

Abstract
Elevated mammalian target of rapamycin (mTOR) signaling has been found in Alzheimer's disease (AD) patients and is linked to diabetes and aging, two known risk factors for AD. However, whether hyperactive mTOR plays a role in the cognitive deficits associated with AD remains elusive. Here, we genetically reduced mTOR signaling in the brains of Tg2576 mice, a widely used animal model of AD. We found that suppression of mTOR signaling reduced amyloid-β deposits and rescued memory deficits. Mechanistically, the reduction in mTOR signaling led to an increase in autophagy induction and restored the hippocampal gene expression signature of the Tg2576 mice to wild-type levels. Our results implicate hyperactive mTOR signaling as a previous unidentified signaling pathway underlying gene-expression dysregulation and cognitive deficits in AD. Furthermore, hyperactive mTOR signaling may represent a molecular pathway by which aging contributes to the development of AD.
Keywords: Aβ, amyloid-β, autophagy, plaques, tangles, tau
Introduction
Alzheimer's disease (AD) is the most common neurodegenerative disorder and the sixth leading cause of death in the United States. Currently, it is estimated that 26 million people worldwide are living with AD and by 2050 the number of people with this disorder can reach 100 million (Thies et al., 2013). AD is characterized by progressive cognitive deficits associated with the build-up of amyloid-β (Aβ) and neurofibrillary tangles. The majority (>95%) of AD cases are sporadic and of unknown etiology, whereas the remaining cases are caused by mutations in one of three genes, presenilin 1 and 2, and amyloid precursor protein (Querfurth and LaFerla, 2010). The single major risk factor for AD is aging; however, little is known as to how the aging process facilitates the development of AD. Nevertheless, molecular changes that occur in the brain as a function of age may facilitate the development of AD.
Overwhelming evidence shows that reducing the activity of the mammalian target of rapamycin (mTOR) increases lifespan and health-span in several genetically different species (Lamming et al., 2013). mTOR is a ubiquitously expressed protein kinase that plays a key role in regulating protein synthesis and cell growth by phosphorylating downstream proteins such as p70 S6 kinase and eukaryotic initiation factor eIF4E binding protein (4E-BP; Wullschleger et al., 2006). mTOR also is a negative regulator of autophagy induction(Wullschleger et al., 2006). By simultaneously regulating protein synthesis and degradation, mTOR is key in controlling protein homeostasis, a process that is altered in AD and other proteinopathies (Lee et al., 2013). Another known function of mTOR signaling is the regulation of synaptic plasticity and function (Tang et al., 2002; Hoeffer et al., 2008; Hoeffer and Klann, 2010; Santini et al., 2014). Toward this end, mTOR activity is necessary for memory consolidation (Tang et al., 2002; Parsons et al., 2006); however, mTOR hyperactivity is detrimental and causes cognitive deficits in animals and people (Ehninger et al., 2008; Puighermanal et al., 2009; Ehninger, 2013).
Several laboratories have shown that mTOR signaling is upregulated in AD patients and animal models (An et al., 2003; Caccamo et al., 2010; Oddo, 2012). Thus, one might expect that lifestyle choices known to increase mTOR signaling, such as a high-sugar or high-fat diet increase the risk of developing AD. Toward this end, we and others have shown that diabetes increases the risk of developing AD by an mTOR-dependent mechanism (Y.Q. Ma et al., 2013; Orr et al., 2014). In contrast, some of the beneficial effects of caloric restrictions on brain function might be mediated by reduction in mTOR signaling (Speakman and Mitchell, 2011). To fully dissect the role of mTOR in the pathogenesis of AD, we used a genetic approach and selectively ablated one mTOR gene from the forebrain of an animal model of AD. Our results highlight a previously unidentified signaling pathway as a key player in AD pathogenesis and offer a potential therapeutic target for this insidious disorder.
Materials and Methods
Mice.
The Tg2576 and CaMKII-CRE mice were purchased from The Jackson Laboratory. The mTOR floxed mice [Mtortm1.1Clyn Mtor<tm1.1Clyn> Targeted Allele Detail MGI Mouse (MGI:4820819)] were a generous gift from Dr. Christopher Lynch, Penn State University College of Medicine. All mice were housed 4–5 to cage, kept on 12 h light/dark cycle and were given ad libitum access to food and water. For each experiment, an equal number of males and females were used. All animal procedures were approved by The Institutional Animal Care and Use Committee of the Banner Sun Health Research Institute.
Morris water maze.
This test was performed in a circular plastic tank of 1.5 m diameter filled with water kept at 25°C. A platform (14 cm diameter) was kept 1.5 cm under the surface of the water and made invisible to mice by adding white nontoxic paint to the water. The tank was in a room with several extramaze visual cues, which served as reference points for mice. The location of the cues and platform were kept constant throughout the testing period. Mice were trained to find the hidden platform for 5 consecutive days, four training trials per day. Before the first trial of the first day, mice were placed on the platform for 10 s, after which they were placed in the water until they reached the platform for a maximum of 60 s. When a mouse found the platform, it was placed in a warm holding cage for 25 s before starting the next trial. If a mouse failed to find the platform in 60 s, it was gently guided to the platform location and allowed to stay on it for 10 s after which it was placed in the warm holding cage. Extreme care was taken to minimize animal stress during these procedures. Spatial memory was assessed during a 60 s probe trial conducted 24 h after the last training trial. During the probe trials, the platform was removed from the water and mice were allowed to freely swim in the tank for 60 s. The entire test was recorded by a video camera mounted on the ceiling. Data were obtained using specialized tracking software (EthoVision XT, Noldus).
Protein extraction.
Mice were killed by CO2 asphyxiation and their brains removed and sagittally bisected. Half of the brain was dropped-fixed in 4% paraformaldehyde and used for histological and immunohistochemical experiments. The hippocampus and cortex were removed from the other half and stored at −80°C until use. Frozen hippocampi were homogenized using a dounce homogenizer in T-PER buffer (Thermo Scientific) supplemented with 0.7 mg/ml pepstatin A and a mini-protease inhibitor tablet and phosphatase inhibitors. Samples were then centrifuged at 25,000 × g for 30 min at 4°C. The supernatant was stored as soluble fraction and used for Western blot and ELISA experiments. The pellet was homogenized in 70% formic acid and centrifuged as described above. The supernatant of this second centrifugation was stored as insoluble fraction and used for ELISA experiments.
Western blots and ELISA.
Proteins from the soluble fraction were loaded on precast SDS/PAGE gels and run under reducing conditions, after which they were transferred to a nitrocellulose membrane. Membranes were then incubated in a 5% milk solution in T-TBS (0.1% Tween 20, 100 mm Tris, pH 7.5; 150 mm NaCl) 1 h at 25°C, washed and incubated in primary antibody overnight at 4°C. Membranes were washed in T-TBS for 30 min and incubated in goat anti-mouse IRDye 680LT or goat anti-rabbit IRDye 800CW LI-COR secondary antibodies (1:10,000) for 1 h at 25°C. After final washes, membranes were imaged and analyzed using the LI-COR Odyssey. Protein densitometry was calculated by dividing the integrated intensity of the protein of interest by integrated intensity of β-actin loading control. ELISA experiments were conducted using precoated Aβ40 and Aβ42 Invitrogen plates by following the manufacturer's protocol.
Immunohistochemistry.
Fixed hemibrains were sliced into 50-μm-thick free-floating sections using a vibratome. Sections were stored in 0.02% sodium azide in PBS at 4°C until use. On the day of the experiment, sections were washed twice with TBS (100 mm Tris, pH 7.5; 150 mm NaCl) and incubated in 3% H2O2 for 30 min at 25°C to quench endogenous peroxidase activity. Next, sections were washed with TBS-A (100 mm Tris, pH 7.5; 150 mm NaCl; 0.1% Triton X-100) and TBS-B (100 mm Tris, pH 7.5; 150 mm NaCl; 0.1% Triton X-100; 2% BSA) for 15 and 30 min, respectively, to block nonspecific binding. Sections were then incubated overnight at 4°C with the appropriate primary antibody. Subsequently, sections were washed in TBS-A and TBS-B to remove excess antibody and incubated in the suitable secondary antibody for 1 h at 25°C. Signal was enhanced by incubating sections in the avidin-biotin complex (Thermo Scientific) for 1 h at 25°C. Sections were then washed and developed with diaminobenzidine substrate using the avidin–biotin horseradish peroxidase system (Vector Labs). Images were obtained with a digital Zeiss camera and analyzed with ImageJ software. Quantification of staining was achieved using pixilation detection acquired by ImageJ. A threshold was set using a positive control and a standard mean gray area function, which allowed the set software to recognize positive staining and decrease error caused by background staining.
Microarray.
The microarray experiments were performed by the genomic core at the University of Texas Health Science Center at San Antonio. RNA was extracted using the mirVana RNA Isolation kit (Life Technologies), following the manufacturer's instructions. To check for RNA integrity, 1 μl of RNA was run on the Agilent 2100 Bioanalyzer (using the RNA nanochip). Average RIN values were between 8.3 and 9.3. cRNA probes (from 500 ng RNA) were made using the Ambion Illumina TotalPrep RNA Amplification Kit (Life Technologies) following the manufacture's protocol. We then measured cRNA concentration and prepared 150 ng/μl dilutions. Subsequently, we assessed the cRNA size distribution profiles by loading 1 μl onto the Agilent Bioanalyzer. We then loaded 1500 ng cRNA onto 4 Illumina Mouse WG-6 v2 Gene Expression beadchips, hybridized 18 h overnight, washed, and scanned on the Illumina iScan following the manufacturer's protocol. Data were analyzed using GeneSpring.
Antibodies.
From Cell Signaling Technology: mTOR (1:1000), total p70S6K (1:1000), p70S6K Thr389 (1:1000), 4EBP1 (1:1000), 4EBP1 Ser65 (1:1000), β-actin (1:10,000), Atg3 (1:1000), Atg5 (1:1000), Atg7 (1:1000), Atg12 (1:1000), PSD95 (1:1000), ULK-1 (1:1000), and NCAM (1:1000). From Millipore, anti-Aβ42 (1:200) and synaptophysin (1:1000). From Covance, 6E10 (1:3000). From Calbiochem, CT20 (1:3000). From Abcam, DCTN-1 (1:1000).
Statistical Analyses.
All data were analyzed using GraphPad Prism, GraphPad Software. Data were analyzed by one- or two-way ANOVA followed by Bonferroni's post hoc analysis, when applicable. Selective experiments were analyzed by Student's t test, as specified in the results section.
Results
Removing one copy of the mTOR gene decreases hippocampal mTOR signaling
To study the role of mTOR in AD, we genetically and selectively removed one mTOR allele from the brains of the Tg2576 mice, a mouse model of AD (Hsiao et al., 1996). This was accomplished by taking advantage of the Cre-Lox site-specific recombination. Precisely, to obtain brain specific mTOR knock-out mice, we first bred hemizygous Tg2576 mice (Tg2576+/0) with homozygous transgenic mice overexpressing the Cre recombinase enzyme under the control of the neuronal specific CaMKII promoter (CRE+/+). As expected ∼50% of the offspring had the following genotype: Tg2576+/0;Cre+/0 (Fig. 1). These mice were bred with homozygous mTOR floxed mice (mTORfl/fl; Lang et al., 2010; Carr et al., 2012). From these crosses, we obtained mice with four genotypes: (1) Tg2576+/0;mTORfl/wt;Cre+/0; (2) Tg25760/0;mTORfl/wt;Cre+/0; (3) Tg2576+/0;mTORfl/wt;Cre0/0; and (4) Tg25760/0;mTORfl/wt;Cre0/0 (Fig. 1). Notably, all four groups of mice are littermates, thereby reducing the genetic background effects on the phenotype. Using this breeding strategy, we removed a single copy of the mTOR gene in the forebrain of the Tg2576 mice. For simplicity, herein the mice with the Tg2576+/0;mTORfl/wt;Cre+/0 genotype are referred to as APP/mTOR+/− mice, the mice with the Tg25760/0;mTORfl/wt;Cre+/0 genotype as mTOR+/− mice, the mice with the Tg2576+/0;mTORfl/wt;Cre0/0 genotype as APP mice, and the mice with the Tg25760/0;mTORfl/wt; Cre0/0 genotype as CTL mice.
Figure 1.

Schematic representation of the breeding strategy used to remove one copy of the mTOR gene from the forebrain of the Tg2576 mice. Abbreviations: “+” indicates the presence of a transgene (APP or CRE); “0” indicates the lack of such transgene; “fl” indicates the presence of a floxed allele; and “wt” indicates the presence of a wild-type allele.
To determine whether removing one copy of the mTOR gene was sufficient to reduce brain mTOR levels and signaling, we measured mTOR levels by Western blots in the hippocampi of 2- and 12-month-old mice (n = 5/genotype/age). We found that in 2-month-old mice, mTOR levels were significantly different among the four genotypes (p = 0.01; Fig. 2A,B). Specifically, mTOR levels were 47 ± 6% and 52 ± 6% lower in the APP/mTOR+/− and mTOR+/− mice compared with the CTL mice, respectively. 4EBP-1 and p70S6K are two proteins downstream of mTOR, which directly phosphorylates them at Thr389 and Ser65; the levels of phosphorylated 4EBP-1 and p70S6K are used as an indication of activated mTOR signaling (Hay and Sonenberg, 2004; Guertin and Sabatini, 2007). In the hippocampi of 2-month-old mice, the total levels of p70S6K and 4EBP-1 were similar among the four groups. In contrast, we found that the levels of p70S6K phosphorylated at Thr389 and 4EBP-1 phosphorylated at Ser65 were significantly different among the four genotypes as indicated by one-way ANOVA (p < 0.001 for both; Fig. 2A–F). A post hoc test with Bonferroni correction showed that the phosphorylation levels of p70S6K and 4EBP-1 were significantly reduced in the APP/mTOR+/− and mTOR+/− mice compared with CTL and APP mice (Fig. 2D,F). To determine whether the changes in mTOR levels and signaling were maintained in older mice, we repeated these measurements in 12-month-old mice (n = 5/genotype). We found that mTOR levels were 37% and 40% lower in the hippocampi of the APP/mTOR+/− and mTOR+/− mice compared with CTL mice, respectively (Fig. 2G,H). Although these changes did not affect total p70S6K and 4EBP-1, one-way ANOVA indicated that p70S6K levels phosphorylated at Thr389 and 4EBP-1 levels phosphorylated at Ser65 were significantly different among the four groups (p = 0.0003 and 0.0029, respectively; Fig. 2G,I–L). A post hoc test with Bonferroni correction showed that phosphorylated levels of p70S6K were significantly higher in the APP mice compared with CTL mice (p < 0.01). However, the phosphorylated levels of p70S6K in the hippocampi of the APP/mTOR+/− were not statistically different from CTL mice and were significantly reduced compared with the APP mice (p < 0.01; Fig. 2J). Similarly, the phosphorylated levels of 4EBP-1 were significantly reduced in the APP/mTOR+/− mice compared with the APP mice (p < 0.05; Fig. 2L). These data clearly show that removing one copy of the mTOR gene in the brains is sufficient to reduce mTOR levels and signaling. Notably, this approach restored the elevated levels of phospho p70S6K of the APP mice to CTL levels.
Figure 2.

Removing one copy of the mTOR gene decreases mTOR levels and signaling. A, Representative Western blots of proteins extracted from the hippocampi of 2-month-old mice probed with the indicated antibodies. B–F, Quantification of the indicated proteins (n = 5/genotype) showed that mTOR levels and the levels of phosphorylated p70S6K and 4EBP1 were significantly decreased in the hippocampi of mice lacking one copy of the mTOR gene. G, Representative Western blots of proteins extracted from the hippocampi of 12-month-old mice probed with the indicated antibodies. H–L, Quantification of the indicated proteins (n = 5/genotype) showed that even as the mice age, mTOR levels and signaling were significantly lower in mice lacking one copy of the mTOR gene. Data were generated by normalizing the levels of the protein of interest to β-actin loading control. Results presented as means ± SEM and analyzed by one-way ANOVA with Bonferroni's correction; *p < 0.05, **p < 0.005.
Reducing mTOR signaling rescues cognitive deficits in the Tg2576 mice
To evaluate the effects of removing one copy of the gene encoding mTOR from the brains of the Tg2576 mice, we tested 2- and 12-month-old mice in the spatial version of the Morris water maze (MWM), using two independent cohorts of mice. Mice received four training trials per day for 5 consecutive days to learn the location of a hidden platform using extra maze cues. Their performance was analyzed using a mixed-model, repeated-measures ANOVA, with genotype as the categorically fixed effects, days as the numeric covariate, animals as the random effect, and escape latency as the dependent variable. At 2 months of age, we found a significant effect for days (p < 0.0001), indicating that the mice learned the task across sessions (Fig. 3A). However, we found a nonsignificant genotype/day interaction (p > 0.05), indicating that there was no difference in the pace of learning among the four different groups (Fig. 3A). Twenty-four hours after the last training trial, we tested spatial memory by measuring the time mice spent in the target quadrant, the latency to reach the platform location, and the number of platform location crosses over a 60 s probe trial. We found no statistically significant changes in these three measurements among the four different genotypes (Fig. 3B–D). To determine whether the physical performance of the mice may have confounded these results, we measured their swim speed during the probe trials and found that it was not statistically different among the four genotypes (Fig. 3E). This is consistent with data in the literature showing that at this age, the Tg2576 mice are not cognitively impaired (Westerman et al., 2002).
Figure 3.
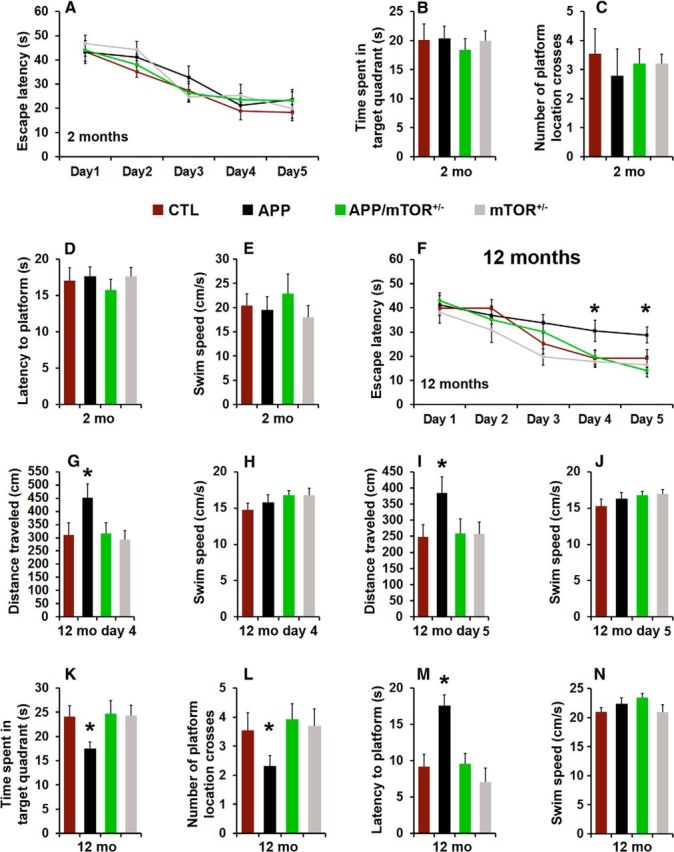
Decreasing mTOR signaling rescues learning and memory deficits. A, Learning curve of 2-month-old mice trained in the spatial reference version of the MWM. Mice were trained to swim to a hidden platform in a tank using extramazal visual cues. All genotypes showed significant improvements over the 5 d of training. Each day represents the average of four training trials. B–D, Reference memory, tested 24 h after the last training trial was not statistically different among the four groups. E, Average swim speed during the probe trials. F, Learning curve of 12-month-old mice in the MWM. All genotypes significantly learned the task; however, at days 4 and 5 the APP mice needed significantly more time to find the hidden platform than the other three genotypes. G, H, Average distance traveled and swim speed of 12-month-old mice during day 4 of training. I, J, Average distance traveled and swim speed of 12-month-old mice during day 5 of training. K–M, Reference memory, tested 24 h after the last training trial was significantly impaired in the APP mice compared with the other three genotypes. Notably, the APP/mTOR+/− mice performed as well as the CTL mice and significantly better than the APP mice. N, Average swim speed during the probe trials was not statistically different among the four genotypes. n = 10/genotype at 2 months of age; n = 14/genotype at 12 months of age. Data are presented as means ± SEM and were analyzed by two-way ANOVA with Bonferroni's correction; *p < 0.05.
As the Tg2576 mice age, they develop progressive cognitive deficits in several behavioral tests, including the MWM (Westerman et al., 2002). At 12 months of age, we found a significant effect for days (p < 0.0001) and genotype (p = 0.0009; Fig. 3F). The day effect indicates that the mice significantly improve their performance across sessions whereas the genotype effect indicates that one or more genotypes are different from each other. A post hoc test with Bonferroni correction showed that at days 4 and 5, the APP mice performed significantly worse than the other three groups (p = 0.039 and 0.009, respectively; Fig. 3F). Notably, the APP/mTOR+/− mice performed as well as the CTL mice across the 5 d of training. To determine whether differences in physical performance could account for the changes in escape latency on days 4 and 5, we measured the average distance traveled and the swim speed of the four groups of mice. Consistent with the escape latency data, the APP mice traveled longer to find the platform than the other groups for both days (p < 0.05; Fig. 3G,I). In contrast, the average swim speed was similar among the different groups for both days (Fig. 3H,J). Twenty-four hours after the last training trial, we conducted probe trials to measure spatial memory. In every measurement, APP mice performed significantly worse than the other three groups (p < 0.01 as calculated by one-way ANOVA; Fig. 3K–M). The swim speed of 12-month-old mice was not statistically significant among the four genotypes (Fig. 3N), clearly indicating that the genotype effects on learning and memory are independent of physical performance. These findings indicate that removing one copy of the mTOR gene from the brains of the Tg2576 mice is sufficient to rescue cognitive deficits.
Reducing mTOR signaling decreases Aβ levels and burden and increases synaptophysin levels
At the end of the behavioral tests, mice were killed and their brains further analyzed. We initially focused on Aβ pathology, as this is the major neuropathological feature of the Tg2576 mice (Kawarabayashi et al., 2001). We immunostained sections from APP and APP/mTOR+/− mice with an Aβ42 specific antibody and found that the Aβ42 immunoreactivity was significantly reduced in the APP/mTOR+/− mice compared with the APP mice (Fig. 4A,B). Quantitative analysis of the overall Aβ load indicated a decrease of 46.77%, which was highly significant (p = 0.0004; Fig. 4C). We next measured Aβ levels by sandwich ELISA and found that soluble Aβ40 and Aβ42 levels were significantly lower in the hippocampi of the APP/mTOR+/− mice compared with APP mice (p < 0.0001 and p = 0.0024, respectively; Fig. 4D,E). Similarly, insoluble Aβ40 and Aβ42 levels were significantly lower in the hippocampi of the APP/mTOR+/− mice compared with the APP mice (p < 0.0001 for both Aβ species; Fig. 4F,G). To determine whether alterations in APP processing could account for the changes in Aβ levels between the APP and the APP/mTOR+/− mice, we measured the levels of full-length APP and its two major C-terminal fragments, C99 and C83 in the hippocampi of 2- and 12-month-old mice. We found that at both ages, full-length APP levels were not different between the two groups (Fig. 5A–C). Similarly, C99 and C83 levels were comparable between APP and APP/mTOR+/− mice at both ages (Fig. 5A,D–G). These results suggest that removing one copy of the mTOR gene is sufficient to reduce Aβ burden and levels without altering APP processing.
Figure 4.
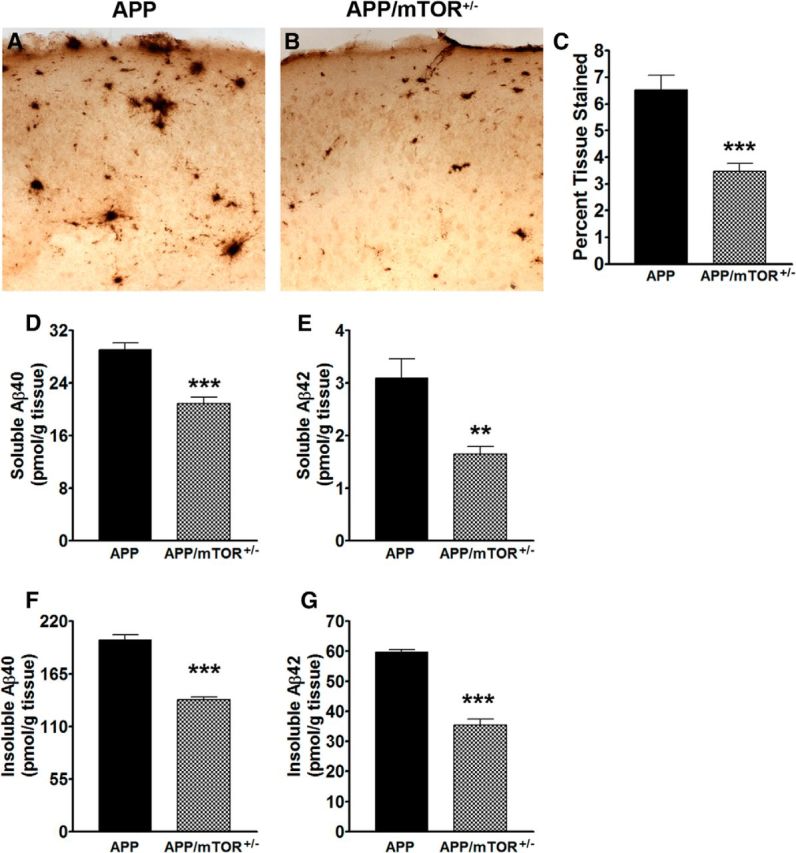
APP/mTOR+/− mice have less Aβ pathology than APP mice. A, B, Representative microphotographs of brain section immunostained with an Aβ42 specific antibody. C, Quantitative analysis of the Aβ immunohistochemistry showed a significant decrease in Aβ load following removal of one copy of the mTOR gene (n = 7/genotype). D, E, Soluble levels of Aβ40 and Aβ42 extracted from frozen hippocampi and measured by sandwich ELISA (n = 8/genotype). F, G, Insoluble levels of Aβ40 and Aβ42 extracted from frozen hippocampi and measured by sandwich ELISA (n = 8/genotype). Data are presented as means ± SEM and were analyzed by t test; **p = 0.002, ***p < 0.0001.
Figure 5.

APP processing is not changed between APP and APP/mTOR+/− mice. A, Representative Western blots of proteins extracted from frozen hippocampi of 2- and 12-month-old mice and probed with the indicated antibodies. B–G, Quantification of the blots shows that the levels of APP, C99 and C83 were not statistically significant between APP/mTOR+/− and APP mice, at any of the age analyzed (n = 5/genotype/time point). Data are presented as means ± SEM and were analyzed by t test.
Synaptic dysfunction is a strong correlate of cognitive deficits in AD and, at least in mouse models, correlates with changes in Aβ. To determine whether the changes in Aβ pathology in the APP/mTOR+/− mice were followed by changes in synaptic markers, we measured the steady-state levels of synaptophysin and PSD95, two commonly used presynaptic and postsynaptic markers, respectively. We found that synaptophysin levels were significantly different among the three groups (p = 0.025 by one-way ANOVA; Fig. 6A,B). Bonferroni's post hoc analysis showed that synaptophysin levels were significantly higher in the APP/mTOR+/− mice compared with APP mice. Notably, synaptophysin levels were similar between CTL and APP/mTOR+/− mice, suggesting that removing one copy of the mTOR gene was sufficient to rescue hippocampal synaptophysin levels. We found a similar trend for PSD95, although the changes did not reach significance (Fig. 6A,C).
Figure 6.

Increased synaptophysin levels in APP/mTOR+/− mice. A, Representative Western blots of protein extracted from 12-month-old CTL, APP and APP/mTOR+/− mice. Blots were probed with the indicated antibodies. B, C, Quantification of the blots shows that synaptophysin levels were restored to CTL levels in the APP/mTOR+/− mice. Although the same trend was apparent for PSD95, the changes were not statistically significant. Data are presented as means ± SEM and were analyzed by one-way ANOVA with Bonferroni correction; *p < 0.05.
Reducing brain mTOR signaling increases autophagy induction
To mechanistically link the reduction in mTOR signaling to cognitive improvements and the decrease in Aβ pathology, we analyzed the two major pathways downstream of mTOR. Specifically, when cell energy levels are high, mTOR facilitates protein translation and transcription; in contrast, when cell energy levels are low, mTOR facilitates autophagy for protein turnover (Wullschleger et al., 2006). Dysregulation in both of these processes has been linked to AD (Nixon and Yang, 2011; Caldeira et al., 2013; Orr and Oddo, 2013). We first measured the levels of four key autophagy related proteins (Atgs), which have been shown to be critical in autophagy induction (Mizushima et al., 1998; Komatsu et al., 2007). We found that Atg3 levels were increased by 75.6% in the hippocampi of APP/mTOR+/− mice compared with APP mice (Fig. 7A,B); however, this change was not statistically significant (p = 0.0509) likely due to the variability within the APP/mTOR+/− mice. In contrast, Atg7 levels were increased by 62.61% in the hippocampi of APP/mTOR+/− mice compared with APP mice (p < 0.006; Fig. 7A,C). Similarly, we found an increase of 62.76 and 76.36% in Atg5 and Atg12 levels, respectively, following the removal of a copy of the mTOR gene (p = 0.01 and p = 0.03 for Atg5 and Atg12, respectively; Fig. 7A,D–E). In the autophagy pathway, once the autophagosome are formed, they fuse with the lysosomes where their cargo is digested (Tanida, 2011). To assess whether increasing autophagy induction was sufficient to facilitate Aβ turnover by the lysosomes, we double-labeled hippocampal sections from APP and APP/mTOR+/− mice with an Aβ42-specific antibody and with Lamp2A, a lysosomal marker. Confocal imaging showed very little colocalization between Aβ and Lamp2A in the APP mice (Fig. 7F). In contrast, the APP/mTOR+/− mice showed a clear and robust colocalization between Aβ and Lamp2A (Fig. 7F), suggesting that upon autophagy induction, Aβ was delivered to the lysosomes for degradation. To better assess the degree of this change, we quantified the number of yellow pixels in a visual field and found a significant 67% increase in the APP/mTOR+/− than the APP mice (Fig. 7G). These data suggest an increase in autophagy induction in the hippocampi of the APP/mTOR+/− mice compared with APP mice.
Figure 7.

Decreasing mTOR signaling increases autophagy induction. A, Representative Western blots of protein extracted from 12-month-old APP and APP/mTOR+/− mice. Blots were probed with the indicated antibodies. B–E, Quantification of the blots shows that the levels of Atg3, Atg5, Atg7 and Atg12 were significantly higher in the APP/mTOR+/− than APP mice (n = 5/genotype). F, Representative microphotographs of hippocampal sections immunostained with the indicated antibodies. G, Semiquantitative analysis showed that the number of yellow pixels (indicating a colocalization between Aβ and the lysosomal protein Lamp2A) was significantly higher in APP/mTOR± mice compared with APP mice (n = 7/genotype). Data are presented as means ± SEM and were analyzed by t test; *p < 0.01, **p < 0.001.
Reducing mTOR signaling restores the gene expression profile of the Tg2576 mice to CTL levels
We next examined global gene expression in the hippocampi of 12-month-old CTL, APP, APP/mTOR+/−, and mTOR+/− mice (n = 6 /genotype) and found that the expression of 266 transcripts was significantly changed among the four groups of mice (one-way ANOVA, p < 0.05; Fig. 8A). A post hoc analysis with Bonferroni correction revealed that 227 of these genes were differentially expressed between APP and APP/mTOR+/− mice (Fig. 8A). The top 20 transcripts showing greatest change in expression between APP and APP/mTOR+/− are summarized in Table 1. To further evaluate the overall structure of the transcriptional changes, we performed a cluster analysis of gene expression levels by clustering all transcripts in an unbiased fashion to create a hierarchical tree. We found that 38 transcripts clustered according to genotype. Specifically, APP/mTOR+/− and CTL mice cluster together, which highlights how reducing mTOR signaling restored the hippocampal gene expression signature of the APP mice to CTL levels (Fig. 8B). To validate the microarray data, we randomly selected three transcripts and measured their equivalent protein levels by Western blots. In all three cases we found that the microarray data closely reflected the Western blots results (Fig. 8C–E). Overall these results clearly indicate that the expression profile of APP mice is altered compared with CTL mice; notably, removing one copy of the mTOR gene was sufficient to reverse the expression of many of the genes altered in the APP mice to CTL levels (Fig. 8).
Figure 8.

Removing one copy of the mTOR gene restores the hippocampal expression profiles of the APP mice to the CTL levels. A, The graph summarizes the expression analysis results from transcripts isolated from frozen hippocampi (n = 6/genotype), indicating the number of genes measured, how many of those were differentially expressed among the four genotypes, and how many of those were differentially expressed between APP and APP/mTOR+/− mice. B, Cluster analysis of samples from all genotypes with a fold-change ≥ 1.8-fold when comparing CTL with APP mice. Each column represents a single sample, whose genotype is color labeled above the cluster diagram. Each row represents a specific transcript, whose identity is listed to the right of the cluster diagram. C–E, Validation of microarray results of three random genes by Western blot. Each graph shows the expression levels of each transcript (relative to CTL) as found by microarray and Western blots. For the Western blots (n = 6/genotype). Data are presented as means ± SEM and were analyzed by one-way ANOVA; *p < 0.05.
Table 1.
Top 20 transcripts showing greatest change in expression between APP and APP/mTOR+/−
| Probe ID | Symbol | Definition | ANOVA p values (among the 4 groups) | FC in APP/mTOR+/− relative to APP | Regulation | FC in APP relative to CTL | Regulation |
|---|---|---|---|---|---|---|---|
| 2900154 | Dctn1 | Dynactin 1 | 0.00105 | 4.66475 | Up | 2.91486 | Down |
| 240025 | Islr2 | Immunoglobulin superfamily containing leucine-rich repeat 2 | 0.00539 | 2.37943 | Up | 1.59996 | Down |
| 2750066 | Rn18s | 18S RNA (Rn18s), noncoding RNA | 0.00001 | 2.3704 | Up | 2.0421 | Down |
| 6510615 | A130022A09Rik | RIKEN cDNA A130022A09 | 0.02492 | 2.36581 | Down | 1.60222 | Up |
| 7050673 | Zxda | Zinc finger, X-linked, duplicated A, noncoding RNA | 0.00042 | 2.13611 | Down | 1.83545 | Up |
| 5080064 | Slc8a2 | Solute carrier family 8 (sodium/calcium exchanger), member 2 | 0.00106 | 2.0633 | Up | 1.67871 | Down |
| 6130482 | C230078M08Rik | RIKEN cDNA C230078M08 gene | 0.00636 | 1.9872 | Up | 1.55682 | Down |
| 840224 | 2610301N02Rik | RIKEN cDNA 2610301N02 gene | 0.00868 | 1.97983 | Down | 1.37266 | Up |
| 3520240 | Rn18s | 18S RNA (Rn18s), noncoding RNA | 0.00001 | 1.971 | Up | 1.7302 | Down |
| 6480044 | Arl4c | ADP-ribosylation factor-like 4C | 0.01214 | 1.96944 | Down | 1.75136 | Up |
| 3870132 | 6330415B21Rik | RIKEN cDNA 6330415B21 gene | 0.0343 | 1.9576 | Down | 1.1986 | Up |
| 1780707 | 9530053J19Rik | RIKEN cDNA 9530053J19 gene | 0.02255 | 1.93924 | Down | 1.44845 | Up |
| 4890670 | Iqsec2 | IQ motif and Sec7 domain 2, transcript variant 2 | 0.00452 | 1.93587 | Up | 1.2717 | Down |
| 1710735 | LOC381219 | Hypothetical LOC381219 | 0.00735 | 1.93024 | Down | 1.33752 | Up |
| 650189 | 3632431M01Rik | Transmembrane and coiled coil domains 1 | 0.04541 | 1.92024 | Down | 1.05674 | Up |
| 5570097 | 5330431K02Rik | RIKEN cDNA 5330431K02 gene | 0.02922 | 1.91527 | Down | 1.3845 | Up |
| 1780037 | BC030476 | cDNA sequence BC030476 | 0.01214 | 1.91476 | Down | 1.52354 | Up |
| 1940356 | 6030424L16Rik | RIKEN cDNA 6030424L16 gene | 0.01271 | 1.91042 | Down | 1.59773 | Up |
| 4040768 | Rn18s | 18S RNA (Rn18s), noncoding RNA | 0.000004 | 1.90669 | Up | 1.68662 | Down |
| 4540102 | BC030499 | cDNA sequence BC030499 | 0.01747 | 1.90504 | Down | 1.53121 | Up |
FC, Fold-change.
Discussion
The data presented here unambiguously show that reduction of mTOR levels and signaling has beneficial effects on AD-like pathology developed by the Tg2576 mice. Indeed, we reported that APP/mTOR+/− mice performed significantly better than APP mice and as well as CTL mice in a spatial learning and memory task. This cognitive improvement was associated with a reduction in Aβ pathology and profound changes in the hippocampal transcriptional signature.
Several laboratories have reported that mTOR hyperactivity is increased in postmortem human brains compared with age-matched controls (Chang et al., 2002; An et al., 2003; Peel and Bredesen, 2003; Griffin et al., 2005; Pei and Hugon, 2008; Pei et al., 2008). Consistently, we have shown that soluble Aβ accumulation is sufficient to increase mTOR activity and signaling in vivo (Caccamo et al., 2011). Along these lines, here we show that mTOR is hyperactive in the hippocampi of 12-month-old APP mice, which are characterized by widespread Aβ accumulation. Notably, mTOR hyperactivity has been linked to increased tau levels and phosphorylation in drosophila and mice (Khurana et al., 2006; Caccamo et al., 2013; Tang et al., 2013). Thus, mTOR might represent a molecular link between Aβ accumulation and tau pathology. Further, overwhelming evidence shows that reducing mTOR signaling increases lifespan and health-span in a variety of organisms, including mammals (Harrison et al., 2009; Selman et al., 2009). Given that aging is by far the strongest risk factor for AD, we hypothesize that one way by which aging facilitates the development of AD is by altering mTOR activity. Consistent with this hypothesis, we and others have shown that rapamycin, an mTOR inhibitor, increases lifespan in genetically heterogeneous mice and decreases AD-like pathology in two independent animal models of AD (Harrison et al., 2009; Caccamo et al., 2010; Spilman et al., 2010; Majumder et al., 2011).
In adult brains, mTOR regulates several protein-dependent events that are critical for learning and memory (Hoeffer et al., 2008; Klann and Sweatt, 2008; Hoeffer and Klann, 2010; Graber et al., 2013). However, strong evidence also shows that mTOR hyperactivity is detrimental for normal brain function (Ehninger et al., 2008; Cao et al., 2009; Puighermanal et al., 2009; Zeng et al., 2009; Ehninger, 2013). For example, clinical mutations in the tuberous sclerosis complex lead to mTOR hyperactivity and learning disabilities in affected patients (Ehninger, 2013). These findings have been replicated in animal models (Ehninger et al., 2008). Consistently, low doses of rapamycin rescue memory deficits associated with cannabinoids consumption (Puighermanal et al., 2009). Overall, there might be a window for mTOR signaling that is optimal for learning and memory and that alterations leading to an excessive increase or decrease in mTOR signaling outside such optimal window may have detrimental effects on learning and memory (Inoki et al., 2005; Cao et al., 2009; Zeng et al., 2009). Given the above mentioned evidence, our experiments were purposely designed to remove only one copy of the mTOR gene from the brain of APP mice. Notably, we showed that this approach reduced but, not abolished mTOR signaling. Indeed, at 12 months of age, mTOR signaling was similar between APP/mTOR+/− and CTL mice; in other words, we successfully reduced the hyperactive mTOR signaling of the APP mice to CTL levels. We postulate that major reductions in brain mTOR signaling might have detrimental effects on learning and memory.
It is not surprising that the transcriptome of AD patients is altered compared with age-matched controls (Caldeira et al., 2013). Nevertheless, detailed analyses of these experiments have led to critical insights into the disease pathogenesis. Some of the transcriptome changes seen in AD brains have been replicated in mouse models. Along these lines, the transcriptome of the Tg2576 mice has also been found to be significantly altered compared with age-matched control mice (Reddy et al., 2004; George et al., 2010; Tan et al., 2013). Here we show that reducing mTOR signaling restored the expression pattern of 227 transcripts. Importantly, the expression of these transcripts was altered in the APP brains compared with CTL, but was restored to CTL levels when one copy of the mTOR gene was removed from the hippocampi of the APP mice. It is tempting to speculate that these changes in the hippocampal transcriptome significantly contribute to the cognitive improvement seen in the APP/mTOR+/− mice. These results are highly consistent with a recent report showing that reducing the activity of the eukaryotic initiation factor 2 α-subunit (eIF2α) ameliorates AD-like pathology and memory deficits associated with Aβ accumulation (T. Ma et al., 2013). Notably, eIF2α is a downstream effector of mTOR (Tuval-Kochen et al., 2013). Together these results argue that reducing mTOR signaling might be beneficial for AD.
To summarize, we provide compelling evidence clearly indicating a primary role of mTOR in AD pathogenesis and suggest a possible way by which aging increases the development of this disorder. Further, the data presented here point to mTOR as a potential therapeutic target for this disorder. The clinical implication of these findings is profound as there are several known compounds, some FDA-approved, known to reduce mTOR signaling.
Footnotes
This work was supported by grants to S.O. by the National Institutes of Health (R01 AG037637). We thank Dr Christopher Lynch, Penn State University College of Medicine, for kindly donating the mTOR floxed mice; Dr Carolina Livi, University of Texas Health Science Center at San Antonio, for her assistance with the gene expression experiments; and Darren Shaw and Lauren Hartman for superb technical assistance.
The authors declare no competing financial interests.
References
- An WL, Cowburn RF, Li L, Braak H, Alafuzoff I, Iqbal K, Iqbal IG, Winblad B, Pei JJ. Up-regulation of phosphorylated/activated p70 S6 kinase and its relationship to neurofibrillary pathology in Alzheimer's disease. Am J Pathol. 2003;163:591–607. doi: 10.1016/S0002-9440(10)63687-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Caccamo A, Majumder S, Richardson A, Strong R, Oddo S. Molecular interplay between mammalian target of rapamycin (mTOR), amyloid-beta, and tau: effects on cognitive impairments. J Biol Chem. 2010;285:13107–13120. doi: 10.1074/jbc.M110.100420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Caccamo A, Maldonado MA, Majumder S, Medina DX, Holbein W, Magrí A, Oddo S. Naturally secreted amyloid-beta increases mammalian target of rapamycin (mTOR) activity via a PRAS40-mediated mechanism. J Biol Chem. 2011;286:8924–8932. doi: 10.1074/jbc.M110.180638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Caccamo A, Magrí A, Medina DX, Wisely EV, López-Aranda MF, Silva AJ, Oddo S. mTOR regulates tau phosphorylation and degradation: implications for Alzheimer's disease and other tauopathies. Aging cell. 2013;12:370–380. doi: 10.1111/acel.12057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Caldeira GL, Ferreira IL, Rego AC. Impaired transcription in Alzheimer's disease: key role in mitochondrial dysfunction and oxidative stress. J Alzheimers Dis. 2013;34:115–131. doi: 10.3233/JAD-121444. [DOI] [PubMed] [Google Scholar]
- Cao R, Li A, Cho HY. mTOR signaling in epileptogenesis: too much of a good thing? J Neurosci. 2009;29:12372–12373. doi: 10.1523/JNEUROSCI.3486-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carr TD, DiGiovanni J, Lynch CJ, Shantz LM. Inhibition of mTOR suppresses UVB-induced keratinocyte proliferation and survival. Cancer prevention research. 2012;5:1394–1404. doi: 10.1158/1940-6207.CAPR-12-0272-T. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chang RC, Wong AK, Ng HK, Hugon J. Phosphorylation of eukaryotic initiation factor-2alpha (eIF2alpha) is associated with neuronal degeneration in Alzheimer's disease. Neuroreport. 2002;13:2429–2432. doi: 10.1097/00001756-200212200-00011. [DOI] [PubMed] [Google Scholar]
- Ehninger D. From genes to cognition in tuberous sclerosis: implications for mTOR inhibitor-based treatment approaches. Neuropharmacology. 2013;68:97–105. doi: 10.1016/j.neuropharm.2012.05.015. [DOI] [PubMed] [Google Scholar]
- Ehninger D, Han S, Shilyansky C, Zhou Y, Li W, Kwiatkowski DJ, Ramesh V, Silva AJ. Reversal of learning deficits in a Tsc2± mouse model of tuberous sclerosis. Nat Med. 2008;14:843–848. doi: 10.1038/nm1788. [DOI] [PMC free article] [PubMed] [Google Scholar]
- George AJ, Gordon L, Beissbarth T, Koukoulas I, Holsinger RM, Perreau V, Cappai R, Tan SS, Masters CL, Scott HS, Li QX. A serial analysis of gene expression profile of the Alzheimer's disease Tg2576 mouse model. Neurotoxicity research. 2010;17:360–379. doi: 10.1007/s12640-009-9112-3. [DOI] [PubMed] [Google Scholar]
- Graber TE, McCamphill PK, Sossin WS. A recollection of mTOR signaling in learning and memory. Learn Mem. 2013;20:518–530. doi: 10.1101/lm.027664.112. [DOI] [PubMed] [Google Scholar]
- Griffin RJ, Moloney A, Kelliher M, Johnston JA, Ravid R, Dockery P, O'Connor R, O'Neill C. Activation of Akt/PKB, increased phosphorylation of Akt substrates and loss and altered distribution of Akt and PTEN are features of Alzheimer's disease pathology. J Neurochem. 2005;93:105–117. doi: 10.1111/j.1471-4159.2004.02949.x. [DOI] [PubMed] [Google Scholar]
- Guertin DA, Sabatini DM. Defining the role of mTOR in cancer. Cancer cell. 2007;12:9–22. doi: 10.1016/j.ccr.2007.05.008. [DOI] [PubMed] [Google Scholar]
- Harrison DE, Strong R, Sharp ZD, Nelson JF, Astle CM, Flurkey K, Nadon NL, Wilkinson JE, Frenkel K, Carter CS, Pahor M, Javors MA, Fernandez E, Miller RA. Rapamycin fed late in life extends lifespan in genetically heterogeneous mice. Nature. 2009;460:392–395. doi: 10.1038/nature08221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hay N, Sonenberg N. Upstream and downstream of mTOR. Genes Dev. 2004;18:1926–1945. doi: 10.1101/gad.1212704. [DOI] [PubMed] [Google Scholar]
- Hoeffer CA, Klann E. mTOR signaling: at the crossroads of plasticity, memory and disease. Trends Neurosci. 2010;33:67–75. doi: 10.1016/j.tins.2009.11.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoeffer CA, Tang W, Wong H, Santillan A, Patterson RJ, Martinez LA, Tejada-Simon MV, Paylor R, Hamilton SL, Klann E. Removal of FKBP12 enhances mTOR-raptor interactions, LTP, memory, and perseverative/repetitive behavior. Neuron. 2008;60:832–845. doi: 10.1016/j.neuron.2008.09.037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hsiao K, Chapman P, Nilsen S, Eckman C, Harigaya Y, Younkin S, Yang F, Cole G. Correlative memory deficits, Abeta elevation, and amyloid plaques in transgenic mice. Science. 1996;274:99–102. doi: 10.1126/science.274.5284.99. [DOI] [PubMed] [Google Scholar]
- Inoki K, Corradetti MN, Guan KL. Dysregulation of the TSC-mTOR pathway in human disease. Nat Genet. 2005;37:19–24. doi: 10.1038/ng1494. [DOI] [PubMed] [Google Scholar]
- Kawarabayashi T, Younkin LH, Saido TC, Shoji M, Ashe KH, Younkin SG. Age-dependent changes in brain, CSF, and plasma amyloid (beta) protein in the Tg2576 transgenic mouse model of Alzheimer's disease. J Neurosci. 2001;21:372–381. doi: 10.1523/JNEUROSCI.21-02-00372.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khurana V, Lu Y, Steinhilb ML, Oldham S, Shulman JM, Feany MB. TOR-mediated cell-cycle activation causes neurodegeneration in a Drosophila tauopathy model. Curr Biol. 2006;16:230–241. doi: 10.1016/j.cub.2005.12.042. [DOI] [PubMed] [Google Scholar]
- Klann E, Sweatt JD. Altered protein synthesis is a trigger for long-term memory formation. Neurobiol Learn Mem. 2008;89:247–259. doi: 10.1016/j.nlm.2007.08.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Komatsu M, Wang QJ, Holstein GR, Friedrich VL, Jr, Iwata J, Kominami E, Chait BT, Tanaka K, Yue Z. Essential role for autophagy protein Atg7 in the maintenance of axonal homeostasis and the prevention of axonal degeneration. Proc Natl Acad Sci U S A. 2007;104:14489–14494. doi: 10.1073/pnas.0701311104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lamming DW, Ye L, Sabatini DM, Baur JA. Rapalogs and mTOR inhibitors as anti-aging therapeutics. J Clin Invest. 2013;123:980–989. doi: 10.1172/JCI64099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lang CH, Frost RA, Bronson SK, Lynch CJ, Vary TC. Skeletal muscle protein balance in mTOR heterozygous mice in response to inflammation and leucine. Am J Physiol Endocrinol Metab. 2010;298:E1283–1294. doi: 10.1152/ajpendo.00676.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee MJ, Lee JH, Rubinsztein DC. Tau degradation: the ubiquitin-proteasome system versus the autophagy-lysosome system. Prog Neurobiol. 2013;105:49–59. doi: 10.1016/j.pneurobio.2013.03.001. [DOI] [PubMed] [Google Scholar]
- Ma T, Trinh MA, Wexler AJ, Bourbon C, Gatti E, Pierre P, Cavener DR, Klann E. Suppression of eIF2alpha kinases alleviates Alzheimer's disease-related plasticity and memory deficits. Nat Neurosci. 2013;16:1299–1305. doi: 10.1038/nn.3486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ma YQ, Wu DK, Liu JK. mTOR and tau phosphorylated proteins in the hippocampal tissue of rats with type 2 diabetes and Alzheimer's disease. Mol Med Rep. 2013;7:623–627. doi: 10.3892/mmr.2012.1186. [DOI] [PubMed] [Google Scholar]
- Majumder S, Richardson A, Strong R, Oddo S. Inducing autophagy by rapamycin before, but not after, the formation of plaques and tangles ameliorates cognitive deficits. PloS one. 2011;6:e25416. doi: 10.1371/journal.pone.0025416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mizushima N, Noda T, Yoshimori T, Tanaka Y, Ishii T, George MD, Klionsky DJ, Ohsumi M, Ohsumi Y. A protein conjugation system essential for autophagy. Nature. 1998;395:395–398. doi: 10.1038/26506. [DOI] [PubMed] [Google Scholar]
- Nixon RA, Yang DS. Autophagy failure in Alzheimer's disease-locating the primary defect. Neurobiology of disease. 2011;43:38–45. doi: 10.1016/j.nbd.2011.01.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oddo S. The role of mTOR signaling in Alzheimer disease. Front Biosci (Schol Ed) 2012;4:941–952. doi: 10.2741/s310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Orr ME, Oddo S. Autophagic/lysosomal dysfunction in Alzheimer's disease. Alzheimer's Res Ther. 2013;5:53. doi: 10.1186/alzrt217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Orr ME, Salinas A, Buffenstein R, Oddo S. Mammalian target of rapamycin hyperactivity mediates the detrimental effects of a high sucrose diet on Alzheimer's disease pathology. Neurobiol Aging. 2014;35:1233–1242. doi: 10.1016/j.neurobiolaging.2013.12.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parsons RG, Gafford GM, Helmstetter FJ. Translational control via the mammalian target of rapamycin pathway is critical for the formation and stability of long-term fear memory in amygdala neurons. J Neurosci. 2006;26:12977–12983. doi: 10.1523/JNEUROSCI.4209-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peel AL, Bredesen DE. Activation of the cell stress kinase PKR in Alzheimer's disease and human amyloid precursor protein transgenic mice. Neurobiology of disease. 2003;14:52–62. doi: 10.1016/S0969-9961(03)00086-X. [DOI] [PubMed] [Google Scholar]
- Pei JJ, Hugon J. mTOR-dependent signalling in Alzheimer's disease. J Cell Mol Med. 2008;12:2525–2532. doi: 10.1111/j.1582-4934.2008.00509.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pei JJ, Björkdahl C, Zhang H, Zhou X, Winblad B. p70 S6 kinase and tau in Alzheimer's disease. J Alzheimers Dis. 2008;14:385–392. doi: 10.3233/jad-2008-14405. [DOI] [PubMed] [Google Scholar]
- Puighermanal E, Marsicano G, Busquets-Garcia A, Lutz B, Maldonado R, Ozaita A. Cannabinoid modulation of hippocampal long-term memory is mediated by mTOR signaling. Nat Neurosci. 2009;12:1152–1158. doi: 10.1038/nn.2369. [DOI] [PubMed] [Google Scholar]
- Querfurth HW, LaFerla FM. Alzheimer's disease. N Engl J Med. 2010;362:329–344. doi: 10.1056/NEJMra0909142. [DOI] [PubMed] [Google Scholar]
- Reddy PH, McWeeney S, Park BS, Manczak M, Gutala RV, Partovi D, Jung Y, Yau V, Searles R, Mori M, Quinn J. Gene expression profiles of transcripts in amyloid precursor protein transgenic mice: up-regulation of mitochondrial metabolism and apoptotic genes is an early cellular change in Alzheimer's disease. Hum Mol Genet. 2004;13:1225–1240. doi: 10.1093/hmg/ddh140. [DOI] [PubMed] [Google Scholar]
- Santini E, Huynh TN, Klann E. Mechanisms of translation control underlying long-lasting synaptic plasticity and the consolidation of long-term memory. Prog Mol Biol Transl Sci. 2014;122:131–167. doi: 10.1016/B978-0-12-420170-5.00005-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Selman C, Tullet JM, Wieser D, Irvine E, Lingard SJ, Choudhury AI, Claret M, Al-Qassab H, Carmignac D, Ramadani F, Woods A, Robinson IC, Schuster E, Batterham RL, Kozma SC, Thomas G, Carling D, Okkenhaug K, Thornton JM, Partridge L, et al. Ribosomal protein S6 kinase 1 signaling regulates mammalian life span. Science. 2009;326:140–144. doi: 10.1126/science.1177221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Speakman JR, Mitchell SE. Caloric restriction. Mol Aspects Med. 2011;32:159–221. doi: 10.1016/j.mam.2011.07.001. [DOI] [PubMed] [Google Scholar]
- Spilman P, Podlutskaya N, Hart MJ, Debnath J, Gorostiza O, Bredesen D, Richardson A, Strong R, Galvan V. Inhibition of mTOR by rapamycin abolishes cognitive deficits and reduces amyloid-beta levels in a mouse model of Alzheimer's disease. PloS one. 2010;5:e9979. doi: 10.1371/journal.pone.0009979. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tan L, Wang X, Ni ZF, Zhu X, Wu W, Zhu LQ, Liu D. A systematic analysis of genomic changes in Tg2576 mice. Molecular neurobiology. 2013;47:883–891. doi: 10.1007/s12035-012-8384-4. [DOI] [PubMed] [Google Scholar]
- Tang SJ, Reis G, Kang H, Gingras AC, Sonenberg N, Schuman EM. A rapamycin-sensitive signaling pathway contributes to long-term synaptic plasticity in the hippocampus. Proc Natl Acad Sci U S A. 2002;99:467–472. doi: 10.1073/pnas.012605299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tang Z, Bereczki E, Zhang H, Wang S, Li C, Ji X, Branca RM, Lehtiö J, Guan Z, Filipcik P, Xu S, Winblad B, Pei JJ. Mammalian target of rapamycin (mTor) mediates tau protein dyshomeostasis: implication for Alzheimer disease. J Biol Chem. 2013;288:15556–15570. doi: 10.1074/jbc.M112.435123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tanida I. Autophagy basics. Microbiol Immunol. 2011;55:1–11. doi: 10.1111/j.1348-0421.2010.00271.x. [DOI] [PubMed] [Google Scholar]
- Thies W, Bleiler L, Alzheimer's Association 2013 Alzheimer's disease facts and figures. Alzheimers Dement. 2013;9:208–245. doi: 10.1016/j.jalz.2013.02.003. [DOI] [PubMed] [Google Scholar]
- Tuval-Kochen L, Paglin S, Keshet G, Lerenthal Y, Nakar C, Golani T, Toren A, Yahalom J, Pfeffer R, Lawrence Y. Eukaryotic initiation factor 2alpha, a downstream effector of mammalian target of rapamycin, modulates DNA repair and cancer response to treatment. PloS one. 2013;8:e77260. doi: 10.1371/journal.pone.0077260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Westerman MA, Cooper-Blacketer D, Mariash A, Kotilinek L, Kawarabayashi T, Younkin LH, Carlson GA, Younkin SG, Ashe KH. The relationship between Abeta and memory in the Tg2576 mouse model of Alzheimer's disease. J Neurosci. 2002;22:1858–1867. doi: 10.1523/JNEUROSCI.22-05-01858.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wullschleger S, Loewith R, Hall MN. TOR signaling in growth and metabolism. Cell. 2006;124:471–484. doi: 10.1016/j.cell.2006.01.016. [DOI] [PubMed] [Google Scholar]
- Zeng LH, Rensing NR, Wong M. The mammalian target of rapamycin signaling pathway mediates epileptogenesis in a model of temporal lobe epilepsy. J Neurosci. 2009;29:6964–6972. doi: 10.1523/JNEUROSCI.0066-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]