

Abstract
Cancer diagnosis and classification have traditionally been based on the assessment of morphology by microscopy. However, the histological classification system is challenging and demand for genetic information is increasing in the era of targeted and personalized molecular therapy. Recently accumulated comprehensive genomic data could be used to provide a molecular cancer classification alongside the histological classification. This study identified a 19 gene signature able to classify endometrial cancers into the two major histological subtypes, endometrioid and serous. In addition, when the genomic classifier was applied to endometrioid adenocarcinoma of high grade (EM-HG), a subset (23.6%, 25/106) was predicted to be similar to serous tumors at the molecular level. In analyses of multiple cancers, the classification model may also be applicable to ovarian cancers.
Endometrial cancer is the most common gynecological malignancy in Western Europe and the USA, and is the seventh most common cancer in women worldwide1,2. The incidence of endometrial cancer has increased steadily in correlation with the current epidemic of obesity3,4. Endometrial cancers are classified by their histologies. A dualistic model classifying endometrial cancers into type I and type II is widely accepted to explain the pathogenesis5,6.
Type I endometrial cancers comprise the majority of endometrial cancers. They occur on a background of unopposed estrogen overstimulation, have endometrioid histology resembling a normal endometrial gland, and are usually diagnosed as low-grade endometrioid adenocarcinoma (EM-LG). By contrast, type II endometrial cancers exhibit non-endometrioid histology, such as serous adenocarcinoma (EM-Serous), and are frequently associated with TP53 mutations and an aggressive clinical course. Despite the differences in pathobiology and tumor behavior between the two types of endometrial cancers, differential diagnosis using the current histological classification system is frequently challenging7,8. In particular, this classification scheme does not always provide a clear distinction of tumor type in cases of high-grade endometrioid adenocarcinoma (EM-HG), referred to as FIGO (International Federation of Gynecology and Obstetrics) grade 3 endometrioid adenocarcinoma6,8. The distinction between type I and type II endometrial cancer is important because different treatments are recommended for each type, and a different clinical course is observed.
Recently, comprehensive genomic data, including whole exome sequencing, RNA sequencing, and large-scale copy number alteration (CNA), have become available for various cancers. These genomic data could be used to provide a molecular cancer classification or identify molecular cancer markers, and, in this era of personalized cancer therapy, to provide practical methods to build bridges between genomics and clinical practice.
In this study, we built classification models to predict the two major histologies of endometrial cancer using whole exome sequencing data, RNA sequencing data, and global copy number data obtained from the TCGA database (http://tcga-data.nci.nih.gov): type I endometrial cancers, i.e., tumors with endometrioid histology, and type II endometrial cancers, i.e., tumors with serous histology. These classification models were then compared to identify the best predictive model with the highest accuracy. The selected classification model was verified using an independent external data set, and the classification model was then applied to EM-HG, mixed-type and multiple cancers, including ovarian serous adenocarcinoma (Ov-Serous) and eight non-gynecological cancers.
Results
Classification model building using expression data for endometrial carcinomas
To build a classification model that is able to discriminate between endometrioid histology and serous histology in endometrial cancer, low-grade (FIGO grades 1 and 2) endometrioid adenocarcinoma (EM-LG, N = 184) and endometrial serous adenocarcinoma (EM-Serous, N = 52) were defined as binary endpoints. The results of 10-fold cross-validation (CV) using all nine classifiers showed high performances (permutation p-value < 0.001) with high sensitivities, specificities, positive predictive value (PPV), and negative predictive value (NPV), irrespective of classifiers (Fig. 1a). Ten-fold cross-validated receiver operating characteristic (ROC) curves showed high area under curve (AUC) values of up to 0.988 (Fig. 1b). Ten-fold cross-validated probabilities for prediction of histologies between EM-LG and EM-Serous were distinctively distributed into two patterns (toward 100% for the serous type or 0% for the endometrioid type) that were clearly correlated with the actual histological types using classification methods such as Prediction Analysis for Microarrays (PAM) (Fig. 1c), Bayesian compound covariates (Fig. 1d), and Adaboost classifiers (Fig. 1e). In the gene selection from the full data set, seven genes were selected by classical classifiers, eight genes by PAM, and 12 genes by Adaboost from the full data set (Fig. 1f). Of these, two genes, CLDN6 and KIAA1324, overlapped in all classification methods (Fig. 1f), suggesting that they are the most important in the morphogenesis of endometrial cancer. Detailed statistics are described for each gene in Supplementary Tables 1–3.
Figure 1. Classification modeling using expression and copy number data for histological subtypes of endometrial cancer.

(a) All nine classifiers showed a high performance, with high sensitivity, specificity, positive predicted value (PPV), negative predicted value (NPV), and (b) AUC values. (c) Ten-fold cross-validated probabilities for each class and misclassification error rates are shown for the PAM method. (d) Other methods such as the Bayesian compound covariate and (e) the Adaptive boosting method also showed the distinctive two probability patterns for endometrioid or serous histologies. (f) Gene lists selected by PAM, Adaptive boosting, and the remaining seven classifiers (classical classifiers) are shown. (g) Performance of the modeling using actual copy number data.
Classification model building using mutation and/or CNA data
We also built classification models using CNA data and/or mutation data. For CNA data, two models were built with the two possible data types: actual copy number data (continuous data) and GISTIC results (binary data). When the actual copy number data classification model was built, a relatively lower sensitivity was present across all classifiers, except for PAM and Adaboost, compared to the expression data classification model (Fig. 1g). For the GISTIC or mutation classification models, reduced sensitivity was also observed (Fig. 2). However, for models combining mutation and GISTIC data, sensitivity increased to 0.83 (permutation p-value < 0.001) but remained lower than the sensitivity observed for the expression data model. Overall, the distribution patterns between EM-LG and EM-Serous were well separated, as shown by the principal component (PC) 1 and PC 2 (Fig. 2). The selected genes for mutation and CNA data are summarized in Supplementary Table 4. Specifically, two mutated genes were selected: PTEN mutations for endometrioid tumors (+beta value) and TP53 mutations for serous tumors (−beta value). For the CNAs, ten genes exhibiting amplification, ABHD16B, BCL7C, BRD4, CCNE1, DNM2, FOSL2, GALK1, PGAP3, TERC, and ZMYND8, were selected and all indicated the serous type (−beta value). There was a significant correlation between the models that used the expression data and the models that used mutation + CNA data (p-value = 0.003; Supplementary Table 5).
Figure 2. Classification modeling using mutation and binary copy number alteration data for the histological subtypes of endometrial cancer.

Distribution patterns by principal component (PC) and accuracy are shown for each model using mutation or copy number variation (CNV), or mutation + CNV, respectively.
External validation of the models using expression data
The expression data classification model showed the best performance. We focused on this classification model since producing expression data is easier and cheaper than detecting mutations and CNAs. The established classification model using expression data was applied to an external independent data set generated using a microarray containing 63 EM-LG and 12 EM-Serous. High sensitivity and specificity were obtained for most classifiers, although two classifiers including 3-Nearest Neighbors and Support Vector Machine showed high error rate for serous (Fig. 3a). For each individual sample, detailed prediction results using the classification models are shown according to the different classifiers (Fig. 3b). The high correlations between the results predicted by the classification model and the original histology are presented. To compare the classifiers, we also applied hierarchical and non-negative matrix factorization (NMF) consensus clustering using 19 selected genes. Consensus clustering analysis showed lower specificity than the classifiers (Fig. 3c). In this analysis, the samples were divided into two groups (k = 2). All samples with EM-Serous histology were found in one group; however, this group also contained many samples with EM-LG histology (Fig. 3d).
Figure 3. External validation of the expression data model.

(a) High sensitivities and specificities were identified using an independent external validation set. (b) The predicted type and original histology of each sample are demonstrated. (c) Sensitivities and specificities and (d) subgrouping using consensus clustering methods.
Application of the model to EM-HG and mixed endometrial carcinoma
This classification model is able to distinguish endometrioid from serous histology in endometrial cancers with a high performance and accuracy. We applied this classification model to EM-HG and endometrial cancers that were originally classified as mixed histology. When the model was applied to samples with mixed histology (N = 10), three (30%) samples were classified as endometrioid and the remaining seven (70%) were classified as serous (Fig. 4a). When we reviewed the histology for the three available tumors, the histology of the sample predicted as serous was consistent with it being of serous histology, and the sample predicted as endometrioid showed predominantly endometrioid histology by hematoxylin and eosin (H&E) staining (Fig. 4a, H&E slides, upper and mid). One sample predicted as serous showed mixed pattern of serous and endometrioid histology (Fig. 4a, H&E slide, lower). The classification model was also applied to EM-HG, as it can be uncertain whether EM-HG belongs to the endometrioid or serous type of tumor. Of the 106 samples with EM-HG histology, the classifiers predicted that 25 (23.6%) samples were serous and the remaining 81 (76.4%) were endometrioid (Fig. 4b). Although the 106 samples were originally diagnosed as endometrioid type with FIGO grade 3, a subset of samples were reclassified as serous, suggesting that the reclassified samples are more similar to EM-Serous than endometrioid tumors at the molecular level. When the clustering analysis was performed using all endometrial cancers, including samples with EM-LG, EM-HG, and EM-Serous histology, EM-HG samples within the EM-LG cluster tended to be predicted as endometrioid, and EM-HG samples within the EM-Serous cluster tended to be predicted as serous (Fig. 4c).
Figure 4. Application of the classification model to mixed endometrial carcinoma and high-grade endometrioid adenocarcinoma (EM-HG).

(a) In the mixed endometrial carcinoma, the predicted type and reviewed histology are well correlated. (b) In the EM-HG, a subset of samples were predicted as serous tumors and (c) the model prediction tended to be correlated with clustering using a whole gene expression pattern.
Application of the model to multiple cancers
Finally, we applied the classification model to nine cancer types. The median values of the probabilities were around 50%, although colorectal cancers tended to be classified as endometrioid as opposed to serous. Most ovarian serous adenocarcinomas were predicted to be serous with high probabilities (Figure 5a). When the clustering analysis was performed for nine cancer types (N = 3766), some cancer types even derived from the same organ were clustered differently, irrespective of tumor origin; however, endometrial cancers and ovarian cancers clustered together (Fig. 5b), suggesting that our model can also be applied to ovarian cancer.
Figure 5. Application of the model to multiple cancer types.
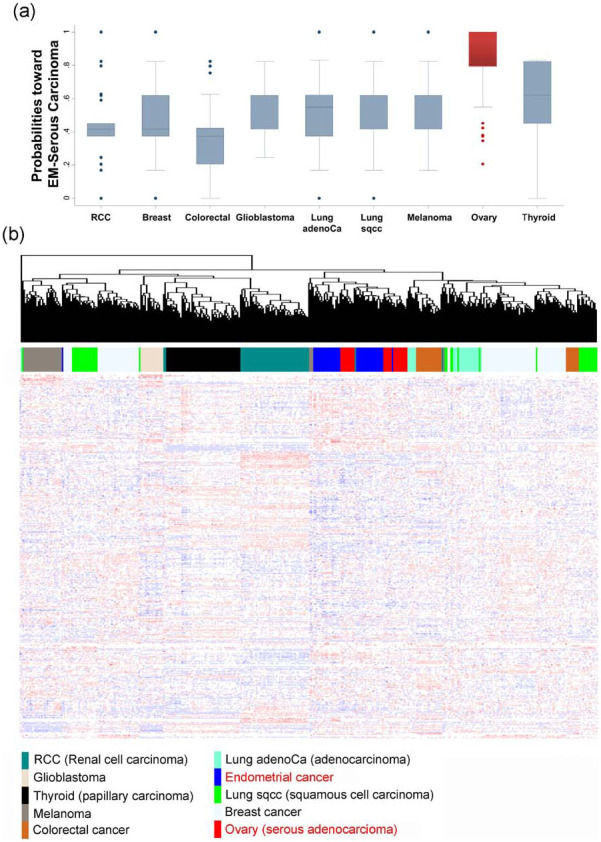
(a) When the established model was applied to nine types of cancers, most ovarian serous adenocarcinomas were predicted as being of serous histology with high probabilities. (b) When clustering was performed (N = 3766), some cancer types of even the same organs failed to cluster, indicating heterogeneity, however, endometrial cancers and ovarian cancers clustered together.
Discussion
In this study, we constructed a classification model using mutation data, CNA data and expression data to distinguish between histological subtypes of endometrial cancer. Recent high-throughput sequencing technology has generated an enormous amount of data for somatic mutations and CNAs in a range of cancer types. The development of models that use somatic mutation data or CNA data is an effective clinical use of genomic data. In our previous study, we made a predictive model using a somatic mutation profile to predict patient survival in ovarian cancer9. In this study, we showed that classification models using mutation and/or discrete copy number data are effective and applicable. These models, using binary data, show high performance, although sensitivity is lower than that of the expression data model. The lower sensitivity of the mutation or copy number data models is probably due to the low frequency of mutations in most genes and to the high frequency of CNAs in EM-Serous but not in EM-LG tumors10. Among the mutations, only the well-known mutations PTEN and TP53 were included in the classification model: PTEN mutations occur frequently in endometrioid adenocarcinoma, and TP53 mutations frequently occur in serous adenocarcinoma.
Our study suggests that expression patterns using up to 19 genes are able to classify endometrial cancer into two subgroups: endometrioid and serous. Of the 19 genes, six genes (L1CAM, CLDN6, RSPO4, PLAG1, FCHO1, and DHCR24) were elevated in EM-Serous, and the remaining 13 genes (PIGR, IHH, TFF3, SPDEF, KIAA1324, C9ORF152, FOXA2, CNGA1, HHAT, UQCR11, MDM2, SORBS2, and PPAP2C) were elevated in EM-LG. Among them, CLDN6 and KIAA1324 were consistently selected in all nine classifiers, suggesting that these genes are the most important in the morphogenesis of endometrial cancer. KIAA1324 is induced by estrogen and is a good endometrial biomarker associated with a hyperestrogenic state and estrogen-related type I endometrial adenocarcinoma11. The two types of endometrial adenocarcinomas can be distinguished by claudin 1 (CLDN1) and claudin 2 (CLDN2)12. Therefore, the related gene CLDN6 may also be a useful marker to discriminate between the two groups.
In this study, a high error rate was present in predicting the serous type of cancer in a few classifiers. Possible reasons include the following: 1) certain classifiers, such as Support Vector Machine, may be inappropriate for binary classification schemes in this data set; 2) unequal distribution of samples between the endometrioid and serous types (fewer serous than endometrioid tumors); and 3) model building relied on RNA sequencing data (TCGA data set), which have a larger dynamic range of expression than the independent microarray data used for validation. Another possible reason is potentially flawed histology since serous tumors may be confused with high-grade endometrioid carcinoma. In addition, different molecular profiles may be present in the same tumor, with increased heterogeneity in high-grade tumors such as serous carcinomas. In this study, we used nine classifiers for classification modeling. Among them, two classifiers, 3-Nearest Neighbors and Support Vector Machine, showed a high error rate for serous cancers. The remaining seven classifiers showed zero or very low error rates for prediction of the serous type, which suggests that the cause of the error rate lies with a few classifiers rather than with the histology or the data. Therefore, the use of several classifiers may be important to minimize misclassification in clinical application. In addition, correlation between histologic and molecular classification is important to lead to the correct diagnosis in clinical application.
When the model was applied to the ten mixed-type histologies, three (30%) samples were classified as endometrioid and the remaining seven (70%) were classified as serous. Although we did not review the histology in all cases, one case predicted as endometrioid showed predominantly endometrioid histology and two cases predicted as serous contained serous areas, which suggests that the classification model correlates with histology. In this study, one of the interesting findings is a discrepancy between histologic diagnosis and classification by modeling in EM-HG, 23.6% of which were classified as serous. Many samples were reclassified as serous using the classification model, which suggests that a subset of EM-HG may be more similar to the EM-Serous type than to the endometrioid type at the molecular level. This finding also suggests that EM-HGs may be more molecularly and histologically heterogeneous than initially thought.
We performed clustering analysis for multiple cancers and found that the cancers did not cluster by tumor origin. This suggests that classification according to molecular features is more effective for treatment. We also found that endometrial cancers and ovarian cancers were closely linked and clustered together. Therefore, our model may be applicable to the classification of ovarian cancers having histological subtypes of serous and endometrioid adenocarcinoma.
In summary, we created endometrial cancer classification models using different platforms and validated the models. A model using a 19 gene signature was able to classify endometrial cancers into the two major histological subtypes. Classification models using genomic data may complement histology in establishing diagnoses, and this study also suggests that using multiple classifiers could be important to minimize misclassification in clinical application. In the era of targeted molecular therapy, it is potentially important to report molecular classification predictions alongside histological classifications.
Methods
Genomic data
For endometrial cancer modeling, the following genomic data were used.
Expression data: mRNA expression data were derived from the RNA seqV2 RESM for endometrial cancer (N = 370), breast cancer (N = 914), colorectal cancer (N = 243), glioblastoma (N = 153), lung adenocarcinoma (N = 230), lung squamous cell carcinoma (N = 347), melanoma (N = 282), renal cell carcinoma (N = 480), Ov-Serous (N = 261), and thyroid papillary carcinoma (N = 486). The data were normalized using quantile normalizations with log2 transformations.
Mutation data: for modeling, all observed somatic mutations across all sequenced cases (N. = 232) and mutated genes, as determined by a merger of the MutSig v2.0 and MutSigCV v0.9 (Q-value ≤ 0.1) test results10, were used. There are 29 genes in the list of genes significantly mutated in endometrial cancer (Supplementary Table 6).
Copy number data: segmented copy number data generated using an Affymetrix Single Nucleotide Polymorphism (SNP) 6.0 array (N = 492) were used. The downloaded segmented copy number data were analyzed with GISTIC2.013,14 to identify significant focal CNAs. The thresholds for significant focal CNAs were as follows: amplification and deletion threshold, 0.1; cap-values, 1.5; broad length cut-off, 0.7; confidence level, 0.95; joint segment size, 4; level peel-off, 1; and maximum sample segments, 2000. Details of each of these parameters have been previously described14. For modeling using CNA data, both actual copy number data and discrete copy number results determined by GISTIC were used. For the discrete copy number results with high-level amplifications or homozygous deletions, a one-tail Wilcoxon signed-rank test was used to filter the cases in which mRNA expression values were significantly higher or lower in amplified or homozygously deleted samples versus diploid samples.
Classification modeling and internal validation
The methods used to build the classification models using continuous variables, such as expression data or actual copy number data, were as follows: compound covariate predictors, diagonal linear discriminant analysis, 1-Nearest Neighbor, 3-Nearest Neighbors, Nearest centroid, Support Vector Machine (SVM), Bayesian compound covariates, class prediction using PAM15, and the Adaptive boosting (Adaboost) method16. Genes with significant differences between the two classes (t-test, p < 0.001) and genes in which the fold-difference between the two classes exceeded 30 were used to fit the classification model. For binary outcome data, such as mutation data and the discrete CNV data from GISTIC, we used a classification method combining Fisher's exact test and 1-norm SVM17 to predict the binary response using mutation and/or CNA data. We selected the significant genes (Fisher exact test p < 0.001) before fitting the classification model. A chi-square test was used for testing the association between the original and predicted responses. To evaluate the predictive performance of the classification models, 10-fold CV procedure was used as follows:
Step 1. The total data were randomly divided into ten equally sized subsets.
Step 2. A single subset was used as the validation data, and the remaining nine subsets were used as training data.
Step 3. The significant genes (t-test, p < 0.001 for continuous data, or Fisher exact test, p < 0.001 for binary data) were selected from the training set.
Step 4. The classification method was applied to the selected genes and a classification model was fitted.
Step 5. A fitted classification model was applied to the validation data and the responses were predicted.
Step 6. Steps 3–5 were repeated ten times.
Step 7. The chi-square p-value was calculated using the original and predicted responses.
To remove the overfitting bias of the 10-fold CV, we calculated a permutation p-value, as in Simon et al.18 and Pang and Jung19, as follows: 1) the naive chi-square p-value (P0) was computed from the 10-fold CV procedure for the original data, 2) the chi-square p-value (Pb) was computed from the 10-fold CV procedure for the b-th permuted data (b = 1, …, B), and 3) a permutation 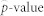 was calculated using the equation
was calculated using the equation 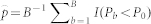 .
.
Measurement of the accuracy of the predictive model
Cross-validated ROC curves and AUC values were used. The performance measurements used were sensitivity (the probability that the EM-Serous would be correctly predicted as EM-Serous), specificity (the probability that the EM-LG would be correctly predicted as EM-LG), PPV (the probability that a sample predicted as EM-Serous actually belongs to EM-Serous), and NPV (the probability that a sample predicted as EM-LG actually belongs to EM-LG). In addition, normalized gene expression data from 91 stage I endometrial cancers derived from the Affymetrix Human Genome U133 Plus 2.020 were used for external validation.
Microscopy imaging data
Three available histological images of mixed-type endometrial cancer (TCGA-AX-A1CR, TCGA-BK-A0CA, and TCGA-D1-A0ZZ) were obtained from Berkeley Cancer Morphometric Data (http://tcga.lbl.gov/biosig/tcgadownload.do).
Consensus clustering for expression data
A consensus hierarchical and NMF clustering with iterative feature selection was performed. Consensus clustering is a resampling-based procedure that repeatedly samples a sample subset and then uses clustering to find intrinsic groupings21,22. Consensus clustering records the proportion of resamplings in which pairs of tumors were in the same clusters21,22. NMF is an algorithm based on decomposition by parts that can reduce the dimension of the data; it is also an efficient method for the identification of distinct molecular patterns and provides a powerful method for class discovery23. These algorithms have been previously described23,24.
Statistical analysis and data mining
Modeling, data analysis, and data mining were performed using the BRB array tool25 and R-program (version 2.14.2; www.r-project.org). Consensus clustering analysis was performed using GenePattern from the Broad Institute with the “ConsensusClustering” and “NMFConsensus” modules and pipelines26. Statistical analyses for association tests were performed using Stata/IC statistical software (version 12; StataCorp, TX) or the R-program (version 2.14.2; www.r-project.org).
Author Contributions
C.O.S. and I.S. designed the study, provided ideas and materials, performed the analysis, designed and manipulated the figures and tables, and wrote the manuscript. All authors reviewed the manuscript.
Supplementary Material
Supplementary Tables 1-6
References
- Ferlay J. et al. Estimates of worldwide burden of cancer in 2008: GLOBOCAN 2008. Int J Cancer 127, 2893–2917, http://dx.doi.org/10.1002/ijc.25516 (2010). [DOI] [PubMed] [Google Scholar]
- Sorosky J. I. Endometrial cancer. Obstet Gynecol 120, 383–397, http://dx.doi.org/10.1097/AOG.0b013e3182605bf1 (2012). [DOI] [PubMed] [Google Scholar]
- Siegel R., Ward E., Brawley O. & Jemal A. Cancer statistics, 2011: the impact of eliminating socioeconomic and racial disparities on premature cancer deaths. CA Cancer J Clin 61, 212–236 (2011). [DOI] [PubMed] [Google Scholar]
- Westin S. N. & Broaddus R. R. Personalized therapy in endometrial cancer: challenges and opportunities. Cancer Biol Ther 13, 1–13 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bokhman J. V. Two pathogenetic types of endometrial carcinoma. Gynecol Oncol 15, 10–17 (1983). [DOI] [PubMed] [Google Scholar]
- Zannoni G. F., Scambia G. & Gallo D. The dualistic model of endometrial cancer: the challenge of classifying grade 3 endometrioid carcinoma. Gynecol Oncol 127, 262–263, 10.1016/j.ygyno.2011.09.036 (2012). [DOI] [PubMed] [Google Scholar]
- Voss M. A. et al. Should grade 3 endometrioid endometrial carcinoma be considered a type 2 cancer-a clinical and pathological evaluation. Gynecol Oncol 124, 15–20 (2012). [DOI] [PubMed] [Google Scholar]
- Park J. Y. et al. Poor prognosis of uterine serous carcinoma compared with grade 3 endometrioid carcinoma in early stage patients. Virchows Arch 462, 289–296, http://dx.doi.org/10.1007/s00428-013-1382-8 (2013). [DOI] [PubMed] [Google Scholar]
- Sohn I. & Sung C. O. Predictive modeling using a somatic mutational profile in ovarian high grade serous carcinoma. PLoS One 8, e54089, 10.1371/journal.pone.0054089 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- The Cancer Genome Atlas Research Network. Integrated genomic characterization of endometrial carcinoma. Nature 497, 67–73, 10.1038/nature12113 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deng L. et al. Identification of a novel estrogen-regulated gene, EIG121, induced by hormone replacement therapy and differentially expressed in type I and type II endometrial cancer. Clin Cancer Res 11, 8258–8264, 10.1158/1078-0432.ccr-05-1189 (2005). [DOI] [PubMed] [Google Scholar]
- Sobel G. et al. Claudin 1 differentiates endometrioid and serous papillary endometrial adenocarcinoma. Gynecol Oncol 103, 591–598, 10.1016/j.ygyno.2006.04.005 (2006). [DOI] [PubMed] [Google Scholar]
- Beroukhim R. et al. Assessing the significance of chromosomal aberrations in cancer: methodology and application to glioma. Proc Natl Acad Sci U S A 104, 20007–20012, 10.1073/pnas.0710052104 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mermel C. H. et al. GISTIC2.0 facilitates sensitive and confident localization of the targets of focal somatic copy-number alteration in human cancers. Genome Biol 12, R41, 10.1186/gb-2011-12-4-r41 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tibshirani R., Hastie T., Narasimhan B. & Chu G. Diagnosis of multiple cancer types by shrunken centroids of gene expression. Proc Natl Acad Sci U S A 99, 6567–6572, 10.1073/pnas.082099299 (2002). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Freund Y. & Schapire R. E. Game theory, on-line prediction and boosting. Proceedings of the ninth annual conference on computational learning theory. New York: ACM, 325–332, doi>10.1145/238061.238163 (1996). [Google Scholar]
- Zhu J., Rosset S., Hastie T. & Tibshirani R. Advances in Neural Information Processing Systems 16. (MIT Press, Cambridge, MA, USA, 2004). [Google Scholar]
- Simon R. M., Subramanian J., Li M. C. & Menezes S. Using cross-validation to evaluate predictive accuracy of survival risk classifiers based on high-dimensional data. Brief Bioinform 12, 203–214, 10.1093/bib/bbr001 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pang H. & Jung S. H. Sample size considerations of prediction-validation methods in high-dimensional data for survival outcomes. Genet Epidemiol 37, 276–282, 10.1002/gepi.21721 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Day R. S. et al. Identifier mapping performance for integrating transcriptomics and proteomics experimental results. BMC Bioinformatics 12, 213, 10.1186/1471-2105-12-213 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lei Z. et al. Identification of Molecular Subtypes of Gastric Cancer With Different Responses to PI3-Kinase Inhibitors and 5-Fluorouracil. Gastroenterology 145, 554–565, 10.1053/j.gastro.2013.05.010 (2013). [DOI] [PubMed] [Google Scholar]
- Monti S., Tamayo P., Mesirov J. & Golub T. Consensus clustering: a resampling-based method for class discovery and visualization of gene expression microarray data. Machine Learning 52, 91–118 (2003). [Google Scholar]
- Brunet J. P., Tamayo P., Golub T. R. & Mesirov J. P. Metagenes and molecular pattern discovery using matrix factorization. Proc Natl Acad Sci U S A 101, 4164–4169, 10.1073/pnas.0308531101 (2004). [DOI] [PMC free article] [PubMed] [Google Scholar]
- The Cancer Genome Atlas Research Network. Integrated genomic analyses of ovarian carcinoma. Nature 474, 609–615, 10.1038/nature10166 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Simon R. et al. Analysis of gene expression data using BRB-ArrayTools. Cancer Inform 3, 11–17 (2007). [PMC free article] [PubMed] [Google Scholar]
- Reich M. et al. GenePattern 2.0. Nat Genet 38, 500–501, 10.1038/ng0506-500 (2006). [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Tables 1-6