

Abstract
Acute intermittent hypoxia (AIH; three 5-min hypoxic episodes) causes a form of phrenic motor facilitation (pMF) known as phrenic long-term facilitation (pLTF); pLTF is initiated by spinal activation of Gq protein-coupled 5-HT2 receptors. Because α1 adrenergic receptors are expressed in the phrenic motor nucleus and are also Gq protein-coupled, we hypothesized that α1 receptors are sufficient, but not necessary for AIH-induced pLTF. In anesthetized, paralyzed, and ventilated rats, episodic spinal application of the α1 receptor agonist phenylephrine (PE) elicited dose-dependent pMF (10 and 100 μM, P < 0.05; but not 1 μM). PE-induced pMF was blocked by the α1 receptor antagonist prazosin (1 mM; −20 ± 20% at 60 min, −5 ± 21% at 90 min; n = 6). Although α1 receptor activation is sufficient to induce pMF, it was not necessary for AIH-induced pLTF because intrathecal prazosin (1 mM) did not alter AIH-induced pLTF (56 ± 9% at 60 min, 78 ± 12% at 90 min; n = 9). Intravenous (iv) prazosin (150 μg/kg) appeared to reduce pLTF (21 ± 9% at 60 min, 26 ± 8% at 90 min), but this effect was not significant. Hypoglossal long-term facilitation was unaffected by intrathecal prazosin, but was blocked by iv prazosin (−4 ± 14% at 60 min, −13 ± 18% at 90 min), suggesting different LTF mechanisms in different motor neuron pools. In conclusion, Gq protein-coupled α1 adrenergic receptors evoke pMF, but they are not necessary for AIH-induced pLTF.
Keywords: spinal cord, α1 receptors, plasticity, respiratory motor neuron
mechanisms of respiratory motor plasticity induced by acute intermittent hypoxia (AIH) have been extensively investigated in recent years (8, 21). In particular, AIH elicits phrenic long-term facilitation (pLTF), a progressive and persistent increase in phrenic motor output lasting more than 1 h following AIH [reviewed in (8)]. Multiple signaling molecules have been identified in the cellular cascade leading to pLTF and to other forms of phrenic motor facilitation (pMF; a general term that includes pLTF) (8). AIH-induced pLTF (three 5 min hypoxic episodes and three 5 min normoxic intervals) requires spinal serotonin type 2 (5-HT2) receptor activation (4, 10, 18), new synthesis of brain-derived neurotrophic factor (3), and activation of its high-affinity receptor, tropomyosin-related kinase receptor B (TrkB) (3). This signaling pathway to pMF has been referred to as the Q pathway because it requires activation of Gq protein-coupled 5-HT2 metabotropic receptors.
Although 5-HT2 receptors by initiating the Q pathway are important for pLTF following AIH, less is known about the involvement of other receptors that can elicit similar signaling cascades, such as α1 adrenergic receptors. Both 5-HT2 and α1 receptors are Gq protein-coupled metabotropic receptors whose signaling cascades often involve inosotol 1,4,5-triphosphate (IP3)/protein kinase C (PKC)/calcium (11). The 5-HT2 receptor antagonist ketanserin blocks pLTF when applied before but not after AIH, suggesting that 5-HT2 receptor activation is necessary for the induction but not maintenance of AIH-induced pLTF (10). Furthermore, episodic activation of 5-HT2A or 5-HT2B receptors with selective agonists is sufficient to elicit pMF (18). Although the widely held view is that 5-HT2 receptors are necessary for AIH-induced pLTF (8, 20, 24), the use of ketanserin as a specific 5-HT receptor antagonist casts some doubt because 1) ketanserin is also a potent inhibitor of α1 receptors (6, 26, 30); 2) α1 receptors are also Gq-coupled metabotropic receptors; 3) α1 receptors are expressed on motoneurons throughout the spinal cord (29); and 4) α1 receptor activation elicits in vitro hypoglossal long-term facilitation (LTF) (25). Thus we tested the hypothesis that α1 receptors are necessary and sufficient for AIH-induced pLTF. In a preliminary report, spinal activation of α1 adrenergic receptors elicited pMF (8); however, it is not known whether α1 adrenergic receptors play any role in AIH-induced pLTF.
Norepinephrine (NE, a natural α1 receptor ligand), via α1 receptors, plays an important role in multiple forms of neuroplasticity. For example, NE modulates hippocampal synaptic plasticity (17), and α1 receptors are involved in the induction of long-term depression (31), long-term potentiation (9), and cortical synaptic depression (35). In the respiratory system, episodic upper airway obstruction triggers vagus-dependent hypoglossal (but not phrenic) LTF through an α1 receptor-dependent, 5-HT-independent mechanism (34). On the basis of these studies, we hypothesized that NE and α1 receptors are both necessary and sufficient for AIH-induced pLTF.
METHODS
All experiments were approved by the Animal Care and Use Committee at the School of Veterinary Medicine, University of Wisconsin-Madison, and conformed to the policies of the National Institutes of Health Guide for the Care and Use of Laboratory Animals. Experiments were performed on 3- to 4-mo-old (300–400 g) Harlan 218a male Sprague-Dawley rats. Rats were housed in 12-h light/dark cycles, with food and water ad libitum.
Drugs and materials.
Phenylephrine (PE, α1 receptor agonist) and prazosin (α1 receptor antagonist) were purchased from Sigma-Aldrich (St. Louis, MO). PE was made in artificial cerebrospinal fluid (aCSF) made fresh weekly containing (in mM) 120 NaCl, 3 KCl, 2 CaCl, 2 MgCl, 23 NaHCO3, and 10 glucose. Prazosin was dissolved in 20% DMSO (Fort Dodge Animal Health, Fort Dodge, IA).
Experimental groups.
For the first investigation on the effects of PE-induced pMF, rats were divided into five groups. Three separate groups of rats received PE (three 5-μl intrathecal boluses separated by 5 min; more details below) at 100 (n = 7), 10 (n = 7), or 1 (n = 8) μM. A fourth group received three 5-μl intrathecal boluses of vehicle (20% DMSO; n = 8), whereas the fifth group (n = 8) received both prazosin (1 mM) and PE (10 μM). In the second investigation, rats were divided into two groups; one group received intrathecal prazosin (1 mM; n = 9) prior to AIH, whereas the other received vehicle (20% DMSO; n = 7). Finally, in the last investigation, prazosin was given intravenously (150 μg/kg iv; n = 6), and changes in both phrenic and hypoglossal nerve activity were monitored after AIH.
Electrophysiological experiments.
Electrophysiological measurements have been described previously (2, 4, 18). In brief, rats were induced with isoflurane, tracheotomized, and pump-ventilated (Small Animal Ventilator 683; Harvard Apparatus, Holliston, MA). Rats were maintained on 3.5–4% isoflurane (50–60% oxygen) for the remainder of the surgical procedure, and then were slowly converted to urethane anesthesia (1.8 g/kg iv; Sigma-Aldrich). Temperature was maintained throughout the experiment with a heated surgical table, and the inspired O2 concentration was monitored using a fuel-cell O2 sensor (TED 60T; Teledyne Analytical Instruments, City of Industry, CA). Anesthetic level was monitored by assessing blood pressure and phrenic nerve responses to toe pinch. Approximately 1 h after beginning the surgical procedure, an infusion (1.5–3 ml/h iv) was initiated with a solution consisting of lactated Ringer (5–20 ml), Hetastarch (2.5–10 ml of 0.6% hetastarch in 0.9% sodium chloride), and sodium bicarbonate (1 ml, 8.4% solution). The infusion rate was adjusted to maintain blood volume and acid-base balance.
Surgical procedure.
From a ventral approach, the vagus nerves were isolated and cut bilaterally in the midcervical region, and a catheter was inserted into the right femoral artery to enable blood pressure monitoring and blood sampling throughout experiments. Blood samples were analyzed for Po2, Pco2, pH, and base excess using a blood gas analyzer (ABL 800; Radiometer, Copenhagen, Denmark). Blood samples (62.5 μl in a heparinized plastic catheter) were drawn before and during the first hypoxic episode when applicable, and at 15, 30, 60, and 90 min after drug/hypoxia to maintain baseline physiological conditions (see below).
From a dorsal approach, the left hypoglossal and phrenic nerves were isolated, and a rectal temperature probe was inserted to monitor temperature throughout the experiment. The nerves were cut distally, desheathed, and placed on bipolar silver recording electrodes submerged in mineral oil. During the 1-h stabilization period after conversion to urethane anesthetic, the rats were paralyzed with pancuronium bromide (1 mg/rat).
The dorsal spinal cord over cervical segment 2 (C2) was exposed via laminectomy. One or two (as indicated) silicone catheters were primed with the relevant drugs and inserted 2 mm through a small incision in the dura so that the catheter tip was just rostral to C4. One catheter was inserted for all studies, except when prazosin and PE were both applied. PE (1, 10, or 100 μM) or vehicle (20% DMSO in aCSF) were applied in three 5-μl boluses, separated by 5 min. Prazosin (1 mM) was injected as a 9-μl bolus 20 min prior to AIH or PE. Intravenous prazosin was given at a dose of 150 μg/kg 20 min prior to AIH.
Nerve activity was amplified (gain ×10 K), band pass-filtered (100 Hz to 10 kHz) (A-M Systems, Carlsberg, WA), and integrated (absolute value, time constant 50 ms; Powerlabs 830; AD Instruments, Colorado Springs, CO) or passed through a moving average (time constant 50 ms; CWE 821 filter; Pavnter, Ardmore, PA). The signal was digitized, recorded continuously, and analyzed using Powerlabs 830 (version 7.2.2, AD Instruments) or Windaq (DATAQ instruments, Akron, OH). Baseline nerve activity was established with FiO2 at approximately 50–60% (yielding PaO2 >180 mmHg) with CO2 added to the inspired gas with a balance of nitrogen. End-tidal CO2 was monitored and maintained throughout the experiment using a flow-through capnograph (Respironics, Andover, MA). The apneic threshold of each animal was determined by progressively lowering inspired CO2 until phrenic nerve discharge ceased. Inspired CO2 was then slowly increased until phrenic nerve activity returned (i.e., recruitment threshold). End-tidal Pco2 was then set 2–3 mmHg above the recruitment threshold for phrenic nerve activity. Once phrenic nerve activity was stable, an arterial blood sample was taken to establish baseline conditions (PaO2, PaCO2, pH, base excess); these conditions were maintained throughout the experiment. Rats then received drug treatment and/or AIH (three times, 5 min of 10.5% O2 separated by 5 min of normoxia). Data were included in the analysis only if they complied with previously established criteria (2, 4): 1) PaO2 during baseline and recovery periods was >180 mmHg; 2) PaO2 during hypoxia was between 35 and 45 mmHg; and 3) PaCO2 remained within 1.5 mmHg of baseline throughout the posthypoxia period. Phrenic or hypoglossal nerve amplitude was evaluated for 1 min before each blood sample. Upon completion of the experiment, rats were humanely overdosed with urethane.
Data analysis.
Peak integrated nerve amplitude was averaged for 30 cycles immediately prior to each recorded data point [baseline; first hypoxic episode (where relevant); and 15, 30, 60, and 90 min post-AIH/drug], where no noise was evident and amplitude appeared representative of that occurring at that time point. At all time points, integrated nerve burst amplitude was normalized to baseline amplitude as a percent change from baseline. Analysis focused on amplitude data because we previously documented in an extensive metaanalysis that frequency LTF is small in this experimental preparation with this AIH protocol (4a). Statistical comparisons were made between all time points using a one-way (drug treatment factor) or two-way (drug treatment and time factors) repeated measures ANOVA as indicated in Results with a Fishers LSD post hoc test to identify individual differences (Sigma Stat version 11; Systat Software, San Jose, CA). Analysis of the hypoxic response was performed using data from min 2 of the first 5-min hypoxic episode, and compared using a Student's t-test. Differences were considered significant if P < 0.05. All values are expressed as means ± 1 SE.
RESULTS
Spinal α1 receptor activation elicits pMF.
Intrathecal PE (10 or 100 μM; three 5-μl injections at 5-min intervals) induced a long-lasting increase in integrated phrenic nerve burst amplitude (i.e., pMF) lasting at least 90 min postinjection. Slight alterations in temperature, PaO2, PaCO2, pH, and mean arterial pressure (MAP) were evident over the course of experiments (Table 1), but all values were within ranges specified in methods.
Table 1.
Physiological variables for Sprague-Dawley 218a rats during electrophysiological experiments after PE (100, 10, 1 μM), prazosin, or vehicle + AIH
| Time | Treatment group | Temperature | PaO2 | PaCO2 | pH | MAP |
|---|---|---|---|---|---|---|
| Baseline | 100 μM PE, n = 7 | 37.7 ± 0.1a | 288.4 ± 9.3 | 47.3 ± 1.0 | 7.323 ± 0.010 | 130.4 ± 10.6 |
| 10 μM PE, n = 7 | 37.7 ± 0.1 | 313.0 ± 9.82 | 46.8 ± 1.3 | 7.342 ± 0.014 | 132.8 ± 5.0 | |
| 1 μM PE, n = 8 | 37.6 ± 0.11 | 308.0 ± 10 | 47.7 ± 0.6 | 7.330 ± 0.006 | 128.1 ± 4.3 | |
| Prazosin + PE, n = 8 | 37.8 ± 0.1b | 308.5 ± 5.4 | 47.1 ± 0.5 | 7.334 ± 0.006 | 94.4 ± 9.73 | |
| Vehicle + AIH, n = 8 | 38.0 ± 0.1 | 303.3 ± 4.3 | 48.4 ± 0.7 | 7.346 ± 0.006 | 120.1 ± 3.3 | |
| Hx | 100 μM PE | |||||
| 10 μM PE | ||||||
| 1 μM PE | ||||||
| Prazosin + PE | ||||||
| Vehicle + AIH | 38.0 ± 0.1 | 41.1 ± 1.8 | 49.2 ± 0.6 | 7.326 ± 0.008 | 102.5 ± 5.1 | |
| 60 min | 100 μM PE | 37.7 ± 0.1 | 294.1 ± 4.3 | 47.0 ± 0.9 | 7.317 ± 0.008 | 114.1 ± 13.2a |
| 10 μM PE | 37.7 ± 0.1 | 311.3 ± 5.6 | 48.6 ± 0.8d | 7.333 ± 0.018 | 125.0 ± 5.2f | |
| 1 μM PE | 37.6 ± 0.11 | 314.6 ± 6.61 | 47.6 ± 0.9 | 7.337 ± 0.009 | 119.2 ± 7.1h | |
| Prazosin + PE | 37.5 ± 0.1b | 314.5 ± 6.0 | 47.5 ± 0.8 | 7.351 ± 0.001 | 94.8 ± 10.13 | |
| Vehicle + AIH | 37.9 ± 0.1 | 289.0 ± 5.8 | 47.7 ± 0.7 | 7.349 ± 0.006 | 99.1 ± 4.4c,3 | |
| 90 min | 100 μM PE | 37.7 ± 0.1 | 302.5 ± 6.1 | 48.6 ± 1.0 | 7.308 ± 0.013 | 110.9 ± 9.8e |
| 10 μM PE | 37.5 ± 0.1a | 304.7 ± 5.7 | 47.1 ± 1.3 | 7.339 ± 0.021 | 121.4 ± 5.9 | |
| 1 μM PE | 37.5 ± 0.1 | 312.9 ± 7.11 | 48.0 ± 0.7 | 7.334 ± 0.007 | 112.9 ± 7.0g | |
| Prazosin + PE | 37.6 ± 0.1 | 310.2 ± 8.0 | 47.7 ± 0.7 | 7.356 ± 0.011b | 91.0 ± 7.93 | |
| Vehicle + AIH | 37.7 ± 0.1c | 290.0 ± 5.6 | 48.8 ± 0.6 | 7.344 ± 0.005a | 92.1 ± 2.7c,3 |
AIH, acute intermittent hypoxia; Hx, hypoxia; MAP, mean arterial pressure; PE, phenylephrine.
P < 0.05 different from baseline 100 μM PE within parameter;
P < 0.05 different from baseline prazosin + PE within temperature;
P < 0.05 different from baseline vehicle + AIH within temperature;
P < 0.05 different from baseline and 90 min 10 μM PE within PaCO2;
P < 0.001 different from all other time points of 100 μM PE within MAP;
P < 0.05 different from baseline 10 μM PE within MAP;
P < 0.01 different from baseline 1 μM PE within MAP;
P < 0.05 different from baseline 1 μM PE within MAP;
P < 0.05 different from vehicle + AIH within parameter within time point;
P < 0.05 different from 100 μM PE within time point;
P < 0.05 different from 10 μM PE within time point.
After PE injections (10 and 100 μM; Fig. 1, B and C, respectively), phrenic nerve burst amplitude ramped progressively, although no significant changes were evident after 1 μM PE (Fig. 1A). The lowest PE dose (1 μM) did not significantly (P > 0.1; n = 8) change phrenic burst amplitude from baseline at 60 (6 ± 10% change from baseline) or 90 min (18 ± 11% change from baseline) postinjection (Fig. 2). Phrenic burst amplitude after 1 μM PE was significantly lower than after 10 μM PE (60 min, P = 0.042) or 100 μM PE (60 min, P = 0.03; 90 min, P = 0.044). PE evoked increases in phrenic burst amplitude from baseline after 10 μM PE (37 ± 7% at 60 min, P = 0.003; 34 ± 9% at 90 min, P = 0.007; n = 7) and 100 μM PE injections (40 ± 20% at 60 min, P = 0.002; 49 ± 19% at 90 min, P < 0.001; n = 7) (Fig. 2). Phrenic burst amplitudes for 10 and 100 μM PE doses were not significantly different from each other at 60 (P = 0.887) or 90 min (P = 0.331) postinjection (Fig. 2).
Fig. 1.

Representative phrenic neurograms from five rats receiving episodic phenylephrine (PE, an α1 receptor agonist), acute intermittent hypoxia (AIH), or prazosin (an α1 receptor antagonist) before PE. Intrathecal applications of low-dose PE (1 μM) (A) did not alter phrenic amplitude (Phr). Intrathecal applications of higher PE doses (10 μM) (B) and (100 μM) (C) separated by 5 min caused phrenic motor facilitation (pMF), similar to phrenic long-term facilitation (pLTF) induced by AIH [three 5-min periods of hypoxia (Hx1, Hx2, and Hx3, respectively) separated by normoxia, and 20% DMSO vehicle preapplication] (D). E: PE-induced pMF was blocked by preapplication of prazosin (1 mM) 20 min prior to episodic PE (10 μM).
Fig. 2.
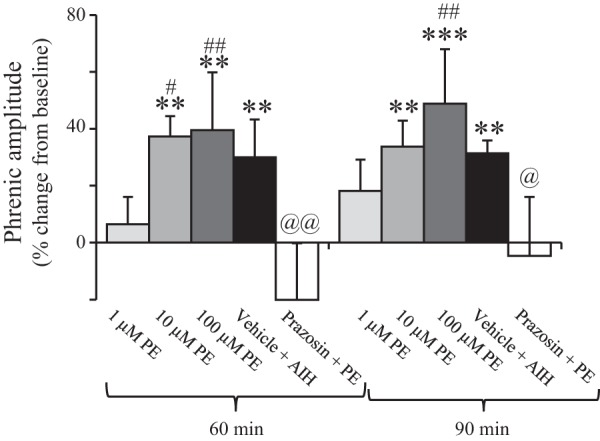
Group data show that high concentrations of PE induce pMF at 60 and 90 min, which were similar to AIH-induced pLTF and blocked by prazosin (an α1 receptor antagonist). Both 10 (n = 7) and 100 (n = 7) μM PE significantly increased phrenic nerve amplitude at 60 and 90 min compared with baseline (**P < 0.01; ***P < 0.001) and were not different from vehicle + AIH (n = 8)-induced pLTF at either time point. Low doses of PE (1 μM; n = 8) did not evoke pMF at any time point and pMF was significantly reduced compared with 10 and 100 μM PE (#P < 0.05; ##P < 0.01). Preapplication of prazosin (1 mM; n = 6) caused a significant reduction in pMF induced by PE (10 μM) at 60 and 90 min (@@P < 0.01; @P < 0.05, two-way repeated measures ANOVA, Fishers LSD post hoc).
As expected, vehicle-treated AIH rats (n = 8) had significantly increased amplitude at both 60 (30 ± 13% change from baseline, P = 0.01) and 90 min (31 ± 4% change from baseline, P = 0.007), indicating pLTF (Figs. 1D and 2). Phrenic amplitude responses to PE at any dose were not significantly different from the pLTF magnitude evoked by vehicle + AIH at 60 or 90 min (P > 0.05). Thus PE induces pMF similar in magnitude to AIH-induced pLTF.
We confirmed that the PE-induced pMF results from α1 receptor activation by demonstrating that pretreatment with prazosin (1 mM, intrathecal) blocked PE (10 μM)-induced pMF (Figs. 1E and 2). The 10 μM PE dose was used because the pMF magnitude was similar to that evoked by AIH, and the higher PE dose caused changes in hypoglossal activity (data not shown), consistent with drug distribution to the brainstem. Hypoglossal activity serves as an internal control for unintended drug distribution in this preparation (4, 18). As evident in the phrenic neurogram (Fig. 1E) and group data (Fig. 2), pMF was diminished when prazosin was applied 20 min prior to episodic PE (n = 6). At 60 min postinjection, phrenic nerve burst amplitude was −20 ± 20% change from baseline and was significantly reduced vs. 10 μM PE (P < 0.001) or vehicle + AIH (P = 0.002). At 90 min postinjection, it was −5 ± 21% change from baseline, and was significantly reduced vs. 10 μM PE alone (P = 0.019) or vehicle + AIH (P = 0.023). There was an overall prazosin treatment effect vs. 10 μM PE (P = 0.007) and vehicle + AIH (P = 0.001).
Overall, PE evokes pMF, and this pMF was inhibited by prazosin, confirming the involvement of α1 receptors in these effects. Because the magnitude of PE-induced pMF is not significantly less than pLTF, α1 receptor activation is sufficient to induce AIH-induced pLTF.
A1 receptor activation is not necessary for AIH-induced pLTF.
Intrathecal prazosin (1 mM, α1 receptor antagonist) applied 20 min prior to AIH decreased MAP during baseline conditions (Table 2). Despite this initial decrease, the overall blood pressure change during the experiment was 11.1 ± 4.0 mmHg, comparable to the vehicle + AIH group (11.4 ± 6.1 mmHg). Because blood pressure changes between groups were similar, blood pressure effects are unlikely to account for the observed results. Although PaO2 decreased during hypoxic episodes, there was no significant difference between baseline and 60 or 90 min post-AIH (Table 2). Small but significant changes in pH were evident during hypoxia and 15 min post-AIH, but had recovered by 60 and 90 min (Table 2). All other measured physiological parameters were within normal limits.
Table 2.
Physiological variables for Sprague-Dawley 218a rats during electrophysiological experiments with prazosin or vehicle + AIH
| Time | Treatment group | Temperature | PaO2 | PaCO2 | pH | MAP |
|---|---|---|---|---|---|---|
| Baseline | Prazosin + AIH, n = 9 | 37.7 ± 0.2 | 302.4 ± 9.3 | 48.8 ± 1.1 | 7.337 ± 0.008 | 98.5 ± 5.8 |
| Vehicle + AIH, n = 7 | 37.7 ± 0.2 | 297.7 ± 15.7 | 49.1 ± 1.7 | 7.342 ± 0.013 | 123.8 ± 7.0 | |
| Hx | Prazosin + AIH | 37.5 ± 0.2 | 40.2 ± 1.5 | 48.5 ± 1.4 | 7.318 ± 0.009 | 72.5 ± 8.4 |
| Vehicle + AIH | 37.6 ± 0.2 | 38.8 ± 1.5 | 50.9 ± 1.4 | 7.329 ± 0.014 | 100.9 ± 5.82 | |
| 60 min | Prazosin + AIH | 37.8 ± 0.17 | 291.2 ± 10.8 | 49.6 ± 1.8 | 7.372 ± 0.005 | 84.3 ± 6.33 |
| Vehicle + AIH | 37.8 ± 0.2 | 280.3 ± 10.0 | 49.6 ± 2.1 | 7.345 ± 0.001 | 114.9 ± 5.9 | |
| 90 min | Prazosin + AIH | 37.5 ± 0.22a | 301.3 ± 14.2b | 49.2 ± 1.3 | 7.353 ± 0.007 | 87.4 ± 5.31 |
| Vehicle + AIH | 37.6 ± 0.2 | 291.0 ± 12.7 | 50.0 ± 2.0 | 7.342 ± 0.012 | 112.2 ± 7.6 |
AIH, acute intermittent hypoxia; Hx, hypoxia; MAP, mean arterial pressure.
P < 0.05 different from 60 min prazosin + AIH;
P < 0.05 different from all other time points within parameter;
P < 0.05 different from baseline prazosin + AIH within MAP;
P < 0.05 different from 60 min vehicle + AIH within MAP.
Intrathecal prazosin had no significant effect on pLTF (Fig. 3). Representative phrenic neurograms illustrate that prazosin pretreatment did not change the response during hypoxic episodes, or following AIH (i.e., pLTF) vs. vehicle controls (Fig. 3). The short-term hypoxic response was not altered by prazosin (prazosin n = 9, 105 ± 26%; vehicle n = 7, 110 ± 17%; P = 0.397; Fig. 4A;). Similarly, pLTF was apparent at 15 min (26 ± 7% change from baseline, P = 0.003), 30 min (30 ± 7%, P < 0.001), 60 min (56 ± 9%, P < 0.001), and 90 min (78 ± 12%, P < 0.001) for prazosin-treated rats (n = 9; Fig. 4B). Vehicle-treated rats (n = 7) expressed normal pLTF at 15 min (23 ± 12% change from baseline, P = 0.019), 30 min (33 ± 13%, P = 0.001), 60 min (56 ± 9%, P < 0.001), and 90 min (68 ± 7%, P < 0.001) (Fig. 4B). There were no significant differences in nerve burst amplitude between treatment groups (prazosin vs. vehicle) at 15 (P = 0.804), 30 (P = 0.815), 60 (P = 0.997), or 90 min (P = 0.725) post-AIH (Fig. 4B). The absence of any prazosin effect on the short-term hypoxic phrenic responses or pLTF suggests that α1 receptor activation is not necessary for AIH-induced pLTF.
Fig. 3.
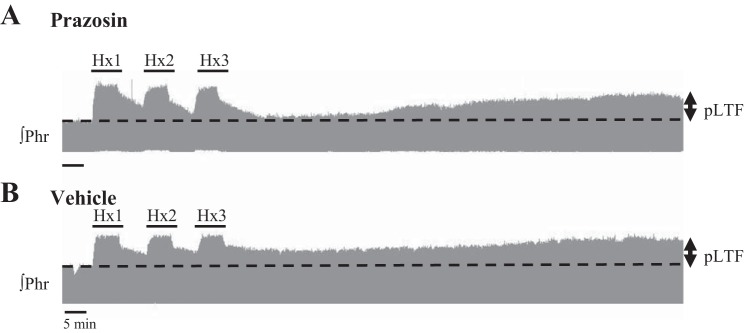
Representative phrenic neurograms from two rats receiving either intrathecal preapplication of prazosin (an α1 receptor antagonist, 1 mM) (A) or vehicle (20% DMSO) (B) 20 min prior to AIH [three, 5 min hypoxic episodes (Hx1, Hx2, Hx3) separated by normoxia]. Prazosin did not alter expression of pLTF. Time controls (n = 2, data not shown) did not show facilitation.
Fig. 4.
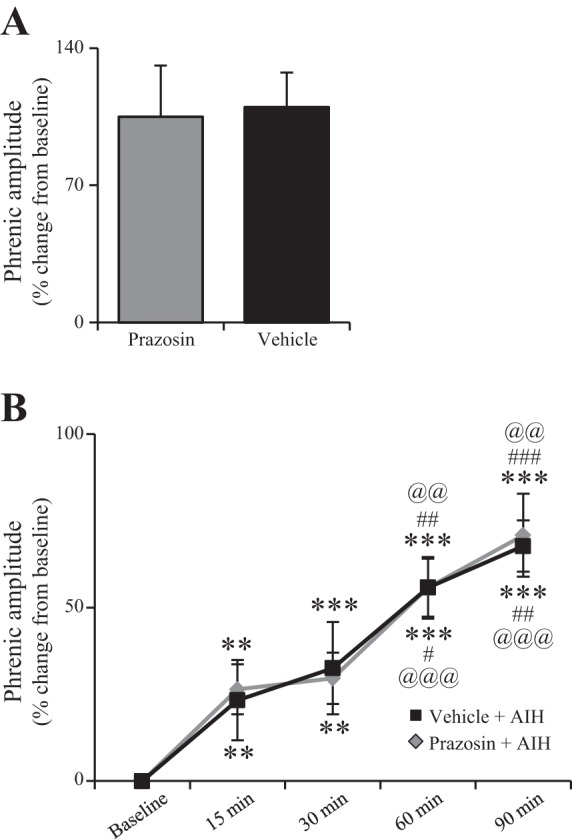
Intrathecal preapplication of prazosin (1 mM; n = 9) did not affect the hypoxia responses (A) (t-test) nor the magnitude of AIH-induced pLTF (B) compared with intrathecal vehicle preapplication (20% DMSO) + AIH (n = 7). Thus α1 receptor activation is not necessary for AIH-induced pLTF (***P < 0.001, **P < 0.01 significantly different from baseline; ###P < 0.001, ##P < 0.01, #P < 0.05 significantly different from 30 min; @@@P < 0.001, @@P < 0.01 significantly different from 15 min; two-way repeated measures ANOVA, Fishers LSD post hoc test). In (B), significance symbols relative to vehicle data appear below data lines; significance symbols relative to prazosin appear above data lines.
As expected, hypoglossal LTF (n = 9) was unaffected by intrathecal prazosin (change from baseline: 19 ± 16% at 15 min, 32 ± 22% at 30 min, 49 ± 24% at 60 min, 61 ± 28% at 90 min; data not shown). Hypoglossal activity in the prazosin + AIH group was not significantly different from that of the vehicle + AIH group (P = 0.554). Significant hypoglossal (XII) LTF was still evident at 60 min (P < 0.001) and 90 min (P < 0.001) after intrathecal prazosin and after intrathecal vehicle at 90 min (P = 0.004). Thus it is unlikely intrathecal prazosin reached brainstem neurons at effective concentrations because it abolishes hypoglossal LTF when applied systemically (25) (Fig. 5).
Fig. 5.
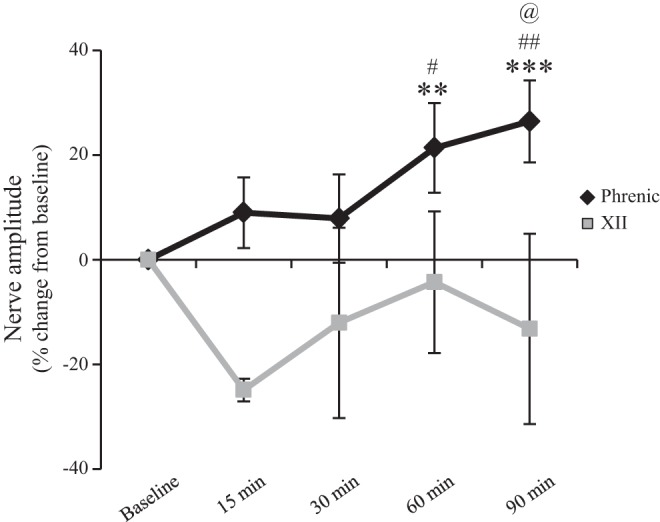
Intravenous preapplication of prazosin (150 μg/kg; n = 6) did not alter development of AIH-induced pLTF (black line), but did significantly inhibit development of hypoglossal (XII) LTF (gray line). Thus α1 receptor activation was not necessary for AIH-induced pLTF, but it was for hypoglossal LTF (***P < 0.001, **P < 0.01 significantly different from baseline; ##P < 0.01, #P < 0.05 significantly different from 30 min; @P < 0.05 significantly different from 15 min; one-way repeated measures ANOVA Fishers LSD post hoc test).
Systemic prazosin administration (150 μg/kg iv) had no significant effect on AIH-induced pLTF. For example, phrenic nerve burst amplitude (% change from baseline) after prazosin + AIH (n = 6) progressively increased (9 ± 7% at 15 min, 8 ± 9% at 30 min, 21 ± 9% at 60 min, P = 0.003; 26 ± 8% at 90 min, P < 0.001; Fig. 5).
In contrast to pLTF, hypoglossal LTF was not evident after systemic prazosin pretreatment (Fig. 5). Hypoglossal nerve amplitude (% change from baseline) after AIH was −25 ± 2% at 15 min, −12 ± 18% at 30 min, −4 ± 14% at 60 min, and −13 ± 18% at 90 min, and was not significantly different from baseline at any time point (n = 6; P > 0.05). Thus distinct mechanisms may give rise to LTF in the hypoglossal vs. phrenic motor pools.
MAP dropped to 46–79 mmHg after iv prazosin administration (66 ± 2 mmHg, n = 6). In an attempt to control for this low blood pressure before AIH, we conducted experiments during which blood was withdrawn to induce a similar hypotension (n = 6). However, this treatment had negative effects on other physiologic parameters (such as nerve instability and changes in base excess) and made this approach unsuitable as a control experiment.
DISCUSSION
Contrary to our original hypothesis, we found that spinal α1 receptor activation is sufficient to evoke pMF, but it is not necessary for AIH-induced pLTF. Additionally, we confirm that systemic α1 receptor activation is necessary for AIH-induced hypoglossal LTF (25). In contrast, spinal 5-HT2 receptor activation is both necessary (4, 10) and sufficient (18) for AIH-induced pLTF. Thus although α1 receptors have considerable potential to elicit spinal respiratory motor plasticity, they do not appear to be critical in the experimental model of AIH-induced pLTF studied here. We suggest possible scenarios in which α1 receptor-dependent respiratory plasticity may be important.
Spinal α1 receptors elicit phrenic motor facilitation.
Understanding the diversity of cellular mechanisms capable of giving rise to different forms of respiratory motor plasticity is important both from a biological perspective as we attempt to understand when and where these different mechanisms come into play in the life of an animal (8), and as a guide in the development of new therapeutic approaches to treat clinical disorders that compromise breathing (23). We have come to appreciate that multiple neurotransmitter/neuromodulatory systems are capable of giving rise to phenotypically similar forms of pMF. For example, spinal 5-HT receptor activation (5-HT2A or 5-HT2B) gives rise to pMF by activating Gq protein-coupled metabotropic receptors (18). Here we confirm a previous preliminary report that spinal activation of Gq protein-coupled α1 receptors also elicits pMF (8) (Figs. 1 and 2). To confirm that PE-evoked pMF occurred through activation of α1 receptors, an α1 receptor antagonist (prazosin) was applied prior to episodic PE, and this pretreatment successfully blocked pMF. Thus PE induces pMF via α1 receptor activation.
This is the first demonstration that a Gq protein-coupled receptor other than 5-HT receptors evokes pMF. Given the convergent signaling pathways of Gq protein-coupled receptors, we hypothesized that activation of multiple, distinct Gq-coupled receptors would lead to pMF via similar intracellular mechanisms. Α1 receptors are most often Gq protein-coupled, although some subtypes can be Gi protein-coupled (16). Knowledge is still limited regarding the downstream intracellular signaling pathways following α1 receptor activation, but it likely extends beyond Gq-activated PKC, and may be dependent on the α1 receptor subtype activated (i.e., α1a, α1b, α1d) (see Ref. 7 for a review). Overall, the predominant action of α1 receptor activation is an increase in intracellular calcium (15), which may be involved in pMF and/or pLTF. This receptor can also signal through mitogen-activated protein kinases such as extracellular regulated kinase (ERK) and c-Jun NH2-terminal kinase (JNK) (27), or phosphatidylinositol 3-kinase (PI3) (22). Several of these molecules play important roles in pMF and pLTF (13). On the basis of available evidence, we cannot conclusively demonstrate that the mechanism of α1 receptor-induced pMF is the same as 5-HT-induced pMF because such detailed mechanistic analysis was beyond the scope of the current study. Regardless, our findings are consistent with the hypothesis that multiple Gq-coupled metabotropic receptors elicit pMF via similar cellular mechanisms.
Neither spinal nor systemic α1 receptor activation is necessary for AIH-induced pLTF.
An unexpected finding was that spinal α1 receptor activation is not necessary for pLTF induced by AIH, whereas it is sufficient to elicit pMF (Figs. 3 and 4). This conclusion is based on the observations that intrathecal prazosin near the phrenic motor nucleus blocks PE-induced pMF, but the same treatment had no significant effect on AIH-induced pLTF.
Because prazosin blocks PE-induced hypoglossal LTF in neonatal in vitro rhythmic slice preparations (bath applied) and AIH-induced hypoglossal LTF (iv) in adult anesthetized rats (25), we wondered whether α1 receptor activation is necessary for pLTF elicited by AIH. However, like the intraspinal prazosin injections, iv prazosin had no significant effect on AIH-induced pLTF (despite causing a profound change in blood pressure). Thus α1 receptor activation in the spinal cord is not necessary for AIH-induced pLTF, unlike AIH-induced hypoglossal LTF.
Hypoglossal LTF was monitored after intrathecal and iv prazosin. As expected, hypoglossal LTF was unaffected by intrathecal prazosin (data not shown), demonstrating that prazosin was not diffusing to the brain at effective concentrations after intraspinal injections (4, 18). On the other hand, iv prazosin blocked hypoglossal LTF, replicating findings of a previous study (25) (Fig. 5). However, the large drop in blood pressure after iv prazosin limits conclusions from the iv experiments, because phrenic and hypoglossal LTF were absent in rats subjected to a similar drop in blood pressure by blood withdrawal (data not shown). Thus we are uncertain whether iv α1 receptor antagonism blocked hypoglossal LTF directly via direct effects on the hypoglossal motor nucleus, or whether this was a nonspecific result of hypotension. In an earlier study, hypotension resulting from blood withdrawal did not abolish hypoglossal LTF (25), suggesting that the influence of this factor requires further investigation.
Differing sensitivities to iv α1 receptor antagonism between phrenic and hypoglossal LTF provide another example in which mechanisms giving rise to LTF differ between hypoglossal and phrenic motor pools in important aspects (5). Whereas AIH-induced hypoglossal LTF may require α1 receptor activation (or is hypotension sensitive), pLTF does not. Further studies are needed to verify these differences. Our findings provide an interesting comparison with a recent study by Tadjalli et al. (34), who report that episodic upper airway occlusions (independent from hypoxemia) elicit long-lasting, vagal-dependent hypoglossal motor facilitation that requires α1, but not 5-HT2, receptor activation. Similar plasticity was not observed in phrenic motor output, suggesting that NE signaling is more effective at promoting hypoglossal vs. phrenic motor facilitation in multiple conditions.
The role of α1 receptor activation in respiratory motor plasticity.
Because spinal α1 receptor activation is sufficient to elicit pMF but is not necessary for AIH-induced pLTF, these receptors likely elicit phrenic motor plasticity in physiological conditions other than AIH. Conditions necessary to cause α1 receptor-dependent pMF are not yet known, but it appears that AIH is not sufficient to trigger α1 receptor activation near the phrenic motor nucleus and elicit facilitation. We suspect that these receptors come into play when the triggering stimulus is either severe (such as profound hypoxemia) or unrelated to hypoxia. For example, the fight-or-flight response may trigger greater (or phasic) NE release, promoting phrenic motor plasticity when an animal must remain alert, either flee or defend itself, or facilitating other task-related behavioral responses (1, 14).
An alternative possibility is that α1 receptor activation plays only a modulatory role in phrenic motor plasticity. For example, whereas NE does not induce hippocampal LTP, it facilitates LTP induction through both β and α1 receptors at previously potentiated synapses (17).
Because both 5-HT and NE are potent stimuli to motor neurons (12, 28), coactivation of 5-HT and α1 receptors in response to specific stimuli (e.g., severe hypoxia or stress in fight-or-flight situations) may augment respiratory plasticity. Cooperative signaling is important for other forms of plasticity (such as memory encoding) and may explain why α1 receptor activation can induce pMF, but is not involved in AIH-induced pLTF. Interactions between 5-HT and α1 receptors are important for long-term potentiation (33); conversely, subthreshold coactivation of muscarinic and NE receptors leads to long-term depression (32). Neverova et al. (25) showed that the 5-HT receptor antagonist methysergid reduced but did not inhibit PE-induced hypoglossal LTF in vitro, suggesting that both 5-HT and NE receptor activation are needed for full expression of in vitro hypoglossal LTF. Although we cannot rule out that 5-HT and α1 receptor coactivation plays an important role in respiratory motor plasticity, it is unlikely that such interactions underlie AIH-induced pLTF.
Given the data presented here, we cannot conclude that α1 receptors are playing any role in AIH-induced pLTF. However, our data raise interesting possibilities for further exploration regarding cooperative signaling and interactions between receptors in other forms of respiratory motor plasticity.
Conclusion.
Here we provide evidence that α1 receptor activation near the phrenic motor nucleus is sufficient to elicit pMF, but that α1 receptors nevertheless do not play a critical role in AIH-induced pLTF. Our findings are consistent with those from Neverova et al. (25) demonstrating involvement of α1 receptors in AIH-induced hypoglossal LTF. We also confirm the hypothesis that multiple Gq protein-coupled metabotropic receptors with different ligands elicit pMF, supporting our concept that multiple transmitter systems use similar cell signaling cascades to elicit pMF (i.e., the Q pathway), and that multiple, distinct cellular cascades are capable of eliciting phenotypically similar pMF (8). Understanding the implications of these diverse means of initiating respiratory motor plasticity, and the interactions between distinct pathways, is essential for our basic understanding of when and where respiratory plasticity is important in the life of an animal. Furthermore, by understanding the diversity of signaling pathways capable of eliciting motor plasticity, we may be able to develop novel therapies to treat disorders leading to respiratory insufficiency.
GRANTS
Support for this study was provided by National Heart, Lung, and Blood Institute Grants HL-80209, HL-69064, and HL-111598 to P.M.M and N.L.N.; by a Parker B. Francis Foundation grant to P.M.M. and N.L.N.; and by a Craig H. Neilsen Foundation grant to S.V.
DISCLOSURES
No conflicts of interest, financial or otherwise, are declared by the author(s).
AUTHOR CONTRIBUTIONS
Author contributions: A.G.H., P.M.M., and G.S.M. conception and design of research; A.G.H., P.M.M., S.V., N.L.N., and E.A.D. performed experiments; A.G.H., P.M.M., S.V., N.L.N., and E.A.D. analyzed data; A.G.H., P.M.M., and G.S.M. interpreted results of experiments; A.G.H. prepared figures; A.G.H. drafted manuscript; A.G.H., P.M.M., S.V., N.L.N., E.A.D., and G.S.M. edited and revised manuscript; A.G.H., P.M.M., S.V., N.L.N., E.A.D., and G.S.M. approved final version of manuscript.
REFERENCES
- 1.Aston-Jones G, Cohen JD. Adaptive gain and the role of the locus coeruleus-norepinephrine system in optimal performance. J Comp Neurol 493: 99–110, 2005 [DOI] [PubMed] [Google Scholar]
- 2.Bach KB, Mitchell GS. Hypoxia-induced long-term facilitation of respiratory activity is serotonin dependent. Respir Physiol 104: 251–260, 1996 [DOI] [PubMed] [Google Scholar]
- 3.Baker-Herman TL, Fuller DD, Bavis RW, Zabka AG, Golder FJ, Doperalski NJ, Johnson RA, Watters JJ, Mitchell GS. BDNF is necessary and sufficient for spinal respiratory plasticity following intermittent hypoxia. Nat Neurosci 7: 48–55, 2004 [DOI] [PubMed] [Google Scholar]
- 4.Baker-Herman TL, Mitchell GS. Phrenic long-term facilitation requires spinal serotonin receptor activation and protein synthesis. J Neurosci 22: 6239–6246, 2002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4a.Baker-Herman TL, Mitchell GS. Determinants of frequency long-term facilitation following acute intermittent hypoxia in vagotomized rats. Respir Physiol Neurobiol: 162: 8–17, 2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Baker-Herman TL, Strey KA. Similarities and differences in mechanisms of phrenic and hypoglossal motor facilitation. Respir Physiol Neurobiol 179: 48–56, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Centurion D, Mehotra S, Sanchez-Lopez A, Gupta S, MaassenVanDenBrink A, Villalon CM. Potential vascular alpha1-adrenoceptor blocking properties of an array of 5-HT receptor ligands in the rat. Eur J Pharmacol 535: 234–242, 2006 [DOI] [PubMed] [Google Scholar]
- 7.Cotecchia S. The alpha1-adrenergic receptors: diversity of signaling networks and regulation. J Recept Signal Transduct Res 30: 410–419, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Dale-Nagle EA, Hoffman MS, MacFarlane PM, Mitchell GS. Multiple pathways to long-lasting phrenic motor facilitation. Adv Exp Med Biol 669: 225–230, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Doze VA, Papay RS, Goldenstein BL, Gupta MK, Collette KM, Nelson BW, Lyons MJ, Davis BA, Luger EJ, Wood SG, Haselton JR, Simpson PC, Perez DM. Long-term alpha1A-adrenergic receptor stimulation improves synaptic plasticity, cognitive function, mood, and longevity. Mol Pharmacol 80: 747–758, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Fuller DD, Zabka AG, Baker TL, Mitchell GS. Phrenic long-term facilitation requires 5-HT receptor activation during but not following episodic hypoxia. J Appl Physiol 90: 2001–2006, 2001 [DOI] [PubMed] [Google Scholar]
- 11.Hannon J, Hoyer D. Molecular biology of 5-HT receptors. Behav Brain Res 195: 198–213, 2008 [DOI] [PubMed] [Google Scholar]
- 12.Heckman CJ, Mottram C, Quinlan K, Theiss R, Schuster J. Motoneuron excitability: the importance of neuromodulatory inputs. Clin Neurophysiol 120: 2040–2054, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Hoffman MS, Nichols NL, Macfarlane PM, Mitchell GS. Phrenic long-term facilitation after acute intermittent hypoxia requires spinal ERK activation but not TrkB synthesis. J Appl Physiol 113: 1184–1193, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Holdefer RN, Jacobs BL. Phasic stimulation of the locus coeruleus: effects on activity in the lateral geniculate nucleus. Exp Brain Res 100: 444–452, 1994 [DOI] [PubMed] [Google Scholar]
- 15.Hwa J, Graham RM, Perez DM. Identification of critical determinants of alpha 1-adrenergic receptor subtype selective agonist binding. J Biol Chem 270: 23189–23195, 1995 [DOI] [PubMed] [Google Scholar]
- 16.Jensen BC, O'Connell TD, Simpson PC. Alpha-1-adrenergic receptors: targets for agonist drugs to treat heart failure. J Mol Cell Cardiol 51: 518–528, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Katsuki H, Izumi Y, Zorumski CF. Noradrenergic regulation of synaptic plasticity in the hippocampal CA1 region. J Neurophysiol 77: 3013–3020, 1997 [DOI] [PubMed] [Google Scholar]
- 18.MacFarlane PM, Vinit S, Mitchell GS. Serotonin 2A and 2B receptor-induced phrenic motor facilitation: differential requirement for spinal NADPH oxidase activity. Neuroscience 178: 45–55, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Mahamed S, Mitchell GS. Is there a link between intermittent hypoxia-induced respiratory plasticity and obstructive sleep apnoea? Exp Physiol 92: 27–37, 2007 [DOI] [PubMed] [Google Scholar]
- 21.Mateika JH, Sandhu KS. Experimental protocols and preparations to study respiratory long term facilitation. Respir Physiol Neurobiol 176: 1–11, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Michelotti GA, Price DT, Schwinn DA. Alpha 1-adrenergic receptor regulation: basic science and clinical implications. Pharmacol Ther 88: 281–309, 2000 [DOI] [PubMed] [Google Scholar]
- 23.Mitchell GS. Respiratory plasticity following intermittent hypoxia: a guide for novel therapeutic approaches to ventilatory control disorders. In: Genetic Basis for Respiratory Control Disorders, edited by Gaultier C. New York: Springer, 2007, p. 291–311 [Google Scholar]
- 24.Mitchell GS, Baker TL, Nanda SA, Fuller DD, Zabka AG, Hodgeman BA, Bavis RW, Mack KJ, Olson EB., Jr. Invited review: intermittent hypoxia and respiratory plasticity. J Appl Physiol 90: 2466–2475, 2001 [DOI] [PubMed] [Google Scholar]
- 25.Neverova NV, Saywell SA, Nashold LJ, Mitchell GS, Feldman JL. Episodic stimulation of alpha1-adrenoreceptors induces protein kinase C-dependent persistent changes in motoneuronal excitability. J Neurosci 27: 4435–4442, 2007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Orallo F, Rosa E, Garcia-Ferreiro T, Campos-Toimil M, Cadavid MI, Loza MI. Cardiovascular effects of ketanserin on normotensive rats in vivo and in vitro. Gen Pharmacol 35: 95–105, 2000 [DOI] [PubMed] [Google Scholar]
- 27.Ramirez MT, Sah VP, Zhao XL, Hunter JJ, Chien KR, Brown JH. The MEKK-JNK pathway is stimulated by alpha1-adrenergic receptor and ras activation and is associated with in vitro and in vivo cardiac hypertrophy. J Biol Chem 272: 14057–14061, 1997 [DOI] [PubMed] [Google Scholar]
- 28.Rekling JC, Funk GD, Bayliss DA, Dong XW, Feldman JL. Synaptic control of motoneuronal excitability. Physiol Rev 80: 767–852, 2000 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Roudet C, Savasta M, Feuerstein C. Normal distribution of alpha-1-adrenoceptors in the rat spinal cord and its modification after noradrenergic denervation: a quantitative autoradiographic study. J Neurosci Res 34: 44–53, 1993 [DOI] [PubMed] [Google Scholar]
- 30.Sathi ZS, Anisuzzaman AS, Morishima S, Suzuki F, Tanaka T, Yoshiki H, Muramatsu I. Different affinities of native alpha1B-adrenoceptors for ketanserin between intact tissue segments and membrane preparations. Eur J Pharmacol 584: 222–228, 2008 [DOI] [PubMed] [Google Scholar]
- 31.Scheiderer CL, Dobrunz LE, McMahon LL. Novel form of long-term synaptic depression in rat hippocampus induced by activation of alpha 1 adrenergic receptors. J Neurophysiol 91: 1071–1077, 2004 [DOI] [PubMed] [Google Scholar]
- 32.Scheiderer CL, Smith CC, McCutchen E, McCoy PA, Thacker EE, Kolasa K, Dobrunz LE, McMahon LL. Coactivation of M(1) muscarinic and alpha1 adrenergic receptors stimulates extracellular signal-regulated protein kinase and induces long-term depression at CA3–CA1 synapses in rat hippocampus. J Neurosci 28: 5350–5358, 2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Tachibana K, Matsumoto M, Togashi H, Kojima T, Morimoto Y, Kemmotsu O, Yoshioka M. Milnacipran, a serotonin and noradrenaline reuptake inhibitor, suppresses long-term potentiation in the rat hippocampal CA1 field via 5-HT1A receptors and alpha 1-adrenoceptors. Neurosci Lett 357: 91–94, 2004 [DOI] [PubMed] [Google Scholar]
- 34.Tadjalli A, Duffin J, Peever J. Identification of a novel form of noradrenergic-dependent respiratory motor plasticity triggered by vagal feedback. J Neurosci 30: 16886–16895, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Trevino M, Frey S, Kohr G. Alpha-1 adrenergic receptors gate rapid orientation-specific reduction in visual discrimination. Cereb Cortex 22: 2529–2541, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]