed model of TGFβ signaling modulation of oligodendrogenesis during the SCWM myelination. TGFβ signaling in OPs controls the canonical downstream TGFβ-R effectors, SMAD2/3/4. Upon TGFβ-R's activation, the heteromeric SMAD2/3/4 complex establishes a nuclear localization, and cooperates with FoxO1 and Sp1 to modulate the transcription of c-myc and p21. Upon binding regulatory sequences, SMAD2/3/4, FoxO1, and Sp1 cooperate to repress the transcription of c-myc and activate p21 transcription, allowing OPs to withdraw from the cell cycle and progress down OL differentiation and maturation.
The CDK inhibitors p21/Cip1, p27/Kip1, and p57/Kip2 are key intrinsic factors in regulating OP cell-cycle progression and development (Zezula et al., 2001; Tokumoto et al., 2002). While p27 is required for withdrawal from the cell cycle, p21 is vital for the initiation of the differentiation program following growth arrest in OPs (Calver et al., 1998; Tokumoto et al., 2002). Thus, the p21 transcriptional upregulation observed following TGFβ signaling activation seems to be an essential step to induce OP differentiation, as forced overexpression of p21 in the fl/fl background was sufficient to rescue the deficits in OP cell cycle arrest and restore OL differentiation. Therefore, our data indicate an essential function of TGFβ signaling in inducing p21 expression and governing the timing of OP differentiation. Moreover, it has recently been described that TGFβ signaling modulates p53 expression, both at the transcriptional and translational level (Li et al., 2013; López-Diaz et al., 2013). In addition, it is widely accepted that p21 is the main effector of p53's growth arrest function in several cell types (Gartel and Tyner, 1999, Radhakrishnan et al., 2006). Therefore, we cannot exclude that TGFβ modulates p53 expression in OL lineage cells, and that the observed modulation of p21 expression by TGFβ signaling in OPs may depend, at least in part, on p53 expression and/or activity. From the data presented in this study, we conclude that TGFβ signaling controls the timing of cell cycle exit in OPs during development, ultimately coordinating postnatal myelination of the SCWM in a cell-autonomous fashion.
Because of the relevance and impact of TGFβ signaling manipulation on OL development and CNS myelination, the interaction of this novel signaling pathway with other extracellular factors like PDGFα (Calver et al., 1998), thyroid hormone (Billon et al., 2001), or BMPs (Samanta and Kessler, 2004; Cheng et al., 2007) still needs to be elucidated. Among these factors, PDGFα is considered to be a key extracellular factor in maintaining OP proliferation, in turn, inhibiting differentiation. This is supported by the evidence that inhibition of PDGFRα signaling prompts the onset of OL differentiation (Raff et al., 1988; Hart et al., 1989). As PDGFRα signaling involves the regulation of p21 expression, it is reasonable to speculate that the anti-mitotic effect of TGFβ1 counters PDGFRα-induced OP proliferation by modulating the expression of p21 and other anti-mitotic genes. Similarly, BMP signaling inhibits OL differentiation and promotes astrogliogenesis, in part by modulating the expression of p21 in the opposite direction as found to be regulated by TGFβ signaling (Samanta and Kessler, 2004). Moreover, TGF-β and BMP signaling pathways have been shown to exert synergistic and antagonistic effects in different cell types (Yew et al., 2005; Keller et al., 2011), strongly suggesting an interaction of both pathways in OL lineage cells. However, further investigations are needed to determine the interaction of TGFβ signaling with these and other extracellular signaling regulators of OL differentiation.
The process of OP differentiation and OL development is strongly modulated by astroglial and microglial cells under several pathological situations (Hiremath et al., 1998; Kotter et al., 2011). However, during postnatal development, the contribution of glial cells does not seem to play a crucial role in this process. Our experiments using fl/fl mice in conjunction with TGFβ1 treatment demonstrates that while other neural cell types (i.e., astrocytes and microglia) express functional TGFβ receptors and may respond to TGFβ1 administration, they do not compensate for the deficit in SCWM myelination in fl/fl mice. This evidence demonstrates the autonomous effect of TGFβ signaling on OL lineage cells to drive oligodendrogenesis and CNS myelination during postnatal development. However, it is still unclear which cell type provides the source of TGFβ ligands that drives OP cell cycle exit and differentiation. While, under certain pathological conditions, TGFβ ligands have been reported in the CNS in astroglial and microglial cells, as well as in infiltrating lymphocytes (Mirshafiey and Mohsenzadegan, 2009; Meoli et al., 2011), the contribution of these or other cell types to the TGFβ signaling-mediated modulation of CNS myelination during postnatal development still needs to be elucidated.
In summary, our study identifies an autonomous and crucial role of the TGFβ signaling pathway during postnatal oligodendrogenesis and CNS myelination, opening up new avenues in developing treatments for demyelinating diseases.
Footnotes
This work was supported by National Institutes of Health 4R00NS057944-03 (A.A.) and 1R01MH099384-01 (A.A.); National Multiple Sclerosis Society (J.P.); and Ministerio de Educacion, Spain, EDU/3495/2010 (J.P.). We thank the National Cancer Institute for the TGFβ-RIIfl/fl mice. We are grateful to H. Collognato, S. Tsirka, and M. Frohman for critically reading this manuscript and to all our colleagues at the Pharmacology Department at SUNY, Stony Brook, for discussion and support. We also thank I. Galve-Roperh for critically reading this manuscript (Complutense University of Madrid).
The authors declare no competing financial interests.
References
- Aguirre A, Dupree JL, Mangin JM, Gallo V. A functional role for EGFR signaling in myelination and remyelination. Nat Neurosci. 2007;10:990–1002. doi: 10.1038/nn1938. [DOI] [PubMed] [Google Scholar]
- Aguirre A, Rubio ME, Gallo V. Notch and EGFR pathway interaction regulates neural stem cell number and self-renewal. Nature. 2010;467:323–327. doi: 10.1038/nature09347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Billon N, Tokumoto Y, Forrest D, Raff M. Role of thyroid hormone receptors in timing oligodendrocyte differentiation. Dev Biol. 2001;235:110–120. doi: 10.1006/dbio.2001.0293. [DOI] [PubMed] [Google Scholar]
- Calver AR, Hall AC, Yu WP, Walsh FS, Heath JK, Betsholtz C, Richardson WD. Oligodendrocyte population dynamics and the role of PDGF in vivo. Neuron. 1998;20:869–882. doi: 10.1016/S0896-6273(00)80469-9. [DOI] [PubMed] [Google Scholar]
- Cheng X, Wang Y, He Q, Qiu M, Whittemore SR, Cao Q. Bone morphogenetic protein signaling and olig1/2 interact to regulate the differentiation and maturation of adult oligodendrocyte precursor cells. Stem Cells. 2007;25:3204–3214. doi: 10.1634/stemcells.2007-0284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chytil A, Magnuson MA, Wright CV, Moses HL. Conditional inactivation of the TGF-beta type II receptor using Cre:Lox. Genesis. 2002;32:73–75. doi: 10.1002/gene.10046. [DOI] [PubMed] [Google Scholar]
- García-Campmany L, Martí E. The TGFbeta intracellular effector SMAD3 regulates neuronal differentiation and cell fate specification in the developing spinal cord. Development. 2007;134:65–75. doi: 10.1242/dev.02702. [DOI] [PubMed] [Google Scholar]
- Gartel AL, Tyner AL. Transcriptional regulation of the p21((WAF1/CIP1)) gene. Exp Cell Res. 1999;246:280–289. doi: 10.1006/excr.1998.4319. [DOI] [PubMed] [Google Scholar]
- Hart IK, Richardson WD, Bolsover SR, Raff MC. PDGF and intracellular signaling in the timing of oligodendrocyte differentiation. J Cell Biol. 1989;109:3411–3417. doi: 10.1083/jcb.109.6.3411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hiremath MM, Saito Y, Knapp GW, Ting JP, Suzuki K, Matsushima GK. Microglial/macrophage accumulation during cuprizone-induced demyelination in C57BL/6 mice. J Neuroimmunol. 1998;92:38–49. doi: 10.1016/S0165-5728(98)00168-4. [DOI] [PubMed] [Google Scholar]
- Howng SY, Avila RL, Emery B, Traka M, Lin W, Watkins T, Cook S, Bronson R, Davisson M, Barres BA, Popko B. ZFP191 is required by oligodendrocytes for CNS myelination. Genes Dev. 2010;24:301–311. doi: 10.1101/gad.1864510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang H, Zhao XF, Zheng K, Qiu M. Regulation of the timing of oligodendrocyte differentiation: mechanisms and perspectives. Neurosci Bull. 2013;29:155–164. doi: 10.1007/s12264-013-1314-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johns LD, Sriram S. Experimental allergic encephalomyelitis: neutralizing antibody to TGF beta1 enhances the clinical severity of the disease. J Neuroimmunol. 1993;47:1–7. doi: 10.1016/0165-5728(93)90278-7. [DOI] [PubMed] [Google Scholar]
- Keller B, Yang T, Chen Y, Munivez E, Bertin T, Zabel B, Lee B. Interaction of TGFβ and BMP signaling pathways during chondrogenesis. PLoS One. 2011;6:e16421. doi: 10.1371/journal.pone.0016421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kotter MR, Stadelmann C, Hartung HP. Enhancing remyelination in disease–can we wrap it up? Brain. 2011;134:1882–1900. doi: 10.1093/brain/awr014. [DOI] [PubMed] [Google Scholar]
- Kuruvilla AP, Shah R, Hochwald GM, Liggitt HD, Palladino MA, Thorbecke GJ. Protective effect of transforming growth factor beta 1 on experimental autoimmune diseases in mice. Proc Natl Acad Sci U S A. 1991;88:2918–2921. doi: 10.1073/pnas.88.7.2918. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li MO, Flavell RA. TGF-beta: a master of all T cell trades. Cell. 2008;134:392–404. doi: 10.1016/j.cell.2008.07.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li MO, Wan YY, Sanjabi S, Robertson AK, Flavell RA. Transforming growth factor- beta regulation of immune responses. Annu Rev Immunol. 2006;24:99–146. doi: 10.1146/annurev.immunol.24.021605.090737. [DOI] [PubMed] [Google Scholar]
- Li W, Song AP, Zhao F, Hu YM, Hua M. A novel human TINP1 gene promotes cell proliferation through inhibition of p53 and p21 expression. Oncol Rep. 2013;30:1848–1852. doi: 10.3892/or.2013.2647. [DOI] [PubMed] [Google Scholar]
- López-Díaz FJ, Gascard P, Balakrishnan SK, Zhao J, Del Rincon SV, Spruck C, Tlsty TD, Emerson BM. Coordinate transcriptional and translational repression of p53 by TGF-β1 impairs the stress response. Mol Cell. 2013;50:552–564. doi: 10.1016/j.molcel.2013.04.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Massagué J, Gomis RR. The logic of TGFbeta signaling. FEBS Lett. 2006;580:2811–2820. doi: 10.1016/j.febslet.2006.04.033. [DOI] [PubMed] [Google Scholar]
- Massagué J, Blain SW, Lo RS. TGF-β signaling in growth control, cancer, and heritable disorders. Cell. 2000;103:295–309. doi: 10.1016/S0092-8674(00)00121-5. [DOI] [PubMed] [Google Scholar]
- Massagué J, Seoane J, Wotton D. SMAD transcription factors. Genes Dev. 2005;19:2783–2810. doi: 10.1101/gad.1350705. [DOI] [PubMed] [Google Scholar]
- McKinnon RD, Piras G, Ida JA, Jr, Dubois-Dalcq M. A role for TGF-beta in oligodendrocyte differentiation. J Cell Biol. 1993;121:1397–1407. doi: 10.1083/jcb.121.6.1397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meoli EM, Oh U, Grant CW, Jacobson S. TGF-β signaling is altered in the peripheral blood of subjects with multiple sclerosis. J Neuroimmunol. 2011;230:164–168. doi: 10.1016/j.jneuroim.2010.10.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miron VE, Boyd A, Zhao JW, Yuen TJ, Ruckh JM, Shadrach JL, van Wijngaarden P, Wagers AJ, Williams A, Franklin RJ, ffrench-Constant C. M2 microglia and macrophages drive oligodendrocyte differentiation during CNS remyelination. Nat Neurosci. 2013;16:1211–1218. doi: 10.1038/nn.3469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mirshafiey A, Mohsenzadegan M. TGF-beta as a promising option in the treatment of multiple sclerosis. Neuropharmacology. 2009;56:929–936. doi: 10.1016/j.neuropharm.2009.02.007. [DOI] [PubMed] [Google Scholar]
- Misumi S, Kim TS, Jung CG, Masuda T, Urakawa S, Isobe Y, Furuyama F, Nishino H, Hida H. Enhanced neurogenesis from neural progenitor cells with G1/S-phase cell cycle arrest is mediated by transforming growth factor beta1. Eur J Neurosci. 2008;28:1049–1059. doi: 10.1111/j.1460-9568.2008.06420.x. [DOI] [PubMed] [Google Scholar]
- Palazuelos J, Ortega Z, Díaz-Alonso J, Guzmán M, Galve-Roperh I. CB2 cannabinoid receptors signal neural progenitor cell proliferation through PI3K/Akt/mTORC1. J Biol Chem. 2012;287:1198–1209. doi: 10.1074/jbc.M111.291294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qi Y, Cai J, Wu Y, Wu R, Lee J, Fu H, Rao M, Sussel L, Rubenstein J, Qiu M. Control of oligodendrocyte differentiation by the Nkx2.2 homeodomain transcription factor. Development. 2001;128:2723–2733. doi: 10.1242/dev.128.14.2723. [DOI] [PubMed] [Google Scholar]
- Racke MK, Dhib-Jalbut S, Cannella B, Albert PS, Raine CS, McFarlin DE. Prevention and treatment of chronic relapsing experimental allergic encephalomyelitis by transforming growth factor-beta 1. J Immunol. 1991;146:3012–3017. [PubMed] [Google Scholar]
- Radhakrishnan SK, Gierut J, Gartel AL. Multiple alternate p21 transcripts are regulated by p53 in human cells. Oncogene. 2006;25:1812–1815. doi: 10.1038/sj.onc.1209195. [DOI] [PubMed] [Google Scholar]
- Raff MC, Lillien LE, Richardson WD, Burne JF, Noble MD. Platelet-derived growth factor from astrocytes drives the clock that times oligodendrocyte development in culture. Nature. 1988;333:562–565. doi: 10.1038/333562a0. [DOI] [PubMed] [Google Scholar]
- Relucio J, Menezes MJ, Miyagoe-Suzuki Y, Takeda S, Colognato H. Laminin regulates postnatal oligodendrocyte production by promoting oligodendrocyte progenitor survival in the subventricular zone. Glia. 2012;60:1451–1467. doi: 10.1002/glia.22365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rivers LE, Young KM, Rizzi M, Jamen F, Psachoulia K, Wade A, Kessaris N, Richardson WD. PDGFRA/NG2 glia generate myelinating oligodendrocytes and piriform projection neurons in adult mice. Nat Neurosci. 2008;11:1392–1401. doi: 10.1038/nn.2220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rowitch DH, Lu QR, Kessaris N, Richardson WD. An ‘oligarchy’ rules neural development. Trends Neurosci. 2002;25:417–422. doi: 10.1016/S0166-2236(02)02201-4. [DOI] [PubMed] [Google Scholar]
- Samanta J, Kessler JA. Interactions between ID and OLIG proteins mediate the inhibitory effects of BMP4 on oligodendroglial differentiation. Development. 2004;131:4131–4142. doi: 10.1242/dev.01273. [DOI] [PubMed] [Google Scholar]
- Siegel PM, Massagué J. Cytostatic and apoptotic actions of TGF-beta in homeostasis and cancer. Nat Rev Cancer. 2003;3:807–821. doi: 10.1038/nrc1208. [DOI] [PubMed] [Google Scholar]
- Stolt CC, Rehberg S, Ader M, Lommes P, Riethmacher D, Schachner M, Bartsch U, Wegner M. Terminal differentiation of myelin-forming oligodendrocytes depends on the transcription factor Sox10. Genes Dev. 2002;16:165–170. doi: 10.1101/gad.215802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tokumoto YM, Apperly JA, Gao FB, Raff MC. Posttranscriptional regulation of p18 and p27 Cdk inhibitor proteins and the timing of oligodendrocyte differentiation. Dev Biol. 2002;245:224–234. doi: 10.1006/dbio.2002.0626. [DOI] [PubMed] [Google Scholar]
- Vergeli M, Mazzanti B, Ballerini C, Gran B, Amaducci L, Massacesi L. Transforming growth factor-beta 1 inhibits the proliferation of rat astrocytes induced by serum and growth factors. J Neurosci Res. 1995;40:127–133. doi: 10.1002/jnr.490400114. [DOI] [PubMed] [Google Scholar]
- Weng Q, Chen Y, Wang H, Xu X, Yang B, He Q, Shou W, Chen Y, Higashi Y, van den Berghe V, Seuntjens E, Kernie SG, Bukshpun P, Sherr EH, Huylebroeck D, Lu QR. Dual-mode modulation of SMAD signaling by SMAD-interacting protein Sip1 is required for myelination in the central nervous system. Neuron. 2012;73:713–728. doi: 10.1016/j.neuron.2011.12.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yew KH, Hembree M, Prasadan K, Preuett B, McFall C, Benjes C, Crowley A, Sharp S, Tulachan S, Mehta S, Tei E, Gittes G. Cross-talk between bone morphogenetic protein and transforming growth factor signaling is essential for exendin-4-induced insulin-positive differentiation of AR42J cells. J Biol Chem. 2005;280:32209–32217. doi: 10.1074/jbc.M505465200. [DOI] [PubMed] [Google Scholar]
- Yuan X, Chittajallu R, Belachew S, Anderson S, McBain CJ, Gallo V. Expression of the green fluorescent protein in the oligodendrocyte lineage: a transgenic mouse for developmental and physiological studies. J Neurosci Res. 2002;70:529–545. doi: 10.1002/jnr.10368. [DOI] [PubMed] [Google Scholar]
- Zezula J, Casaccia-Bonnefil P, Ezhevsky SA, Osterhout DJ, Levine JM, Dowdy SF, Chao MV, Koff A. p21cip1 is required for the differentiation of oligodendrocytes independently of cell cycle withdrawal. EMBO Rep. 2001;2:27–34. doi: 10.1093/embo-reports/kve008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu X, Bergles DE, Nishiyama A. NG2 cells generate both oligodendrocytes and gray matter astrocytes. Development. 2008;135:145–157. doi: 10.1242/dev.004895. [DOI] [PubMed] [Google Scholar]