

Abstract
Adenovirus inundates the productively infected cell with linear, double-stranded DNA and an abundance of single-stranded DNA. The cellular response to this stimulus is antagonized by the adenoviral E1B and E4 early genes. A mutant group C adenovirus that fails to express the E1B-55K and E4orf3 genes is unable to suppress the DNA-damage response. Cells infected with this double-mutant virus display significant morphological heterogeneity at late times of infection and frequently contain fragmented nuclei. Nuclear fragmentation was due to the translocation of apoptosis inducing factor (AIF) from the mitochondria into the nucleus. The release of AIF was dependent on active poly(ADP-ribose) polymerase-1 (PARP-1), which appeared to be activated by viral DNA replication. Nuclear fragmentation did not occur in AIF-deficient cells or in cells treated with a PARP-1 inhibitor. The E1B-55K or E4orf3 proteins independently prevented nuclear fragmentation subsequent to PARP-1 activation, possibly by altering the intracellular distribution of PAR-modified proteins.
Keywords: Adenovirus, Apoptosis inducing factor, Poly(ADP-ribose) polymerase-1, E1B-55K, E4orf3, Nuclear morphology
INTRODUCTION
Entry of adenovirus into a host cell and the replication of the linear, double-stranded DNA viral genome inundates the cell with what may be sensed as double-stranded DNA breaks, regions of single-stranded DNA, and stalled replication forks (Karen et al., 2009; Nichols et al., 2009; Shepard and Ornelles, 2004). Adenovirus dampens the host DNA-damage response (DDR) by the action of viral proteins encoded in the E1B and E4 early genes (Turnell and Grand, 2012; Weitzman et al., 2004). One mechanism of signaling DNA damage proceeds through the phosphorylation of latent cellular proteins (Karran, 2000). Among the first responders to DNA damage is the sensor protein complex Mre11/Rad50/Nbs1 (MRN) (Williams et al., 2010). Once localized to sites of DNA damage, the MRN complex recruits apical kinases related to the phosphoinositol-3'-kinase, including the DNA-dependent protein kinase (DNA-PK), ataxia telangiectasia mutated protein (ATM), and ATM- and Rad3-related protein (ATR) (Durocher and Jackson, 2001; Nam and Cortez, 2011). ATM is most closely associated with the double-stranded DDR through checkpoint kinase 2 (Chk2), ATR is associated with the single-stranded DDR though checkpoint kinase 1, and DNA-PK is associated with the repair of double-stranded breaks through non-homologous end-joining (NHEJ) (Karran, 2000; Turnell and Grand, 2012). Once active, these apical kinases activate distal kinases, scaffolding proteins such as H2AX, and effector proteins such as p53 that direct DNA repair (Khanna and Jackson, 2001), cell cycle arrest (Zhou and Elledge, 2000), or cell death (Biss and Xiao, 2012). Adenovirus inhibits multiple steps of the DDR pathway. The MRN complex is inhibited by the adenoviral E1B-55K/E4orf6 protein complex that targets Mre11 for degradation and by the adenovirus serotype 5-specific E4orf3-mediated sequestration of MRN protein members into nuclear tracks and cytoplasmic aggresomes (Araujo et al., 2005; Stracker et al., 2002; Stracker et al., 2005). Inactivation of the MRN complex blocks signaling through the kinases ATM (Carson et al., 2003; Mathew and Bridge, 2008) and ATR (Carson et al., 2009). The ATR arm of the DDR is targeted during adenovirus serotype 12 infections by E4orf6-mediated degradation of TOPBP1 independently of E1B-55K (Blackford et al., 2010) and by the E1B-55K-associated protein 5 (E1B-AP5) and its ability to promote phosphorylation of ATR substrates. The NHEJ arm of the DDR is targeted by E4orf6 in complex with E1B-55K through the degradation of DNA ligase IV (Baker et al., 2007) and independently of E1B-55K by disrupting the association of XRCC4 with DNA ligase IV, thus precluding binding to DNA (Jayaram et al., 2008a; Jayaram et al., 2008b). E4orf6 also inhibits protein phosphatase 2A (PP2A) leading to the sustained phosphorylation of DNA-PK and H2AX with hyperactivation of the DDR in response to ionizing radiation (Hart et al., 2007; Hart et al., 2005). The master regulator of cell survival, p53, is inhibited at the transcriptional level by the E1B-55K protein alone (Hartl et al., 2008; Martin and Berk, 1998; Yew and Berk, 1992), through degradation by the E1B-55K/E4orf6 complex (Querido et al., 2001; Querido et al., 1997), and through the inhibitory methylation of p53-target promoters by the E4orf3 protein (Soria et al., 2010). Cells infected with an E4-deleted adenovirus show evidence of a strong DDR, including activation of non-homologous end-joining resulting in concatenation of the viral genome (Weiden and Ginsberg, 1994). Genome concatenation is believed to contribute to the inhibition of late protein synthesis in some E4-mutant adenoviral infections (Jayaram and Bridge, 2005). Defects in viral DNA replication in the absence of E4 adenoviral proteins are due to the Nbs1-dependent, Rad50-stabilized binding of Mre11 to viral DNA (Mathew and Bridge, 2007, 2008). Cells infected with a virus bearing deletions in the E1B-55K and E4orf3 genes also show robust DNA damage signaling and typically die as quickly as cells infected with single-mutant viruses (Shepard and Ornelles, 2004).
The phosphorylation-mediated DNA damage signaling to p53 has been studied extensively in the context of a viral infection (reviewed in Turnell and Grand, 2012); however, signaling also proceeds through the activation of poly(ADP-ribose) polymerase-1 (PARP-1). PARP-1 activation occurs in response to DNA-damage and promotes the addition of poly(ADP-ribose) (PAR) to PARP-1 itself, the ribosylation of cellular proteins such as histones, and the accumulation of free PAR chains (Halldorsson et al., 1978; Wang et al., 2009a). PARP-1 automodification and the localization of PAR at sites of DNA damage recruit DNA signaling and repair proteins and leads to PAR-modification of these proteins (Haince et al., 2007; Li and Yu, 2013; Sousa et al., 2012; Wang et al., 2012). Not all forms of DNA damage activate PARP-1 to an equivalent extent. In neuronal cells, cell death associated with PARP-1 activation tends to result from excitotoxic signals or in response to the DNA-alkylating agent N-methyl-N'-nitro-N-nitrosoguanidine (MNNG) (Andrabi et al., 2006; Yu et al., 2003; Yu et al., 2002). PARP-1 activation is also required for apoptosis inducing factor (AIF) to translocate to the nucleus (Yu et al., 2002).
Translocation of AIF into the nucleus can lead to profound changes to the integrity of the cell nucleus and the survival of the cell. AIF provokes a caspase-independent form of cell death accompanied by PARP-1-dependent nuclear fragmentation (Susin et al., 1999; Yu et al., 2002). The mechanism by which active PARP-1 signals for death and facilitates AIF release from the mitochondria remains unclear. Studies have demonstrated that free PAR chains are sufficient to induce caspase-independent cell death (Andrabi et al., 2006). Still other studies indicate that AIF itself must be PAR ribosylated in order to be released from the mitochondria and to translocate to the nucleus (Wang et al., 2002). Translocation of AIF to the nucleus was found in irradiated cells radiosensitized by the E4orf6 protein of adenovirus and AIF was found to be required for this radiosensitization (Hart et al., 2007). Perhaps this interaction of AIF with an adenoviral protein suggests the existence of a relationship between adenovirus and caspase-independent death signaling, much in the same way that adenovirus inhibits caspase-dependent apoptosis (Burgert et al., 2002; Degenhardt et al., 2000).
We report here that adenovirus-infected cells that fail to express the E1B-55K and E4orf3 proteins show striking heterogeneity in cellular morphology and nuclear integrity. The emergence of this heterogeneity unmasks the importance of these two adenoviral proteins in orchestrating the apparent uniformity seen in a typical adenoviral infection. Viral genome replication was found to activate PARP-1 and, when signaling was unhindered, showed similarities to excitotoxic cell death. Either the E1B-55K or E4orf3 proteins were sufficient to prevent translocation of AIF from the mitochondria to the nucleus and to prevent nuclear fragmentation. The mechanism by which the E1B-55K and E4orf3 proteins prevent AIF translocation remains to be determined, although evidence is presented to suggest that these proteins alter the distribution of PAR-modified proteins, thus preventing nuclear fragmentation.
RESULTS
Striking morphological heterogeneity among cells infected with the E1B-55K/E4orf3 double-mutant virus
Adherent HeLa cells in a subconfluent monolayer exhibit the characteristic morphology of epithelial cells where each cell adopts an irregular polygonal shape (Fig. 1A, mock). After 72 h, cells infected with the E1B-55K single-mutant virus (Fig. 1A, ΔE1B-55K) were loosely attached to the substrate and appeared uniformly round. The loosely-attached rounded cells showed a median diameter of 11 μm (Fig. 1B, ΔE1B-55K). Cells infected with either the wild-type or the E4orf3 single-mutant viruses appeared similar to E1B-55K single-mutant virus-infected cells (data not shown) and the detached rounded cells were of similar size to the E1B-55K single-mutant virus-infected cells (Fig. 1B). By contrast, cells infected by the E1B-55K/E4orf3 double-mutant virus included detached and rounded cells as well as adherent, polygonal cells (Fig.1A, ΔE1B-55K/ΔE4orf3). These attached cells were infected because over 99% of similarly infected cells stained for an early adenovirus protein (data not shown). The diameter of the detached and rounded double-mutant virus-infected cells varied considerably with a median diameter of 16 μm and an interquartile range nearly twice that of the wild-type and single-mutant virus-infected cells (Fig. 1B). These cells also appeared significantly larger than wild-type, E4orf3- and E1B-55K-mutant virus-infected cells (p = 0.01, 0.001 and 0.0002, respectively). The heterogeneous morphology of double-mutant virus-infected cells was confirmed by flow cytometry. Forward scatter reflects the volume of the cell while side scatter reflects properties such as the presence of cytoplasmic granules, nuclear shape, and membrane roughness. Although there was no appreciable difference in cell volume, side scatter increased for all infected cells and the heterogeneity in this measurement was especially pronounced in cells infected with the double-mutant virus (Fig. 1C).
Figure 1.

HeLa cells infected with the E1B-55K/E4orf3 double-mutant virus are heterogeneous in appearance at late times of infection. (A) HeLa cells were mock-infected or infected at an MOI of 10 with the E1B-55K-deleted virus (ΔE1B-55K) or E1B-55K/E4orf3 double-mutant virus (ΔE1B-55K/ΔE4orf3). Representative images captured by phase-contrast microscopy at 72 hpi are shown. (B) HeLa cells were infected with the indicated viruses and the apparent diameters of detached cells were determined as described in the Materials and Methods. The mean diameters of 120 to 250 cells are summarized in the box and whiskers plot. Differences among mean cell diameters from the three independent experiments were significant (p = 0.008, one-way ANOVA allowing for unequal variances among groups.) Application of the t-test with correction for multiple comparisons indicated that cells infected with the E1B-55K/E4orf3 double-mutant virus were significantly larger than other virus-infected cells (p < 0.01). (C) HeLa cells were infected at an MOI of 10 with the indicated viruses and live cells were analyzed for size (Forward Scatter) and intracellular or surface perturbations (Side Scatter) by flow cytometry. The percentage of cells found in the upper left quadrant is indicated in each panel. (D) HeLa cells were mock-infected or infected with wild-type or double-mutant viruses at an MOI of 10 and processed for scanning electron microscopy at 72 hpi.
The striking variability in cell shape determined by phase contrast microscopy and flow cytometry was evident in the morphology of the plasma membrane visualized by scanning electron microscopy. Mock-infected cells were flat, polygonal and displayed numerous microvilli (Fig. 1D, mock). Wild-type virus-infected cells, which were uniformly round in appearance, displayed numerous small blebs and ruffles. Very few microvilli were evident in these cells (Fig 1D, wild-type). By contrast, the representative image of E1B-55K/E4orf3 double-mutant virus-infected cells illustrates the heterogeneity in cell shape and the variability in membrane morphology (Fig. 1D, ΔE1B-55K/ΔE4orf3). Some cells appeared smooth with few membrane blebs and appeared similar to wild-type virus-infected cells. Additional cells contained small blebs that were larger in extent and protruded further from the membrane than in wild-type virus-infected cells. Some cells appeared to retain long thin cytoplasmic processes attached to the substrate. Still other cells appeared to have fragmented and possibly damaged membranes showing a granular texture (Fig. 1D, ΔE1B-55K/ΔE4orf3). It should be noted that although all infected cells displayed a substantial cytopathic effect, over 90% of the cells infected with the single-mutant viruses and over 75% of the cells infected with the double-mutant virus remained viable by Trypan blue dye-exclusion at 72 hpi (data not shown and see Shepard and Ornelles, 2004). These results indicate that, even though the cells were infected at a high multiplicity, expression of the E1B-55K and E4orf3 proteins contributes to the uniform appearance of adenovirus-infected cells at late times of infection.
Unusual nuclear morphology among cells infected with the E1B-55K/E4orf3 double-mutant virus
In addition to the perturbations seen at the whole cell level and the plasma membrane, cells infected with the E1B-55K/E4orf3 double-mutant virus show aberrant nuclear morphology. This aberrant nuclear morphology was first observed at approximately 54 hpi and reached a steady-state level by 72 hpi (data not shown). The nuclear lamina was visualized in infected HeLa cells 72 hpi by immunofluorescence microscopy and DNA was simultaneously visualized by staining with 4',6-diamidino-2-phenylindole (DAPI). Most cells infected with the double-mutant adenovirus displayed a non-uniform distribution of DNA surrounded by the nuclear lamina similar to that observed in wild-type and single-mutant virus-infected cells (Fig. 2A, B and C). The nuclei of approximately 10% of the double-mutant virus-infected cells contained highly condensed chromatin surrounded by diffuse lamina staining (Fig. 2D, E and F). These nuclei superficially resemble a metaphase nucleus but lack the characteristic bipolar symmetry (Tanenbaum and Medema, 2010). The remaining 15–20% of cells contains nuclei of an unusual and distinctly fragmented nature (Fig. 2G, H and I). The DNA in these fragmented nuclei was partitioned into approximately 5 to 25 discrete bodies surrounded by an apparently intact nuclear lamina. Confocal microscopy confirmed that many of these discrete DAPI-stained bodies were separated from each other and surrounded by nuclear lamina. These lamina-bound blebs of nuclear material differ from the fragmented nuclei seen in apoptotic cells because proteolytic cleavage of lamin and the subsequent disruption of nuclear lamina occur prior to apoptotic nuclear fragmentation (Lazebnik et al., 1995).
Figure 2.
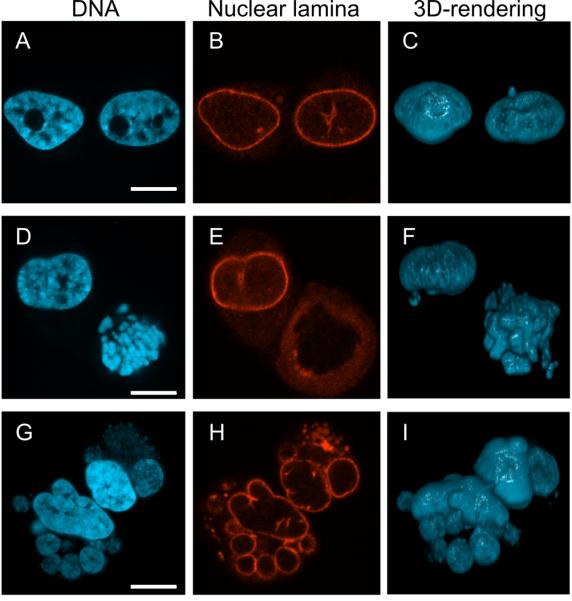
HeLa cells infected with the E1B-55K/E4orf3 double-mutant virus exhibit unusual nuclear morphology. HeLa cells were infected with the double-mutant virus at an MOI of 10 and stained at 72 hpi with DAPI to visualize DNA (A, D, G) or an autoimmune serum recognizing the nuclear lamina (B, E, H). Each row represents a pair of cells imaged by confocal microscopy with the three dimensional distribution of DNA represented in the rendering in panels C, F, and I. Scale bar indicates 10 μm.
Nuclear fragmentation by the E1B-55K/E4orf3 double-mutant virus occurs in tumorigenic and non-tumorigenic cell lines
The nuclear fragmentation elicited by the E1B-55K/E4orf3 double-mutant virus resembles fragmentation associated with caspase-independent programmed cell death. Because the pathways governing growth, survival and death in tumor cells often differ from those in benign cells (Hsieh et al., 2012; Sette et al., 2012; Zeng et al., 2012), we evaluated additional cell lines for their susceptibility to nuclear fragmentation by the double-mutant virus. In both cervical cancer-derived HeLa cells and prostate cancer-derived PC3 cells, the frequency of cells with fragmented nuclei after infection with wild-type and single-mutant adenoviruses was the same as mock-infected cells (Fig. 3A). By contrast, more cells with fragmented nuclei (8–15%) were seen after infection with the double-mutant virus.
Figure 3.

The E1B-55K/E4orf3 double-mutant virus elicits nuclear fragmentation in multiple cell lines. The viruses indicated were used at an MOI of 10 to infect (A) tumorigenic HeLa and PC3 cells and (B) non-tumorigenic hTERT RPE-1 and MCF10A cells. Cells were stained for DNA at 72 hpi and the fraction of cells containing fragmented nuclei determined. Results from a representative experiment of at least three experiments are shown. Error bars indicate the upper 95% exact binomial confidence interval for the representative experiment.
Similar results were obtained with two non-tumorigenic, immortal cell lines. Approximately 5 to 15% of the nuclei of human telomerase-expressing retinal pigment epithelial cells (hTERT RPE-1) and mammary epithelial MCF10A cells contained fragmented nuclei 72 h after infection with the double-mutant virus. As observed for tumor-derived cells, the frequency of fragmented nuclei was less than 1% for non-infected cells and cells infected with the wild-type or single-mutant viruses (Fig. 3B). Although the frequency of nuclear fragmentation varied among cell lines, the increase appears to be unrelated to the tumorigenic nature of the cell line. The increase in fragmentation was significant for double-mutant virus-infected cells compared to all other infected cells among HeLa, hTERT RPE-1, and MCF10A cells and for double-mutant compared to E4orf3-mutant virus-infected PC3 cells.
AIF is released from the mitochondria independent of other mitochondrial proteins
The fragmented nuclei in double-mutant virus-infected cells appear similar to those observed in cells dying by a caspase-independent mechanism linked to the release of apoptosis-inducing factor (AIF) from the mitochondria (Lorenzo et al., 1999). However, adenovirus-infected cells do not exhibit classical markers of apoptosis, most likely because the E1B-19K protein inhibits mitochondrial permeabilization by binding to Bax and Bak and preventing both Bax and Bak hetero- and homo-oligomerization (Han et al., 1998; Sundararajan and White, 2001). The localization of AIF and another mitochondrial protein, COX IV, was evaluated at 72 hpi. In mock-infected cells, AIF was restricted to the mitochondria and, at the resolution afforded by fluorescence microscopy, co-localized with COX IV in elongated structures spanning the cytoplasm (Fig. 4A). Although most cells infected with the wild-type or single-mutant viruses detached by 72 hpi and showed a thin rim of cytoplasm about the swollen nucleus, AIF and COX IV remained closely associated in the cytoplasm (data not shown) and AIF remained excluded from the nucleus as seen with the representative cells infected with the E1B-55K-mutant virus (Fig. 4B, ΔE1B-55K). By contrast, AIF was found outside of the mitochondria and in the nuclei of a subset of cells infected with the double-mutant virus. The coincidence of AIF and DNA only occurred in cells showing nuclear fragmentation, as seen with the representative cells (Fig. 4B, ΔE1B-55K/ΔE4orf3, fragmented).
Figure 4.

AIF is found outside the mitochondria in cells with fragmented nuclei. HeLa cells were infected with the indicated viruses at an MOI of 10, processed for immunofluorescence at 72 hpi, and imaged by confocal microscopy. Representative images are shown in panels A and B. The scale bar indicates 10 μm. The virus identified as ΔpTP bears a deletion in the preterminal protein gene and is unable to direct viral DNA replication. (A) Staining for AIF and the mitochondrial marker COX IV is coincident in the mock-infected cells. (B) AIF remains in the cytoplasm in mock-infected cells and cells infected with the E1B-55K-mutant virus. Nuclear translocation of AIF is seen only in cells with fragmented nuclei following infection with the E1B-55K/E4orf3 double-mutant virus. (C) The coincidence of AIF and COX IV was determined in at least 50 cells and quantified with the Pearson's correlation coefficient. Cells infected with the double-mutant virus were stratified into those with intact nuclei and those with fragmented nuclei. (D) Manders' coefficients of overlap were determined to quantify the proportion of COX IV staining that is coincident with AIF staining (red symbols) and the proportion of AIF staining that is coincident with COX IV staining (green symbols).
The association of AIF and COX IV was quantified with at least 50 confocal images of infected cells stained for AIF and COX IV. Double-mutant virus-infected cells were further stratified into those with intact or fragmented nuclei. Pearson's correlation coefficient provides a global measure of the association of staining for AIF and COX IV. For all cells containing intact nuclei, AIF and COX IV showed a strong positive correlation, with a median value of 0.95. More than 75% of the cells analyzed had a value greater than 0.9. By contrast, the median Pearson's correlation coefficient for double-mutant virus-infected cells with fragmented nuclei was 0.77 and varied over a wider range among individual cells (Fig. 4C). These results show a significant (p < 0.0001) separation of staining for COX IV and AIF in cells with fragmented nuclei. These images were also used to determine modified Manders' coefficients of overlap, which quantifies the proportion of one signal above an automatically determined threshold that is coincident with the other signal. This pair of coefficients provides a directional measure of how frequently one fluorophore coincides with the other. For mock-infected cells and cells infected with the wild-type or single-mutant viruses or with a replication-defective viral mutant deleted of the pre-terminal protein gene, nearly all the AIF signal was coincident with COX IV signal. Because COX IV is restricted to mitochondria, this indicates that most of the AIF remained in the mitochondria in these cells. A slightly smaller fraction of the COX IV signal was coincident with AIF, suggesting that some COX IV staining occurred in areas without AIF. Similar Manders' coefficients were measured for both COX IV and AIF in double-mutant virus-infected cells with intact nuclei (Fig. 4D). However, the Manders' coefficients for double-mutant virus-infected cells with fragmented nuclei revealed the basis for a reduced Pearson's correlation coefficient seen in Fig. 4C. The fraction of COX IV coincident with AIF was modestly reduced. By contrast, the fraction of AIF signal that overlapped COX IV signal was significantly reduced in cells containing fragmented nuclei compared to all other conditions (p < 0.0001). This indicates that a significant portion of AIF was found in areas of the cell devoid of COX IV staining. This measurement supports the visual evidence indicating that some AIF remained in the mitochondria but that a portion was released from the mitochondria and often entered the nucleus. In double-mutant virus-infected cells with fragmented nuclei, AIF also colocalized less with cytochrome c, which was found in discrete structures mimicking COX IV staining (data not shown). These results further show that AIF escapes the mitochondria in the presence of the E1B-19K protein, which is a potent inhibitor of mitochondrial permeability.
Nuclear fragmentation is AIF-dependent
Nuclear fragmentation following infection with the E1B-55K/E4orf3 double-mutant virus resembles effects associated with a PARP-1-responsive and caspase-independent mechanism of cell death that is mediated by AIF (Gilliams-Francis et al., 2003; Yu et al., 2002). To determine if the nuclear fragmentation observed here required AIF, AIF-proficient and AIF-deficient HeLa cells were infected and their nuclear morphology was evaluated after 72 h. The ablation of AIF in HeLa cells is evident by the absence of AIF protein detected by immunoblotting (Fig. 5A). Irrespective of AIF status, few uninfected cells or cells infected with the wild-type or single mutant viruses contained fragmented nuclei (<2.5%). However, AIF-deficient cells failed to display the increase in fragmented nuclei seen in AIF-proficient cells after infection with the double-mutant virus (Fig. 5B). This result indicates that AIF is required for the nuclear fragmentation seen in double-mutant viral infections of HeLa cells. Although tumor cell lines often differ in their susceptibility to death by the AIF-dependent pathways (Kanno et al., 2012), we observed similar results using PC3 cells depleted of AIF. The knockdown of AIF in the PC3 cells was comparable to that achieved in HeLa cells and has been previously described (Lewis et al., 2012). Even though fewer PC3 cells contained fragmented nuclei overall, the only cells to show a significant (p < 0.05) increase in fragmented nuclei were AIF-proficient cells infected with the double-mutant virus (Fig. 5C). Additional studies that measured changes in the electrical impedance across the infected cell monolayer over time indicated that AIF also contributed to the heterogeneity observed at the whole-cell level in double-mutant virus-infected cells and that this whole-cell heterogeneity was suppressed by either E1B-55K or E4orf3 (data not shown). This requirement for the E1B-55K and E4orf3 proteins to suppress AIF-mediated nuclear fragmentation and AIF-dependent morphological heterogeneity reveals a new relationship between AIF and adenovirus infection.
Figure 5.
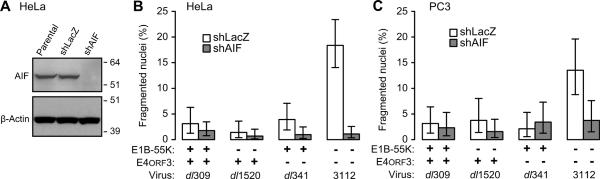
Nuclear fragmentation is AIF-dependent. (A) Protein lysates from identical numbers of HeLa cells (Parental) or HeLa cells stably expressing an shRNA targeting bacterial beta galactosidase (shLac) or AIF (shAIF) were analyzed by immunoblotting for AIF and β-actin. The approximate migration of molecular weight standards (kDa) is indicated on the right. In panels B and C, the viruses indicated were used at an MOI of 10 to infect AIF-proficient (shLacZ) and AIF-deficient (shAIF) (B) HeLa and (C) PC3 cells. Cells were stained for DNA at 72 hpi and the fraction of cells containing fragmented nuclei determined. Results from a representative experiment of at least three experiments are shown. Error bars indicate the 95% exact binomial confidence intervals for the representative experiment.
PARP-1 is active during adenoviral infections
PARP-1 is thought to facilitate AIF release from the mitochondria during caspase-independent cell death (Wang et al., 2009a; Wang et al., 2012). Consequently, we evaluated PARP-1 activity in adenovirus-infected cells by measuring the overall PAR-ribosylation. Quantitative immunofluorescence data obtained from all infected cells at 72 hpi showed increased functional PARP-1 activity, as measured by an increase in PAR-ribosylation compared to mock-infected cells. The mean fluorescence intensity for PAR-staining in cells infected with the wild-type, single-, or double-mutant viruses was 3.5- to 5-fold greater than that observed in mock-infected cells (Fig. 6A). Application of the t-test to the mean fluorescent intensity with the Holm correction for multiple comparisons confirmed that mock-infected cells contained significantly less PAR-staining than infected cells (p < 0.05) and that the level of PAR staining among infected cells was indistinguishable (p > 0.5, Fig. 6B). These results show that PARP-1 is activated after adenovirus infection irrespective of the status of the E1B-55K and E4orf3 genes.
Figure 6.
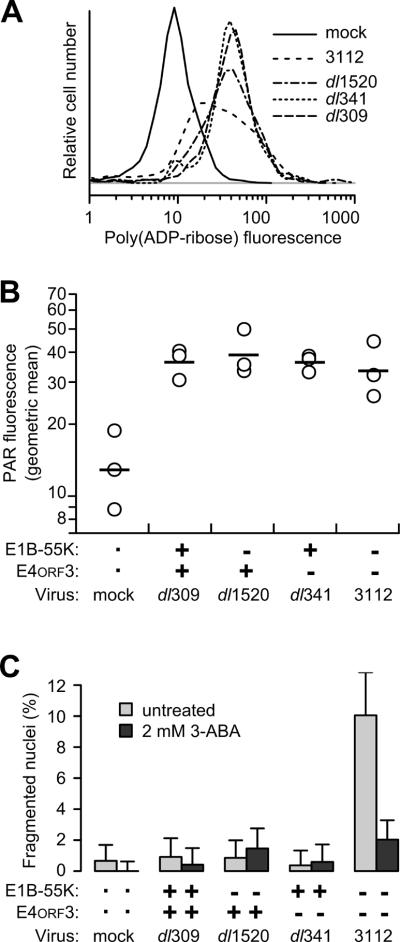
PAR levels increase in adenovirus-infected cells and PARP-1 activity is necessary for nuclear fragmentation in double-mutant virus-infected cells. HeLa cells were infected with the viruses indicated at an MOI of 10. (A) Cells were fixed and stained by indirect immunofluorescence for PAR. The fluorescence intensity of between 100 and 1000 individual cells for each virus was determined by quantitative immunofluorescence microscopy. The log of the total fluorescence intensity is plotted as a density distribution. (B) The mean fluorescence intensity of PAR-stained cells from three independent experiments are plotted with open symbols. The geometric mean for the three experiments is indicated with the horizontal bar. (C) Infected cells were treated with PARP-1-inhibitor 3-aminobenzamide. Cells were stained for DNA at 72 hpi and the fraction of cells containing fragmented nuclei was determined. Results from a representative experiment of three independent experiments are shown. Error bars indicate the upper limit of the 95% exact binomial confidence interval for the representative experiment.
AIF-dependent nuclear fragmentation is PARP-dependent
Although the increase in PAR-modified proteins shows that PARP-1 is activated in all adenovirus-infected cells, only double-mutant virus-infected cells contain fragmented nuclei (see Fig. 3A). This indicates that PARP-1 activation is not sufficient to induce nuclear fragmentation. To determine if PARP-activation is required for nuclear fragmentation, cells were treated with the PARP inhibitor 3-aminobenzamide (3-ABA) and then infected with wild-type, single-mutant, and double-mutant viruses. At 72 hpi, the nuclear morphology was quantified. The low level of nuclear fragmentation seen in wild-type and single-mutant virus-infected cells was unchanged by the PARP-1 inhibitor. More fragmented nuclei were observed in untreated double-mutant virus-infected cells, as expected. However, the PARP-1 inhibitor prevented nuclear fragmentation in double-mutant virus-infected cells, reducing the levels to that observed in cells infected with the E1B-55K-mutant virus (Fig. 6C). Although it seems reasonable that the PARP-1 inhibitor would block the release of AIF from the mitochondria and translocation into the nucleus, this remains to be determined in adenovirus-infected cells. The PARP-1 inhibitor affected the frequency of fragmented nuclei and not total nuclear morphological heterogeneity including condensed chromatin (data not shown). These results suggest that, although not sufficient, PARP-1 activity is necessary for AIF to promote nuclear fragmentation during adenoviral infections.
The E1B-55K and E4orf3 proteins alter the distribution of PAR-modified proteins
The increase in PAR-modification shows that PARP-1 is activated following adenovirus infection. This raises the question of how the E1B-55K and E4orf3 proteins prevent the abundant PAR chains and PAR-modified proteins from triggering nuclear fragmentation. Another DNA-damage signal that increases in adenovirus-infected cells is the phosphorylation of histone H2AX or γH2AX. It has been suggested that adenovirus disrupts this signaling process by mislocalizing γH2AX rather than preventing the phosphorylation of H2AX (Nichols et al., 2009). In order to determine if PAR-modified proteins are also differentially localized in adenovirus-infected cells, the distribution of PAR was systematically evaluated by fluorescence microscopy at 72 hpi. PAR staining is faint in most mock-infected cells. In the few strongly stained cells, PAR was diffusely distributed throughout the cell as small puncta (Fig. 7A–B). All of these mock-infected cells would have been scored as containing a diffuse pattern as noted in Fig. 7C. A similar diffuse distribution, albeit much brighter, was seen in the majority of virus-infected cells (Figs. 7C, 7D–H). In wild-type and single-mutant virus-infected cells, we observed two additional staining patterns. A clustered pattern consisting of focal concentrations in limited regions of the nucleus was seen in 10–20% of the cells infected with the wild-type or single-mutant viruses (Figs. 7C, 7I–M). A more finely speckled pattern was observe throughout the nucleus of 10–30% of cells when the E1B-55K protein was expressed (Figs. 7C, 7N–R). Cells infected with the double-mutant virus, however, contained only diffusely distributed PAR-modified proteins. The frequency of the three staining patterns was quantified in three experiments. Representative results are summarized in Fig. 7C. These results show that the E1B-55K and E4orf3 proteins alter localization of PAR-modified proteins. However, it remains unclear if the distinct staining pattern seen in a fraction of wild-type or single-mutant virus-infected cells is sufficient to block the PARP-1-dependent signal for nuclear fragmentation.
Figure 7.

The E1B-55K and E4orf3 proteins alter the distribution of PAR or PAR-modified proteins. HeLa cells were mock-infected or infected with the wild-type, single-mutant, or double-mutant viruses and stained for PAR and DNA at 72 hpi. PAR is represented by green and DNA by magenta. (A,B) Representative low-magnification fields of mock-infected cells show faint, diffuse PAR staining throughout the cell. The scale bar indicates 10 μm. (C) Three patterns of PAR-staining were identified in virus-infected cells. The relative frequency of the three patterns of PAR localization are tabulated for approximately 300 cells for each viral infection from one of three representative experiments. (D-H, Diffuse) Representative cells showing a diffusely distributed pattern of PAR staining through cytoplasm and nucleus. (I-M, Clustered) Representative cells displaying focal concentrations of PAR staining within a small portion of the nucleus. (N-R, Speckled) Representative cells showing a more widely distributed pattern of PAR-stained foci throughout a larger portion of the nucleus. The scale bar indicates 10 μm.
Viral genome replication activates PARP-1
The presence of the linear, double-stranded adenovirus DNA genome has been suggested to be sufficient to trigger the DNA damage response (Cherubini et al., 2011; Karen et al., 2009; Nichols et al., 2009), which could promote the activation of PARP-1. However, other consequences of a productive infection include aberrant cellular DNA synthesis (Nichols et al., 2009), increased macromolecular synthesis, and the production of reactive oxygen species, all of which could stimulate pathways that converge on the activation of PARP-1. The potential contribution of these factors to PARP-1 activation was tested.
Adenovirus elicits aberrant cellular DNA synthesis including re-replication, which is the replication of once-replicated segments of the genome without an intervening mitosis (Nichols et al., 2009). Re-replication is especially extensive in cells infected with the E1B-55K-mutant virus (unpublished observations and Cherubini et al., 2006) and leads to double-stranded DNA breaks and regions of single stranded DNA that trigger the DNA-damage response pathway (Hook et al., 2007; Jones and Petermann, 2012). Because the adenovirus DNA polymerase is unaffected by a level of aphidicolin that blocks cellular DNA synthesis (Kwant and van der Vliet, 1980), double-mutant virus-infected cells were treated with a concentration of aphidicolin sufficient to suppress cellular but not viral DNA replication before measuring nuclear fragmentation (Fig. 8A). Aphidicolin reduced the frequency of infected cells with greater than 4N DNA content from approximately 30% to less than 2% (data not shown), confirming that the increase in DNA content was due to cellular DNA synthesis. Nonetheless, aphidicolin had no effect on the number of fragmented nuclei seen in double-mutant virus-infected cells, demonstrating that aberrant cellular DNA synthesis is not the likely trigger for PARP-dependent nuclear fragmentation.
Figure 8.

Adenoviral genome replication triggers PARP-1 activation. HeLa cells infected at an MOI of 10 with the indicated viruses were treated with (A) a concentration of aphidicolin (0.08 μM) at the time of infection sufficient to inhibit cellular DNA replication but not viral DNA replication, (B) 100 μg per ml of the protein synthesis inhibitor cycloheximide at the indicated times post-infection, or (C) the broad spectrum antioxidant and free radical scavenger N-acetylcysteine (20 mM) at various times before or after infection. Cells were stained for DNA and the fraction of cells with fragmented nuclei was determined at 72 hpi. Results from representative experiments of at least three independent experiments are shown. Error bars indicate the 95% exact binomial confidence interval for the representative experiment. (D) HeLa cells were infected with the indicated viruses, including an adenovirus mutant unable to direct viral DNA replication (ΔpTP). Protein lysates were collected at 72 hpi in the presence of protease and phosphatase inhibitors and material from identical numbers of cells analyzed by immunoblotting for PAR-ribosylated cellular proteins indicated by square brackets. The approximate migration of protein standards (in kDa) is shown on the right. Arrowheads on the left indicate the position of the hexon (upper) and penton base (lower) adenovirus capsid proteins determined by separately stained blots. (E) HeLa cells were infected with the indicated viruses, including the ΔpTP virus. The cells were fixed and stained by indirect immunofluorescence for PAR at 72 hpi. The fluorescence intensity of 300 to 1500 cells for each infection was determined by quantitative microscopy as described in the Materials and Methods. Results from a representative experiment of three are shown.
In addition to aberrant cellular DNA synthesis, double-mutant virus-infected cells continue to synthesize host proteins at late times of infection (Shepard and Ornelles, 2004). The continual synthesis of both cellular and viral proteins could be a metabolic stress that triggers PARP-1 activation. Alternatively, the failure to block host protein synthesis at late times of infection could permit the synthesis of antiviral effector proteins that contribute to PARP-mediated nuclear fragmentation. To determine if sustained protein synthesis is required for PARP-mediated nuclear fragmentation, mock-infected or E1B-55K/E4orf3 double-mutant virus-infected cells were treated cycloheximide at various times post infection before being evaluated for nuclear fragmentation at 72 hpi. Although the number of fragmented nuclei in non-infected cells increased with longer exposures to cycloheximide in an AIF-independent manner (data not shown), the frequency of cells with fragmented nuclei among the infected cells remained approximately constant (Fig. 8B). This result suggests that although the prolonged inhibition of protein synthesis can affect nuclear integrity in non-infected cells, the AIF-dependent nuclear fragmentation observed in the virus-infected cells does not require ongoing protein synthesis.
Reactive oxygen species (ROS) can promote PARP activation (Ame et al., 1999). Because the protein VI-dependent, pH-independent escape of adenovirus from the endosome (Wiethoff et al., 2005) triggers ROS production through cathepsin B-mediated mitochondrial destabilization in macrophages (McGuire et al., 2011), we used N-acetylcysteine, a pan-reactive ROS inhibitor to determine if the production of ROS contributed to the activation of PARP and nuclear fragmentation. The ROS inhibitor had no impact on nuclear fragmentation measured at 72 hpi whether added prior to, shortly after, or long after infection with the double-mutant virus (Fig. 8C). This result suggests that virus-induced reactive oxygen production does not trigger nuclear fragmentation.
Replication of the double-stranded adenovirus genome leads to numerous double-stranded DNA ends with a free 3'-hydroxyl and abundant single-stranded DNA. These structures also have been postulated to trigger a potent DNA damage response (Shepard and Ornelles, 2004; Turnell and Grand, 2012). To determine if viral genome replication activates PARP-1, cells were infected with wild-type, single-, and double-mutant viruses, all of which direct equivalent levels of viral DNA replication (Shepard and Ornelles, 2004). These viruses were compared to a virus bearing a deletion in the pre-terminal protein gene (H5wt300ΔpTP) that is therefore unable to direct the synthesis of new viral DNA. Cells infected with H5wt300ΔpTP contained no more fragmented nuclei than mock-infected cells, which was expected because this viruses expresses both E1B-55K and E4ORF3 (data not shown). Cellular lysates were collected and the levels of PAR-ribosylated proteins were analyzed by immunoblot (Fig. 8D). Automodification of PARP-1 with PAR is a hallmark of PARP-1 activation and is recognized as heavy, but indistinct, PAR staining in the higher molecular weight region. These results seem to show an increase in PAR-ribosylation in wild-type, single-, and double-mutant viral infections over that of mock and H5wt300ΔpTP viral infections, but the indistinct nature of PAR staining makes this difficult to definitively measure. Intriguingly, lysates from wild-type and single-mutant virus-infected cells contained prominent PAR-modified proteins that comigrated with the late viral hexon and penton base proteins (Fig. 8D). Immunoprecipitation of the late viral proteins followed by immunoblotting for PAR revealed PAR-ribosylation of the penton protein, but not the hexon protein recovered by immunoprecipitation (data not shown). The intensity of PAR staining within individual cells was used to provide a measure of PAR ribosylation during viral infections. Quantitative immunofluorescence showed an increase in PAR staining in cells infected with each virus studied here except the replication-defective virus H5wt300ΔpTP. PAR fluorescence intensity in H5wt300ΔpTP virus-infected cells was reduced to the background levels seen in mock-infected cells (Fig. 8E). These results support the hypothesis that viral DNA replication triggers a potent host response that activates PARP-1, which in turn could lead to nuclear fragmentation through AIF. Because the activation of PARP-1 does not require late protein synthesis, we propose that that it is the act of viral genome replication per se that activates PARP-1.
In summary, these findings suggest that adenoviral genome replication triggers a cellular DNA-damage response that leads to the activation of PARP-1. Notably, this activation occurs in wild-type virus-infected cells, suggesting that at least a portion of the DNA damaging signaling network is operational. However, in the absence of the viral E1B-55K and E4ORF3 proteins, a subset of cells responds to this signal by releasing AIF from the mitochondria and undergo AIF-dependent nuclear fragmentation.
DISCUSSION
Replication of the double-stranded DNA adenoviral genome provokes a cellular response that includes activation of PARP-1. In the absence of the adenoviral E1B-55K and E4ORF3 proteins, active PARP-1 leads to AIF-dependent nuclear fragmentation. The precise stimulus provided by adenoviral DNA replication that activates PARP-1 is not known. However, it seems likely that the viral genome itself and forms of DNA that arise during viral DNA replication are the likely culprits. Adenoviruses are unique among animal viruses (Challberg and Kelly, 1989) because the viral genome persists as a linear, double-stranded DNA molecule during the course of the infection (Knipe, 2006). The lytically infected cell contains thousands of double-stranded DNA ends, each of which has the 5' terminal phosphate covalently attached to the preterminal protein and a free 3' hydroxy terminal group. One single-stranded DNA genome is generated each time a double-stranded DNA genome is used as a template (Knipe, 2006). These DNA structures can stimulate a robust DNA-damage response (Germann et al., 2010; Peterson and Cote, 2004). We previously reported that viral genome replication leads to widespread phosphorylation of H2AX, suggesting that the infected cell experiences a diffusely distributed genotoxic signal (Nichols et al., 2009). This response may more closely mimic the type of widespread DNA damage caused by alkylating agents, rather than the more localized damage caused by ionizing radiation (IR). At the molecular level, IR causes discrete foci of damaged DNA and triggers the ATM-Chk2 pathway (Khalil et al., 2011; Thompson et al., 2012). Alkylating agents such as MMNG, on the other hand, activate the ATM protein only when PARP-activation is inhibited (Carrozza et al., 2009). Alkylation by MMNG activates the ATR protein to arrest cell cycle progression and to promote mismatch repair (Stojic et al., 2004). Significantly, cells infected with the E1B-55K/E4ORF3 double-mutant virus activate ATR signaling independent of ATM (Carson et al., 2009).
PARP-1 was activated in adenovirus-infected cells irrespective of the status of the E1B-55K and E4ORF3 genes (Fig. 6). The E1B-55K and E4ORF3 proteins most likely act after PARP-1 activation to block the release of AIF from the mitochondrion and its subsequent nuclear translocation (Fig. 4). Active PARP-1 may contribute to the mitochondrial release and nuclear translocation of AIF by several mechanisms (Andrabi et al., 2008; Wang et al., 2009a). Exogenously applied free PAR is cytotoxic and the introduction of neutralizing PAR antibodies into excitotoxic model systems are protective (Andrabi et al., 2006), indicating that PAR alone may be sufficient to cause AIF-dependent cell death. In some cells, the exogenous application of NAD+ or nicotinamide and the systematic depletion of NAD+ promotes AIF translocation from the mitochondria to the nucleus (Alano et al., 2010; Cimadamore et al., 2009), suggesting that the depletion of cellular energy stores due to the consumption of NAD+ during ADP-ribose polymerization contributes to the translocation of AIF. Cytosolic levels of calcium increase and activate the protease calpain after PAR-formation and NAD+ depletion (Pink et al., 2000; Tagliarino et al., 2001). In ex vivo experiments, activated calpains have been shown to cleave purified AIF (Wang et al., 2009b) and to release membrane-bound AIF from purified mitochondria (Ozaki et al., 2009; Polster et al., 2005). In vivo, however, calpain activity was not required for AIF release from the mitochondria and its subsequent nuclear translocation (Wang et al., 2009b). Calpain activation also was required for PARP-1 and AIF-mediated cell death in neuronal cells (Cao et al., 2007; Mizukoshi et al., 2010). The presence of cytotoxic quantities of PAR, NAD+ depletion, calcium release, and calpain activation may play a role in adenovirus-mediated nuclear fragmentation. It is possible that the adenoviral oncoproteins antagonize nuclear fragmentation downstream of PARP-1 activation at any of these steps.
Neither the E4ORF3 nor E1B-55K protein directly interacts with AIF or with factors that are known to facilitate the release of AIF from the mitochondrion such as calpain, tBid, or Bax (Harada et al., 2002). The E1B-55K protein blocks transcription of p53-dependent genes including BAX (Berk, 2005) which could limit pro-death signaling to the mitochondria. AIF-dependent, caspase-independent neuronal cell death requires p53 (Cregan et al., 2002). The death of these cells could be rescued through the inhibition of p53 by the adenovirus E1B-55K protein (Aloyz et al., 1998). However, because nuclear fragmentation in the p53-null PC3 prostate cancer cell line proceeded only in the absence of both E1B-55K and E4ORF3 function (Fig. 3 and data not shown), it seems unlikely that E1B-55K-mediated suppression of p53 is sufficient to block AIF translocation and nuclear fragmentation. Nevertheless, the lack of p53 signaling in the PC3 cells could have contributed to a lower frequency of nuclear fragmentation in these cells. It also is possible that the unfettered DNA-damage response signaling in infected cells lacking the E1B-55K and E4ORF3 proteins leads to an overwhelming stimulus through PARP-1 that promotes AIF release. Free chains of PAR may be more abundant in double-mutant virus-infected cells; however, total levels of PAR, including PAR-modified proteins, were no more abundant in these cells than in wild-type virus-infected cells (Fig. 6). It also is possible that the nature of the signal provoked by the adenovirus genome in the double-mutant virus-infected cells is unique. Despite equivalent levels of viral genome replication directed by the wild-type, single-, and double-mutant viruses, late viral proteins are synthesized at a much reduced rate in cells infected with the double-mutant virus (Shepard and Ornelles, 2004). A key late viral protein that is underrepresented in these cells is protein VII, which protects viral genomes from detection by the host DNA-damage response (Karen and Hearing, 2011; Vayda et al., 1983). The nature of signaling from nascent viral genomes devoid of protein VII could differ from that of protein VII-rich genomes.
Differences in PAR localization in the adenoviral mutant infections (Fig. 7) lead us to suggest that the adenoviral E1B-55K and E4orf3 proteins redirect PARP-1 activity or mislocalize cytotoxic PAR chains or PAR-modified proteins in order to inactivate a key signal for nuclear fragmentation. Aggregates that were intensely stained for PAR were observed in the nuclei of cells infected with the wild-type and single-mutant viruses. Structures of similar appearance were observed at a higher frequency in cells stained for the late adenoviral proteins, suggesting that these aggregates may be composed of late viral proteins and that some contain PAR-modified proteins. By contrast, PAR staining in the double-mutant virus-infected cells was more diffusely distributed through the nucleus and the large PAR-ribosylated aggregates were absent (Fig. 7C). Although some degree of diffuse PAR staining is seen in all viral infections, perhaps the level of aggregation seen in a subset of cells is sufficient to reduce PAR signaling in cells that are predisposed to nuclear fragmentation. Immunoblotting and immunoprecipitation experiments suggest that the late adenoviral penton base protein is PAR-ribosylated (Fig. 8D and data not shown). Although a protein of mobility similar to the hexon protein was PAR-ribosylated, our failure to detect PAR-modified hexon protein following immunoprecipitation may indicate that prominent PAR-modified 110-kDa protein in Fig. 8D is not the hexon protein or that the antibody used for immunoprecipitation failed to recognize PAR-modified hexon protein. The amount of PAR-ribosylated penton base protein in E1B-55K single-mutant virus-infected cells was apparently the same as that in wild-type and E4orf3-mutant virus-infected cells (Fig. 8D). This is unexpected because cells infected with the E1B-55K-mutant virus contain less late viral proteins than wild-type virus-infected cells. Perhaps a fixed amount of the relatively abundant viral penton base protein in cells infected with the wild-type or single-mutant viruses serves as a non-productive substrate for PARP-1. In addition, the molecular meshworks formed by the E1B-55K (Pennella et al., 2010) and E4orf3 (Ou et al., 2012; Patsalo et al., 2012) proteins may restrict the trafficking of PAR-modified proteins within the nucleus and dampen PARP-1 initiated signaling.
The fragmented nuclei in cells infected with the E1B-55K/E4orf3 double-mutant virus are unusual but not unique. These nuclei resemble those observed in PARP-1- and AIF-dependent excitotoxic neuronal cell death (Yu et al., 2003). Excitotoxic stimuli such as glutamate overstimulation (Zhang and Bhavnani, 2006) triggers a form of cell death that differs from that caused by classical inducers of apoptosis, oxidative stress, or the withdrawal of trophic factors in that excitotoxic cell death shows virtually no caspase-3 activity and is accompanied by an increase in cytoplasmic calcium (Diwakarla et al., 2009). Injuries to the brain such as trauma or stroke often trigger an efflux of glutamate leading to nuclear fragmentation that was shown to be AIF- and PARP-1-dependent in mouse models of focal and global brain ischemia (Eliasson et al., 1997; Thal et al., 2011). Similar AIF- and PARP-1-dependent signaling occurs in neurodegenerative disease models for Parkinson's disease and retinitis pigmentosa, in programmed cell death that occurs as embryonic stem cell differentiate into neuroepithelial cells, and in response to the DNA alkylating agent MMNG (Andrabi et al., 2008; Cimadamore et al., 2009; Murakami et al., 2008; Perier et al., 2010; Yamashima et al., 2001; Yu et al., 2002). A productive adenovirus infection activates the cellular DNA-damage response and can drive entry of quiescent cells into the cell cycle. Perhaps this form of overstimulation mimics the synaptic surge that initiates neuronal cell death.
The E4orf6 protein of adenovirus was reported to radiosensitize RKO colon cancer cells (Hart et al., 2007). Stable expression of E4orf6 in the RKO cell line inhibited protein phosphatase 2A. Consequently, two targets of protein phosphatase 2A, histone H2AX and the DNA-dependent protein kinase, remained phosphorylated for prolonged times after exposure to IR. It was postulated that the sustained DNA-damage signaling after IR leads to the hyperactivation of PARP-1 and translocation of AIF to the nucleus. Because phosphorylated H2AX cooperates with AIF to fragment chromatin (Artus et al., 2010), E4orf6-expressing cells suffered increased chromatin damage in response to IR (Hart et al., 2007). These findings led us to test the contribution of E4orf6 to nuclear fragmentation in the context of a viral infection where viral DNA replication would provide the damage signal in place of IR. Instead, we found that infection with a virus deleted of both E4orf3 and E4orf6 still caused nuclear fragmentation, leading us to conclude that, in the context of a viral infection, the presence of E4orf6 is not necessary for AIF-dependent nuclear fragmentation (data not shown).
A hallmark of late stage apoptosis is DNA fragmentation (Wilson, 1998). However, apoptotic DNA fragmentation differs from the nuclear fragmentation triggered by infection with the E1B-55K/E4orf3 double-mutant virus. Apoptotic nuclear bodies are typically composed of cellular DNA and nuclear material pulling away from a central mass. The resulting fragments of nuclear material are not defined by a distinct boundary of nuclear lamina as we report here (Soboloff et al., 2001; Zhao et al., 2006). Indeed, breakdown of the nuclear lamina precedes nuclear fragmentation during apoptosis (Burke, 2001; Lazebnik et al., 1995). In addition, AIF-dependent ex vivo fragmentation causes infrequent DNA cleavage resulting in large fragments of DNA, as opposed to classical apoptotic DNA laddering that occurs in the two-step process involving both AIF and Endo G (Susin et al., 2000; Wang et al., 2002). Pulsed-field and standard gel electrophoresis of infected cell DNA showed no apoptotic DNA laddering in double-mutant virus-infected cells (Shepard and Ornelles, 2004). The failure of the pan-caspase inhibitor zVAD-fmk to change the morphological heterogeneity of E1B-55K/E4orf3 double-mutant virus-infected cells provides additional support for the caspase-independent nature of the adenovirus-induced nuclear fragmentation described here (data not shown). The heterogeneous nature of double-mutant viral infections may reflect stochastic differences in protein expression within a cell population. This could lead to some cells having the requisite amount of one trigger protein to surpass a threshold and engage one pathway over another.
Although the specific mechanisms by which the adenoviral E1B-55K and E4orf3 proteins enforce morphological homogeneity remain unknown, continued signaling after the insult of adenoviral genome replication elicits nuclear fragmentation. This morphologically distinctive nuclear fragmentation depends on AIF and on PARP-1 activation. Forced re-entry of mature, differentiated neuronal cells into the cell cycle causes cell death (Feddersen et al., 1992) that is dependent on AIF (Bonni, 2003; Chung et al., 2011). Since adenoviruses are able to productively infect terminally differentiated cells and force entry into an S-phase-like state, the E1B-55K and E4orf3 proteins may be necessary to suppress forms of cell death that prevail in terminally differentiated cells. The heterogeneous nature of cells infected with the E1B-55K and E4orf3 double mutant-virus suggests that adenovirus triggers diverse pathways of cell death that must be antagonized by equally diverse functions provided by the early adenoviral proteins. Continuing to study the ways by which adenovirus limits heterogeneity may unveil novel relationships between adenovirus and host cellular processes that govern cell death and survival.
MATERIALS AND METHODS
Chemicals
All chemicals used in this study were obtained from Sigma/Aldrich (St. Louis, MO) unless otherwise noted. The PARP-1 inhibitor, 3-aminobenzamide was prepared as a concentrated stock of 200 mM in water with gentle heating and used at a concentration of 2 mM. Aphidicolin was used at a concentration of 0.08 μM to inhibit cellular DNA synthesis. Cycloheximide was used at a concentration of 100 μg per ml from a stock solution of 100 mg per ml in dimethyl sulfoxide to inhibit protein synthesis. N-acetylcysteine was used at a concentration of 20 mM from a stock solution 200 mM in serum-free medium to inhibit the production of reactive oxygen.
Cell lines
Cell culture media, cell culture supplements, and sera were obtained from Invitrogen (Gaithersburg, MD) or Lonza (Hopkinton, MA) through the Tissue Culture and Virus Vector Core Laboratory of the Comprehensive Cancer Center of Wake Forest University. HeLa cells were originally obtained from the American Type Culture Collection (ATCC, Manassas, VA) and maintained in Dulbecco's modified Eagle medium (DMEM) supplemented with 10% newborn calf serum. HeLa cells stably transduced with lentiviral short hairpin RNA-expressing constructs against AIF or LacZ were generated as described (Lewis et al., 2012) and maintained in DMEM supplemented with 10% fetal bovine serum and 1 μg per ml puromycin. PC3 cells, which were originally derived from a metastatic prostatic adenocarcinoma, were stably transduced with lentiviral short hairpin RNA-expressing constructs against AIF or LacZ (Lewis et al., 2012) and maintained in RPMI supplemented with 10% fetal bovine serum. MCF10A cells were maintained in a 1:1 mixture of DMEM and F12 (DMEM/F12) medium supplemented with 5% heat-inactivated horse serum, 2 mM GlutaMAX and 100 U penicillin, 0.1 mg streptomycin, 10 μg insulin, 20 ng epidermal growth factor, 0.5 μg hydrocortisone, and 100 ng cholera toxin per ml. Retinal pigmented epithelial cells immortalized by hTERT (hTERT RPE-1) were obtained from the ATCC and maintained in DMEM/F12 supplemented with 10% FBS and 0.01 mg per ml hygromycin B. All cells were cultured in a 5% CO2 atmosphere at 37°C by passaging twice weekly at a 1:4 dilution for MCF10A cells and at a 1:10 dilution for HeLa, PC3, and hTERT RPE-1 cells. For high-resolution immunofluorescence microscopy, cells were grown on nitric acid-cleaned, sterilized glass coverslips.
Viruses
The virus dl309 was used as the wild-type virus for this study. This virus contains several deletions and a substitution within the E3B region, but displays wild-type characteristics in cultured cells (Jones and Shenk, 1979). The E1B-55K and E4ORF3 doubly-deleted virus 3112 was described previously (Shepard and Ornelles, 2003) and is a recombinant of the E1B-55K-deleted virus dl1520 (Barker and Berk, 1987) and the E4ORF3-deleted dl341 (Sarnow et al., 1982). The replication-deficient virus H5wt300ΔpTP bears a deletion in the terminal protein gene (Schaack, 2005). The H5wt300ΔpTP virus was grown in 293 cells expressing the E2B gene. All other viruses were grown in 293 cells and concentrated virus stocks prepared by sequential centrifugation through CsCl gradients as described previously (Shepard and Ornelles, 2004). Viral stock titers and the relative titers for each cell line were determined by a fluorescent focus assay. Titers determined by plaque assay with 293 cells were found to be identical to titers determined by fluorescent focus assay.
Antibodies
Primary antibodies included a human anti-nuclear antibody derived from autoimmune human serum that was determined to stain the nuclear lamina used at a 1:100 dilution, a mouse monoclonal antibody against COX IV from Abcam (Eugene, OR) used at a 1:1000 dilution, a rabbit polyclonal antibody against AIF (Cell Signaling, Danvers, MA) used at a 1:250 dilution for immunofluorescent microscopy, a mouse monoclonal antibody from Santa Cruz Biotechnology against AIF (sc-13116, Santa Cruz, CA) used for western blotting, a mouse monoclonal antibody against β-Actin (A5316, Sigma/Aldrich, St. Louis, MO) used for western blotting, a polyclonal rabbit anti-PAR antibody (#551813) from BD Pharmingen (San Jose, CA) used at a 1:400 dilution for microscopy and 1:2000 dilution for western blotting, a polyclonal goat anti-PARP-1 (N-20, Santa Cruz, CA) used at a 1:1000 dilution. Secondary antibodies used for immunofluorescence microscopy were anti-mouse or anti-rabbit whole IgG conjugated to Alexa Fluor 488 (AF488) or Alexa Fluor 568 (AF568) from Invitrogen used at 2 μg per ml. Secondary antibodies used for western blot analysis were anti-rabbit or anti-goat conjugated to horseradish peroxidase from Jackson ImmunoResearch Laboratories (West Grove, PA) and used at a concentration of 0.1 μg per ml.
Microscopy
For immunofluorescence microscopy, cells were fixed for 30 min with 2% paraformaldehyde and permeabilized for 5 min with 0.2% Triton X-100 in phosphate-buffered saline at room temperature. All subsequent washes were performed with Tris-buffered saline with BSA, glycine and Tween-20 (TBS-BGT: 0.137 M NaCl, 0.003 M KCl, 0.025 M Tris-Cl [pH 8.0], 0.0015 M MgCl2, 0.5% bovine serum albumin, 0.1% glycine, 0.05% Tween 20, and 0.02% sodium azide). Antibodies used for immunofluorescence were diluted in TBS-BGT supplemented with 10% normal goat serum (Invitrogen). Samples were stained for 90 min with primary antibody and for 30 min with secondary antibody, with washes in between. Samples were mounted with ProLong Gold mounting media (Invitrogen) containing 4′,6-diamidino-2-phenylindole (DAPI). Micrographs were obtained by either epifluorescence or transmitted light microscopy using a Nikon TE300 inverted microscope or by confocal laser scanning microscopy with a Nikon TiE inverted microscope fitted with a Nikon CISi system. A 20×/0.45 NA dry phase-contrast objective was used for live cell imaging. A 100×/1.4 NA magnification oil-immersion objective was used for all other micrographs. Mean apparent cell diameters were determined from live cells photographed with the 20× objective. A distinct phase-refractile ring defined the perimeter of each detached cell. Using the macro language of the open-source image processing software ImageJ (Rasband) and the open source software environment R, a Gaussian blur was applied and the mean diameter of the phase-refractile ring was determined as the mean of the central 14 of 18 measured diameters. To quantify levels of PAR-staining, 12-bit monochromatic images were acquired with a Retiga EX 1350 digital camera (QImaging Corp., Burnaby, British Columbia, Canada) using the 100× objective. The relative brightness and contrast of the images from each experiment were adjusted to fall within the same linear range using mock-infected cells. Fluorescence intensity was measured with the tools available in ImageJ. The modified Manders and Pearson coefficient of correlation was determined with the JACoP plugin for ImageJ (Bolte and Cordelieres, 2006). Samples for scanning electron microscopy were fixed in 2.5% glutaraldehyde in 0.1 M sodium cacodylate buffer and processed by the Wake Forest University Microscopy Core Lab. The samples were imaged using a Philips 515 SEM.
Flow cytometry
HeLa cells were harvested by scraping. Single cells were obtained by passing the harvested cells through a 35 μm cell strainer cap. Physical properties of the cells were determined by analyzing forward and side scatter using a Benton Dickinson (BD) FACS Calibur instrument.
Western blotting
Cells grown and infected in 60-mm diameter culture plates were washed in the presence of protease and phosphatase inhibitors (2 mM EDTA, 1 mM NaF, 1 mM sodium pyrophosphate, 1 mM phenylmethylsulfonyl fluoride, 1 mM Na3VO4, and 2 μM leupeptin), harvested by scraping and suspended in a small volume of one-tenth concentrated PBS. An equal volume of 2X sodium dodecyl sulfate (SDS) protein sample buffer (20% SDS, 0.5 M Tris [pH 6.8], glycerol, 0.01% bromophenol blue, and 5% β-mercaptoethanol) was added. The cell lysate was heated for 5 min at 95°C and sonicated 3× for 20 sec. The lysates were separated by SDS-polyacrylamide gel electrophoresis through 10% or 15% acrylamide gels. The proteins were then electrophoretically transferred to nitrocellulose (Whatman/GE Healthcare) overnight at 4°C. The nitrocellulose was blocked in TBS-BGT containing 5% nonfat dry milk and sodium azide, stained with primary antibodies diluted in TBS-BGT with sodium azide overnight at 4°C, and stained with secondary antibodies diluted in TBS-BGT without sodium azide for 30 min at room temperature. The stained proteins were visualized by a mixture of SuperSignal West Pico and SuperSignal West Femto chemiluminescence substrate from Pierce/ThermoScientific (Rockford, IL) and X-ray film.
Statistical analyses
Differences among mean apparent cell diameters were evaluated by one-way ANOVA allowing for unequal variances among groups and application of the t-test with the Bonferroni correction for multiple comparisons. A corrected p-value less than 0.05 was considered significant. The proportion of cells showing nuclear fragmentation was compared for each pair of viruses by Fisher's exact test. The resulting p-values from this pairwise analysis were compared to a critical value adjusted by Hochberg's step-up procedure to correct for multiple comparisons. P-values less than an adjusted initial critical value (α) of 0.05 were considered significant. The Pearson's and Manders' correlation coefficients, which varied between 0 and 1, were transformed by the logistic function to achieve a normal distribution and then analyzed by the t-test with the Bonferroni correction for multiple comparisons. Adjusted p-values less than 0.05 were considered significant.
HIGHLIGHTS
E1B-55K or E4orf3 prevents nuclear fragmentation
Nuclear fragmentation requires AIF and PARP-1 activity
Adenovirus DNA replication activates PARP-1
E1B-55K or E4orf3 proteins alter the distribution of PAR
ACKNOWLEDGMENTS
Cell culture reagents were provided by the Cell and Virus Vector Core Laboratory and flow cytometry was performed in the flow cytometry core of the Comprehensive Cancer Center of Wake Forest University, which is supported in part by National Cancer Institute grant CA12197. R.L.T. was supported by the Training Program in Immunology and Pathogenesis 5 T32 AI007401 from the National Institutes of Health. This research was supported by grants R01 CA127621 (to D.A.O) from the National Cancer Institute, W81XWH-08-1-0045 (to J.C.W.) from the Department of Defense Prostate Cancer Research Program, and RSG-09-166-01-CCG (to J.C.W.) from the American Cancer Society. The content of this report is solely the responsibility of the authors and does not necessarily represent the official views of the respective funding agencies.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
REFERENCES
- Alano CC, Garnier P, Ying W, Higashi Y, Kauppinen TM, Swanson RA. NAD+ depletion is necessary and sufficient for poly(ADP-ribose) polymerase-1-mediated neuronal death. J. Neurosci. 2010;30:2967–2978. doi: 10.1523/JNEUROSCI.5552-09.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aloyz RS, Bamji SX, Pozniak CD, Toma JG, Atwal J, Kaplan DR, Miller FD. p53 is essential for developmental neuron death as regulated by the TrkA and p75 neurotrophin receptors. J. Cell Biol. 1998;143:1691–1703. doi: 10.1083/jcb.143.6.1691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ame JC, Rolli V, Schreiber V, Niedergang C, Apiou F, Decker P, Muller S, Hoger T, Menissier-de Murcia J, de Murcia G. PARP-2, A novel mammalian DNA damage-dependent poly(ADP-ribose) polymerase. J. Biol. Chem. 1999;274:17860–17868. doi: 10.1074/jbc.274.25.17860. [DOI] [PubMed] [Google Scholar]
- Andrabi SA, Dawson TM, Dawson VL. Mitochondrial and nuclear cross talk in cell death: parthanatos. Ann. N. Y. Acad. Sci. 2008;1147:233–241. doi: 10.1196/annals.1427.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Andrabi SA, Kim NS, Yu SW, Wang H, Koh DW, Sasaki M, Klaus JA, Otsuka T, Zhang Z, Koehler RC, Hurn PD, Poirier GG, Dawson VL, Dawson TM. Poly(ADP-ribose) (PAR) polymer is a death signal. Proc. Natl. Acad. Sci. U. S. A. 2006;103:18308–18313. doi: 10.1073/pnas.0606526103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Araujo FD, Stracker TH, Carson CT, Lee DV, Weitzman MD. Adenovirus type 5 E4orf3 protein targets the Mre11 complex to cytoplasmic aggresomes. J. Virol. 2005;79:11382–11391. doi: 10.1128/JVI.79.17.11382-11391.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Artus C, Boujrad H, Bouharrour A, Brunelle MN, Hoos S, Yuste VJ, Lenormand P, Rousselle JC, Namane A, England P, Lorenzo HK, Susin SA. AIF promotes chromatinolysis and caspase-independent programmed necrosis by interacting with histone H2AX. EMBO J. 2010;29:1585–1599. doi: 10.1038/emboj.2010.43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baker A, Rohleder KJ, Hanakahi LA, Ketner G. Adenovirus E4 34k and E1b 55k oncoproteins target host DNA ligase IV for proteasomal degradation. J. Virol. 2007;81:7034–7040. doi: 10.1128/JVI.00029-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barker DD, Berk AJ. Adenovirus proteins from both E1B reading frames are required for transformation of rodent cells by viral infection and DNA transfection. Virology. 1987;156:107–121. doi: 10.1016/0042-6822(87)90441-7. [DOI] [PubMed] [Google Scholar]
- Berk AJ. Recent lessons in gene expression, cell cycle control, and cell biology from adenovirus. Oncogene. 2005;24:7673–7685. doi: 10.1038/sj.onc.1209040. [DOI] [PubMed] [Google Scholar]
- Biss M, Xiao W. Selective tumor killing based on specific DNA-damage response deficiencies. Cancer Biol. Ther. 2012;13:239–246. doi: 10.4161/cbt.13.5.18921. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blackford AN, Patel RN, Forrester NA, Theil K, Groitl P, Stewart GS, Taylor AM, Morgan IM, Dobner T, Grand RJ, Turnell AS. Adenovirus 12 E4orf6 inhibits ATR activation by promoting TOPBP1 degradation. Proc. Natl. Acad. Sci. U. S. A. 2010;107:12251–12256. doi: 10.1073/pnas.0914605107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bolte S, Cordelieres FP. A guided tour into subcellular colocalization analysis in light microscopy. J. Microsc. 2006;224:213–232. doi: 10.1111/j.1365-2818.2006.01706.x. [DOI] [PubMed] [Google Scholar]
- Bonni A. Neurodegeneration: A non-apoptotic role for AIF in the brain. Curr. Biol. 2003;13:R19–21. doi: 10.1016/s0960-9822(02)01382-9. [DOI] [PubMed] [Google Scholar]
- Burgert HG, Ruzsics Z, Obermeier S, Hilgendorf A, Windheim M, Elsing A. Subversion of host defense mechanisms by adenoviruses. Curr. Top. Microbiol. Immunol. 2002;269:273–318. doi: 10.1007/978-3-642-59421-2_16. [DOI] [PubMed] [Google Scholar]
- Burke B. Lamins and apoptosis: a two-way street? J. Cell Biol. 2001;153:F5–7. doi: 10.1083/jcb.153.3.f5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cao G, Xing J, Xiao X, Liou AK, Gao Y, Yin XM, Clark RS, Graham SH, Chen J. Critical role of calpain I in mitochondrial release of apoptosis-inducing factor in ischemic neuronal injury. J. Neurosci. 2007;27:9278–9293. doi: 10.1523/JNEUROSCI.2826-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carrozza MJ, Stefanick DF, Horton JK, Kedar PS, Wilson SH. PARP inhibition during alkylation-induced genotoxic stress signals a cell cycle checkpoint response mediated by ATM. DNA Repair (Amst) 2009;8:1264–1272. doi: 10.1016/j.dnarep.2009.07.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carson CT, Orazio NI, Lee DV, Suh J, Bekker-Jensen S, Araujo FD, Lakdawala SS, Lilley CE, Bartek J, Lukas J, Weitzman MD. Mislocalization of the MRN complex prevents ATR signaling during adenovirus infection. EMBO J. 2009;28:652–662. doi: 10.1038/emboj.2009.15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carson CT, Schwartz RA, Stracker TH, Lilley CE, Lee DV, Weitzman MD. The Mre11 complex is required for ATM activation and the G2/M checkpoint. EMBO J. 2003;22:6610–6620. doi: 10.1093/emboj/cdg630. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Challberg MD, Kelly TJ. Animal virus DNA replication. Annu. Rev. Biochem. 1989;58:671–717. doi: 10.1146/annurev.bi.58.070189.003323. [DOI] [PubMed] [Google Scholar]
- Cherubini G, Naim V, Caruso P, Burla R, Bogliolo M, Cundari E, Benihoud K, Saggio I, Rosselli F. The FANC pathway is activated by adenovirus infection and promotes viral replication-dependent recombination. Nucleic Acids Res. 2011;39:5459–5473. doi: 10.1093/nar/gkr084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cherubini G, Petouchoff T, Grossi M, Piersanti S, Cundari E, Saggio I. E1B55K-deleted adenovirus (ONYX-015) overrides G1/S and G2/M checkpoints and causes mitotic catastrophe and endoreduplication in p53-proficient normal cells. Cell Cycle. 2006;5:2244–2252. doi: 10.4161/cc.5.19.3263. [DOI] [PubMed] [Google Scholar]
- Chung SH, Calafiore M, Plane JM, Pleasure DE, Deng W. Apoptosis inducing factor deficiency causes reduced mitofusion 1 expression and patterned Purkinje cell degeneration. Neurobiol. Dis. 2011;41:445–457. doi: 10.1016/j.nbd.2010.10.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cimadamore F, Curchoe CL, Alderson N, Scott F, Salvesen G, Terskikh AV. Nicotinamide rescues human embryonic stem cell-derived neuroectoderm from parthanatic cell death. Stem Cells. 2009;27:1772–1781. doi: 10.1002/stem.107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cregan SP, Fortin A, MacLaurin JG, Callaghan SM, Cecconi F, Yu SW, Dawson TM, Dawson VL, Park DS, Kroemer G, Slack RS. Apoptosis-inducing factor is involved in the regulation of caspase-independent neuronal cell death. J. Cell Biol. 2002;158:507–517. doi: 10.1083/jcb.200202130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Degenhardt K, Perez D, White E. Pathways used by adenovirus E1B 19K to inhibit apoptosis. Symp. Soc. Exp. Biol. 2000;52:241–251. [PubMed] [Google Scholar]
- Diwakarla S, Nagley P, Hughes ML, Chen B, Beart PM. Differential insult-dependent recruitment of the intrinsic mitochondrial pathway during neuronal programmed cell death. Cell. Mol. Life Sci. 2009;66:156–172. doi: 10.1007/s00018-008-8490-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Durocher D, Jackson SP. DNA-PK, ATM and ATR as sensors of DNA damage: variations on a theme? Curr. Opin. Cell Biol. 2001;13:225–231. doi: 10.1016/s0955-0674(00)00201-5. [DOI] [PubMed] [Google Scholar]
- Eliasson MJ, Sampei K, Mandir AS, Hurn PD, Traystman RJ, Bao J, Pieper A, Wang ZQ, Dawson TM, Snyder SH, Dawson VL. Poly(ADP-ribose) polymerase gene disruption renders mice resistant to cerebral ischemia. Nat. Med. 1997;3:1089–1095. doi: 10.1038/nm1097-1089. [DOI] [PubMed] [Google Scholar]
- Feddersen RM, Ehlenfeldt R, Yunis WS, Clark HB, Orr HT. Disrupted cerebellar cortical development and progressive degeneration of Purkinje cells in SV40 T antigen transgenic mice. Neuron. 1992;9:955–966. doi: 10.1016/0896-6273(92)90247-b. [DOI] [PubMed] [Google Scholar]
- Germann MW, Johnson CN, Spring AM. Recognition of damaged DNA: structure and dynamic markers. Med. Res. Rev. 2010 doi: 10.1002/med.20226. [DOI] [PubMed] [Google Scholar]
- Gilliams-Francis KL, Quaye AA, Naegele JR. PARP cleavage, DNA fragmentation, and pyknosis during excitotoxin-induced neuronal death. Exp. Neurol. 2003;184:359–372. doi: 10.1016/j.expneurol.2003.07.003. [DOI] [PubMed] [Google Scholar]
- Haince JF, Kozlov S, Dawson VL, Dawson TM, Hendzel MJ, Lavin MF, Poirier GG. Ataxia telangiectasia mutated (ATM) signaling network is modulated by a novel poly(ADP-ribose)-dependent pathway in the early response to DNA-damaging agents. J. Biol. Chem. 2007;282:16441–16453. doi: 10.1074/jbc.M608406200. [DOI] [PubMed] [Google Scholar]
- Halldorsson H, Gray DA, Shall S. Poly (ADP-ribose) polymerase activity in nucleotide permeable cells. FEBS Lett. 1978;85:349–352. doi: 10.1016/0014-5793(78)80489-x. [DOI] [PubMed] [Google Scholar]
- Han J, Modha D, White E. Interaction of E1B 19K with Bax is required to block Bax-induced loss of mitochondrial membrane potential and apoptosis. Oncogene. 1998;17:2993–3005. doi: 10.1038/sj.onc.1202215. [DOI] [PubMed] [Google Scholar]
- Harada JN, Shevchenko A, Shevchenko A, Pallas DC, Berk AJ. Analysis of the adenovirus E1B-55K-anchored proteome reveals its link to ubiquitination machinery. J. Virol. 2002;76:9194–9206. doi: 10.1128/JVI.76.18.9194-9206.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hart LS, Ornelles D, Koumenis C. The adenoviral E4orf6 protein induces atypical apoptosis in response to DNA damage. J. Biol. Chem. 2007;282:6061–6067. doi: 10.1074/jbc.M610405200. [DOI] [PubMed] [Google Scholar]
- Hart LS, Yannone SM, Naczki C, Orlando JS, Waters SB, Akman SA, Chen DJ, Ornelles D, Koumenis C. The adenovirus E4orf6 protein inhibits DNA double strand break repair and radiosensitizes human tumor cells in an E1B-55K-independent manner. J. Biol. Chem. 2005;280:1474–1481. doi: 10.1074/jbc.M409934200. [DOI] [PubMed] [Google Scholar]
- Hartl B, Zeller T, Blanchette P, Kremmer E, Dobner T. Adenovirus type 5 early region 1B 55-kDa oncoprotein can promote cell transformation by a mechanism independent from blocking p53-activated transcription. Oncogene. 2008;27:3673–3684. doi: 10.1038/sj.onc.1211039. [DOI] [PubMed] [Google Scholar]
- Hook SS, Lin JJ, Dutta A. Mechanisms to control rereplication and implications for cancer. Curr. Opin. Cell Biol. 2007;19:663–671. doi: 10.1016/j.ceb.2007.10.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hsieh TH, Tsai CF, Hsu CY, Kuo PL, Lee JN, Chai CY, Wang SC, Tsai EM. Phthalates induce proliferation and invasiveness of estrogen receptor-negative breast cancer through the AhR/HDAC6/c-Myc signaling pathway. FASEB J. 2012;26:778–787. doi: 10.1096/fj.11-191742. [DOI] [PubMed] [Google Scholar]
- Jayaram S, Bridge E. Genome concatenation contributes to the late gene expression defect of an adenovirus E4 mutant. Virology. 2005;342:286–296. doi: 10.1016/j.virol.2005.08.004. [DOI] [PubMed] [Google Scholar]
- Jayaram S, Gilson T, Ehrlich ES, Yu XF, Ketner G, Hanakahi L. E1B 55k-independent dissociation of the DNA ligase IV/XRCC4 complex by E4 34k during adenovirus infection. Virology. 2008a;382:163–170. doi: 10.1016/j.virol.2008.08.045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jayaram S, Ketner G, Adachi N, Hanakahi LA. Loss of DNA ligase IV prevents recognition of DNA by double-strand break repair proteins XRCC4 and XLF. Nucleic Acids Res. 2008b;36:5773–5786. doi: 10.1093/nar/gkn552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jones N, Shenk T. Isolation of adenovirus type 5 host range deletion mutants defective for transformation of rat embryo cells. Cell. 1979;17:683–689. doi: 10.1016/0092-8674(79)90275-7. [DOI] [PubMed] [Google Scholar]
- Jones RM, Petermann E. Replication fork dynamics and the DNA damage response. Biochem. J. 2012;443:13–26. doi: 10.1042/BJ20112100. [DOI] [PubMed] [Google Scholar]
- Kanno T, Gotoh A, Fujita Y, Nakano T, Nishizaki T. A(3) adenosine receptor mediates apoptosis in 5637 human bladder cancer cells by G(q) protein/PKC-dependent AIF upregulation. Cell. Physiol. Biochem. 2012;30:1159–1168. doi: 10.1159/000343306. [DOI] [PubMed] [Google Scholar]
- Karen KA, Hearing P. Adenovirus core protein VII protects the viral genome from a DNA damage response at early times after infection. J. Virol. 2011;85:4135–4142. doi: 10.1128/JVI.02540-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karen KA, Hoey PJ, Young CS, Hearing P. Temporal regulation of the Mre11-Rad50-Nbs1 complex during adenovirus infection. J. Virol. 2009;83:4565–4573. doi: 10.1128/JVI.00042-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karran P. DNA double strand break repair in mammalian cells. Curr. Opin. Genet. Dev. 2000;10:144–150. doi: 10.1016/s0959-437x(00)00069-1. [DOI] [PubMed] [Google Scholar]
- Khalil A, Morgan RN, Adams BR, Golding SE, Dever SM, Rosenberg E, Povirk LF, Valerie K. ATM-dependent ERK signaling via AKT in response to DNA double-strand breaks. Cell Cycle. 2011;10:481–491. doi: 10.4161/cc.10.3.14713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khanna KK, Jackson SP. DNA double-strand breaks: signaling, repair and the cancer connection. Nat. Genet. 2001;27:247–254. doi: 10.1038/85798. [DOI] [PubMed] [Google Scholar]
- Knipe D.M.a.P.M.H. In: Fields Virology. Fifth edition Wolters Kluwer Health, editor. Lippincott Williams & Wilkins; Philadelphia: 2006. p. 3177. [Google Scholar]
- Kwant MM, van der Vliet PC. Differential effect of aphidicolin on adenovirus DNA synthesis and cellular DNA synthesis. Nucleic Acids Res. 1980;8:3993–4007. doi: 10.1093/nar/8.17.3993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lazebnik YA, Takahashi A, Moir RD, Goldman RD, Poirier GG, Kaufmann SH, Earnshaw WC. Studies of the lamin proteinase reveal multiple parallel biochemical pathways during apoptotic execution. Proc. Natl. Acad. Sci. U. S. A. 1995;92:9042–9046. doi: 10.1073/pnas.92.20.9042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lewis EM, Wilkinson AS, Jackson JS, Mehra R, Varambally S, Chinnaiyan AM, Wilkinson JC. The enzymatic activity of apoptosis-inducing factor supports energy metabolism benefiting the growth and invasiveness of advanced prostate cancer cells. J. Biol. Chem. 2012;287:43862–43875. doi: 10.1074/jbc.M112.407650. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li M, Yu X. Function of BRCA1 in the DNA Damage Response Is Mediated by ADP-Ribosylation. Cancer Cell. 2013;23:693–704. doi: 10.1016/j.ccr.2013.03.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lorenzo HK, Susin SA, Penninger J, Kroemer G. Apoptosis inducing factor (AIF): a phylogenetically old, caspase-independent effector of cell death. Cell Death Differ. 1999;6:516–524. doi: 10.1038/sj.cdd.4400527. [DOI] [PubMed] [Google Scholar]
- Martin ME, Berk AJ. Adenovirus E1B 55K represses p53 activation in vitro. J. Virol. 1998;72:3146–3154. doi: 10.1128/jvi.72.4.3146-3154.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mathew SS, Bridge E. The cellular Mre11 protein interferes with adenovirus E4 mutant DNA replication. Virology. 2007;365:346–355. doi: 10.1016/j.virol.2007.03.049. [DOI] [PubMed] [Google Scholar]
- Mathew SS, Bridge E. Nbs1-dependent binding of Mre11 to adenovirus E4 mutant viral DNA is important for inhibiting DNA replication. Virology. 2008;374:11–22. doi: 10.1016/j.virol.2007.12.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McGuire KA, Barlan AU, Griffin TM, Wiethoff CM. Adenovirus type 5 rupture of lysosomes leads to cathepsin B-dependent mitochondrial stress and production of reactive oxygen species. J. Virol. 2011;85:10806–10813. doi: 10.1128/JVI.00675-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mizukoshi S, Nakazawa M, Sato K, Ozaki T, Metoki T, Ishiguro S. Activation of mitochondrial calpain and release of apoptosis-inducing factor from mitochondria in RCS rat retinal degeneration. Exp. Eye Res. 2010;91:353–361. doi: 10.1016/j.exer.2010.06.004. [DOI] [PubMed] [Google Scholar]
- Murakami Y, Ikeda Y, Yonemitsu Y, Onimaru M, Nakagawa K, Kohno R, Miyazaki M, Hisatomi T, Nakamura M, Yabe T, Hasegawa M, Ishibashi T, Sueishi K. Inhibition of nuclear translocation of apoptosis-inducing factor is an essential mechanism of the neuroprotective activity of pigment epithelium-derived factor in a rat model of retinal degeneration. Am. J. Pathol. 2008;173:1326–1338. doi: 10.2353/ajpath.2008.080466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nam EA, Cortez D. ATR signalling: more than meeting at the fork. Biochem. J. 2011;436:527–536. doi: 10.1042/BJ20102162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nichols GJ, Schaack J, Ornelles DA. Widespread phosphorylation of histone H2AX by species C adenovirus infection requires viral DNA replication. J. Virol. 2009;83:5987–5998. doi: 10.1128/JVI.00091-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ou HD, Kwiatkowski W, Deerinck TJ, Noske A, Blain KY, Land HS, Soria C, Powers CJ, May AP, Shu X, Tsien RY, Fitzpatrick JA, Long JA, Ellisman MH, Choe S, O'Shea CC. A structural basis for the assembly and functions of a viral polymer that inactivates multiple tumor suppressors. Cell. 2012;151:304–319. doi: 10.1016/j.cell.2012.08.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ozaki T, Yamashita T, Ishiguro S. Mitochondrial m-calpain plays a role in the release of truncated apoptosis-inducing factor from the mitochondria. Biochim. Biophys. Acta. 2009;1793:1848–1859. doi: 10.1016/j.bbamcr.2009.10.002. [DOI] [PubMed] [Google Scholar]
- Patsalo V, Yondola MA, Luan B, Shoshani I, Kisker C, Green DF, Raleigh DP, Hearing P. Biophysical and functional analyses suggest that adenovirus E4-ORF3 protein requires higher-order multimerization to function against promyelocytic leukemia protein nuclear bodies. J. Biol. Chem. 2012;287:22573–22583. doi: 10.1074/jbc.M112.344234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pennella MA, Liu Y, Woo JL, Kim CA, Berk AJ. Adenovirus E1B 55-kilodalton protein is a p53-SUMO1 E3 ligase that represses p53 and stimulates its nuclear export through interactions with promyelocytic leukemia nuclear bodies. J. Virol. 2010;84:12210–12225. doi: 10.1128/JVI.01442-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perier C, Bove J, Dehay B, Jackson-Lewis V, Rabinovitch PS, Przedborski S, Vila M. Apoptosis-inducing factor deficiency sensitizes dopaminergic neurons to parkinsonian neurotoxins. Ann. Neurol. 2010;68:184–192. doi: 10.1002/ana.22034. [DOI] [PubMed] [Google Scholar]
- Peterson CL, Cote J. Cellular machineries for chromosomal DNA repair. Genes Dev. 2004;18:602–616. doi: 10.1101/gad.1182704. [DOI] [PubMed] [Google Scholar]
- Pink JJ, Planchon SM, Tagliarino C, Varnes ME, Siegel D, Boothman DA. NAD(P)H:Quinone oxidoreductase activity is the principal determinant of beta-lapachone cytotoxicity. J. Biol. Chem. 2000;275:5416–5424. doi: 10.1074/jbc.275.8.5416. [DOI] [PubMed] [Google Scholar]
- Polster BM, Basanez G, Etxebarria A, Hardwick JM, Nicholls DG. Calpain I induces cleavage and release of apoptosis-inducing factor from isolated mitochondria. J. Biol. Chem. 2005;280:6447–6454. doi: 10.1074/jbc.M413269200. [DOI] [PubMed] [Google Scholar]
- Querido E, Blanchette P, Yan Q, Kamura T, Morrison M, Boivin D, Kaelin WG, Conaway RC, Conaway JW, Branton PE. Degradation of p53 by adenovirus E4orf6 and E1B55K proteins occurs via a novel mechanism involving a Cullin-containing complex. Genes Dev. 2001;15:3104–3117. doi: 10.1101/gad.926401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Querido E, Marcellus RC, Lai A, Charbonneau R, Teodoro JG, Ketner G, Branton PE. Regulation of p53 levels by the E1B 55-kilodalton protein and E4orf6 in adenovirus-infected cells. J. Virol. 1997;71:3788–3798. doi: 10.1128/jvi.71.5.3788-3798.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rasband WS. ImageJ. U. S. National Institutes of Health; Bethesda, Maryland, USA: pp. 1997–2012. http://imagej.nih.gov. [Google Scholar]
- Sarnow P, Hearing P, Anderson CW, Reich N, Levine AJ. Identification and characterization of an immunologically conserved adenovirus early region 11,000 Mr protein and its association with the nuclear matrix. J. Mol. Biol. 1982;162:565–583. doi: 10.1016/0022-2836(82)90389-8. [DOI] [PubMed] [Google Scholar]
- Schaack J. Adenovirus vectors deleted for genes essential for viral DNA replication. Front. Biosci. 2005;10:1146–1155. doi: 10.2741/1607. [DOI] [PubMed] [Google Scholar]
- Sette G, Salvati V, Memeo L, Fecchi K, Colarossi C, Di Matteo P, Signore M, Biffoni M, D'Andrea V, De Antoni E, Canzonieri V, De Maria R, Eramo A. EGFR inhibition abrogates leiomyosarcoma cell chemoresistance through inactivation of survival pathways and impairment of CSC potential. PLoS One. 2012;7:e46891. doi: 10.1371/journal.pone.0046891. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shepard RN, Ornelles DA. E4orf3 is necessary for enhanced S-phase replication of cell cycle-restricted subgroup C adenoviruses. J. Virol. 2003;77:8593–8595. doi: 10.1128/JVI.77.15.8593-8595.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shepard RN, Ornelles DA. Diverse roles for E4orf3 at late times of infection revealed in an E1B 55-kilodalton protein mutant background. J. Virol. 2004;78:9924–9935. doi: 10.1128/JVI.78.18.9924-9935.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Soboloff J, Sasaki H, Tsang BK. Follicular stage-dependent tumor necrosis factor alpha-induced hen granulosa cell integrin production and survival in the presence of transforming growth factor alpha in vitro. Biol. Reprod. 2001;65:477–487. doi: 10.1095/biolreprod65.2.477. [DOI] [PubMed] [Google Scholar]
- Soria C, Estermann FE, Espantman KC, O'Shea CC. Heterochromatin silencing of p53 target genes by a small viral protein. Nature. 2010;466:1076–1081. doi: 10.1038/nature09307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sousa FG, Matuo R, Soares DG, Escargueil AE, Henriques JA, Larsen AK, Saffi J. PARPs and the DNA damage response. Carcinogenesis. 2012;33:1433–1440. doi: 10.1093/carcin/bgs132. [DOI] [PubMed] [Google Scholar]
- Stojic L, Mojas N, Cejka P, Di Pietro M, Ferrari S, Marra G, Jiricny J. Mismatch repair-dependent G2 checkpoint induced by low doses of SN1 type methylating agents requires the ATR kinase. Genes Dev. 2004;18:1331–1344. doi: 10.1101/gad.294404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stracker TH, Carson CT, Weitzman MD. Adenovirus oncoproteins inactivate the Mre11-Rad50-NBS1 DNA repair complex. Nature. 2002;418:348–352. doi: 10.1038/nature00863. [DOI] [PubMed] [Google Scholar]
- Stracker TH, Lee DV, Carson CT, Araujo FD, Ornelles DA, Weitzman MD. Serotype-specific reorganization of the Mre11 complex by adenoviral E4orf3 proteins. J. Virol. 2005;79:6664–6673. doi: 10.1128/JVI.79.11.6664-6673.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sundararajan R, White E. E1B 19K blocks Bax oligomerization and tumor necrosis factor alpha-mediated apoptosis. J. Virol. 2001;75:7506–7516. doi: 10.1128/JVI.75.16.7506-7516.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Susin SA, Daugas E, Ravagnan L, Samejima K, Zamzami N, Loeffler M, Costantini P, Ferri KF, Irinopoulou T, Prevost MC, Brothers G, Mak TW, Penninger J, Earnshaw WC, Kroemer G. Two distinct pathways leading to nuclear apoptosis. J. Exp. Med. 2000;192:571–580. doi: 10.1084/jem.192.4.571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Susin SA, Lorenzo HK, Zamzami N, Marzo I, Snow BE, Brothers GM, Mangion J, Jacotot E, Costantini P, Loeffler M, Larochette N, Goodlett DR, Aebersold R, Siderovski DP, Penninger JM, Kroemer G. Molecular characterization of mitochondrial apoptosis-inducing factor. Nature. 1999;397:441–446. doi: 10.1038/17135. [DOI] [PubMed] [Google Scholar]
- Tagliarino C, Pink JJ, Dubyak GR, Nieminen AL, Boothman DA. Calcium is a key signaling molecule in beta-lapachone-mediated cell death. J. Biol. Chem. 2001;276:19150–19159. doi: 10.1074/jbc.M100730200. [DOI] [PubMed] [Google Scholar]
- Tanenbaum ME, Medema RH. Mechanisms of centrosome separation and bipolar spindle assembly. Dev. Cell. 2010;19:797–806. doi: 10.1016/j.devcel.2010.11.011. [DOI] [PubMed] [Google Scholar]
- Thal SE, Zhu C, Thal SC, Blomgren K, Plesnila N. Role of apoptosis inducing factor (AIF) for hippocampal neuronal cell death following global cerebral ischemia in mice. Neurosci. Lett. 2011;499:1–3. doi: 10.1016/j.neulet.2011.05.016. [DOI] [PubMed] [Google Scholar]
- Thompson JW, Graham RM, Webster KA. DNase activation by hypoxia-acidosis parallels but is independent of programmed cell death. Life Sci. 2012;91:223–229. doi: 10.1016/j.lfs.2012.03.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Turnell AS, Grand RJ. DNA viruses and the cellular DNA-damage response. J. Gen. Virol. 2012;93:2076–2097. doi: 10.1099/vir.0.044412-0. [DOI] [PubMed] [Google Scholar]
- Vayda ME, Rogers AE, Flint SJ. The structure of nucleoprotein cores released from adenovirions. Nucleic Acids Res. 1983;11:441–460. doi: 10.1093/nar/11.2.441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang X, Yang C, Chai J, Shi Y, Xue D. Mechanisms of AIF-mediated apoptotic DNA degradation in Caenorhabditis elegans. Science. 2002;298:1587–1592. doi: 10.1126/science.1076194. [DOI] [PubMed] [Google Scholar]
- Wang Y, Dawson VL, Dawson TM. Poly(ADP-ribose) signals to mitochondrial AIF: a key event in parthanatos. Exp. Neurol. 2009a;218:193–202. doi: 10.1016/j.expneurol.2009.03.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Y, Kim NS, Li X, Greer PA, Koehler RC, Dawson VL, Dawson TM. Calpain activation is not required for AIF translocation in PARP-1-dependent cell death (parthanatos) J. Neurochem. 2009b;110:687–696. doi: 10.1111/j.1471-4159.2009.06167.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Z, Wang F, Tang T, Guo C. The role of PARP1 in the DNA damage response and its application in tumor therapy. Front. Med. 2012;6:156–164. doi: 10.1007/s11684-012-0197-3. [DOI] [PubMed] [Google Scholar]
- Weiden MD, Ginsberg HS. Deletion of the E4 region of the genome produces adenovirus DNA concatemers. Proc. Natl. Acad. Sci. U. S. A. 1994;91:153–157. doi: 10.1073/pnas.91.1.153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weitzman MD, Carson CT, Schwartz RA, Lilley CE. Interactions of viruses with the cellular DNA repair machinery. DNA Repair (Amst) 2004;3:1165–1173. doi: 10.1016/j.dnarep.2004.03.018. [DOI] [PubMed] [Google Scholar]
- Wiethoff CM, Wodrich H, Gerace L, Nemerow GR. Adenovirus protein VI mediates membrane disruption following capsid disassembly. J. Virol. 2005;79:1992–2000. doi: 10.1128/JVI.79.4.1992-2000.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Williams GJ, Lees-Miller SP, Tainer JA. Mre11-Rad50-Nbs1 conformations and the control of sensing, signaling, and effector responses at DNA double-strand breaks. DNA Repair (Amst) 2010;9:1299–1306. doi: 10.1016/j.dnarep.2010.10.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilson MR. Apoptosis: unmasking the executioner. Cell Death Differ. 1998;5:646–652. doi: 10.1038/sj.cdd.4400394. [DOI] [PubMed] [Google Scholar]
- Yamashima T, Zhao L, Wang XD, Tsukada T, Tonchev AB. Neuroprotective effects of pyridoxal phosphate and pyridoxal against ischemia in monkeys. Nutr. Neurosci. 2001;4:389–397. doi: 10.1080/1028415x.2001.11747375. [DOI] [PubMed] [Google Scholar]
- Yew PR, Berk AJ. Inhibition of p53 transactivation required for transformation by adenovirus early 1B protein. Nature. 1992;357:82–85. doi: 10.1038/357082a0. [DOI] [PubMed] [Google Scholar]
- Yu SW, Wang H, Dawson TM, Dawson VL. Poly(ADP-ribose) polymerase-1 and apoptosis inducing factor in neurotoxicity. Neurobiol. Dis. 2003;14:303–317. doi: 10.1016/j.nbd.2003.08.008. [DOI] [PubMed] [Google Scholar]
- Yu SW, Wang H, Poitras MF, Coombs C, Bowers WJ, Federoff HJ, Poirier GG, Dawson TM, Dawson VL. Mediation of poly(ADP-ribose) polymerase-1-dependent cell death by apoptosis-inducing factor. Science. 2002;297:259–263. doi: 10.1126/science.1072221. [DOI] [PubMed] [Google Scholar]
- Zeng CW, Zhang XJ, Lin KY, Ye H, Feng SY, Zhang H, Chen YQ. Camptothecin induces apoptosis in cancer cells via microRNA-125b-mediated mitochondrial pathways. Mol. Pharmacol. 2012;81:578–586. doi: 10.1124/mol.111.076794. [DOI] [PubMed] [Google Scholar]
- Zhang Y, Bhavnani BR. Glutamate-induced apoptosis in neuronal cells is mediated via caspase-dependent and independent mechanisms involving calpain and caspase-3 proteases as well as apoptosis inducing factor (AIF) and this process is inhibited by equine estrogens. BMC Neurosci. 2006;7:49. doi: 10.1186/1471-2202-7-49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao CQ, Jiang LS, Dai LY. Programmed cell death in intervertebral disc degeneration. Apoptosis. 2006;11:2079–2088. doi: 10.1007/s10495-006-0290-7. [DOI] [PubMed] [Google Scholar]
- Zhou BB, Elledge SJ. The DNA damage response: putting checkpoints in perspective. Nature. 2000;408:433–439. doi: 10.1038/35044005. [DOI] [PubMed] [Google Scholar]