

Abstract
Purpose
Enhanced structural features of resection-induced intestinal adaptation have been demonstrated following the administration of multiple different growth factors and peptides. Among these, the insulin-like growth factor (IGF) system has been considered to be significant. In this study, we employ mutant mouse strains to directly test the contribution of IGF2 and its enterocyte receptor (IGF1R) toward the adaptation response to massive small bowel resection (SBR).
Methods
IGF2-knockout (IGF2-KO) (n=8) and intestine specific IGF1R-knockout mice (IGF1R-IKO) (n=9) and their wild type (WT) littermates (n=5, n=7, respectively) underwent 50% proximal SBR. At post-operative day 7, structural adaptation was measured as crypt depth and villus height. Rates of enterocyte proliferation and apoptosis were also recorded.
Results
The successful deletion of IGF2 and IGF1R expression in the enterocytes was confirmed by RT-PCR and Western blot, respectively. Normal adaptation occurred in both IGF2-KO and IGF1R-IKO mice after 50% SBR. Post-operative rates of proliferation and apoptosis in both IGF2-KO and IGF1R-IKO mice were no different than their respective controls.
Conclusion
IGF2 and functional IGF1R signaling in enterocytes are both dispensable for resection-induced adaptation responses. The mechanism for IGF-stimulation of intestinal adaptation may involve other ligands or cellular compartments within the intestine.
Keywords: short gut syndrome, intestinal adaptation, insulin-like growth factor 1 receptor, insulin-like growth factor-2
INTRODUCTION
Intestinal adaptation is the compensatory response to massive small bowel resection (SBR). This adaptive process is defined structurally by taller villi and deeper crypts which serve to enhance mucosal surface area for improved absorption and digestion [1]. Insulin-like growth factor 1 (IGF1) and insulin-like growth factor 2 (IGF2) are recognized intestinotrophic factors for the small intestine epithelium [2, 3] and signal through insulin-like growth factor 1 receptor (IGF1R). An important role for the IGF system has been suggested by studies in which infusion of IGF1 is associated with an exaggerated adaptation response [4–8]. No prior studies have reported the effects of IGF2 administration in the context of SBR.
Previously, we showed that when Retinoblastoma (Rb) is deleted specifically in the intestine, villus height is markedly increased [9–11]. In the Rb-deficient intestine with expanded villus height, we found that IGF2 expression was dramatically up-regulated in the enterocytes [12]. By breeding Rb-null mice into an IGF2-deficient background, we found that the hyperplastic mucosal phenotype was reversed. Based upon these observations, we propose the hypothesis that IGF2 and IGF1R signaling regulates important adaptive structural responses to massive SBR.
MATERIALS AND METHODS
Experimental design
All Protocols and experiments were approved by the Washington University Animals Studies Committee (Protocol #20130038) and followed National Institutes of Health animal care guidelines.
Both IGF2 knock out (IGF2-KO) (n=8) and IGF1R intestine-specific knockout (IGF1R-IKO) (n=9) mouse lines and their wild type (WT) littermates (n=5, n=7 respectively) underwent 50% proximal SBR. Intestinal tissue was obtained at the time of resection for baseline histology. At 7 days after SBR, structural adaptation was measured by measuring changes in crypt depth and villus height. Enterocytes were isolated and used for protein and RNA analysis to confirm gene deletion. Rates of crypt cell proliferation and apoptosis were also analyzed.
Animals and small bowel resection
IGF-2 null mice were generously donated by Dr. Carla Kim, (Department of Genetics; Harvard Medical School). C57BL/6 mice (Jackson Laboratory; Bar Harbor, Maine) were used as wild type controls. For IGF1R-IKO mice, we used an intestine specific tamoxifen inducible (Villin Cre-ER (+); IGF1R (f/f)) recombinant system [13]. Wild type littermates (Villin Cre-ER (−); IGF1R (f/f)) were used as controls. Mice were kept in our standard animal holding area with a 12 hour light-dark schedule and small bowel resection was performed between ages 8 to 10 weeks of age.
Villin Cre-ER (+); IGF1R (f/f) mice and their wild type littermates (Villin Cre-ER (−); IGF1R(f/f) were injected with tamoxifen for 3 consecutive days. After the series of intraperitoneal injections, the mice were given a 3–5 day rest period prior to SBR. All mice were fed standard liquid diet (Micro-Stabilized Rodent Liquid Diet LD 101; Purina Mills, St Louis, MO) 24 hours prior to SBR. Mice were given free access to water immediately after surgery and resumed feeding from the standard liquid diet on post-operative day 1. All mice were weighed at the time of resection and also weighed again at the time of harvest on post-operative day 7.
All mice underwent a 50% proximal SBR as previously described [1]. The intestinal resections were done by transecting the bowel approximately 1 to 2 cm distal from the ligament of Treitz and 12 cm proximal from the ileocecal junction with removal of the intervening segment. End-to-end primary anastomosis was done with interrupted 9-0 monofilament sutures. A 2 cm distal segment of the resected bowel was fixed in formalin for baseline histology. Sham operations consisted of transection of the intestine at 12 cm from the ileocecal junction with reanastomosis, but without bowel resection.
Harvest and tissue isolation
All mice were harvested on post-operative day 7 as previously described [9]. The entire small bowel was flushed with ice-cold phosphate buffered saline with protease inhibitors (0.2nM phenylmethysulfonyl fluoride, 5μg/mL aprotinin, 1μM benzamidine, 1mM sodium orthovanadate, and 2μM cantharidin; EMD, Gibbstown, NJ) and excised. A 2 cm segment of bowel distal to the anastomosis was taken starting from 1 cm distal to the anastomosis. This piece of small intestine was fixed in formalin to compare with histology from previously removed bowel. The remainder of the distal segment of bowel was used to isolate crypt and villus enterocyte using our previously published protocol [9]. RNA and protein were isolated for further analysis to verify appropriate genotypes.
Histology
All intestinal sections were cut into two sections each, 50μm apart. Villus height and crypt depth were measured using MetaMorph computer program (Molecular Devices, Dowington, PA). At least 20 villi and crypts were measured per animal. Post-operative and intraoperative crypt depth and villus height were compared to calculate a percentage change to determine the magnitude of resection-induced adaptation.
Cell proliferation
At 90 minutes before harvest, 5-bromodeoxyuridine (BrdU; Zymed Laboratories, Inc, San Francisco, CA) was injected intraperitoneally. Rates of proliferation weredetermined by counting the number of cells staining positive with BrDu divided by the total number of cells in each crypt. A minimum of 20 crypts were counted per histology slide.
Apoptosis index
Each postoperative H&E stained sections were analyzed for apoptotic bodies (pyknotic nuclei, condensed chromatin) [14]. An apoptosis index was measured by counting the number apoptotic bodies found per 50 crypts.
Western blotting
Crypts were isolated and then lysed with sodium dodecyl sulfate (SDS) lysing buffer (50mM Trist-HCL pH 6.8, 2% sodium dodecyl sulfate, 10% glycerol, and 5% mercaptoethanol). The cells were sonicated for 5 seconds and heated in 100°C for 5 minutes. Protein quantification was performed using RC DC kit (Bio-Rad, Hercules, CA). Equal amounts of protein were loaded. Western blot was performed using 8% SDS-PAGE gel probing for IGF1R using anti-IGF1R antibody (Cell Signaling Technology, Danvers, MA) and Lamin B1 using anti-Lamin B1 antibody (Biovision Inc., San Francisco, CA).
RT-PCR
RNA was isolated as previously described [9, 12]. Total RNA was extracted from the intestinal tissue by following the manufacturer’s protocol for the RNAqueous kit (Ambion, Austin Texas). Total RNA concentration was determined spectrophotometrically. The quality of the obtained RNA was evaluated using the Bio-Rad Experion System with an RNA StdSens Chip and reagents (Bio-Rad Laboratories, Richmond, CA). A TaqMan RNA-to Ct 1-Step kit (Applied Biosytems, Foster city, CA) was used per the manufacturer’s protocol to determine relative gene expression directly from the isolated RNA. Equal amounts of RNA were used for real-time PCR with B-actin as an endogenous control and a standard whole bowel sample used as the calibrator. IGF2 gene expression was examined using primers, reagents and with a 750 Fast Real-Time PCR instrument all from Applied Biosystems (Foster City, CA).
Statistical analysis
Student’s t-test was done to compare percent postoperative changes in villus height and crypt depth and also to compare rates of proliferation and apoptosis between groups. A p-value of less than 0.05 was considered to be significant.
RESULTS
Confirmation of enterocyte IGF2 and IGF1R knockout
The successful deletion of IGF1R protein in the small intestine was confirmed by Western blot (Figure 1). Absent IGF2 mRNA expression was confirmed by RT-PCR [12]. The intestine-specific IGF1R knockout mice (IGF1R-IKO) have been characterized previously [15] and neither baseline crypt depth nor villus height were affected. We also verified that there was no phenotypic difference in crypt depth or villus height in unoperated IGF2-KO.
Figure 1.
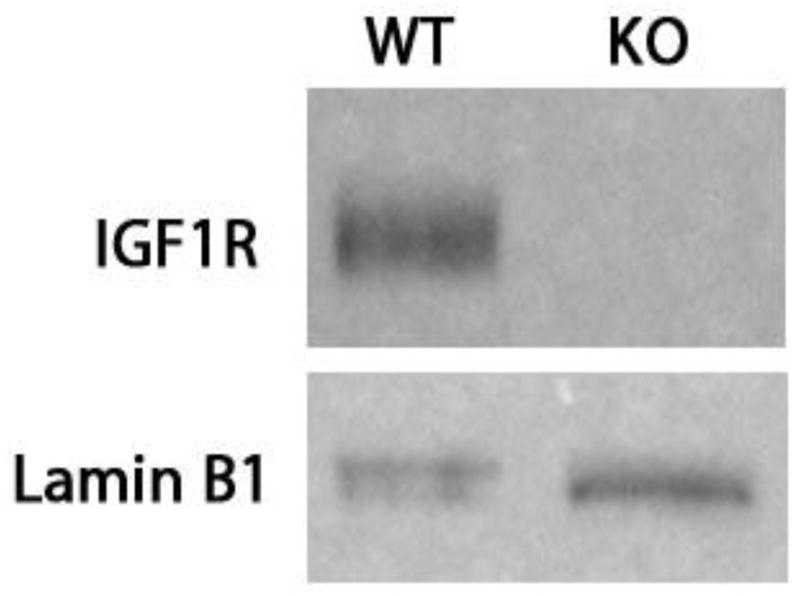
Western blotting confirming disrupted IGF1R expression in the crypt enterocytes. Villin Cre-ER (+); IGF1R (f/f) mice and their wild type littermates (Villin Cre-ER- (−)); IGF1R(f/f)were both injected with Tamoxifen for three days prior to resection. Lamin B1 was used as loading control. Successful deletion of IGF1R intestinal knockout was confirmed with all Villin Cre-ER(+); IGF1R (f/f) mice.
Structural adaptation in IGF2 and IGF1R enterocyte knockout mice
IGF2 null mice had an average 30.0±7.1% increase in crypt depth and an average of 33.7±5.9% increase in villus height after SBR when compared with C57BL/6 average of 23.0±5.2% increase in crypt depth and 48.0±8.5% in villus height (Figure 2A). There was no statistical difference in the degree of adaptation between the C57BL/6 mice and IGF2 nulls. Wild type littermates had equivalent adaptation with an average of 20.7±4.3% versus 23.4±10% increase in crypt depth and an average of 31.5±4.1% versus 34.5±5.1% in villus height respectively (Figure 2B). There was also no statistical significance in the degree of adaptation between intestinal IGFR-nulls and their wild type littermates for crypt depth and villus height respectively (Figure 2B).
Figure 2.
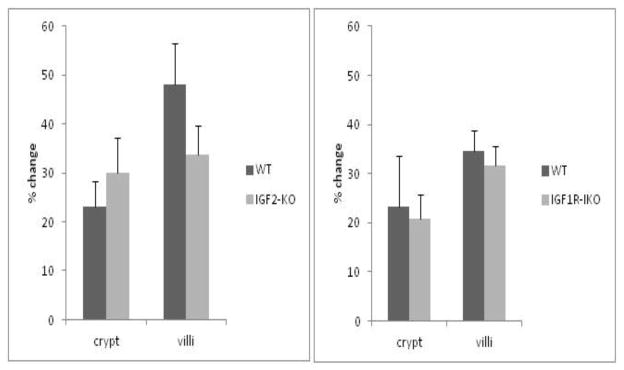
Percentage change in crypt depth and villus height for (A) IGF2-KO and (B) IGF1R-intestinal knockout (IKO) mice after small bowel resection. No differences in postoperative villus or crypt growth were observed between the mutant strains or corresponding wild-type (WT) mice in any parameter.
Rates of enterocyte proliferation in IGF2 and IGF1R intestinal knockout mice
Rates of proliferation in both IGF2-KO and intestinal IGF1R-IKO mice were similar and statistically not different following SBR. IGF2-KO had an average proliferative index of 0.49±0.02 and C57BL/6 had an average of 0.45±0.01 (Figure 3A). Similarly, IGF1R-KO mice had an average proliferative index of 0.49±0.01 compared to their wild type littermates with an average of 0.46±0.03 (Figure 3B).
Figure 3.

Post-operative rates of crypt cell proliferation in (A) IGF2-KO and (B) IGF1R- intestinal knockout (IKO) compared to their wild type (WT) littermates. BrdU was injected 90 minutes prior to harvest. Immunostaining was performed on tissue sections and BrdU- positive stained cells were counted in each crypt. A ratio was calculated as number of cells stained positive divided by total number of cells in the crypts. A minimum of 20 crypts were counted per mouse. No statistical differences were observed between either mouse strains versus their corresponding WT controls.
Rates of apoptosis in IGF2 and IGF1R enterocyte knockout mice
The apoptosis index in both IGF2-KO and IGF1R-IKO’s were not different after SBR when compared with WT mice. IGF2-KO had an average apoptotic index of 0.025±0.02 versus 0.016±0.01 in WT controls (Figure 4A). IGF1R-IKO mice had an apoptotic index of 0.04±0.008 compared with 0.03±0.008 in WT littermates (p=0.34; Figure 4B).
Figure 4.

Post-operative crypt apoptosis index for (A) IGF2-KO and (B) IGF1R- intestinal knockout (IKO) mice compared to their wild type littermates. H&E stained sections of ileum were analyzed for apoptotic bodies. Apoptotic index was calculated as number of apoptotic bodies found per number of crypts. A minimum of 50 crypts were counted per mouse. There were no statistical differences observed between the mutant mice and their corresponding WT strains.
DISCUSSION
IGF1 and IGF2 are known intestinotrophic factors [16]. Exogenous infusion of IGF1 has been shown to increase intestinal mucosal growth and enhance adaptation after SBR [4–8] Furthermore, IGF1 is a key mediator for glucagon-like-peptide-2 (GLP-2), another known intestinotrophic factor [17]. However, IGF2 has not been previously studied in the same context despite being considered to be a significant IGF in human adults [18]. Both IGF1 and IGF2 ligands bind to IGF1R [16, 19, 20]. This background therefore provides the rationale for testing the contribution of the IGF axis toward resection-associated adaptation.
In this study, we selectively disrupted IGF2 globally and IGF1R selectively within the intestinal epithelium to determine the impact of this ligand and receptor on adaptation. We confirmed the enterocyte deletion of IGF2 and IGF1R in the knockout mice and compared the effects of SBR with their respective littermates. We found that both IGF2-KO and IGF1R-IKO mice were able to adapt normally after SBR. There were also no differences in percentage weight loss between IGF2-KO and IGF1R-IKO and their respective wild-type controls after SBR. Furthermore, we observed that the rates of proliferation and apoptosis index were also equivalent.
Our finding of normal adaptation after disrupting IGF2 and IGF1R expression would suggest that the IGF system is dispensable for normal adaptation responses to massive SBR. This would be supported by the lack of significant phenotype in the unoperated intestine of the two knockout strains. On the other hand, it must be considered that the expressions of other (non-IGF) intestinotrophic growth factors are able to compensate for a lack of IGF2 or IGF1R signaling. Double and/or triple knockout mouse strains would be necessary to test the relevance of compensatory activity or expression of other ligands or growth factor receptors.
In the case of IGF2-knockout mice, it is possible that the IGF1R is activated by other IGF ligands [21, 22]. Although not measured in this study, increased compensatory expression of these other factors could therefore have modulated adaptation independent of IGF2. Supportive of this concept are the findings of enhanced adaptation responses to SBR following the exogenous administration of other IGF1R ligands such as IGF1 [4–8] as well as insulin [23–25].
Our finding of normal adaptation in the mice with enterocyte-targeted deletion of IGF1R expression would suggest that neither IGF1R activity in enterocytes nor IGF1 and IGF2 ligand expression are important for resection-induced adaptation. IGF1 and IGF2 are capable of binding to the insulin receptor although with a tenfold lower affinity for IGF2 and a 50- to 100-fold lower affinity binding for IGF1[26]. Therefore, it must be considered that the IGF ligands are capable of governing intestinal adaptation via the insulin receptor.
Since our intestinal IGF1R knockout mice have normal IGF1R activity in all other cells besides enterocytes, it is plausible that the IGF system drives adaptation via non-epithelial cells within the intestine. IGF1 is known to be mitogenic to myofibroblasts [27]. As such, it is possible that intestinal mucosal growth is driven by IGF1R stimulation of subepithelial mesenchymal or immune cells. This paradigm is similar to our findings of impaired resection-induced adaptation responses under multiple conditions of global inhibition of epidermal growth factor receptor (EGFR) signaling [28] but preserved responses in mice with genetically disrupted expression of intestinal epithelial EGFR [29].
We recently found that targeted enterocyte deficiency of Rb was associated with significant intestinal epithelial growth and enhanced expression of epithelial IGF2 [12]. In these mice, the trophic effects of Rb-deficiency were completely reversed by breeding with the same mutant strains (IGF2-KO and IGF1R-IKO) as in the current study. These findings would suggest that the hyperplastic mucosal phenotype associated with Rb deficiency and SBR may occur via different mechanisms. More importantly, these findings underscore the consideration that mucosal surface area expansion can be accomplished by multiple disparate, perhaps overlapping and/or additive signaling pathways. Since this is an important physiologic response, precise molecular dissection of the most important and potent intestinotrophic signaling pathways and most relevant cell compartments will be required to optimize therapeutic interventions in patients who have suffered a massive intestinal loss.
Acknowledgments
This work was supported by National Institutes of Health Grants R01 DK 059288 (Warner), P30DK52574– Morphology and Murine Models Cores of the Digestive Diseases Research Core Center of the Washington University School of Medicine, and the Children’s Surgical Sciences Research Institute of the St. Louis Children’s Hospital Foundation. Dr. Sun is also supported by a Research Fellowship Award through the Association for Academic Surgery Foundation.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Helmrath MA, VanderKolk WE, Can G, et al. Intestinal adaptation following massive small bowel resection in the mouse. J Am Coll Surg. 1996;183:441–449. [PubMed] [Google Scholar]
- 2.Conlon MA, Francis GL, Tomas FM, et al. Continuous 14 day infusion of IGF-II increases the growth of normal female rats, but exhibits a lower potency than IGF-I. J Endocrinol. 1995;144:91–98. doi: 10.1677/joe.0.1440091. [DOI] [PubMed] [Google Scholar]
- 3.Zaina S, Pettersson L, Thomsen AB, et al. Shortened life span, bradycardia, and hypotension in mice with targeted expression of an Igf2 transgene in smooth muscle cells. Endocrinology. 2003;144:2695–2703. doi: 10.1210/en.2002-220944. [DOI] [PubMed] [Google Scholar]
- 4.Burrin DG, Wester TJ, Davis TA, et al. Orally administered IGF-I increases intestinal mucosal growth in formula-fed neonatal pigs. Am J Physiol. 1996;270:R1085–1091. doi: 10.1152/ajpregu.1996.270.5.R1085. [DOI] [PubMed] [Google Scholar]
- 5.Conlon MA, Tomas FM, Owens PC, et al. Long R3 insulin-like growth factor-I (IGF-I) infusion stimulates organ growth but reduces plasma IGF-I, IGF-II and IGF binding protein concentrations in the guinea pig. J Endocrinol. 1995;146:247–253. doi: 10.1677/joe.0.1460247. [DOI] [PubMed] [Google Scholar]
- 6.Dahly EM, Guo Z, Ney DM. IGF-I augments resection-induced mucosal hyperplasia by altering enterocyte kinetics. Am J Physiol Regul Integr Comp Physiol. 2003;285:R800–808. doi: 10.1152/ajpregu.00014.2003. [DOI] [PubMed] [Google Scholar]
- 7.Lemmey AB, Ballard FJ, Martin AA, et al. Treatment with IGF-I peptides improves function of the remnant gut following small bowel resection in rats. Growth Factors. 1994;10:243–252. doi: 10.3109/08977199409010990. [DOI] [PubMed] [Google Scholar]
- 8.Lemmey AB, Martin AA, Read LC, et al. IGF-I and the truncated analogue des-(1-3)IGF-I enhance growth in rats after gut resection. Am J Physiol. 1991;260:E213–219. doi: 10.1152/ajpendo.1991.260.2.E213. [DOI] [PubMed] [Google Scholar]
- 9.Guo J, Longshore S, Nair R, et al. Retinoblastoma protein (pRb), but not p107 or p130, is required for maintenance of enterocyte quiescence and differentiation in small intestine. J Biol Chem. 2009;284:134–140. doi: 10.1074/jbc.M806133200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Haigis K, Sage J, Glickman J, et al. The related retinoblastoma (pRb) and p130 proteins cooperate to regulate homeostasis in the intestinal epithelium. J Biol Chem. 2006;281:638–647. doi: 10.1074/jbc.M509053200. [DOI] [PubMed] [Google Scholar]
- 11.Leinicke JA, Longshore S, Wakeman D, et al. Regulation of retinoblastoma protein (Rb) by p21 is critical for adaptation to massive small bowel resection. J Gastrointest Surg. 2012;16:148–155. doi: 10.1007/s11605-011-1747-8. discussion 155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Choi P, Guo J, Erwin CR, et al. IGF-2 mediates intestinal mucosal hyperplasia in retinoblastoma protein (Rb)-deficient mice. J Pediatr Surg. 2013;48:1340–1347. doi: 10.1016/j.jpedsurg.2013.03.042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.el Marjou F, Janssen KP, Chang BH, et al. Tissue-specific and inducible Cre-mediated recombination in the gut epithelium. Genesis. 2004;39:186–193. doi: 10.1002/gene.20042. [DOI] [PubMed] [Google Scholar]
- 14.Wakeman D, Guo J, Santos JA, et al. p38 MAPK regulates Bax activity and apoptosis in enterocytes at baseline and after intestinal resection. Am J Physiol Gastrointest Liver Physiol. 2012;302:G997–1005. doi: 10.1152/ajpgi.00485.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Rowland KJ, Trivedi S, Lee D, et al. Loss of glucagon-like peptide-2-induced proliferation following intestinal epithelial insulin-like growth factor-1-receptor deletion. Gastroenterology. 2011;141:2166–2175. e2167. doi: 10.1053/j.gastro.2011.09.014. [DOI] [PubMed] [Google Scholar]
- 16.Lund PK. Molecular basis of intestinal adaptation: the role of the insulin-like growth factor system. Ann N Y Acad Sci. 1998;859:18–36. doi: 10.1111/j.1749-6632.1998.tb11108.x. [DOI] [PubMed] [Google Scholar]
- 17.Bortvedt SF, Lund PK. Insulin-like growth factor 1: common mediator of multiple enterotrophic hormones and growth factors. Curr Opin Gastroenterol. 2012;28:89–98. doi: 10.1097/MOG.0b013e32835004c6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.LeRoith D, Roberts CT., Jr The insulin-like growth factor system and cancer. Cancer Lett. 2003;195:127–137. doi: 10.1016/s0304-3835(03)00159-9. [DOI] [PubMed] [Google Scholar]
- 19.Frasca F, Pandini G, Scalia P, et al. Insulin receptor isoform A, a newly recognized, high-affinity insulin-like growth factor II receptor in fetal and cancer cells. Mol Cell Biol. 1999;19:3278–3288. doi: 10.1128/mcb.19.5.3278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Heidegger I, Pircher A, Klocker H, et al. Targeting the insulin-like growth factor network in cancer therapy. Cancer Biol Ther. 2011;11:701–707. doi: 10.4161/cbt.11.8.14689. [DOI] [PubMed] [Google Scholar]
- 21.Belfiore A, Frasca F, Pandini G, et al. Insulin receptor isoforms and insulin receptor/insulin-like growth factor receptor hybrids in physiology and disease. Endocr Rev. 2009;30:586–623. doi: 10.1210/er.2008-0047. [DOI] [PubMed] [Google Scholar]
- 22.Pollak M. The insulin receptor/insulin-like growth factor receptor family as a therapeutic target in oncology. Clin Cancer Res. 2012;18:40–50. doi: 10.1158/1078-0432.CCR-11-0998. [DOI] [PubMed] [Google Scholar]
- 23.Ben Lulu S, Coran AG, Mogilner JG, et al. Oral insulin stimulates intestinal epithelial cell turnover in correlation with insulin-receptor expression along the villus-crypt axis in a rat model of short bowel syndrome. Pediatr Surg Int. 2010;26:37–44. doi: 10.1007/s00383-009-2520-x. [DOI] [PubMed] [Google Scholar]
- 24.Ben Lulu S, Coran AG, Shehadeh N, et al. Oral insulin stimulates intestinal epithelial cell turnover following massive small bowel resection in a rat and a cell culture model. Pediatr Surg Int. 2012;28:179–187. doi: 10.1007/s00383-011-2991-4. [DOI] [PubMed] [Google Scholar]
- 25.Sukhotnik I, Shehadeh N, Shamir R, et al. Oral insulin enhances intestinal regrowth following massive small bowel resection in rat. Dig Dis Sci. 2005;50:2379–2385. doi: 10.1007/s10620-005-3067-x. [DOI] [PubMed] [Google Scholar]
- 26.Blakesley VA, Scrimgeour A, Esposito D, et al. Signaling via the insulin-like growth factor-I receptor: does it differ from insulin receptor signaling? Cytokine Growth Factor Rev. 1996;7:153–159. doi: 10.1016/1359-6101(96)00015-9. [DOI] [PubMed] [Google Scholar]
- 27.Simmons JG, Pucilowska JB, Keku TO, et al. IGF-I and TGF-beta1 have distinct effects on phenotype and proliferation of intestinal fibroblasts. Am J Physiol Gastrointest Liver Physiol. 2002;283:G809–818. doi: 10.1152/ajpgi.00057.2002. [DOI] [PubMed] [Google Scholar]
- 28.Warner BW, Erwin CR. Critical roles for EGF receptor signaling during resection-induced intestinal adaptation. J Pediatr Gastroenterol Nutr. 2006;43 (Suppl 1):S68–73. doi: 10.1097/01.mpg.0000226393.87106.da. [DOI] [PubMed] [Google Scholar]
- 29.Rowland KJ, McMellen ME, Wakeman D, et al. Enterocyte expression of epidermal growth factor receptor is not required for intestinal adaptation in response to massive small bowel resection. J Pediatr Surg. 2012;47:1748–1753. doi: 10.1016/j.jpedsurg.2012.03.089. [DOI] [PMC free article] [PubMed] [Google Scholar]