

Abstract
TP53, one of the most important oncosuppressors, is frequently mutated in cancer. Several p53 mutant proteins escape proteolytic degradation and are highly expressed in an aberrant conformation often acquiring pro-oncogenic activities that promote tumor progression and resistance to therapy. Therefore, it has been vastly proposed that reactivation of wild-type (wt) function(s) from mutant p53 (mutp53) may have therapeutic significance. We have previously reported that Zn(II) restores a folded conformation from mutp53 misfolding, rescuing wild-type (wt) p53/DNA-binding and transcription activities. However, whether Zn(II) affects mutp53 stability has never been investigated. Here we show that a novel Zn(II) compound induced mutp53 (R175H) protein degradation through autophagy, the proteolytic machinery specifically devoted to clearing misfolded proteins. Accordingly, pharmacological or genetic inhibition of autophagy prevented Zn(II)-mediated mutp53H175 degradation as well as the ability of the Zn(II) compound to restore wtp53 DNA-binding and transcription activity from this mutant. By contrast, inhibition of the proteasome failed to do so, suggesting that autophagy is the main route for p53H175 degradation. Mechanistically, Zn(II) restored the wtp53 ability to induce the expression of the p53 target gene DRAM (damage-regulated autophagy modulator), a key regulator of autophagy, leading to autophagic induction. Accordingly, inhibition of wtp53 transactivation by pifithrin-α (PFT-α) impaired both autophagy and mutp53H175 degradation induced by curcumin-based zinc compound (Zn(II)-curc). Viewed together, our results uncover a novel mechanism employed by Zn(II)-curc to reactivate mutp53H175, which involves, at least in part, induction of mutp53 degradation via wtp53-mediated autophagy.
Keywords: p53, autophagy, cloroquine, LC3-II, cancer, Zn(II), DRAM
The tumor-suppressor p53 is a transcription factor that is activated in response to a variety of stress signals.1 P53 tumor-suppressor functions mostly depend on transcriptional activation of target genes involved in cell-cycle arrest, senescence and apoptosis.2 For those reasons, TP53 is the most frequent target for genetic alterations in tumors, being mutated in over 50% of human cancer types and indirectly inactivated in many others. These findings indicate that the presence of a functional p53 pathway is incompatible with neoplastic cell growth.3
The majority of TP53 alterations consists of missense mutations within the DNA-binding domain (DBD) that lead to the synthesis of p53 proteins that have lost the ability to bind to sequence-specific DNA recognition elements.2, 4 Moreover, p53 mutant (mutp53) proteins may acquire a misfolded and partially denatured conformation5 conferring to the proteins high tendency to form micro- and macro-aggregates.3 The result is accumulation of hyperstable mutp53 proteins in tumors. The primary effect of these mutations is the impairment of p53 to activate the typical gene target of wild-type (wt) p53 (that is, Puma, Bax, Noxa, p21 and so on).2, 4 The main outcome of p53 mutations is therefore loss of tumor-suppressor functions. However, the extremely high expression levels of mutp53 in tumors has suggested that these proteins may acquire novel oncogenic functions (gain of function – GOF) that actively contribute to cancer development and progression.6, 7 Misfolded p53 protein aggregates have been implicated in the acquisition of GOF functions because mutp53 can sequester in these aggregates various tumor suppressors including p53 itself, and the family members p63 and p73;8 moreover, mutp53 may contribute to the transcription of MDR1 (multidrug resistant gene)9 and cyclin B2 genes,10 involved in drug resistance and cell proliferation. Consistent with this notion, mice harboring knock-in p53 mutations of the type found in human tumors display the appearance of carcinomas. Stabilization of mutp53 in these animals promotes more aggressive and metastatic tumors that are rarely detected in p53-null mice.11, 12, 13, 14 Therefore, targeting mutp53 for protein degradation, other than mutp53 reactivation by small compounds, is another strategy that may have anticancer activity.15, 16
Abnormally folded proteins are usually targeted for degradation by autophagy. Autophagy is a proteolytic process that is activated during various conditions of cellular stress, including nutrient deprivation or DNA damage to eliminate unfolded proteins or damaged organelles to survive bioenergetic stress and/or induce cell death.17 Autophagy allows cells to sequester cytoplasmic content through the formation of double-membrane vescicles (autophagosomes) and target them for degradation through the fusion of autophagosomes with lysosomes.18 A recent study showed that glucose starvation induces mutp53 protein degradation through autophagy, which renders tumor cells hypersensitive to autophagic cell death.19 This is an interesting finding because whereas the role of the proteasome- and ubiquitin-dependent degradation of wtp53 in unstressed condition, through the E3-ubiquitin ligase MDM2, is well known,20 the molecular mechanisms regulating mutp53 stability/degradation are still elusive.21
Our previous studies showed that zinc supplementation reactivates misfolded/mutant p53 into an active form, competent to DNA-binding and transcriptional activation of wt target genes.22, 23, 24, 25, 26, 27 However, whether zinc supplementation might also affect mutp53 protein levels has never been addressed. Here, we asked whether a novel curcumin-based zinc compound (Zn(II)-curc),28, 29 able to reactivate mutp53 proteins,27 could modify mutp53 stability. We specifically analyzed conformation defective R175H p53 mutant (p53H175) because it is one of the most frequently and vastly studied p53 mutant, which not only exhibits dominant-negative activities over wtp53 but also possesses GOF properties.30 We found that Zn(II)-curc triggered p53H175 protein downregulation. The mechanism of p53H175 downregulation was at protein level, as p53 RNA levels were not affected. Degradation of mutp53H175 protein correlated with autophagy induction that was, in part, dependent on wtp53 reactivation. Thus, inhibition of wtp53 transactivation by pifithrin-α (PFT-α) impaired Zn(II)-curc-induced mutp53 degradation. Furthermore and importantly, blocking of autophagy with pharmacological or genetic inhibitors impaired Zn(II)-curc-induced mutp53 degradation, whereas blocking the proteasome failed to do so. Inhibition of autophagy impaired Zn(II)-induced reactivation of wtp53 functions, including binding to target promoters and transactivation of target genes, rather enhancing the dominant-negative effects of p53H175. Finally, impairment of mutp53 reactivation by autophagy inhibitors correlated with reduction of cancer cell death induced by Zn(II)-curc. Altogether, these data suggest that autophagy has a role in a fully efficient mutant p53H175 reactivation by Zn(II)-curc.
Results
Zn(II)-curc triggers mutp53H175 protein downregulation
To address whether Zn(II)-curc affected mutp53 stability we analyzed subconfluent proliferating SKBR3 cells (carrying R175H p53 gene mutation) compared with HCT116 cells carrying wtp53. As shown in Figure 1a, western immunoblotting revealed a marked decrease in endogenous mutp53H175 protein levels starting 8 h after Zn(II)-curc treatment. On the contrary, Zn(II)-curc was not able to decrease wtp53 levels in HCT116 cells that rather increased during time (Figure 1a), suggesting that Zn(II)-curc may specifically act on mutant p53 reduction. To rule-out cell type-specific effects, we performed similar experiments in H1299 lung cancer cells (p53-null) where we ectopically expressed p53H175 plasmid. In agreement with the results in SKBR3 cells, Zn(II)-curc consistently reduced exogenous mutp53H175 levels (Figure 1b). Of note, the p53 mRNA levels were not modified by Zn(II)-curc (Figure 1c), underlining the hypothesis that Zn(II)-curc was modifying mutp53H175 protein stability. This effect was further analyzed in the presence of the translation inhibitor cycloheximide (CHX). The results show that mutp53H175 protein underwent faster downregulation after Zn(II)-curc treatment, compared with mutp53H175 protein levels in untreated cells (Figure 1d, compare lanes 1 and 2 with lanes 5 and 6, upper panel). The effect was already evident after 3 h, when the mutp53175 levels dropped drastically in the Zn(II)-curc sample, compared to the control, as measured using densitometric analysis (Figure 1d, lower panel). These data strongly suggest that Zn(II)-curc induces mutp53H175 degradation.
Figure 1.

Zn(II)-curc compound triggers mutp53H175 protein degradation. (a) Stability of endogenous p53H175 protein in SKBR3 and HCT116 cells treated with Zn(II)-curc (100 μM) for 4–8–16 h. Protein levels were measured with western blot using antibody to p53 (FL393). A shorter and longer exposure of p53 blot is showing. Anti-β-actin was used as protein loading control. (b) H1299 cells were transiently transfected with p53H175 plasmid (0.1 μg) and 24 h after transfection treated with Zn(II)-curc (100 μM) for 8–16–24 h to monitor p53 stability. Protein levels were measured with western blot using antibody to p53. Anti-β-actin was used as protein loading control. (c) SKBR3 cells were left untreated or treated with Zn(II)-curc (100 μM) for 16 and 24 h before analysis of p53 expression using RT-PCR. β-actin was used as a control for efficiency of RNA extraction and transcription. (d) H1299 cells were transiently transfected with p53H175 plasmid (0.1 μg). Twenty-four hours after transfection cells were treated with Zn(II)-curc (100 μM) for 8 h. Untreated or Zn(II)-curc-treated cells were then incubated with the translation inhibitor CHX (40 μM) for the indicated hours to assess p53 protein levels using western immunoblot as in (a and b). Lower panel: p53 bands were quantitated using densitometry and plotted as relative amount of p53 levels after ZN(II)-curc treatment compared with control
Reactivation of wtp53 transactivation by Zn(II)-curc is involved in mutp53H175 degradation
We previously demonstrated that Zn(II)-curc reactivates p53H175 transactivation that may be blocked by the concomitant use of PFT-α,27 an inhibitor of wtp53 transcriptional activity.31 Here, we found that PFT-α impaired the Zn(II)-curc-induced mutp53H175 degradation (Figure 2a, compare lane 3 with lane 2) re-establishing the mutp53 levels to those of SKBR3-untreated cells (Figure 2a, lane 4 with lane 1); PFT-α alone did not modify mutp53 levels (Figure 2a), strengthening the hypothesis that p53 transactivation was having a role into its one protein degradation. We then tested whether MDM2 had a role in such mutp53H175 degradation. To this end, we used small-interfering RNA (siRNA) interference to deplete MDM2 in SKBR3 cells (Figure 2b, left panel). We found that mutp53H175 protein still underwent degradation after Zn(II)-curc treatment in cells depleted of MDM2 function (Figure 2b, right panel). These results suggest that reactivation of wtp53 transactivation by Zn(II)-curc is involved in mutp53H175 degradation. However, they also suggest that a different mechanism rather than the proteasome pathway is involved in such degradation.
Figure 2.

Reactivation of wtp53 transactivation by Zn(II)-curc is mechanistically involved in mutp53H175 degradation. (a) Stability of endogenous p53H175 protein in SKBR3 cells treated with Zn(II)-curc (100 μM) for 24 h with or without p53 inhibitor PFT-α (30 μM). Protein levels were measured with western blot using antibody to p53. Anti-β-actin was used as protein loading control. (b) SKBR3 cells were depleted of MDM2 function by siRNA interference, as monitored using RT-PCR analysis (left panel) and 36 h after transfection treated with Zn(II)-curc (100 μM) for 16 h or left untreated. p53 protein levels (right panel) were measured using western blot as in a. Anti-β-actin was used as protein loading control
The Zn(II)-curc-induced mutp53H175 degradation depends on autophagy
Having observed that MDM2 was not responsible of mutp53H175 degradation in our setting, we then explored the role of autophagy, a cellular mechanism of unfolded protein degradation within lysosomes.17 To this aim, we first assessed the expression of microtubule-associated protein light chain 3 (LC3) after conversion from LC3-I form to its autophagosome membrane-associated lipidated, activated LC3-II form, and the expression levels of p62, a bona fide autophagic substrate.32 As shown in Figure 3a, parallel to mutp53H175 degradation, Zn(II)-curc increased LC3-II expression and reduced p62 levels. Importantly, the use of chemical inhibitors of autophagic/lysosomal degradation – that is, 3-methyl-adenine (3-MA) and cloroquine (CQ)32 – prevented Zn(II)-curc-induced mutp53H175 degradation, and allowed the accumulation of slowly migrating forms in SDS-PAGE (Figure 3b). By contrast, the proteasome inhibitor MG132 failed to do so (Figure 3b), in agreement with the above-reported results with siRNA for MDM2 (Figure 2b). The presence of a double band for p53 that sometimes we get in western immunoblotting might be the result of post-translational modifications after treatments, although it needs further examination. Similar results were obtained in H1299 cells where we ectopically expressed p53R175H plasmid (Figure 3c). In stark contrast with the effect exerted on mutp53H175, inhibition of autophagy with either 3-MA or CQ in wt p53 carrying cells did not stabilize the wtp53 protein levels, which were instead markedly increased by MG132 treatment (Figure 3d), as expected.20 To further ascertain the role of autophagy in mutp53H175 degradation, we then knocked down using small RNA interference the essential autophagic gene ATG5, one of the members of the ATG family necessary for autophagy because of its role in autophagosome elongation.33 We found that ATG5 knockdown (si-ATG5) in SKBR3 cells completely rescued Zn(II)-curc-induced mutp53H175 degradation (Figure 3e, compare lane 4 with lane 2). Further, the modest, yet reproducible induction of ATG5 protein levels following Zn(II)-curc treatment (Figure 3e, compare lane 2 with lane 1), together with the LC3-II conversion and p62 degradation (Figure 3a), indicated that autophagy was induced during Zn(II)-curc treatment. These data suggest that wtp53 and mutp53 proteins undergo degradation through different mechanisms; thus, autophagy is likely the main pathway for mutp53H175 degradation following Zn(II)-curc administration.
Figure 3.

The Zn(II)-curc-induced mutp53H175 degradation depends on autophagy. (a) SKBR3 cells were treated with Zn(II)-curc (100 μM) for 16 and 24 h. Protein levels were measured with western blot using antibodies to LC3-II, p62 and p53. Anti-β-actin was used as protein loading control. (b) Subconfluent SKBR3 cells were treated with Zn(II)-curc (100 μM) for 8 h either alone or in co-treatment with CQ (25 μM), 3-MA (5 mM) or MG132 (20 μM). Protein levels were measured with western blot using antibody to p53. Anti-β-actin was used as protein loading control. (c) H1299 cells were transiently transfected with p53H175 plasmid (0.1 μg). Twenty-four hours after transfection cells were treated with Zn(II)-curc (100 μM) for 8 h either alone or in co-treatment with CQ (25 μM) or MG132 (20 μM). Protein levels were measured with western blot using antibodies to p53 and LC3-II. Anti-β-actin was used as protein loading control. (d) Subconfluent RKO cells, carrying wtp53, were treated with CQ (25 μM), 3-MA (5 mM) or MG132 (20 μM) for 6 h. Protein levels were measured with western blot using antibody to p53. Anti-β-actin was used as protein loading control. (e) SKBR3 cells were transfected with siRNA for ATG5 (si-ATG5) or with control siRNA (si-ctr). Thirty-six hours after transfection, cells were mock-untreated or treated with Zn(II)-curc (100 μM) for 16 h. Protein levels were measured with western blot using antibodies to p53 and ATG5. Anti-tubulin was used as protein loading control
Zn(II)-curc triggers p53-dependent autophagy
Various forms of p53 mutants have been found to block the extent of autophagic flux.19, 34 Other studies have shown that wtp53 may induce autophagy through different mechanisms including transcriptional activation of autophagy genes such as DRAM (damage-regulated autophagy modulator).35 Therefore, we asked whether, in our model of mutp53H175 reactivation, p53 was responsible of induction of autophagy. In line with the original findings that DRAM is induced by wtp53,35 we found clear DRAM expression in H1299 (p53 null) cells after wtp53 overexpression (Figure 4a) and in RKO (wtp53) cells after induction of DNA damage with cisplatin, whereas such induction was not evident in SKBR3 treated with cisplatin (Figure 4a). Then we tested in vivo p53 binding to DRAM promoter by employing chromatin immunoprecipitation (ChIP) assay. The results show that mutp53 was not bound to DRAM promoter in SKBR3 control cells; on the other hand we found strong enrichment of p53 binding to DRAM promoter after Zn(II)-curc; as a control of specific DRAM binding by p53 we found that p73 failed to bind the promoter both in untreated and Zn(II)-curc-treated cells (Figure 4b). Analyses of DRAM mRNA showed increased gene expression after Zn(II)-curc treatment in SKBR3 cells (Figure 4c); conversely, MDR1, target gene of mutp53, showed a significant decrease after Zn(II)-curc treatment (Figure 4c). Similarly, H1299 cells transfected with p53H175 plasmid showed increased DRAM expression and concomitant decrease in MDR1 expression after Zn(II)-curc treatment (Figure 4d), suggestive of an unbalance of folded/misfolded p53 protein activity in this setting. Importantly, inhibition of wtp53 transcriptional activity by PFT-α completely abolished DRAM expression induced by Zn(II)-curc while rescuing MDR1 expression (Figure 4e). These results suggest that reactivation of wtp53 transactivation by Zn(II)-curc was likely inducing autophagy. To further address this issue, we finally observed that the clear induction of LC3-II conversion following Zn(II)-curc administration was markedly impaired by PFT-α (Figure 4f). Similarly, LC3-II was strongly impaired by p53 depletion with siRNA (Figure 4g), as evidenced with densitometric analysis of western blots. The role of p53 transactivation was finally evaluated with p21 analysis using western blot that showed increased p21 levels after Zn(II)-curc treatment, which were abolished following p53 depletion (Figure 4h). Altogether, these results indicate that Zn(II)-curc triggers autophagy dependent on p53 transactivation.
Figure 4.

Zn(II)-curc triggered p53-dependent autophagy. (a) RKO and SKBR3 cells were treated with cisplatin (cispl, 5 μg/ml) for 16 h, while H1299 cells were transfected with wtp53 plasmid (0.1 μg). After treatment and transfection, cells were collected, RNA was extracted and DRAM mRNA was monitored using RT-PCR analysis. β-actin mRNA was used as internal control. (b) SKBR3 cells (6 × 106) were plated in 150-mm dish and the day after treated with Zn(II)-curc (100 μM) for 16 h before being assayed for ChIP with anti-p53 and anti-p73 antibodies. PCR analyses were performed on the immunoprecipitated DNA using primers specific for DRAM gene promoter. A sample representing linear amplification of the total chromatin (input) was included as control. Additional controls included immunoprecipitation performed with nonspecific immunoglobulins (no Ab). (c) SKBR3 cells were treated with Zn(II)-curc (100 μM) for 16 and 24 h with or without co-treatment with autophagy inhibitor CQ (25 μM) for 24 h before being assayed for RT-PCR of DRAM and MDR1. (d) H1299 cells were transiently transfected with p53H175 plasmid (0.1 μg). Twenty-four hours after transfection, cells were treated with Zn(II)-curc (100 μM) for 24 h before assayed for RT-PCR of DRAM and MDR1. β-actin was used as a control for efficiency of RNA extraction and transcription. (e) SKBR3 cells were treated with Zn(II)-curc (100 μM) 24 h with or without co-treatment with autophagy inhibitor CQ (25 μM) before being assayed for RT-PCR of DRAM and MDR1. (f) SKBR3 cells treated with Zn(II)-curc (100 μM) over a 24-h time course with or without p53 inhibitor PFT-α (30 μM). Protein levels were measured with western blot using antibodies to p53, LC3-II and p21 (h). Anti-β-actin was used as protein loading control. (g) SKBR3 cells were transfected with control or sip53 vector and 36 h after transfection treated with Zn(II)-curc (100 μM) for 16 and 24 h. Protein levels were analyses with western blot using antibodies to p53 and LC3-II. Anti-β-actin was used as protein loading control. P53 and LC3-II levels were measured using densitometric analysis of the autoradiographs and plotted as p53/β-actin and LC3/β-actin ratios, ±S.D.
Autophagy inhibition impairs p53H175 reactivation by Zn(II)-curc
It has been shown that p53H175 exerts a dominant-negative effect on wtp53, preventing wtp53 binding to target gene promoters.30 We previously demonstrated that Zn(II)-curc reactivates mutp53 binding to gene promoters target of wtp53.27 Therefore, we next asked whether the rescue of p53H175 protein levels by autophagy inhibition could impair those p53 functions reactivated by Zn(II)-curc. To this end, the in vivo p53/DNA binding was analyzed in SKBR3 cells with ChIP assay. The results show that mutp53 was not bound to a canonical wtp53 target gene promoter (that is, Puma) in SKBR3 control cells, while the binding was enriched after Zn(II)-curc treatment (Figure 5a); in an opposite way, the binding to gene promoters, target of mutp53 (specifically, MDR1 and cyclin B2),9, 10 was reverted after Zn(II)-curc treatment (Figure 5a), as previously reported.27 In agreement with our hypothesis, CQ co-treatment reverted the p53/DNA-binding activity induced by Zn(II), specifically reducing the p53 recruitment on Puma promoter while strongly increasing the p53 enrichment on MDR1 and cyclin B2 promoters (Figure 5a). In line with these results, luciferase assays performed in H1299 cells transiently transfected with the p53H175 plasmid show that the activity of the Noxa-luc promoter, induced after Zn(II)-curc administration, was inhibited by CQ co-treatment (Figure 5b). Finally, in vivo transcription of endogenous p53 target genes was examined by employing reverse transcription polymerase chain reaction (RT-PCR) analysis. The results show that the induction of wtp53 target genes (that is, Bax, Noxa and Puma) and downregulation of the mutp53 target gene (that is, MDR1), following Zn(II)-curc treatment, was efficiently blocked by the concomitant administration of autophagy inhibitor CQ, both in SKBR3 (Figure 5c) and H1299 cells expressing p53H175 (Figure 5d). These data underline an unbalance of folded–misfolded mutp53 proteins when, respectively, Zn(II)-curc alone or concomitantly to autophagy inhibitor CQ is administrated to mutp53-carrying cells. Such an imbalance reflects the predominant effect of reactivated wtp53 or of mutant p53 proteins, in this setting. Thus, our hypothesis is that zinc addition reactivates wtp53 function by acting on the folding of most (but likely not all) of the mutant p53 proteins within a cell, which then triggers the degradation of the remaining mutant p53 proteins by autophagy. Certainly, additional studies are needed to better understand the timing of such regulatory loop. Viewed as a whole, these data indicate that autophagy inhibition represents an obstacle to ability of Zn(II)-curc to fully reactivate p53R175H mutant.
Figure 5.

Autophagy inhibition impairs p53H175 reactivation by Zn(II)-curc. (a) SKBR3 cells (6 × 106) were plated in a 150-mm dish and the day after treated with Zn(II)-curc (100 μM) for 16 h with or without co-treatment with autophagy inhibitor CQ (25 μM) before being assayed for ChIP with anti-p53 antibody. PCR analyses were performed on the immunoprecipitated DNA using primers specific for wtp53 target gene promoter Puma or for mutp53 target promoters MDR1 and cyclin B2. A sample representing linear amplification of the total chromatin (input) was included as control. Additional controls included immunoprecipitation performed with nonspecific immunoglobulins (no Ab). (b) H1299 cells were co-transfected with Noxa-luc promoter (1 μg) and p53H175 plasmid (0.1 μg). Twenty-four hours after transfection, cells were treated with Zn(II)-curc (100 μM) for 16 h with or without co-treatment with autophagy inhibitor CQ (25 μM). Results, normalized to β-gal activity are the mean±S.D. of three independent experiments performed in duplicate. (c) SKBR3 cells were treated with Zn(II)-curc (100 μM) for 16 and 24 h with or without co-treatment with autophagy inhibitor CQ (25 μM) for 24 h before being assayed for RT-PCR of p53 target genes. (d) H1299 cells were transiently transfected with p53H175 plasmid (0.1 μg). Twenty-four hours after transfection, cells were treated with Zn(II)-curc (100 μM) for 24 h either alone or in co-treatment with CQ (25 μM) before being assayed for RT-PCR as in c. β-actin was used as a control for efficiency of RNA extraction and transcription
Autophagy inhibition reduces cell death induced by Zn(II)-curc in SKBR3 cancer cells
Next, we evaluated the biological outcome of autophagy inhibition in the context of Zn(II)-curc administration. To this end, SKBR3 cells were treated with Zn(II)-curc alone or in combination with the autophagy inhibitor CQ and cell death was monitored. The results show that the Zn(II)-curc-induced cell death was significantly reduced by CQ co-treatment (Figure 6). Altogether, the results suggest that blocking autophagy might in part inhibit the antiproliferative effects of Zn(II)-curc, likely through impairment of mutp53 reactivation.
Figure 6.

Autophagy inhibition reduces Zn(II)-curc-induced cell death. Subconfluent SKBR3 cells were seeded and the day after treated with Zn(II)-curc (100 μM) for 24 h with or without co-treatment with CQ (25 μM). Cell viability was measured by trypan blue exclusion staining and expressed as percentage±S.D. of three independent experiments. *P=0.025
Discussion
Mutant p53 is often highly expressed in tumors because of its increased half-life. Clinical studies have shown that a high level of mutp53 is correlated with more aggressive tumors, resistance to therapies and poorer outcomes.36, 37 Thus, preventing p53 mutant accumulation provides an important chemopreventive and chemotherapeutic strategy, although the reason why mutp53 is not degraded in cancer cells remains unclear. Many studies provide tantalizing evidence that small-molecule drugs can restore the activity of p53 and activate apoptosis in mutp53-carrying tumor cells.15, 16, 38, 39 Moreover, other studies point to target mutant p53 for protein degradation.40 Thus, abrogation of mutant p53 levels by depleting p53 with siRNA has been shown to reduce tumor malignancy, impairing the mutp53 GOF and allowing the apoptotic response to drugs.41, 42 The findings presented in this paper show that Zn(II)-curc induced mutant p53H175 protein degradation and that autophagy is the main proteolytic mechanism involved in such outcome.
Whereas wtp53 protein is normally degraded by E3-ubiquitin ligase MDM2, a key negative regulator of p53,20, 43 the exact identity of the mutant p53 degradation pathways is still undefined.21, 44 Here we found that MDM2 and the proteasome pathway were not the mechanisms of Zn(II)-curc-induced mutp53H175 degradation, rather autophagy was the main route involved in such outcome. This is in agreement with a previous study suggesting that the regulation of the stability of mutant p53 differs from that of wtp53.44 Thus, in our hands, inhibition of proteasome by MG132 or depletion of MDM2 by siRNA did not modify mutp53H175 degradation by Zn(II)-curc. On the contrary, inhibition of autophagy by 3-MA, CQ or siRNA for ATG5 efficiently rescued mup53H175 degradation by Zn(II)-curc. Our results are in accord with the study showing that autophagy, induced, for instance, by glucose restriction triggers mutp53 degradation sensitizing tumor cells to autophagic cell death.19, 21, 45
Autophagy is a key proteolytic system that acts as an alternative to the proteasome pathway and is particularly devoted to clearing cellular misfolded proteins or protein aggregates.17 Previously, it has been shown that the DBD of p53 is conformationally unstable and that the majority of hotspot disease mutants such as R175H, R282W and R248Q further destabilize the DBD.5 Consequently, a proportion of these mutants are at least partially unfolded and therefore inactive.46 Various forms of p53 mutants have been found to block the extent of autophagic flux especially when localized in the cytoplasm.34 On the other hand, different studies have shown that wtp53 may have a role in inducing autophagy through, for instance, activation of AMPK kinase/mTOR signaling47 or transcriptional induction of autophagy genes such as DRAM.35
How is autophagy induced in our setting? We previously demonstrated that Zn(II)-curc reactivates p53 DNA binding and transactivation by likely acting on protein misfolding, although the exact mechanism of such reactivation is still elusive.27 The rationale for the use of Zn(II) as a reactivating p53 molecule is rather compelling and stems from the fact that p53 contains one single zinc ion near the DNA-binding interface, which is necessary for the thermodynamic stability of the DBD and is needed for wtp53 oncosuppressor function.48 Importantly, certain mutp53 proteins, including p53H175, are prone to the loss of DBD-bound Zn(II) and this in turn promotes unfolding and aggregation.49 Therefore, the specific benefit of Zn(II) administration consists in its ability to reactivate mutp53 function by chaperoning zinc.26 Our original results were indeed confirmed by a recent study showing the positive effect of a thiosemicarbazone compound in promoting the refolding of p53H175, specifically because of its zinc-chelating properties50 and by another study showing that Zn(II) chelators may inhibit p53-mediated apoptosis by inducing denaturation of wtp53 as well as inhibition of transcription-dependent apoptotic pathways.51 Having established that Zn(II)-curc reactivates mutp53, here we found that such reactivation was responsible for triggering autophagy. Thus, mutp53 reactivation induced LC3-II conversion and increased the transcription of DRAM, a wtp53 target gene critical for the induction of apoptosis.35 Inhibiting p53 transactivation with PFT-α impaired both Zn(II)-curc-induced autophagy and mutp53 degradation. Similarly, p53 depletion with siRNA strongly impaired the LC3-II induction, compared with control cells treated with Zn(II)-curc. In agreement with our hypothesis that autophagy degraded mutp53H175 protein, inhibition of autophagy, by both pharmacological and genetic means, impaired p53H175 protein degradation by Zn(II)-curc as well as reverted the Zn(II)-curc-induced p53/DNA binding and transactivation of wtp53 target genes. These findings strongly indicate that reactivation of wtp53 transactivation by Zn(II)-curc was mechanistically involved in mutp53H175 degradation by wtp53-induced autophagy and that, in turn, autophagy inhibition represented an obstacle to the ability of Zn(II)-curc to fully reactivate p53H175 mutant. Thus, our hypothesis is that zinc addition reactivates wtp53 function by acting on the folding of most (but likely not all) of the mutant p53 proteins within a cell, which then triggers the degradation of the remaining mutant p53 proteins by p53-induced autophagy (Figure 7). Only the control of the balance between folded–misfolded p53 proteins may favor wt over mutant p53 functions. Certainly, additional studies are needed to strengthen our hypothesis and better understand the timing of such regulatory loop. Moreover, our results also highlight how p53-dependent autophagy was responsible in a regulatory loop to induce mutp53H175 degradation. It would be interesting in future studies to evaluate whether other p53-reactivating compounds may behave similarly to Zn(II)-curc and induce mutp53 degradation through autophagy.
Figure 7.
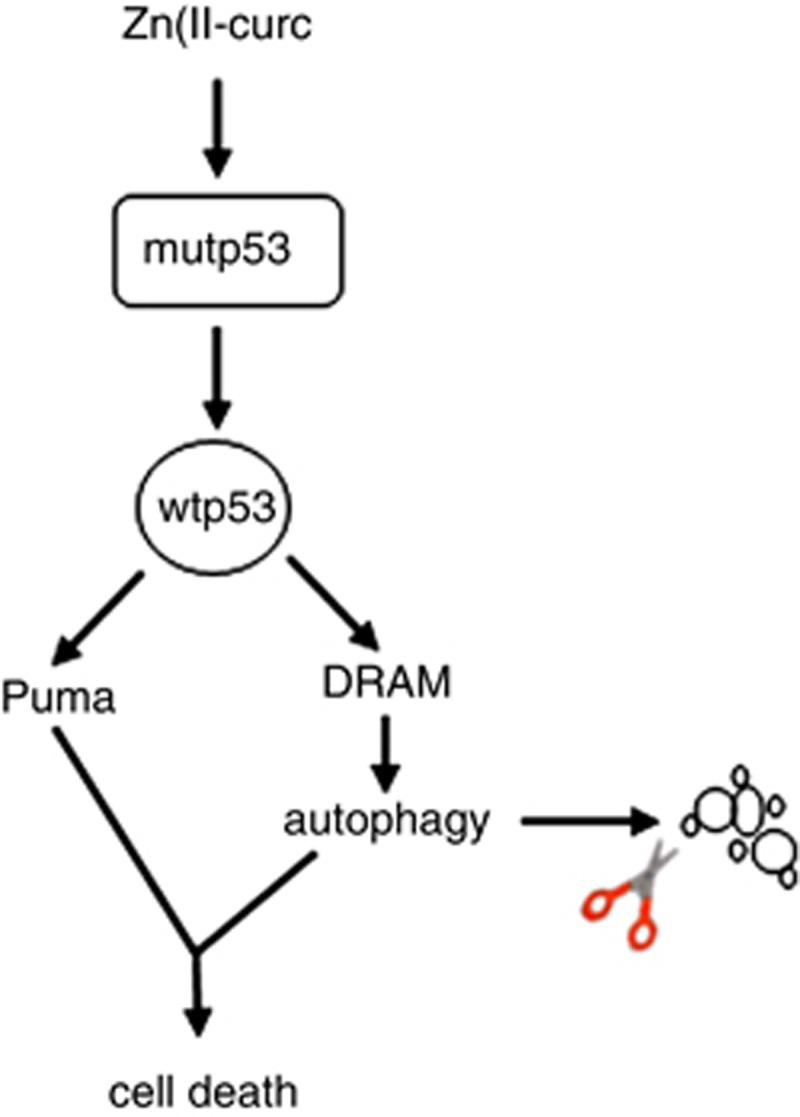
Proposed model of Zn(II)-curc-induced mutp53 degradation. The scheme combines the Zn(II)-curc-induced mutp53 reactivation27 with the p53-induced autophagy through DRAM,35 leading to cell death and autophagy-mediated mutp53 degradation. Zn(II)-curc addition reactivates wtp53 transactivation (that is, Puma) by acting on the folding of most (but likely not all) of the mutant p53 proteins within a cell; wtp53 transactivation of DRAM induces autophagy, which then triggers the degradation of the remaining mutant p53 proteins
The role of autophagy in cancer and in response to therapies is somehow controversial; however, some studies show that stimulation of autophagy may be detrimental to cancer cells by inducing cell death and that inhibition of autophagy may lead to enhanced tumor growth.52 Here, our data are consistent with the idea that autophagy acts as a tumor barrier shedding new light on the molecular mechanisms that regulate mutp53H175 stability and consequently oncosuppressor functions. Whether or not additional p53 mutants other than p53H175 are regulated in this way remains to be determined. These data also suggest a greater caution in the use of autophagy inhibitors as antitumor drug,53 specially in the context of tumors carrying p53H175 mutant. In conclusion, our results demonstrate that Zn(II)-curc induced degradation of mutant p53H175 protein levels through autophagy. As p53 mutants have been proposed as targets for chemotherapy, conditions that can promote their degradation along with reactivation of p53 oncosuppressor functions may have therapeutic advantages as is the case of Zn(II)-curc compound.
Materials and Methods
Chemicals, cell lines and culture conditions
Unless otherwise indicated, media, antibiotics and supplements for cell culture were purchased from Gibco-Life Technologies (Carlsbad, CA, USA), plasticware from Corning B.V. Life Sciences (Schiphol Rijk, The Netherlands) and chemicals from Sigma-Aldrich (St. Louis, MO, USA). The following reagents were used: Zn(II)-curc28 dissolved in DMSO and used at 100 μM; the early inhibitor of autophagosome formation 3-MA was dissolved in dH2O and used at 5 mM; the inhibitor of autophagic protein degradation CQ was dissolved in dH2O and used at 25 μM; the proteasome inhibitor MG132 was dissolved in DMSO and used at 20 μM; the p53 inhibitor PFT-α (ENZO Life Sciences, Lausen, Switzerland) was dissolved in DMSO and used at 30 μM; cisplatin (Teva, Pharma Italia, Italy) was used at 5 μg/ml and CHX, the inhibitor of protein synthesis was dissolved in dH2O and diluted into the medium to a final concentration of 40 μg/ml. The human colon cancer RKO (wtp53), breast cancer SKBR3 (expressing p53R175H mutation) and lung cancer H1299 (p53 null) cells were maintained in RPMI-1640 medium, whereas the human colon cancer HCT116 (wtp53) cells were maintained in DMEM, all supplemented with 10% heat-inactivated fetal bovine serum plus glutamine and antibiotics in a humidified atmosphere with 5% CO2 at 37 °C.
RNA interference, transfections and luciferase assay
siRNAs specific for human ATG5 and MDM2 as well as siRNA unrelated to the human genome were purchased from Dharmacon (Thermo Scientific, Milan, Italy); pSuper vector was used for p53 depletion (sip53).22 Transient transfections were carried out in SKBR3 cells by means of the LipofectaminePlus method (Invitrogen, Life Technologies). H1299 cells were transfected with wtp53 or p53H175 expression vector by means of the N,N-bis-(2-hydroxyethyl)-2-amino-ethanesulphonic acid-buffered saline version of the calcium phosphate procedure.54 The plasmid reporter used was the luciferase reporter gene driven by the p53-dependent natural promoter Noxa (Noxa-luc; kindly provided by T. Taniguchi, University of Tokyo, Japan). Transfection efficiency was normalized with the use of a co-transfected β-galactosidase (β-gal) plasmid. Luciferase activity was assayed on whole-cell extract and the luciferase values were normalized to β-gal activity and protein content and expressed as relative luciferase unit (RLU).
Immunoblotting
Total cell extracts were prepared by incubation in lysis buffer (50 mM Tris-HCl, pH 7.5, 150 mM NaCl, 5 mM EDTA, 150 mM KCl, 1 mM dithiothreitol, 1% Nonidet P-40) and a mix of protease inhibitors. Equal amount of cell lysates were separated according to molecular weight on 10–18% SDS-polyacrylamide gels and electrotransferred to polyvinylidene difluoride membranes (Millipore Corporation, Billerica, MA, USA). Unspecific binding sites were blocked by incubating membranes for 1 h in 0.05% Tween-20 (v/v in TBS) supplemented with 5% non-fat powdered milk or bovine serum albumin, followed by overnight incubation at 4 °C with primary antibodies specific for: ATG5 and LC3B (Sigma-Aldrich), p53 (FL393), p21 and p62 (Santa Cruz Biotechnology, Dallas, TX, USA). Equal lane loading was monitored by probing membranes with antibodies specific for β-actin (Calbiochem, San Diego, CA, USA) or tubulin (Immunological Sciences, Rome, Italy). Primary antibodies were detected with appropriate horseradish peroxidase-labeled secondary antibodies (Bio-Rad, Hercules, CA, USA). Enzymatic signals were visualized using chemoluminescence (ECL Detection system, GE Healthcare, Milan, Italy).
RNA isolation and RT-PCR analysis
Cells were harvested in TRIzol Reagent (Invitrogen) and total RNA was isolated following the manufacturer's instructions. cDNA was synthesized from 2 μg of total RNA with MuLV reverse transcriptase kit (Applied Biosystems, Foster City, CA, USA). Semiquantitative PCR was carried out by using Hot-Master Taq polymerase (Eppendorf S.r.l., Milan, Italy) with 2 μl cDNA reaction and gene-specific oligonucleotides (Supplementary Table S1) under conditions of linear amplification. PCR products were run on a 2% agarose gel and visualized with ethidium bromide staining using UV light. The housekeeping β-actin gene was used as a control for efficiency of RNA extraction and transcription.
ChIP assay
ChIP analysis was carried out essentially as described.55 Briefly, protein complexes were crosslinked to DNA in living cells by adding formaldehyde directly to the cell culture medium at 1% final concentration. Chromatin extracts containing DNA fragments with an average size of 500 bp were incubated overnight at 4 °C with milk shaking using polyclonal anti-p53 antibody (FL393, Santa Cruz Biotechnology) and affinity-purified rabbit anti-p73 antibody (lot A300-126A-2, Bethyl Laboratories Inc.). Before use, protein G (Pierce, Thermo Scientific) was blocked with 1-μg/μl sheared herring sperm DNA and 1-μg/μl bovine serum albumin for 3 h at 4 °C and then incubated with chromatin and antibodies for 2 h at 4 °C. PCR was performed with HOT-MASTER Taq (Eppendorf) using 2 μl of immunoprecipitated DNA and promoter-specific primers for human Puma, MDR1, DRAM, and cyclin B2 promoters (Supplementary Table S1). Immunoprecipitation with nonspecific immunoglobulins (IgG; Santa Cruz Biotechnology) was performed as negative controls. The amount of precipitated chromatin measured in each PCR was normalized with the amount of chromatin present in the input of each immunoprecipitation. PCR products were run on a 2% agarose gel and visualized with ethidium bromide staining using UV light.
Measurement of cell proliferation and viability
Cell numbers were determined in duplicate at different time points. Cell counts were performed using a hemocytometer by adding trypan blue to equal volume of cell suspension. The percentage of cell viability, as blue/total cells, was assayed by scoring 200 cells per well three times.
Statistical procedures
All experiments, unless otherwise indicated, were performed in triplicate instances, yielding comparable results. All experimental results were expressed as the arithmetic mean and S.D. of measurements was shown. Paired or unpaired (as appropriate) two-tailed Student's t-test was used for statistical significance of the differences between treatment groups. Statistical analysis was performed using analysis of variance at 5% (P<0.05) or 1% (P<0.01).
Acknowledgments
This study was funded by the Italian Association for Cancer Research (AIRC) Grant (IG11377) to GD. We thank MP Gentileschi for technical support and G Piaggio for sharing reagents.
Glossary
- β-gal
β-galactosidase
- ChIP
chromatin immunoprecipitation
- CHX
cycloheximide
- CQ
cloroquine
- DBD
DNA-binding domain
- DRAM
damage-regulated autophagy modulator
- GOF
gain of oncogenic function
- LC3
microtubule-associated protein light chain 3
- MDR1
multidrug resistance
- mutp53
mutant p53
- PFT-α
pifithrin-α
- RLU
relative luciferase unit
- RT-PCR
reverse transcription-polymerase chain reaction
- siRNA
small-interfering RNA
- wtp53
wild-type p53
The authors declare no conflict of interest.
Footnotes
Supplementary Information accompanies this paper on Cell Death and Disease website (http://www.nature.com/cddis)
Edited by A Stephanou
Supplementary Material
References
- Vousden KH, Lane DP. P53 in health and disease. Nat Rev Mol Cell Biol. 2007;8:275–283. doi: 10.1038/nrm2147. [DOI] [PubMed] [Google Scholar]
- Vousden KH, Prives C. Blinded by the light: the growing complexity of p53. Cell. 2009;137:413–431. doi: 10.1016/j.cell.2009.04.037. [DOI] [PubMed] [Google Scholar]
- Muller PAJ, Vousden KH. P53 mutations in cancer. Nat Cell Biol. 2013;15:2–8. doi: 10.1038/ncb2641. [DOI] [PubMed] [Google Scholar]
- Olivier M, Hollstein M, Hainaut P. TP53 mutations in human cancers: origins, consequences, and clinical use. Cold Spring Harb Perspect Biol. 2010;2:a001008. doi: 10.1101/cshperspect.a001008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Joerger AC, Fersht AR. Structural biology of the tumor suppressor p53 and cancer-associated mutants. Adv Cancer Res. 2007;97:1–23. doi: 10.1016/S0065-230X(06)97001-8. [DOI] [PubMed] [Google Scholar]
- Brosh R, Rotter V. When mutants gain new powers: news from the mutant p53 field. Nat Rev Cancer. 2009;9:701–713. doi: 10.1038/nrc2693. [DOI] [PubMed] [Google Scholar]
- Freed-Pastor WA, Prives C. Mutant p53: one name, many proteins. Genes Dev. 2012;26:1268–1286. doi: 10.1101/gad.190678.112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu J, Reumers J, Couceiro JR, De Smet F, Gallardo R, Rudyak S, et al. Gain of function of mutant p53 by coaggregation with multiple tumor suppressors. Nat Chem Biol. 2011;7:285–295. doi: 10.1038/nchembio.546. [DOI] [PubMed] [Google Scholar]
- Sampath J, Sun D, Kidd VJ, Grenet J, Gandhi A, Shapiro LH, et al. Mutant p53 cooperates with ETS and selectively up-regulates human MDR1 not MRP1. J Biol Chem. 2001;276:39359–39367. doi: 10.1074/jbc.M103429200. [DOI] [PubMed] [Google Scholar]
- Di Agostino S, Strano S, Emiliozzi V, Zerbini V, Mottolese M, Sacchi A, et al. Gain of function of mutant p53: The mutant p53/NF-Y protein complex reveals an aberrant transcriptional mechanism of cell cycle regulation. Cancer Cell. 2006;10:191–202. doi: 10.1016/j.ccr.2006.08.013. [DOI] [PubMed] [Google Scholar]
- Lang GA, Iwakuma T, Suh YA, Liu G, Rao VA, Parant JM, et al. Gain of function of a p53 hot spot mutation in a mouse model of Li-Fraumeni syndrome. Cell. 2004;119:861–872. doi: 10.1016/j.cell.2004.11.006. [DOI] [PubMed] [Google Scholar]
- Olive KP, Tuveson DA, Ruhe ZC, Yin B, Willis NA, Bronson RT, et al. Mutant p53 gain of function in two mouse models of Li-Fraumeni syndrome. Cell. 2004;119:847–860. doi: 10.1016/j.cell.2004.11.004. [DOI] [PubMed] [Google Scholar]
- Hanel W, Marchenko N, Xu S, Xiaofeng YS, Weng W, Moll U. Two hot spot mutant p53 mouse models display differential gain of function in tumorigenesis. Cell Death Differ. 2013;7:898–909. doi: 10.1038/cdd.2013.17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jackson JG, Lozano G. The mutant p53 mouse as a pre-clinical model. Oncogene. 2013;32:4325–4330. doi: 10.1038/onc.2012.610. [DOI] [PubMed] [Google Scholar]
- Wiman KG. Pharmacological reactivation of mutant p53: from protein structure to the cancer patient. Oncogene. 2010;29:4245–4252. doi: 10.1038/onc.2010.188. [DOI] [PubMed] [Google Scholar]
- Brown CJ, Cheok CF, Verma CS, Lane DP. Reactivation of p53: from peptides to small molecules. Trends Pharmacol Sci. 2011;32:53–62. doi: 10.1016/j.tips.2010.11.004. [DOI] [PubMed] [Google Scholar]
- Mizushima N. Autophagy: process and function. Genes Dev. 2007;21:2861–2873. doi: 10.1101/gad.1599207. [DOI] [PubMed] [Google Scholar]
- Glick D, Barth S, Mcleod KF. Autophagy: cellular and molecular mechanisms. J Pathol. 2010;221:3–12. doi: 10.1002/path.2697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rodriguez OC, Choudhury S, Kolukula V, Vietsch EE, Catania J, Preet A, et al. Dietary downregulation of mutant p53 levels via glucose restriction: mechanisms and implications for tumor therapy. Cell Cycle. 2012;11:4436–4446. doi: 10.4161/cc.22778. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haupt Y, Maya R, Kazaz A, Oren M. Mdm2 promotes the rapid degradation of p53. Nature. 1997;387:296–299. doi: 10.1038/387296a0. [DOI] [PubMed] [Google Scholar]
- Choudhury S, Kolukula VK, Preet A, Albanese C, Avantaggiati ML. Dissecting the pathways that destabilize mutant p53: the proteasome or autophagy. Cell Cycle. 2013;12:1022–1029. doi: 10.4161/cc.24128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Puca R, Nardinocchi L, Gal H, Rechavi G, Amariglio N, Domany E, et al. Reversible dysfunction of wild-type p53 following homeodomain-interacting protein kinase-2 knockdown. Cancer Res. 2008;68:3707–3714. doi: 10.1158/0008-5472.CAN-07-6776. [DOI] [PubMed] [Google Scholar]
- Puca R, Nardinocchi L, Bossi G, Sacchi A, Rechavi G, Givol D, et al. Restoring wtp53 activity in HIPK2 depleted MCF7 cells by modulating metallothionein and zinc. Exp Cell Res. 2009;315:67–75. doi: 10.1016/j.yexcr.2008.10.018. [DOI] [PubMed] [Google Scholar]
- Puca R, Nardinocchi L, Porru M, Simon AJ, Rechavi G, Leonetti C, et al. Restoring p53 active conformation by zinc increases the response of mutant p53 tumor cells to anticancer drugs. Cell Cycle. 2011;10:1679–1689. doi: 10.4161/cc.10.10.15642. [DOI] [PubMed] [Google Scholar]
- Margalit O, Simon AJ, Yakubov E, Puca R, Yosepovich A, Avivi C, et al. Zinc supplementation augments in vivo antitumor effect of chemotherapy by restoring p53 function. Int J Cancer. 2012;131:562–568. doi: 10.1002/ijc.26441. [DOI] [PubMed] [Google Scholar]
- D'Orazi G, Givol D. p53 reactivation: the link to zinc. Cell Cycle. 2012;11:2581–2582. doi: 10.4161/cc.21020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garufi A, Trisciuoglio D, Porru M, Leonetti C, Stoppacciaro A, D'Orazi V, et al. A fluorescent curcumin-based Zn(II)-complex reactivates mutant (R175H and R273H) p53 in cancer cells. J Exp Clin Cancer Res. 2013;32:72. doi: 10.1186/1756-9966-32-72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pucci D, Bellini T, Crispini A, D'Agnano I, Liguori PF, Garcia-Orduña P, et al. DNA binding and cytotoxicity of fluorescent curcumin-based Zn(II) complexes. Med Chem Commun. 2012;3:462–468. [Google Scholar]
- Pucci D, Crispini A, Mendiguchía BS, Pirillo S, Ghedini M, Morelli S, et al. Improving the bioactivity of Zn(II)-curcumin based complexes. Dalton Trans. 2013;42:9679–9687. doi: 10.1039/c3dt50513h. [DOI] [PubMed] [Google Scholar]
- Willis A, Jung EJ, Wakefield T, Chen X. Mutant p53 exerts a dominant negative effect by preventing wild-type p53 from binding to the promoter of its target genes. Oncogene. 2004;23:2330–2338. doi: 10.1038/sj.onc.1207396. [DOI] [PubMed] [Google Scholar]
- Komarov PG, Komarova EA, Kondratov RV, Christov-Tselkov K, Coon JS, Chernov MV, et al. A chemical inhibitor of p53 that protects mice from the side effects of cancer therapy. Science. 1999;285:1733–1737. doi: 10.1126/science.285.5434.1733. [DOI] [PubMed] [Google Scholar]
- Barth S, Glick D, Macleod KF. Autophagy: assays and artifacts. J Pathol. 2010;221:117–124. doi: 10.1002/path.2694. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mizushima N, Yoshimori T, Ohsumi Y. The role of Atg proteins in autophagosome formation. Annu Rev Cell Dev Biol. 2011;27:107–132. doi: 10.1146/annurev-cellbio-092910-154005. [DOI] [PubMed] [Google Scholar]
- Morselli E, Tasdemir E, Maiuri MC, Galluzzi L, Kepp O, Criollo A, et al. Mutant p53 protein localized in the cytoplasm inhibits autophagy. Cell Cycle. 2008;7:3056–3061. doi: 10.4161/cc.7.19.6751. [DOI] [PubMed] [Google Scholar]
- Crighton D, Wilkinson S, O'Prey J, Syed N, Smith P, Harrison PR, et al. DRAM, a p53-induced modulator of autophagy, is critical for apoptosis. Cell. 2006;126:121–134. doi: 10.1016/j.cell.2006.05.034. [DOI] [PubMed] [Google Scholar]
- Petitjean A, Achatz MI, Borresen-Dale AL, Hainaut P, Olivier M. TP53 mutations in human cancers: functional selection and impact on cancer prognosis and outcomes. Oncogene. 2007;26:2157–2165. doi: 10.1038/sj.onc.1210302. [DOI] [PubMed] [Google Scholar]
- Morton JP, Timpson P, Karim SA, Ridgway RA, Athineos D, Doyle B, et al. Mutant p53 drives metastasis and overcomes growth arrest/senescence in pancreatic cancer. Proc Natl Acad Sci USA. 2010;107:246–251. doi: 10.1073/pnas.0908428107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bullock AN, Fersht AR. Rescuing the function of mutant p53. Nat Rev Cancer. 2001;1:68–76. doi: 10.1038/35094077. [DOI] [PubMed] [Google Scholar]
- Bykov VJ, Lamber JM, Hainaut P, Wiman KG. Mutant p53 rescue and modulation of p53 redox state. Cell Cycle. 2009;8:16. doi: 10.4161/cc.8.16.9382. [DOI] [PubMed] [Google Scholar]
- Yan W, Zhang Y, Zhang J, Liu S, Cho J, Chen X. Mutant p53 protein is targeted by arsenic for degradation and plays a role in arsenic-mediated growth suppression. J Biol Chem. 2011;286:17478–17486. doi: 10.1074/jbc.M111.231639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bossi G, Lapi E, Strano S, Rinaldo C, Blandino G, Sacchi A. Mutant p53 gain of function: reduction of tumor malignancy of human cancer cells through abrogation of mutant p53 expression. Oncogene. 2006;25:304–309. doi: 10.1038/sj.onc.1209026. [DOI] [PubMed] [Google Scholar]
- Bossi G, Marampon F, Maor-Aloni R, Zani B, Rotter V, Oren M, et al. Conditional RNA interference in vivo to study mutant p53 oncogenic gain of function on tumor malignancy. Cell Cycle. 2008;7:1870–1879. doi: 10.4161/cc.7.12.6161. [DOI] [PubMed] [Google Scholar]
- Alarcon-Vargas D, Ronai Z. p53-Mdm2: the affair that never ends. Carcinogenesis. 2002;23:541–547. doi: 10.1093/carcin/23.4.541. [DOI] [PubMed] [Google Scholar]
- Lukashchuk N, Vousden KH. Ubiquitination and degradation of mutant p53. Mol Cell Biol. 2007;27:8284–8295. doi: 10.1128/MCB.00050-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang X, Di Pasqua AJ, Govind S, McCracken E, Hong C, Mi L, et al. Selective depletion of mutant p53 by cancer chemopreventive isothiocyanates and its structure-activity relationships. J Med Chem. 2011;54:809–816. doi: 10.1021/jm101199t. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gannon JV, Greaves R, Iggo R, Lane DP. Activating mutations in p53 produce a common conformational effect. A monoclonal antibody specific for the mutant form. EMBO J. 1990;9:1595–1602. doi: 10.1002/j.1460-2075.1990.tb08279.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jing K, Song KS, Shin S, Kim N, Jeong S, Oh HR, et al. Docosahexaenoic acid induces autophagy through p53/AMPK/mTOR signaling and promotes apoptosis in human cancer cells harboring wild-type p53. Autophagy. 2011;7:1348–1358. doi: 10.4161/auto.7.11.16658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Butler JS, Loh SN. Structure, function and aggregation of the zinc-free form of the p53 DNA binding domain. Biochemistry. 2003;42:2396–2403. doi: 10.1021/bi026635n. [DOI] [PubMed] [Google Scholar]
- Loh SN. The missing zinc: p53 misfolding and cancer. Metallomics. 2010;2:442–449. doi: 10.1039/c003915b. [DOI] [PubMed] [Google Scholar]
- Yu X, Vazquez A, Levine AJ, Carpizo DR. Allele-specific p53 mutant reactivation. Cancer Cell. 2012;21:614–625. doi: 10.1016/j.ccr.2012.03.042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morita A, Ariyasu S, Ohya S, Takahashi I, Wang B, Tanaka K, et al. Evaluation of Zinc (II) chelators for inhibiting p53-mediated apoptosis. Oncotarget. 2013;4:2439–2450. doi: 10.18632/oncotarget.1535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bhutia SK, Mukhopadhyay S, Sinha N, Das DN, Panda PK, Patra SK, et al. Autophagy: cancer's friend or foe. Adv Cancer Res. 2013;118:61–95. doi: 10.1016/B978-0-12-407173-5.00003-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Amaravadi RK, Lippincott-Schwartz J, Yin XM, Weiss WA, Takebe N, Timmer W, et al. Principles and current strategies for targeting autophagy for cancer treatment. Clin Cancer Res. 2011;17:654–666. doi: 10.1158/1078-0432.CCR-10-2634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen C, Okayama H. High-efficiency transformation of mammalian cells by plasmid DNA. Mol Cell Biol. 1987;7:2754–2756. doi: 10.1128/mcb.7.8.2745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nardinocchi L, Puca R, Sacchi A, Rechavi G, Givol D, D'Orazi G. Targeting hypoxia in cancer cells by restoring homeodomain-interacting protein kinase-2 and p53 activity and suppressing HIF-1alpha. PLoS One. 2009;4:e6819. doi: 10.1371/journal.pone.0006819. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.