

Abstract
X-linked myotubular myopathy is a congenital myopathy caused by deficiency of myotubularin. Patients often present with severe perinatal weakness, requiring mechanical ventilation to prevent death from respiratory failure. We recently reported that an activin receptor type IIB inhibitor produced hypertrophy of type 2b myofibers and modest increases of strength and life span in the severely myopathic Mtm1δ4 mouse model of X-linked myotubular myopathy. We have now performed a similar study in the less severely symptomatic Mtm1 p.R69C mouse in hopes of finding greater treatment efficacy. Activin receptor type IIB inhibitor treatment of Mtm1 p.R69C animals produced behavioral and histological evidence of hypertrophy in gastrocnemius muscles but not in quadriceps or triceps. The ability of the muscles to respond to activin receptor type IIB inhibitor treatment correlated with treatment-induced increases in satellite cell number and several muscle-specific abnormalities of hypertrophic signaling. Treatment-responsive Mtm1 p.R69C gastrocnemius muscles displayed lower levels of phosphorylated ribosomal protein S6 and higher levels of phosphorylated eukaryotic elongation factor 2 kinase than were observed in Mtm1 p.R69C quadriceps muscle or in muscles from wild-type littermates. Hypertrophy in the Mtm1 p.R69C gastrocnemius muscle was associated with increased levels of phosphorylated ribosomal protein S6. Our findings indicate that muscle-, fiber type-, and mutation-specific factors affect the response to hypertrophic therapies that will be important to assess in future therapeutic trials.
X-linked myotubular myopathy (XLMTM) is a severe form of congenital myopathy with an estimated incidence of 1:50,000 male births that most often presents with severe perinatal weakness and respiratory failure.1,2 Many patients with XLMTM die within the first year of life despite the use of mechanical ventilation, and no treatments approved by the Food and Drug Administration are available. XLMTM is caused by mutations in the gene that encodes myotubularin (MTM1), which is a phosphoinositide phosphatase thought to be involved in endosomal trafficking, cytoskeletal organization, apoptosis, and/or maintenance of the sarcoplasmic reticulum/T-tubular system within myofibers.3–8 Muscle biopsies from patients with XLMTM display excessively small fibers with increased numbers of fibers that contain central nuclei and central aggregation of organelles.9 Although the number of centrally nucleated fibers bears little relationship to a patient's prognosis, there is a clear correlation between the degree of fiber smallness at birth and the severity of the patients' disease.10 Two murine models of myotubularin deficiency are used, the severely symptomatic Mtm1δ4 (also referred to as Mtm1 knockout in prior studies3,11,12) and the moderately symptomatic Mtm1 p.R69C mice,13 both of which display weakness and myofiber smallness and similar pathology to that seen in XLMTM.
Because of the relationship between myofiber size and symptomatic severity in patients with XLMTM and in Mtm1δ4 mice, we had previously hypothesized that correction of myofiber smallness in myotubularin deficiency would greatly improve strength. Inhibitors of myostatin or nonfunctional decoys of its receptor, the activin type IIB receptor (ActRIIB), can be used to inhibit this negative regulator of myofiber size, leading to myofiber hypertrophy. Myostatin binds to (and signals through) the ActRIIB to activate the transforming growth factor-β pathway, which prevents progression through the cell cycle and down-regulates several key processes related to myofiber hypertrophy.14,15 We recently reported a trial of ActRIIB-mFC in Mtm1δ4 mice, which produced 17% extension of life span, with transient increases in weight, forelimb grip strength, myofiber size, and myofiber hypertrophy restricted to type 2b myofibers in Mtm1δ4 animals.12 Interestingly, ActRIIB-mFc produces hypertrophy in all muscle fiber types in wild-type (WT) mice,12,16 which suggests that myotubularin deficiency interferes with the activation of hypertrophic pathways in oxidative fibers.
We hypothesized that the transience of the therapeutic effects observed in treated Mtm1δ4 mice may have been related to the severity of the disease, so we have now repeated this study in the less severely affected Mtm1 p.R69C mouse.13 Surprisingly, treatment of Mtm1 p.R69C mice did not produce significant increases in animal weight or grip strength, and treatment-induced myofiber hypertrophy was only observed in the Mtm1 p.R69C gastrocnemius muscles. The ability of these muscles to respond to ActRIIB-mFC treatment correlated with treatment-induced increases in satellite cell number and several muscle-specific abnormalities of hypertrophic signaling. The main difference between treatment-responsive (gastrocnemius) and treatment-resistant (quadriceps) muscles in Mtm1 p.R69C mice was related to low levels of phosphorylated ribosomal protein 6 (p-rpS6) and high levels of eukaryotic elongation factor 2 kinase (eEF2K) in the treatment-responsive gastrocnemius muscle that were not observed in other Mtm1 p.R69C muscles or in WT mice. rpS6 and eEF2K are terminal signaling molecules of the insulinlike growth factor-1/Akt and extracellular signal-related kinase (ERK) pathways that are involved in the fine-tuning of global protein synthesis, with a role in the determination of cell size that remains unclear (reviewed in Meyuhas17). Our findings indicate that the response to hypertrophic agents does not always correlate with activities of known hypertrophic pathways, such as the Akt pathway, but unexpectedly varies both by muscle type and fiber type and in XLMTM is affected by the nature of the Mtm1 mutation. These results highlight that there is much we still do not understand about the control of muscle size and emphasize the importance of evaluating multiple muscle and fiber types in future trials of hypertrophic therapies.
Materials and Methods
Live Animal Studies
All studies were performed with approval from the Institutional Animal Care and Use Committee at Boston Children's Hospital. Genotyping of MTM1/C57BL6 (Mtm1δ4) and Mtm1 p.R69C/C57BL6 (Mtm1 p.R69C) mice was performed as previously described.12,13,18 Male WT and Mtm1 p.R69C mice were given intraperitoneal injections twice per week with a soluble ActRIIB-mFC (alias RAP-031; Acceleron Pharma, Cambridge, MA) at a dose of 20 mg/kg or an equivalent volume of Tris-buffered saline (the vehicle used with ActRIIB-mFc), as previously described12 from 14 days until 6 months of life (MOL). Animals were sacrificed at 6 MOL because of a plateau in observable therapeutic effects. Animals were weighed twice per week during the treatment period. Forelimb grip strength was measured weekly with the use of a Chatillon grip force meter (Columbus Instruments, Columbus, OH) by placing the animal on a horizontal grid and allowing it to pull away from the experimenter by using only its fore limbs. The maximum of three independent measurements, with a 1-minute recovery period between measurements, was used for subsequent statistical analysis. To evaluate antigravity hanging performance, animals were tested weekly by placing the animal on a rigid mesh surface, inverting the surface at a height of approximately 40 cm above their cage, and recording the amount of time necessary for the animal to fall back into the cage. Animals that did not fall within 60 seconds were lowered back into their cages. The maximum of three independent measurements, with a 1-minute recovery period between measurements, was used for subsequent statistical analysis.
Pathological Evaluation and Tissue Collection
Animals were euthanized with carbon dioxide followed by cervical dislocation, per the regulations of the Institutional Animal Care and Use Committee at Children's Hospital Boston. Animals were photographed intact and after removal of the skin from the torso and limbs. The quadriceps, gastrocnemius, triceps, soleus, extensor digitorum longus, and diaphragm muscles were removed, weighed, and frozen in liquid nitrogen-cooled isopentane.
Histological Evaluation
Myofiber Analysis
Cross sections (8 μm) of isopentane-frozen quadriceps, gastrocnemius, or triceps muscles were taken midway down the length of the muscle and stained with H&E for evaluation with the use of an Olympus BX53 microscope with an Olympus DP72 camera and cellSens Standard software version 1.5 (Olympus, Center Valley, PA). Fiber size was determined through measurements of MinFeret diameter, as we have previously used,12 because this measurement offers a myofiber size measurement that is relatively independent of fiber orientation.19 Overall fiber size and the number of internally nucleated fibers were manually measured with micrographs of two ×100 magnification fields (using a 10× objective lens) of H&E-stained slides (range, 259 to 1113 fibers; n = 4 to 5 animals quantified per treatment group). To evaluate fiber type-specific responses, 8 μm frozen transverse sections of quadriceps muscle were double-stained with rabbit anti-dystrophin antibodies (ab15277; Abcam, Cambridge, MA) and mouse monoclonal antibodies against myosin heavy chain type 1 (Skeletal, Slow, clone NOQ7.5.4D; Sigma-Aldrich, St. Louis, MO), type 2a (clone SC-71; Developmental Studies Hybridoma Bank, Iowa City, IA), or type 2b (clone BF-F3; Developmental Studies Hybridoma Bank). Secondary antibodies included fluorescein isothiocyanate-conjugated anti-mouse IgG (Sigma-Aldrich) or IgM (dilution 1:100; Sigma-Aldrich) and Alexa Fluor-conjugated anti-rabbit IgG (Molecular Probes, Carlsbad, CA). Because of variation in the number and type of oxidative fibers (type 1 and 2a fibers) in mouse muscle, quantification was performed by evaluating the type 2b myosin-positive (glycolytic) and type 2b myosin-negative (oxidative) populations on a whole-slide scan of one quadriceps and one gastrocnemius muscle from four vehicle-treated WT mice, four ActRIIB-mFc–treated WT mice, six vehicle-treated Mtm1 p.R69C mice, and six ActRIIB-mFc–treated Mtm1 p.R69C mice. MinFeret diameter was evaluated by using a novel automated technique developed by Dr. Lin Yang that correlated extremely well with our manual measurements.12 The automated image quantification algorithm began with ridge detection to enhance the muscle fiber boundaries, followed by robust seed detection that was based on concave area identification to find initial seeds for muscle fibers. The final muscle fiber boundaries were automatically delineated with a gradient vector flow deformable model. After each fiber was automatically and accurately delineated, the MinFeret diameter was calculated by finding the shortest axis along the segmented contour of each muscle fiber. The number of muscle fibers was determined with scanned slides that had been stained with anti-dystrophin antibodies. A software analysis algorithm was developed with Visiomorph software version 3.4.3.0 (Visiopharm, Medicon Valley, Denmark) to allow the distinction between black pixels inside myofibers from the white/gray pixels (in grayscale) in areas of dystrophin staining. Bayesian classification was used to segment the image, and postprocessing steps were applied related to shape and area to disregard counted areas that did not correspond to myofibers. Fiber count analysis was performed on muscles from four to six animals per treatment group for both the quadriceps and gastrocnemius muscles.
Satellite Cell Quantification
The quantification of satellite cells was performed with immunoperoxidase staining for Pax7 (Developmental Studies Hybridoma Bank) and biotin-SP–conjugated donkey anti-mouse IgG Fab fragment, as previously described.8 Images were quantified by counting the number of Pax7+ cells within a field and dividing by the total number of fibers (range, 1117 to 3631 fibers; n = 3 animals quantified per treatment group).
Hypertrophic Pathway Studies
Muscle tissues from the quadriceps, gastrocnemius, triceps, and elsewhere in the fore and hind limbs from WT and Mtm1 p.R69C mice at 6 MOL or WT and Mtm1δ4 mice at 6 weeks of life (WOL) were frozen at the time of necropsy and stored at −80°C until analysis. Frozen muscles were crushed in liquid nitrogen and homogenized with lysing buffer (Cell Signaling Technology, Danvers, MA) that contained protease inhibitor (Roche, Basel, Switzerland) and phosphatase inhibitor (Roche). Western blot procedures were performed as previously described.20 Transferred proteins were probed with antibodies against a variety of antigens. Antibodies that recognize the following antigens were all obtained from Cell Signaling Technologies: Akt (4691), phospho-Akt (Thr308; 2965), phospho-Akt (Ser473; 4060), S6 ribosomal protein (2217), phospho-S6 ribosomal protein (Ser240/244; 5364), ERK (4695), phospho-ERK (ERK 1/2, Thr202/204; 4370), p70-S6 kinase (S6K; 2708), phospho–p70-S6K (Thr421/Ser424; 9204), p90-ribosomal S6 kinase (RSK)1/2/3 (9355), phospho–p90-RSK (Ser380; 9335), phospho–4E-BP1(Thr37/46; 2855), and phospho-eEF2K (Ser366; 3691). Other antibodies used for Western blot studies recognize myostatin (MAB788; R&D Systems, Minneapolis, MN), ActRIIB (ab76940; Abcam), and glyceraldehyde-3-phosphate dehydrogenase (GAPDH; G8795; Sigma-Aldrich) and were visualized with chemiluminescent horseradish peroxidase antibody detection reagent (Denville Scientific, Metuchen, NJ). Adequacy of transfer was determined by Ponceau S staining. Quantification of protein levels normalized to GAPDH was performed with ImageJ software version 1.45s (NIH, Bethesda, MD). Samples from four animals per treatment group were used for analysis. Two additional Mtm1 p.R69C/vehicle and Mtm1 p.R69C/ActRIIB-mFc animals were added to the quantitative evaluation of total and p-rpS6 (based on results from replicate gels) to further establish the degree of interanimal variability.
Statistical Analysis
Statistical evaluation was performed with Prism software version 4 software (GraphPad, Inc., San Diego, CA) or by statisticians at the Quantitative Health Sciences Section at the Medical College of Wisconsin. For statistical analysis of animal weight, forelimb grip strength, and antigravity hanging performance, analyses of variance were performed with Bonferroni posttests. For muscle weight and average myofiber diameter measurements, one-way analyses of variance with Bonferroni posttests were performed. For statistical analysis of Western blot data, t-tests were used to compare protein expression between different genotypes or treatments, and paired t-tests were used to compare protein expression between the gastrocnemius and quadriceps muscles of the same genotype and treatment group.
Results
Weight Studies
In animals that received vehicle injections, Mtm1 p.R69C animals were distinguishable from age-matched WT animals on the basis of weight at 7 WOL (P < 0.05) (Figure 1A). These differences increased as the animals became older because of continued weight gain in WT animals, in contrast to the relative weight plateau seen after 7 WOL in Mtm1 p.R69C animals. ActRIIB-mFc–treated WT mice showed significant weight gains in comparison with vehicle-treated animals at 6 WOL (P < 0.05) that continued to increase with age. ActRIIB-mFC–treated Mtm1δ4 animals displayed a consistent slight increase (averaging 1 to 2 g) in weight that did not reach statistical significance and did not show the continued increases observed in WT mice.
Figure 1.
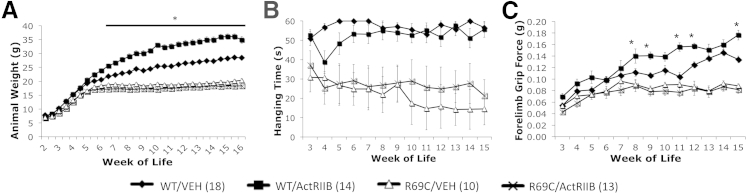
Behavioral findings in vehicle- and ActRIIB-mFC–treated mice. A: Body weight of VEH- and ActRIIB-treated WT and R69C mice. B: Antigravity hanging performance of VEH- and ActRIIB-treated mice. WT and R69C mice were significantly different after 3 weeks of age. C: Forelimb grip force of VEH- and ActRIIB-treated mice. No significant differences were seen for R69C/VEH compared with R69C/ActRIIB animals. Data are expressed as means ± SEM of (n) animals for each treatment group. ∗P < 0.05 (A and C). ActRIIB, ActRIIB-mFC; R69C, Mtm1 p.R69C; VEH, vehicle.
Antigravity Hanging Performance
WT mice displayed the ability to hang for up to 60 seconds (the maximum period tested) after 3 WOL (Figure 1B). Treatment of WT mice with ActRIIB-mFC did not affect antigravity hanging performance. Vehicle-treated Mtm1 p.R69C animals exhibited significantly impaired hanging performance at 4 WOL (P < 0.05), which, although variable, did not degenerate into the complete inability for antigravity hanging that we observed in Mtm1δ4 mice.12 Treatment with ActRIIB-mFC did not significantly improve the antigravity hanging performance of Mtm1 p.R69C mice.
Forelimb Grip Strength
Forelimb grip strength measurements were consistently lower in Mtm1 p.R69C mice than in WT mice after 12 WOL (P < 0.05) (Figure 1C). ActRIIB-mFC–treated WT animals also showed an increase in grip strength as the treatment period progressed (P < 0.01); however, treatment with ActRIIB-mFC did not measurably improve forelimb grip strength performance of Mtm1 p.R69C mice.
Gross Evaluation
Mtm1 p.R69C mice can be easily distinguished from WT mice by gross and histological examination.13 Mtm1 p.R69C mice are smaller than age-matched WT mice and have proportionately smaller muscles (data not shown). At 6 MOL (ie, after 5.5 months of treatment), ActRIIB-mFC treatment of WT animals produced significant increases in mass of the quadriceps, triceps, and gastrocnemius muscles (P < 0.05), but only the gastrocnemius muscle of treated Mtm1 p.R69C mice showed significant treatment-induced increases in mass (P < 0.05) (Figure 2A).
Figure 2.

Histological evaluation of VEH- and ActRIIB-mFC–treated mice. A: Weights of individual muscles in treated and untreated animals at 6 MOL. B: Myofiber size of VEH- and ActRIIB-mFC–treated WT and R69C mice, as measured by manual MinFeret diameter measurements of H&E-stained slides. C: H&E-stained transverse sections from the quadriceps and gastrocnemius muscles of VEH- and ActRIIB-mFC–treated WT and Mtm1δ4 mice. D and E: Automatic morphometric measurements of MinFeret diameter were made for fiber type, and frequency histograms for the quadriceps (D) and gastrocnemius (E) muscles are shown. Measurements from VEH-treated mice are shown with black bars and measurements from ActRIIB-mFc–treated mice are shown with white bars. Glycolytic fibers represent the population of fibers that is positive for type 2b myosin, and oxidative fibers represent the population of fibers that was negative for type 2b myosin. Data are expressed as means ± SEM. Quantifications were performed on whole-slide scans of the entire muscle on four animals per treatment group. ∗P < 0.05. Scale bar = 50 μm (C). R69C, Mtm1 p.R69C; VEH, vehicle.
Histological Examination
Myofiber Analysis
As previously described,13 quadriceps muscle fibers of vehicle-treated Mtm1 p.R69C mice are smaller than those found in age-matched WT mice, with mean fiber diameters of 24 ± 0.53 and 51 ± 1.27 μm, respectively, by 6 MOL (shown as means ± SEM; P < 0.05) (Figure 2). Although central nucleation is not a major pathological feature in this animal model, muscles of Mtm1 p.R69C mice showed increased numbers of centrally nucleated fibers in comparison with WT muscles (WT/vehicle quadriceps = 1.43% ± 0.8%, R69C/vehicle quadriceps = 5.3% ± 1.0%, WT/vehicle gastrocnemius = 0.8% ± 0.3%, and R69C/vehicle gastrocnemius = 4.9% ± 1.4% internally nucleated fibers; P < 0.05), and the number of internally nucleated fibers did not significantly increase or decrease after ActRIIB-mFc treatment. Treatment of WT animals with ActRIIB-mFC increased myofiber size by 6 MOL, as evidenced by a 5-μm (10%) increase in mean fiber diameter and an increased percentage of large fibers within the gastrocnemius and quadriceps muscles (Figure 2). Immunostaining for oxidative (type 1 or 2a) or glycolytic (type 2b) myosin subtypes within these muscles indicated that all types of WT muscle fibers experienced hypertrophy in response to ActRIIB-mFC treatment, as evidenced by a rightward shift in histograms of fiber size (Figure 2, D and E). In contrast, the gastrocnemius, but not quadriceps, muscles from ActRIIB-mFC–treated Mtm1 p.R69C animals showed marked hypertrophy in only a subpopulation of muscle fibers compared with their vehicle-treated counterparts, with a 10-μm (32%) increase in mean fiber diameter in the gastrocnemius muscle (P < 0.05) and no significant increase in the quadriceps muscle. Hypertrophy in treated Mtm1 p.R69C mice was noted only in the glycolytic type 2b fibers, similar to what was seen in our prior study of Mtm1δ4 mice (Figure 2, D and E).12 The proportion of type 1, 2a, and 2b fibers was highly variable between individual animals, but no significant differences were noted for genotype or treatment group (data not shown). In addition, counts of myofiber number within the gastrocnemius muscle indicated no significant difference between WT and Mtm1 p.R69C mice (WT/vehicle gastrocnemius = 5898 ± 222 fibers, R69C/vehicle gastrocnemius = 5743 ± 425 fibers; shown as means ± SEM; P = 0.56), or between vehicle- and ActRIIB-mFC–treated mice of either genetic background (WT/ActRIIB-mFc gastrocnemius = 6432 ± 496 fibers, R69C/ActRIIB-mFc gastrocnemius = 6391 ± 543 fibers). Similar results were observed on fiber counts in the quadriceps muscle (WT/vehicle quadriceps = 4821 ± 412 fibers, R69C/vehicle quadriceps = 6290 ± 249 fibers, WT/ActRIIB-mFc quadriceps = 5294 ± 299 fibers, and R69C/ActRIIB-mFc quadriceps = 6976 ± 557 fibers), where, despite a baseline difference between WT/vehicle and R69C/vehicle specimens (possibly because of differences in sampling; P < 0.05), no change was found in fiber number after treatment of WT (P = 0.37) or R69C (P = 0.30) mice. These results are consistent with a muscle type-restricted histological improvement of Mtm1 p.R69C mice after ActRIIB-mFC treatment, which accounts for the observed increases in gastrocnemius weight at the same stage.
Satellite Cell Quantification
The restriction of hypertrophy to certain muscles in Mtm1 p.R69C mice suggests factors affecting treatment efficacy may be differentially distributed among certain muscles. Because we have recently described abnormalities of satellite cell number in the Mtm1δ4 model of myotubularin deficiency,8 we evaluated satellite cell number in the gastrocnemius and quadriceps muscles of vehicle- and ActRIIB-mFc–treated Mtm1 p.R69C mice with the use of immunoperoxidase staining for Pax7 to determine whether treatment responsiveness in these muscles correlated with satellite cell behavior. Similar to what was seen in Mtm1δ4 mice,12 the gastrocnemius and quadriceps muscles of vehicle-treated Mtm1 p.R69C mice both displayed severe depletion of satellite cells at 6 MOL (<2% of fibers with one or more associated satellite cells in Mtm1 p.R69C mice versus >5% of fibers with associated satellite cells in WT mice in both muscles; P < 0.05) (Figure 3). However, ActRIIB-mFc treatment differentially increased satellite cell numbers in Mtm1 p.R69C gastrocnemius versus quadriceps muscles. In WT muscles (both gastrocnemius and quadriceps) and in the gastrocnemius muscles of Mtm1 p.R69C mice, ActRIIB-mFc treatment was associated with a higher number of satellite cells per fiber (77% increase in satellite cells/fiber in WT/ActRIIB quadriceps, 87% increase in satellite cells/fiber in WT/ActRIIB gastrocnemius, and 52% increase in satellite cells/fiber in R69C/ActRIIB gastrocnemius; P < 0.05) (Figure 3). By contrast, no such increase was observed in the quadriceps muscles of ActRIIB-mFc–treated Mtm1 p.R69C mice (18% decrease in satellite cells/fiber in R69C/ActRIIB quadriceps) (Figure 3).
Figure 3.

Pax7+ satellite cell quantification in VEH- and ActRIIB-treated mice. A: Immunostaining results for Pax7 in the gastrocnemius muscle of VEH- and ActRIIB-treated WT and R69C mice at 6 MOL. B: Quantification of Pax7+ cells in the quadriceps and gastrocnemius muscles of treated and untreated mice, expressed as the number of satellite cells observed divided by the number of fibers in the evaluated images. Data are expressed as means ± SEM. ∗P < 0.05. Scale bar = 50 μm. ActRIIB, ActRIIB-mFC; R69C, Mtm1 p.R69C; VEH, vehicle.
Hypertrophic Pathway Studies
Differential activation of hypertrophic pathways within different muscles is a possible explanation for the muscle-specific treatment efficacy observed in Mtm1 p.R69C mice. Endogenous amounts of the activin type 2B receptor were variable but equivalent in the gastrocnemius and quadriceps muscles of Mtm1 p.R69C mice (Figure 4). Treatment with ActRIIB-mFc variably increased activin type 2B receptor levels in all muscles, but we were unable to determine whether this was because of actual up-regulation of the receptor or cross-reactivity with injected ActRIIB-mFc (Figure 4).
Figure 4.

Expression of myostatin and activin receptor type IIB in VEH- and ActRIIB-treated mice. A: Western blot analysis shows the relative expression of myostatin and the activin type IIB receptor in the quadriceps and gastrocnemius muscles of VEH- and ActRIIB-treated WT and R69C mice at 6 MOL. GAPDH staining is used as a basis for loading and quantification studies. The numbers under each set of two lanes refer to individual mice from the study. B: Quantification of myostatin and activin type IIB receptor expression in the quadriceps and gastrocnemius muscles of VEH- and ActRIIB-treated WT and R69C mice. Expression is normalized to the GAPDH expression for each lane. These values were obtained from gels in which the WT and R69C samples were run in parallel on the same gels and immunoblotted on the same membranes. Data are expressed as means ± SEM. ∗P < 0.05. ActRIIB, ActRIIB-mFC; G, gastrocnemius; Q, quadriceps; R69C, Mtm1 p.R69C; VEH, vehicle.
Endogenous myostatin levels tended to be inherently lower in both Mtm1 p.R69C muscles than in WT muscles, but they were lowest in Mtm1 p.R69C quadriceps. ActRIIB-mFc treatment tended to reduce myostatin levels further in all muscles, but none of these effects was statistically significant. Because myostatin inhibits hypertrophic signaling (Figure 5), the fact that myostatin levels showed a trend higher in Mtm1 p.R69C gastrocnemius versus quadriceps muscles with and without treatment led us to look at other signaling pathways to account for the differential sensitivity of gastrocnemius muscle to ActRIIB-mFc.
Figure 5.

Simplified signaling pathway for muscle hypertrophy. Elements of the Akt and ERK pathways evaluated in the present study are shown.
As noted above, treatment-related hypertrophy was restricted to glycolytic (type 2b) myofibers in Mtm1 p.R69C mice, which is the same pattern that we previously encountered after treatment of Mtm1δ4 mice with ActRIIB-mFc.12 Because the insulinlike growth factor-1/Akt pathway is responsible for hypertrophy of type 2b myofibers, and previous work has established a link between the myostatin pathway and insulinlike growth factor-1/Akt pathway (reviewed in Han et al21), we focused on several elements of the Akt pathway to evaluate whether baseline levels or signal activation behaved in a muscle-specific fashion (Figures 5 and 6). Given the observed pattern of treatment efficacy, we hypothesized that the most relevant signaling molecule(s) would show differential expression or phosphorylation when comparing the gastrocnemius and quadriceps muscles from vehicle- and ActRIIB-treated Mtm1 p.R69C mice (Figure 6, A and B). Levels of total and p-Akt kinase were similar in the quadriceps and gastrocnemius muscles of these mice, but two proteins associated with the terminal steps of the Akt pathway, rpS6 and eEF2K, did show significantly different levels of phosphorylation. Specifically, rpS6 showed low levels of phosphorylation in vehicle-treated Mtm1 p.R69C gastrocnemius muscles and considerably higher levels of phosphorylation in Mtm1 p.R69C quadriceps muscles in five of the six animals tested (Figure 6), whereas the levels of p-eEF2K were consistently higher in gastrocnemius muscles. High levels of p-rpS6 and p-eEF2K were also seen in age-matched WT muscles (both gastrocnemius and quadriceps) (Figure 6). Treatment of Mtm1 p.R69C mice showed a trend toward increased rpS6 phosphorylation in the gastrocnemius muscle (P = 0.06), but produced no change in the already high level of p-rpS6 in the quadriceps muscle (Figure 6). Total levels of P70/S6K, the upstream kinase for both rpS6 and EF2K, were higher in ActRIIB-treated gastrocnemius muscles, and p-P70/S6K showed a trend toward increased levels in ActRIIB-treated gastrocnemius muscles, as well. Taken together, these results suggest a differential effect at the level of P70/S6K localization or relative substrate utilization. With respect to eEF2K, a higher level of p-eEF2K was seen in the Mtm1 p.R69C gastrocnemius muscles than in the quadriceps muscles (P < 0.05) (Figure 6), but this did not change with ActRIIB-mFc treatment.
Figure 6.

Akt pathway activation in VEH- and ActRIIB-treated mice. A: Western blot analysis shows the relative expression of Akt, p-Akt (at Ser473 and Thr308), total P70/S6K, p-P70/S6K (at Thr421/Ser424), rpS6, p-rpS6 (at Ser240/244), p-4EBP1 (at Thr37/46), and p-eEF2K (at Ser366) in the quadriceps and gastrocnemius muscles of VEH- and ActRIIB-treated WT and R69C mice at 6 MOL. GAPDH staining is used as a basis for loading and quantification studies. The numbers under each set of two lanes refer to individual mice from the study. B: Quantification of the Western blot analysis results shown in A. Expression is normalized to the GAPDH expression for each lane. These values were obtained from gels in which the WT and R69C samples were run in parallel on the same gels and immunoblotted on the same membranes. Data are expressed as means ± SEM. ∗P < 0.05. ActRIIB, ActRIIB-mFC; G, gastrocnemius; Q, quadriceps; R69C, Mtm1 p.R69C; VEH, vehicle.
We similarly evaluated Akt signaling intermediates in gastrocnemius and quadriceps muscles from the ActRIIB-responsive Mtm1δ4 mice (Supplemental Figure S1). Frozen muscles from Mtm1δ4 and WT mice at 6 WOL (the strain and time point used in our prior published trial) showed variable p-rpS6 levels within and between animals. Although the same trends were apparent in the muscles from the two different mutant strains, the muscle type-specific differences in p-rpS6 and p-eEF2K levels in Mtm1δ4 muscles were not statistically significant (Supplemental Figure S1). In side-by-side comparisons (not shown), the amount of p-rpS6 in WT and Mtm1δ4 animals was considerably higher than that seen in the Mtm1 p.R69C gastrocnemius muscle, suggesting that the low levels of p-rpS6 in this muscle represent the true signaling abnormality.
Because rpS6 can alternatively be phosphorylated through the ERK pathway via RSK/P90, the levels of total and p-ERK and RSK/P90 were also evaluated (Figure 7). No significant differences were found in the baseline levels of upstream kinases of the Akt or ERK pathways which would explain the low levels of p-rpS6 observed in the Mtm1 p.R69C gastrocnemius muscle (Figures 5, 6, and 7).
Figure 7.

ERK pathway activation in VEH- and ActRIIB-treated mice. A: Western blot analysis shows the relative expression of total ERK, p-ERK, total RSK/P90, and p-RSK/P90 in the quadriceps and gastrocnemius muscles of (VEH- and ActRIIB-treated WT and R69C mice at 6 MOL. GAPDH staining is used as a basis for loading and quantification studies. The numbers under each set of two lanes refer to individual mice from the study. B: Quantification of the Western blot results shown in A. Expression is normalized to the GAPDH expression for each lane. These values were obtained from gels in which the WT and R69C samples were run in parallel on the same gels and immunoblotted on the same membranes. Data are expressed as means ± SEM. ∗P < 0.05. ActRIIB, ActRIIB-mFC; G, gastrocnemius; Q, quadriceps; R69C, Mtm1 p.R69C; VEH, vehicle.
Vehicle-treated WT animals displayed no significant differences in hypertrophic pathway activation compared with the gastrocnemius and quadriceps muscles from the same animal (Figures 6 and 7). Treatment of WT mice with ActRIIB-mFc produced a significant increase in the total levels of Akt and P70/S6K, but no significant differences in the phosphorylation of these proteins.
Discussion
Because of the relationship between myofiber size and symptomatic severity in patients with XLMTM and in myotubularin-deficient mice, we had previously hypothesized that correction of myofiber smallness in myotubularin deficiency would greatly improve strength. Our initial study that used ActRIIB-mFc in the severely symptomatic Mtm1δ4 mouse model of XLMTM suggested that inhibition of myostatin and ActRIIB by using this drug produced generalized increases in animal mass, muscle mass, and type 2b myofiber size with slight increases in forelimb grip strength and survival.12 In the present study, we performed a similar trial on the less severely symptomatic Mtm1 p.R69C mouse model13 to determine the potential of this treatment strategy in less severely symptomatic patients with XLMTM. Surprisingly, we report here that ActRIIB-mFc is less effective as a treatment for Mtm1 p.R69C mice than for Mtm1δ4 mice and that treatment-induced hypertrophy was restricted to specific muscles within Mtm1 p.R69C animals. After evaluating several factors that may vary between individual muscles, we have found that the inability to respond to ActRIIB-mFc therapy was associated with reduced numbers of satellite cells in the muscle. In addition, we found abnormalities of hypertrophic signaling in the treatment-responsive Mtm1 p.R69C gastrocnemius muscle that were not present in the treatment-resistant Mtm1 p.R69C quadriceps muscle or in WT muscles, including low levels of p-rpS6 and high levels of p-eEF2k. Although the relationship between each of these factors and treatment efficacy remains unclear, this study identifies several potentially useful therapeutic biomarkers for this strategy and highlights the importance of evaluating multiple muscles and myofiber types when assessing treatment efficacy, especially when whole-animal effects are not apparent.
A comparison of our two trials that used ActRIIB-mFc in myotubularin-deficient mice highlights several key differences between treatment responses in Mtm1δ4 and Mtm1 p.R69C mice. Mtm1δ4 and Mtm1 p.R69C show considerable differences in phenotypic severity, which is thought to be due to the total absence of myotubularin in Mtm1δ4 mice in comparison with a small amount of functional mutant myotubularin in Mtm1 p.R69C mice.13 In Mtm1δ4 mice, ActRIIB-mFc treatment produced significant increases in total body mass, which corresponded to increases in the mass of all major muscles measured (quadriceps, gastrocnemius, and triceps).12 Histologically, this corresponded to marked hypertrophy of a subset of fibers, and this hypertrophy was restricted to a large subpopulation of the glycolytic (type 2b) myofiber population. In the present trial, these patterns of hypertrophy (including increased muscle size and hypertrophy restricted to the type 2b fiber population) were virtually identical in the gastrocnemius muscles of Mtm1 p.R69C mice but were absent in the other major muscles evaluated histologically (quadriceps and triceps). Because no apparent morphological differences were found between the gastrocnemius, quadriceps, and triceps muscles in untreated animals, we deduced that some factor must affect treatment efficacy that could vary between individual muscles of the same animal. We chose to evaluate satellite cell expression and hypertrophic signaling in the quadriceps and gastrocnemius muscles because we would least expect variation between individual muscles of the hind limb.
The restriction of treatment responsiveness to type 2b myofibers in our prior study that used Mtm1δ4 mice suggested that the Akt pathway may be involved in the effectiveness of ActRIIB-mFc in myotubularin-deficient mice, so we evaluated the activation of this pathway in the context of our present results. Recent studies have shown that siRNA-mediated silencing of myotubularin results in inhibition of Akt phosphorylation in HeLa cells and primary human skeletal muscle myotubes.22 By contrast, Akt and phospho-Akt levels are reported to be both increased (such that the phospho-Akt/Akt ratio is unaffected) in myotubularin-deficient mice.23 In our study, the levels of p-Akt were similar in WT mice compared with Mtm1 p.R69C mice (Figure 6), and evaluations of tissue from 6-week-old Mtm1δ4 mice and their WT littermates also showed similar or increased levels of p-Akt in Mtm1δ4 muscle (Supplemental Figure S1). These data indicate that the ability to p-Akt is intact in these two myotubularin-deficient mouse models.
Although we were able to document the activation of elements of the Akt pathway in treatment-responsive muscles, it was surprising that the pattern of activation was different in WT mice compared with Mtm1 p.R69C mice. Treated WT mice, but not their Mtm1 p.R69C littermates, displayed significant increases in total Akt, p-Akt at Thr308, and total P70/S6K in response to treatment with ActRIIB-mFc. Prior work with myostatin inhibition to promote myofiber hypertrophy has reported the involvement of the Akt pathway in myofiber hypertrophy induced by myostatin inhibition in myotubes24 and various mouse models.25–27 However, because the response to ActRIIB/myostatin inhibition is not affected in Akt-deficient mice,28 Akt itself is not an absolute requirement for hypertrophy in this setting.
The key abnormalities of hypertrophic signaling found in Mtm1 p.R69C mice were low levels of p-rpS6 and high levels of p-eEF2K in the treatment-responsive gastrocnemius muscle in comparison with the treatment-resistant quadriceps muscle (Figure 6). The amount of p-rpS6 in the Mtm1 p.R69C gastrocnemius muscle is also lower than that seen in age-matched WT muscles and muscles collected from Mtm1δ4 and WT mice at 6 WOL (Supplemental Figure S1). Evidence suggests that activation of S6K through the Akt pathway activates two opposing systems related to global protein synthesis (reviewed in Meyuhas17). Phosphorylation of EF2K and/or eukaryotic initiation factor 4B (eIF4B) produce increases in global protein synthesis, whereas phosphorylation of rpS6 produces a decrease in global protein phosphorylation,29 and together these molecules are thought to fine-tune protein synthesis after Akt pathway activation. The relationship of rpS6 phosphorylation and cell size has primarily been elucidated in the context of the lack of rpS6 phosphorylation, whereby the inability to phosphorylate rpS6 has been associated with decreased cell size (in a variety of cell types, including muscle) and decreased muscle strength.30 For treatment responsiveness in Mtm1 p.R69C mice, we did not identify signaling abnormalities that explained the limitation of treatment efficacy in most muscles, but it is possible that the low levels of p-rpS6 and the capacity to increase p-rpS6 with ActRIIB-mFc therapy would explain why the treatment was more effective in the gastrocnemius muscle. The relationships between eIF4B and EF2 and treatment efficacy in our study are unclear. eIF4B was not detectable with the use of Western blot assays and p-EF2, whereas showing higher levels in the Mtm1 p.R69C gastrocnemius muscle than the quadriceps muscle (Figure 6) was not seen at higher levels in treated animals.
Although we found that muscles that experienced hypertrophy in response to ActRIIB-mFc treatment had higher satellite cell numbers than were seen in vehicle-treated muscles, the relationships among myostatin inhibition, satellite cell number, and hypertrophy remain unclear. Several recent studies have established that satellite cells are not necessary for hypertrophy in the context of myostatin inhibition,31,32 although other studies have implicated satellite cell proliferation as an element of hypertrophy after myostatin inhibition.33 There is, however, more agreement with the idea that myostatin can affect satellite cell biology, and myostatin inhibition has been shown to increase satellite cell proliferation in the context of dexamethasone treatment,34 muscle regeneration in diabetic mice,35 and satellite cell number in chronic kidney disease.36 In the context of our present findings, we observed a relationship between treatment responsiveness and satellite cell number, but whether the satellite cells directly contributed to the hypertrophic response is less clear. Given that the treatment window here extended from 2 WOL (when approximately 10% of fibers are associated with satellite cells) to 6 MOL (when the number of fibers associated with satellite cells has generally decreased to approximately 5%), it is not clear whether ActRIIB-mFc treatment produced satellite cell proliferation or prevented the decrease in satellite cell numbers that is normally observed with maturation. Significantly, we observed a similar depletion of satellite cells in Mtm1 p.R69C muscles and Mtm1δ4 muscle,8 and this depletion occurred to an equivalent extent when comparing the vehicle-treated Mtm1 p.R69C quadriceps and gastrocnemius muscles at 6 MOL. These data suggest that the differential efficacy of ActRIIB-mFc treatment was not a consequence of satellite cell number differences between these muscles in the nontreated state. However, the data do suggest that the microenvironmental factors that are permissive or restrictive of hypertrophy in the Mtm1 p.R69C quadriceps muscle can also exert effects on satellite cell proliferation. As such, quantification of Pax7+ satellite cells may be a useful biomarker of treatment efficacy or processes interfering with treatment efficacy in future studies that involve myostatin inhibition.
Overall, this study establishes that the treatment responsiveness of myotubularin-deficient mice cannot be predicted on the basis of symptomatic severity and that the response to ActRIIB-mFc treatment varies considerably between individual muscles from Mtm1 p.R69C mice. Our findings indicate that ActRIIB-mFc has limited usefulness as a sole therapeutic agent in the treatment of myotubularin deficiency, although it may be useful in combination with gene3 and enzyme replacement37 therapies for XLMTM that are currently under development. The muscle-specific pattern of hypertrophy observed here also provides a cautionary lesson about the evaluation of treatment efficacy at the whole-animal level. Clearly, the commonly accepted hypertrophic pathways are more complex than currently appreciated, because the interactions between level of myotubularin deficiency, muscle type, and fiber type lead to unpredictable outcomes. Although the mechanism(s) responsible for the limitation of treatment efficacy in Mtm1 p.R69C mice remain unclear, the associations described here among myofiber hypertrophy, p-rpS6, p-eEF2K, and satellite cell numbers provide useful biomarkers for the evaluation of treatment efficacy in myotubularin deficiency or when inhibiting myostatin/ActRIIB signaling in other disease states.
Acknowledgments
We thank Drs. Vandana Gupta, Pankaj Agrawal, and Behzad Moghadaszadeh for helpful discussions and scientific advice and Tara C. Smith for her technical assistance on protein isolation and Western blot studies.
Footnotes
Supported by NIH/National Institute of Arthritis and Musculoskeletal and Skin Diseases grants R01 AR044345 (A.H.B.) and K08 AR059750-01 (M.W.L.), and Loan Repayment ProgramL40 AR057721-01 (M.W.L.), Muscular Dystrophy Association grant MDA201302 (A.H.B.), the Where There's a Will There's a Cure Foundation (A.H.B.), and the Joshua Frase Foundation (A.H.B.).
Disclosures: J.L.L. is a former employee of Acceleron Pharma, Inc., S.P. is a current employee of Acceleron Pharma, Inc., and both have equity in Acceleron Pharma, Inc., which manufactures the soluble activin type IIB receptor (ActRIIB)-mFC used in this study.
Supplemental Data
Akt pathway activation in untreated WT and Mtm1δ4 mice at 1.5 MOL. A: Western blot analysis shows the relative expression of Akt, p-Akt (at Ser473), rpS6, p-rpS6 (at Ser240/244), and p-eEF2K (at Ser366) in the quadriceps and gastrocnemius muscles of WT and Mtm1δ4 mice at 1.5 MOL. GAPDH staining is used as a basis for loading and quantification studies. The numbers under each set of two lanes refer to individual mice from the study. B: Quantification of the Western blot results shown in A. Expression is normalized to the GAPDH expression for each lane. The values were obtained from the exposure shown in A, and the results were repeated three times. Data are expressed as means ± SD. G, gastrocnemius; Q, quadriceps.
References
- 1.Heckmatt J.Z., Sewry C.A., Hodes D., Dubowitz V. Congenital centronuclear (myotubular) myopathy. A clinical, pathological and genetic study in eight children. Brain. 1985;108:941–964. doi: 10.1093/brain/108.4.941. [DOI] [PubMed] [Google Scholar]
- 2.Jungbluth H., Wallgren-Pettersson C., Laporte J. Centronuclear (myotubular) myopathy. Orphanet J Rare Dis. 2008;3:26. doi: 10.1186/1750-1172-3-26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Buj-Bello A., Fougerousse F., Schwab Y., Messaddeq N., Spehner D., Pierson C.R., Durand M., Kretz C., Danos O., Douar A.M., Beggs A.H., Schultz P., Montus M., Denefle P., Mandel J.L. AAV-mediated intramuscular delivery of myotubularin corrects the myotubular myopathy phenotype in targeted murine muscle and suggests a function in plasma membrane homeostasis. Hum Mol Genet. 2008;17:2132–2143. doi: 10.1093/hmg/ddn112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Cao C., Backer J.M., Laporte J., Bedrick E.J., Wandinger-Ness A. Sequential actions of myotubularin lipid phosphatases regulate endosomal PI(3)P and growth factor receptor trafficking. Mol Biol Cell. 2008;19:3334–3346. doi: 10.1091/mbc.E08-04-0367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Dowling J.J., Vreede A.P., Low S.E., Gibbs E.M., Kuwada J.Y., Bonnemann C.G., Feldman E.L. Loss of myotubularin function results in T-tubule disorganization in zebrafish and human myotubular myopathy. PLoS Genet. 2009;5:e1000372. doi: 10.1371/journal.pgen.1000372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Laporte J., Hu L.J., Kretz C., Mandel J.L., Kioschis P., Coy J.F., Klauck S.M., Poustka A., Dahl N. A gene mutated in X-linked myotubular myopathy defines a new putative tyrosine phosphatase family conserved in yeast. Nat Genet. 1996;13:175–182. doi: 10.1038/ng0696-175. [DOI] [PubMed] [Google Scholar]
- 7.Hnia K., Tronchere H., Tomczak K.K., Amoasii L., Schultz P., Beggs A.H., Payrastre B., Mandel J.L., Laporte J. Myotubularin controls desmin intermediate filament architecture and mitochondrial dynamics in human and mouse skeletal muscle. J Clin Invest. 2011;121:70–85. doi: 10.1172/JCI44021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Lawlor M.W., Alexander M.S., Viola M.G., Meng H., Joubert R., Gupta V., Motohashi N., Manfready R.A., Hsu C.P., Huang P., Buj-Bello A., Kunkel L.M., Beggs A.H., Gussoni E. Myotubularin-deficient myoblasts display increased apoptosis, delayed proliferation, and poor cell engraftment. Am J Pathol. 2012;181:961–968. doi: 10.1016/j.ajpath.2012.05.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Pierson C.R., Tomczak K., Agrawal P., Moghadaszadeh B., Beggs A.H. X-linked myotubular and centronuclear myopathies. J Neuropathol Exp Neurol. 2005;64:555–564. doi: 10.1097/01.jnen.0000171653.17213.2e. [DOI] [PubMed] [Google Scholar]
- 10.Pierson C.R., Agrawal P.B., Blasko J., Beggs A.H. Myofiber size correlates with MTM1 mutation type and outcome in X-linked myotubular myopathy. Neuromuscul Disord. 2007;17:562–568. doi: 10.1016/j.nmd.2007.03.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Al-Qusairi L., Weiss N., Toussaint A., Berbey C., Messaddeq N., Kretz C., Sanoudou D., Beggs A.H., Allard B., Mandel J.L., Laporte J., Jacquemond V., Buj-Bello A. T-tubule disorganization and defective excitation-contraction coupling in muscle fibers lacking myotubularin lipid phosphatase. Proc Natl Acad Sci U S A. 2009;106:18763–18768. doi: 10.1073/pnas.0900705106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Lawlor M.W., Read B.P., Edelstein R., Yang N., Pierson C.R., Stein M.J., Wermer-Colan A., Buj-Bello A., Lachey J.L., Seehra J.S., Beggs A.H. Inhibition of activin receptor type IIb increases strength and lifespan in myotubularin-deficient mice. Am J Pathol. 2011;178:784–793. doi: 10.1016/j.ajpath.2010.10.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Pierson C.R., Dulin-Smith A.N., Durban A.N., Marshall M.L., Marshall J.T., Snyder A.D., Naiyer N., Gladman J.T., Chandler D.S., Lawlor M.W., Buj-Bello A., Dowling J.J., Beggs A.H. Modeling the human MTM1 p.R69C mutation in murine Mtm1 results in exon 4 skipping and a less severe myotubular myopathy phenotype. Hum Mol Genet. 2012;21:811–825. doi: 10.1093/hmg/ddr512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.McCroskery S., Thomas M., Maxwell L., Sharma M., Kambadur R. Myostatin negatively regulates satellite cell activation and self-renewal. J Cell Biol. 2003;162:1135–1147. doi: 10.1083/jcb.200207056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Joulia-Ekaza D., Cabello G. Myostatin regulation of muscle development: molecular basis, natural mutations, physiopathological aspects. Exp Cell Res. 2006;312:2401–2414. doi: 10.1016/j.yexcr.2006.04.012. [DOI] [PubMed] [Google Scholar]
- 16.Cadena S.M., Tomkinson K.N., Monnell T.E., Spaits M.S., Kumar R., Underwood K.W., Pearsall R.S., Lachey J.L. Administration of a soluble activin type IIB receptor promotes skeletal muscle growth independent of fiber type. J Appl Physiol. 2010;109:635–642. doi: 10.1152/japplphysiol.00866.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Meyuhas O. Physiological roles of ribosomal protein S6: one of its kind. Int Rev Cell Mol Biol. 2008;268:1–37. doi: 10.1016/S1937-6448(08)00801-0. [DOI] [PubMed] [Google Scholar]
- 18.Buj-Bello A., Laugel V., Messaddeq N., Zahreddine H., Laporte J., Pellissier J.F., Mandel J.L. The lipid phosphatase myotubularin is essential for skeletal muscle maintenance but not for myogenesis in mice. Proc Natl Acad Sci U S A. 2002;99:15060–15065. doi: 10.1073/pnas.212498399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Brooke M.H., Engel W.K. The histographic analysis of human muscle biopsies with regard to fiber types. 4. Children's biopsies. Neurology. 1969;19:591–605. doi: 10.1212/wnl.19.6.591. [DOI] [PubMed] [Google Scholar]
- 20.Wattanasirichaigoon D., Swoboda K.J., Takada F., Tong H.Q., Lip V., Iannaccone S.T., Wallgren-Pettersson C., Laing N.G., Beggs A.H. Mutations of the slow muscle alpha-tropomyosin gene, TPM3, are a rare cause of nemaline myopathy. Neurology. 2002;59:613–617. doi: 10.1212/wnl.59.4.613. [DOI] [PubMed] [Google Scholar]
- 21.Han H.Q., Zhou X., Mitch W.E., Goldberg A.L. Myostatin/activin pathway antagonism: molecular basis and therapeutic potential. Int J Biochem Cell Biol. 2013;45:2333–2347. doi: 10.1016/j.biocel.2013.05.019. [DOI] [PubMed] [Google Scholar]
- 22.Razidlo G.L., Katafiasz D., Taylor G.S. Myotubularin regulates Akt-dependent survival signaling via phosphatidylinositol 3-phosphate. J Biol Chem. 2011;286:20005–20019. doi: 10.1074/jbc.M110.197749. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Al-Qusairi L., Prokic I., Amoasii L., Kretz C., Messaddeq N., Mandel J.L., Laporte J. Lack of myotubularin (MTM1) leads to muscle hypotrophy through unbalanced regulation of the autophagy and ubiquitin-proteasome pathways. FASEB J. 2013;27:3384–3394. doi: 10.1096/fj.12-220947. [DOI] [PubMed] [Google Scholar]
- 24.Morissette M.R., Cook S.A., Buranasombati C., Rosenberg M.A., Rosenzweig A. Myostatin inhibits IGF-I-induced myotube hypertrophy through Akt. Am J Physiol Cell Physiol. 2009;297:C1124–C1132. doi: 10.1152/ajpcell.00043.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Hulmi J.J., Oliveira B.M., Silvennoinen M., Hoogaars W.M., Ma H., Pierre P., Pasternack A., Kainulainen H., Ritvos O. Muscle protein synthesis, mTORC1/MAPK/Hippo signaling, and capillary density are altered by blocking of myostatin and activins. Am J Physiol Endocrinol Metab. 2013;304:E41–E50. doi: 10.1152/ajpendo.00389.2012. [DOI] [PubMed] [Google Scholar]
- 26.Kalista S., Schakman O., Gilson H., Lause P., Demeulder B., Bertrand L., Pende M., Thissen J.P. The type 1 insulin-like growth factor receptor (IGF-IR) pathway is mandatory for the follistatin-induced skeletal muscle hypertrophy. Endocrinology. 2012;153:241–253. doi: 10.1210/en.2011-1687. [DOI] [PubMed] [Google Scholar]
- 27.Welle S., Burgess K., Mehta S. Stimulation of skeletal muscle myofibrillar protein synthesis, p70 S6 kinase phosphorylation, and ribosomal protein S6 phosphorylation by inhibition of myostatin in mature mice. Am J Physiol Endocrinol Metab. 2009;296:E567–E572. doi: 10.1152/ajpendo.90862.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Goncalves M.D., Pistilli E.E., Balduzzi A., Birnbaum M.J., Lachey J., Khurana T.S., Ahima R.S. Akt deficiency attenuates muscle size and function but not the response to ActRIIB inhibition. PLoS One. 2010;5:e12707. doi: 10.1371/journal.pone.0012707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Ruvinsky I., Sharon N., Lerer T., Cohen H., Stolovich-Rain M., Nir T., Dor Y., Zisman P., Meyuhas O. Ribosomal protein S6 phosphorylation is a determinant of cell size and glucose homeostasis. Genes Dev. 2005;19:2199–2211. doi: 10.1101/gad.351605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Ruvinsky I., Katz M., Dreazen A., Gielchinsky Y., Saada A., Freedman N., Mishani E., Zimmerman G., Kasir J., Meyuhas O. Mice deficient in ribosomal protein S6 phosphorylation suffer from muscle weakness that reflects a growth defect and energy deficit. PLoS One. 2009;4:e5618. doi: 10.1371/journal.pone.0005618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Lee S.J., Huynh T.V., Lee Y.S., Sebald S.M., Wilcox-Adelman S.A., Iwamori N., Lepper C., Matzuk M.M., Fan C.M. Role of satellite cells versus myofibers in muscle hypertrophy induced by inhibition of the myostatin/activin signaling pathway. Proc Natl Acad Sci U S A. 2012;109:E2353–E2360. doi: 10.1073/pnas.1206410109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Wang Q., McPherron A.C. Myostatin inhibition induces muscle fibre hypertrophy prior to satellite cell activation. J Physiol. 2012;590:2151–2165. doi: 10.1113/jphysiol.2011.226001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Gilson H., Schakman O., Kalista S., Lause P., Tsuchida K., Thissen J.P. Follistatin induces muscle hypertrophy through satellite cell proliferation and inhibition of both myostatin and activin. Am J Physiol Endocrinol Metab. 2009;297:E157–E164. doi: 10.1152/ajpendo.00193.2009. [DOI] [PubMed] [Google Scholar]
- 34.Dong Y., Pan J.S., Zhang L. Myostatin suppression of Akirin1 mediates glucocorticoid-induced satellite cell dysfunction. PLoS One. 2013;8:e58554. doi: 10.1371/journal.pone.0058554. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Jeong J., Conboy M.J., Conboy I.M. Pharmacological inhibition of myostatin/TGF-beta receptor/pSmad3 signaling rescues muscle regenerative responses in mouse model of type 1 diabetes. Acta Pharmacol Sin. 2013;34:1052–1060. doi: 10.1038/aps.2013.67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Zhang L., Rajan V., Lin E., Hu Z., Han H.Q., Zhou X., Song Y., Min H., Wang X., Du J., Mitch W.E. Pharmacological inhibition of myostatin suppresses systemic inflammation and muscle atrophy in mice with chronic kidney disease. FASEB J. 2011;25:1653–1663. doi: 10.1096/fj.10-176917. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Lawlor M.W., Armstrong D., Viola M.G., Widrick J.J., Meng H., Grange R.W., Childers M.K., Hsu C.P., O'Callaghan M., Pierson C.R., Buj-Bello A., Beggs A.H. Enzyme replacement therapy rescues weakness and improves muscle pathology in mice with X-linked myotubular myopathy. Hum Mol Genet. 2013;22:1525–1538. doi: 10.1093/hmg/ddt003. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Akt pathway activation in untreated WT and Mtm1δ4 mice at 1.5 MOL. A: Western blot analysis shows the relative expression of Akt, p-Akt (at Ser473), rpS6, p-rpS6 (at Ser240/244), and p-eEF2K (at Ser366) in the quadriceps and gastrocnemius muscles of WT and Mtm1δ4 mice at 1.5 MOL. GAPDH staining is used as a basis for loading and quantification studies. The numbers under each set of two lanes refer to individual mice from the study. B: Quantification of the Western blot results shown in A. Expression is normalized to the GAPDH expression for each lane. The values were obtained from the exposure shown in A, and the results were repeated three times. Data are expressed as means ± SD. G, gastrocnemius; Q, quadriceps.