

Abstract
Pancreatic ductal adenocarcinoma (PDA) has an aggressive natural history and is resistant to therapy. The receptor for advanced glycation end-products (RAGE) is a pattern recognition receptor for many damage associated molecular pattern (DAMP) molecules. RAGE is overexpressed in both human and murine models of PDA as well as most advanced epithelial neoplasms. The immunosuppressive nature of the PDA micro-environment is facilitated, in part, by the accumulation of regulatory immune cell infiltrates such as myeloid-derived suppressor cells (MDSCs). To study the role of RAGE expression in the setting of mutant Ras-promoted pancreatic carcinogenesis (KC), a triple transgenic model of spontaneous murine PDA in a RAGE-null background (KCR) was generated. KCR mice had markedly delayed pancreatic carcinogenesis and a significant diminution of MDSCs compared to KC mice at comparable time points post weaning. While RAGE was not required for the development or suppressor activity of MDSCs, its absence was associated with temporally limited pancreatic neoplasia and altered phenotype and function of the myeloid cells. In lieu of MDSCs, KCR animals at comparable time points exhibited mature CD11b+Gr1−F4/80+ cells which were not immunosuppressive in vitro. KCR mice also maintained a significantly less suppressive milieu evidenced by marked decreases in CCL22 in relation to CXCL10 and diminished serum levels of IL-6.
Keywords: Rodent, Monocytes/Macrophages, Inflammation, Tolerance/Suppression/Anergy, Tumor Immunity, Transgenic/Knockout Mice
INTRODUCTION
Pancreatic ductal adenocarcinoma (PDA) is a largely lethal disease. The incidence and mortality rates in the United States are nearly identical, and patients who achieve a 5 year survival from time of diagnosis are rare (6%) (1, 2). Histopathologically, PDA progresses in a conserved fashion beginning with the emergence of malignant precursor lesions termed pancreatic intraepithelial neoplasia or PanIN lesions which progressively become more severely hyper- and dysplastic and finally culminate in frank invasive cancer (3, 4). While immunotherapies such as cellular and peptide-based vaccines, monoclonal antibodies against tumor signaling molecules, and recombinant cytokines have exhibited moderate successes in many tumor types, pancreatic cancer patients have yet to benefit substantially from these strategies (5). A significant obstacle to immunotherapeutic intervention is the immunosuppressive pancreatic tumor micro-environment (6). Immune suppression is facilitated by soluble factors such as TGFβ, IL-10, VEGF, and FasL derived from both the tumor and stromal compartments, the downregulation of NK cell surface ligands such as MICA/B in tumor tissue, and a marked increase in regulatory immune cell infiltrate consisting of regulatory T cells (Tregs), tumor associated macrophages (TAMs), and myeloid derived suppressor cells or MDSCs (6-8).
MDSCs are a recently identified heterogeneous population of hematopoietic cells of myeloid lineage (9, 10). While normally present in relatively low quantities in the peripheral blood of healthy individuals, PDA patients exhibit high frequencies of these cells in the peripheral blood (11). Furthermore, MDSCs traffic to tumor tissue where they directly inhibit anti-tumor immune effector cells through a variety of mechanisms including depriving T cells of nutrients via arginase-I production and tryptophan and cysteine depletion, interfering with trafficking by inducing the downregulation of CD62L and L-selectin, and upregulating reactive nitrogen and oxygen species (NOS and ROS) (9, 10, 12, 13). MDSCs further support immune suppression in the tumor micro-environment by promoting reparative wound healing and angiogenesis and facilitating the recruitment of regulatory T cells (14).
Identifying MDSCs based on cell surface marker expression in cancer patients has been difficult given the phenotypic heterogeneity. Several subsets have been characterized as CD11b+CD14+, CD11b+CD33+, and CD11b+CD15+, with the latter primarily being recruiting to primary tumor tissue in PDA patients (9, 15). In mice, however, these cells are readily and reliably identified by the co-surface expression of CD11b and Gr1 and can be further characterized based on the expression of IL-4Rα and limited expression of co-stimulatory molecules such as CD80 (10, 16). They accumulate in the spleen, blood, and tumor in a variety of murine tumor models (17, 18). In a spontaneous KrasG12D-driven transgenic model of PDA that recapitulates the histopathology of human PDA with high fidelity (termed “KC” mice), MDSC accumulate both systemically (measured in the spleen) and locally in pancreas tissue (17).
The receptor for advanced glycation end-products (RAGE) is an MHC class III encoded protein, characterized as a damage associated molecular pattern (DAMP) molecule receptor. It serves as the cognate receptor for the prototypical DAMP, high-mobility group box 1 (HMGB1) and several S100 proteins including S100A8 and S100A9 (19, 20). RAGE-mediated signaling plays a role in the pathogenesis of epithelial derived cancers such as PDA by activating key survival pathways such as autophagy in cancer cells as well as propagating and sustaining pro-tumor host inflammatory responses (20-23). While normal pancreatic ductal epithelial cells do not routinely stain positively for RAGE, with the emergence and progression of pancreatic neoplasia, RAGE is markedly upregulated and overexpressed (22). The contribution of RAGE to intratumoral MDSC accumulation was first demonstrated in RAGE−/− mice in an inducible skin cancer model (24). In addition, mice deficient for the RAGE ligand, S100A9, exhibit a significant reduction in the incidence and burden of colitis-associated colorectal tumors and show demonstrable decreases in intratumoral and splenic MDSC frequency (18, 25, 26). To determine if RAGE plays a role in the accumulation of MDSCs during pancreatic carcinogenesis, we have backcrossed RAGE-null mice into the KC strain. The resultant Pdx1-Cre:KrasG12D/+: Rage−/− mice are termed “KCR”. We demonstrate here that the targeted ablation of RAGE in the emerging Kras-driven tumor microenvironment limits development of PanIN lesions and the associated accumulation of MDSCs.
MATERIALS AND METHODS
Mouse strains
Wild-type C57BL/6 mice were purchased from Taconic (Hudson, NY, USA). RAGE knockout (Rage−/−.GFP) mice (SVEV129×C57BL/6) were obtained from Dr. Angelika Bierhaus (27) as a kind gift. Pdx-1-Cre and KrasG12D/+ transgenic mice were obtained from the MMHCC/NCI Mouse Repository. The genotypes Pdx1-Cre:KrasG12D/+ (termed KC) and Rage−/− were crossed to generate Pdx1-Cre:KrasG12D/+: Rage−/− mice (termed KCR). Genomic and recombination screens were done by polymerase chain reaction and analysis of GFP expression (data not shown).
Flow Cytometry
Flow cytometric analysis was performed on the C6 flow cytometer (Accuri Cytometers, Ann Arbor, MI, USA) instrument provided by the University of Pittsburgh Cancer Institute Flow and Imaging Cytometry core facility and analyzed using FlowJo software (Tree Star Inc, Ashland, OR, USA). Murine spleens were homogenized through a 70μm nylon filter (BD Biosciences, San Jose, CA, USA) and washed with PBS. Red blood cells were lysed with Red Blood Cell Lysing Buffer (Sigma-Aldrich, St. Louis, MO, USA) according to the manufacturer’s instructions. Single cell suspensions were derived from the pancreas by mechanical separation and collagenase digestion (Sigma-Aldrich, St. Louis, MO, USA). The resulting single cell suspensions were then stained with the following fluorescently labeled antibodies: CD11b, CD11c, F4/80, Gr1, IL-4Rα and corresponding isotype controls (all from BD Biosciences, San Jose, CA, USA). In cases, the cells were then permeabilized with 0.2% Triton-X and stained with antibodies to intracellular iNOS/NOS Type II and Arginase-I (BD Biosciences, San Jose, CA, USA). During analysis viable cells were identified via forward and side scatter and gated accordingly. Phenotyping of MDSC cell populations in KC and KCR mice (Figure 3) were based on a CD11c+Gr1+ gate.
Figure 3. RAGE is dispensable for the differentiation and suppressor activity of MDSC.
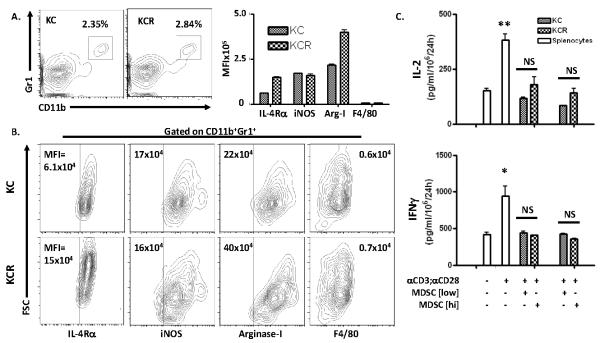
(A) Representative flow cytometry diagrams of splenocytes from 20 week old KC (n=2) and KCR (n=2) mice stained for co-surface expression of MDSC markers CD11b and Gr1. (A, B) Flow diagrams of splenocytes gated for CD11b and Gr1 positivity and stained for surface expression of IL-4Rα and F4/80, and intracellular expression of iNOS/NOS type II and arginase-I from KC and KCR mice. (A) Graphical representation of the MFIs of each marker (+/− SD). (B) MFI values illustrated on diagrams. Demarcations based on isotype controls (data not shown). (C) MDSCs from mice in panels A and B were magnetically separated and co-cultured with bulk wild-type splenocytes stimulated with αCD3/αCD28 activator beads at rations of 1:1 (MDSC [low]) and 2:1 (MDSC [hi]). Graphs depict IL-2 and IFNγ levels detected in the resultant supernatants by ELISA (+/− SEM, *p<0.05, **p<0.01, NS = not significant).
Histology
Harvested pancreatic tissue was formalin fixed (Sigma-Aldrich, St. Louis, MO, USA) and stained with hematoxylin and eosin and mounted onto glass slides by the University of Pittsburgh Department of Pathology. Images were visualized and captured at a magnification of 10X by Nikon Eclipse E800 fluorescent microscope under bright field settings (Melville, NY, USA).
Co-culture assays
4×105 Bulk wild-type splenocytes were obtained as previously mentioned from C57/BL6 mice. To activate effector splenocytes, cells were incubated with T-activator CD3/CD28 Dynabeads (Dynal AS, Oslo, Norway) at a ratio of 1:1 according to the manufacturer’s instructions. For co-culture conditions, splenocytes from KC and KCR mice were incubated with αGr1 or αCD11b microbeads and magnetically separated using MACS LS separation columns (all from Miltenyi Biotec, Leiden, The Netherlands) according to the manufacturer’s instructions and purity assayed by flow cytometry. 4×105 (termed “low”) or 8×105 (termed “hi”) Gr1+ or CD11b+ cells were incubated with bulk WT splenocytes for 96 hours. The resulting supernatants were harvested for analysis.
In-vitro differentiation of MDSCs from murine bone marrow
MDSCs were generated in-vitro using the following method. Fresh bone marrow flushed from the femurs and tibias of mice was resuspended in media supplemented with G-CSF (100ng/ml) and GM-CSF (250 U/ml) and cultured for 3 days. On day 4, 80 ng/ml of IL-13 was added to the culture for an additional 24 hours (28). All cytokines were from eBioscience, San Diego, CA, USA. Harvested cells routinely were 50% CD11b+Gr1+ and were magnetically sorted as previously mentioned for functional analysis.
ELISA analysis
Supernatants from co-culture experiments or serum from murine peripheral blood (obtained via direct cardiac puncture) was assayed for detectable levels of IL-2, IFNγ, and IL-6 by ELISA Ready-SET-Go! Kits (eBioscience, San Diego, CA, USA) according the manufacturer’s instructions.
Quantitative PCR
Pancreatic tissue from 35 week old mice was harvested and snap frozen in liquid nitrogen. Messenger RNA was then isolated and analyzed for the relative quantity (RQ) of CCL22, CXCL10 mRNA and then normalized to a control housekeeping gene, HPRT1 and displayed as a ratio. Analysis was performed with StepOne software (Applied Biosystems, Foster City, CA, USA)
Statistical analysis
Data are expressed as means +/− SEM of at least two independent experiments performed in triplicate. Statistical analysis was performed using a two-tailed student’s T test. P values below 0.05 were assigned statistical significance.
RESULTS
Targeted ablation of RAGE attenuates development of high grade PanIN lesions in KC mice
To determine if RAGE contributes to pancreatic carcinogenesis, 20 week and 35 week old WT, RAGE-null, KC, and KCR mice were sacrificed and their pancreata harvested and stained with H & E for visualization of PanIN lesion formation. At 20 weeks, both the KC and KCR strains show the occurrence of atypical ducts. However, glandular and islet cells remain largely intact, and both strains exhibit a paucity of fibrotic and inflammatory tissue (Figure 1A). At 35 weeks of age KC mice exhibit extensive atypical ductal morphology within pancreatic tissue, the emergence of low and high grade PanIN lesions, and severe fibrogenesis indicative of emergent pancreatic carcinogenesis. In KC mice in which RAGE has been chromosomally ablated (KCR mice), the frequency of hyperplastic and dysplastic ductal epithelium is substantially decreased and the integrity of glands and beta cells in the islets is largely retained (Figure 1B). Furthermore, we have previously reported that these mice have a significantly enhanced survival rate (22). These findings suggest that RAGE directly or indirectly contributes to the pathogenesis of early pancreatic carcinogenesis.
Figure 1. KCR mice exhibit mark decreases in PanIN lesion development and progression.
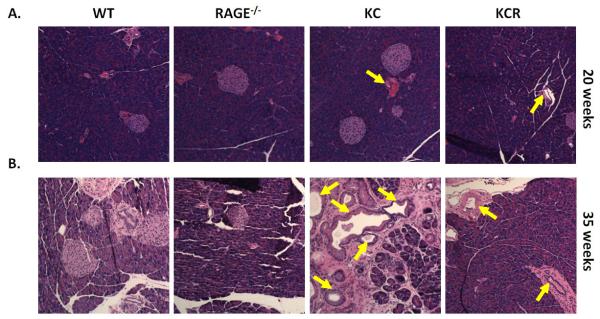
(A and B) H & E staining of pancreatic tissue sections from wild-type, RAGE-null, KC, and KCR mice harvested at (A) 20 weeks of age and (B) 35 weeks of age. PanIN lesions are designated with arrows.
RAGE promotes splenic MDSC accumulation during Kras-mediated pancreatic carcinogenesis
MDSCs accumulate within the spleens of KC mice as they progress towards ductal adenocarcinoma (17). To determine if the integrity of the RAGE signaling pathway is an important factor in this observation, WT, RAGE-null, KC, and KCR mice were sacrificed at 35 weeks of age at which point animals expressing mutant Kras display evidence of pancreatic carcinogenesis manifested by the presence of low and high grade PanIN lesions within the pancreatic ducts (Figure 1B). Spleens from these mice were processed into single-cell suspensions for flow cytometric analysis of immune cell frequencies. Both WT and RAGE-null animals exhibit comparable basal levels of CD11b+Gr1+ MDSCs ranging from 3-4% (Figure 2A) in the absence of apparent pathology. As has been previously documented, Kras-driven pancreatic carcinogenesis resulted in a significant increase in splenic MDSC frequency to 20-35% (17). Interestingly, KCR mice fail to accumulate MDSCs with a relative paucity of CD11b+Gr1+ cells (Figure 2A and B). This is significantly less than the KC strain (p=0.0225) at comparable times. However other cells of myeloid-lineage such as myeloid dendritic cells (DCs) identified by the co-expression of CD11b and CD11c are elevated in both KC and KCR mice to similar extents, suggesting that the reduction in splenic MDSCs in the KCR strain is restricted to CD11b+Gr1+ cells.
Figure 2. RAGE ablation is associated with limited splenic MDSC accumulation during Ras-mediated pancreatic carcinogenesis.

(A) Representative flow cytometry diagrams of splenocytes stained for co-surface expression of MDSC markers CD11b and Gr1 and myeloid DC markers CD11b and CD11c from 35 week old wild-type (n=2), RAGE-null (n=2), KC (n=8), and KCR (n=5) mice. Note the boxes denoted MDSCs are distinct from the boxes denoting DC populations. (B) Scatter plots depicting the frequencies of splenic MDSCs identified by expression of CD11b, Gr1, and IL-4Rα and frequencies of splenic DCs of myeloid origin (CD11b+CD11c+) in mice from panel A. (C) Wild-type, RAGE-null, KC, and KCR mice were sacked at 20 (n=2 per strain), 24 (n=2 per strain), 26 (n=2 per strain), and 35 weeks (n=2 WT and RAGE-null, n=8 KC, n=5 KCR). The kinetics of splenic MDSC accumulation with the progression of pancreatic neoplasia (KC and KCR mice) is depicted.
To observe how the kinetics of MDSC accumulation are affected by the presence or absence of RAGE, mice from all strains were sacked at 20, 24, 26, and 35 weeks of age and their splenocytes assayed for MDSC frequency. As precursor lesions develop in the pancreas, the KC mice begin to accumulate high levels of MDSCs at 26 weeks of age. This is significantly elevated when compared with KCR mice (p=0.049) at the same time point. This becomes further accentuated by 35 weeks of age (p=0.023) (Figure 2C). The absence of RAGE is associated with a significant lack of MDSC accumulation over time in these animals. These findings suggest that RAGE is critical for in vivo MDSC accumulation during pancreatic carcinogenesis. Of note, a study by Connolly et al using the Kras-driven pancreatic neoplasia model with the additional p48 mutation found that splenic CD11b+Gr1+ expansion was not evident until 9 months of age and that MDSCs isolated from non-tumor tissue did not exhibit a suppressive phenotype (29).
RAGE is dispensable for the differentiation and suppressor activity of MDSC
The failure of MDSCs to accumulate within the spleens of KCR mice could be due to a requirement for RAGE to develop MDSCs within the bone marrow compartment. To determine if RAGE is critical for either the differentiation of MDSCs from hematopoietic progenitor cells or their immune inhibitory activity, both KC and KCR mice were sacrificed at 20 weeks of age and their spleens harvested for MDSC phenotyping and functional analysis. At 20 weeks of age, both KC and KCR mice exhibit small but measurable CD11b+Gr1+ MDSC populations as assessed by flow cytometry. These cells are present at levels comparable to WT and RAGE−/− animals (2-3%) (Figure 3A). These cells were stained for cell surface expression of the MDSC marker, IL-4Rα and the mature macrophage marker, F4/80 which is absent or expressed at low levels on MDSCs, which typically display an immature myeloid phenotype (18, 25). These cells were also stained for intracellular expression of iNOS/NOS Type II and arginase-I which facilitate the suppressive function of MDSCs on effector and helper T cells (30, 31). CD11b+Gr1+ cells from both the KC and KCR strains expressed IL-4Rα, iNOS/NOS Type II, and arginase-I (Figure 3B) Interestingly, MDSCs derived from the spleens of KCR mice exhibited higher expression of both IL-4Rα and arginase-I than those derived from KC mice (Figure 3A and B). As expected, MDSCs from neither strain expressed F4/80 (Figure 3B).
To confirm that these cells which phenotypically resembled MDSCs were capable of suppressing T cell activity and to determine if RAGE was required for suppressor function, they were isolated by magnetically activated cell sorting based on their positivity of Gr1 and evaluated in an in vitro co-culture assay. Bulk WT splenocytes were stimulated with αCD3;αCD28 activator beads alone or in a co-culture with the isolated MDSCs from both the KC and KCR strains at a ratio of 1:1 (“MDSC [low]”) or 1:2 (“MDSC [hi]”). After 96 hours of co-culture, the resultant supernatants were harvested and analyzed by ELISA for levels of the proliferative T cell survival cytokine, IL-2 and the Th1-polarizing cytokine IFNγ as indicators of relative T cell proliferation and activity. As expected, stimulation with αCD3;αCD28 activator beads induced significant secretion of both IL-2 (p<0.01) and IFNγ (p<0.05) from WT splenocytes (Figure 3C). When co-cultured with MDSCs at either ratio isolated from KC or KCR mice, the secretion of these cytokines was inhibited and remained at non-stimulated levels. Importantly, there was no significant difference between MDSCs derived from either strain in terms of their suppressive capacity (Figure 3C). To further validate the lack of a requirement for RAGE in MDSC hematopoiesis, MDSCs were generated in vitro from both non-tumor bearing WT and RAGE−/− mice. CD11b+Gr1+ cells were generated to equal extents (50%) in marrow from both strains (Figure S1A). Furthermore, these cells were sorted and evaluated for suppressor function and previously described. Cells from both strains were equally suppressive (Figure S1B). These findings suggest that RAGE is not critical for the development of MDSCs in vivo or their ability to exert suppressor function in vitro. Moreover, the failure of KCR mice to accumulate splenic MDSCs during pancreatic carcinogenesis is not due to an impaired ability to generate functioning MDSCs.
Splenic myeloid cells derived from KCR mice exhibit a mature phenotype and do not exhibit suppressor function
A lack of MDSC accumulation during pancreatic carcinogenesis in KCR mice could be due to a failure in MDSC recruitment. Alternatively, the absence of RAGE could result in the induction of the differentiation of MDSCs from an immature (Gr1+) to a mature (Gr1−) phenotype or a failure to prevent such a differentiation from occurring. To investigate the latter hypothesis, 35 week old WT, RAGE-null, KC, and KCR mice were sacked and their splenocytes analyzed for expression of CD11b and the mature macrophage marker, F4/80 by flow cytometry. In both WT and RAGE-null mice, populations of CD11b+F4/80+ and CD11b+F4/80− cells are discernable (Figure 4A). In the KC strain, as expected, there is a significant increase in the frequencies of CD11b+F4/80− cells which are mostly Gr1+ MDSCs (Figure 4A and B). Interestingly, compared with the KC strain, the majority of cells in the KCR spleen which stained positive for CD11b now expressed F4/80, suggesting these cells now exhibited a mature myeloid phenotype (Figure 4A and B) (32).
Figure 4. Splenic myeloid cells derived from KCR mice exhibit a mature phenotype and do not exhibit suppressor function.

(A) Representative flow cytometry diagrams of splenocytes from 35 week old KC (n=6) and KCR (n=5) mice stained for the cell surface expression of the myeloid-lineage marker CD11b and the mature macrophage marker F4/80. (B) Scatter plot depicting the frequencies of splenic myeloid cells exhibiting immature phenotypes identified by the expression of CD11b and the lack of F4/80 expression in mice from panel A. (C) CD11b+ splenocytes from mice in panels A and B were magnetically separated and co-cultured with bulk wild-type splenocytes stimulated with αCD3/αCD28 activator beads at ratios of 1:1 (CD11b+ [low]) and 2:1 (CD11b+ [hi]). Graphs depict IL-2 and IFNg levels detected in the resultant supernatants by ELISA (+/− SEM, *p<0.05).
To determine if these CD11b+F4/80+ cells which comprised the majority of CD11b+ cells in the KCR spleen had lost their capacity to suppress effector immune cells, they were positively selected based on the expression of CD11b by MACS. CD11b+ cells from KC spleens were also isolated to compare with the F4/80+-enriched cells derived from the KCR mice. Again these cells were co-cultered in vitro with bulk WT splenocytes that were stimulated with αCD3;αCD28 activator beads at 2 ratios for 96 hours as described previously for Figure 3. Resultant supernatants were then harvested and analyzed by ELISA for secreted IL-2 and IFNγ As expected, CD11b+ myeloid cells derived from the KC strain which were enriched for Gr1+F4/80− cells inhibited the secretion of both IL-2 and IFNγ(Figure 4C). In contrast, myeloid cells isolated from the KCR strain at the same time point and enriched for Gr1−F4/80+ cells failed to suppress activated splenocyte cytokine secretion significantly (p<0.05). This suggests that in lieu of the MDSCs that accumulate in KC mice with the progression of pancreatic neoplasia, in the absence of RAGE, myeloid cells are either recruited or differentiated into mature F4/80+ cells which lack suppressor activity and could contribute to both the paucity of “classical” MDSCs and the inhibited occurrence of malignant pancreatic lesions.
RAGE promotes a regulatory milieu within the emerging pancreatic tumor micro-environment
The presence or absence of host regulatory immune cell infiltrate in the pancreatic tumor micro-environment is in part regulated by factors present both within tumor tissue and circulating in the peripheral blood (33, 34). To investigate whether there were differences in the tumor micro-environments of both KC and KCR mice that might contribute to both the failure to accumulate MDSCs in the absence of RAGE and the influx of mature, non-suppressive myeloid cells in KCR mice (Figure 4), pancreatic tissue was harvested from 35 week old WT, RAGE-null, KC, and KCR mice and analyzed for mRNA levels of chemokines known to attract either immunoregulatory (CCL22) or immunostimulatory (CXCL10) immune cells by quantitative PCR (35, 36). To determine what the relative levels of mRNA of these chemokines are in relation to each other, the ratio of CCL22 to CXCL10 was obtained. As expected, KC mice demonstrate a significant increase in CCL22:CXCL10 indicating a pronounced regulatory chemokine milieu in the pancreata of these mice. In the absence of RAGE, this ratio is significantly diminished (p=0.037), suggested that the emerging tumor micro-environments of these animals is substantially less likely to recruit regulatory infiltrate (Figure 5A).
Figure 5. RAGE is associated with a regulatory milieu within the emerging pancreatic tumor micro-environment.

(A) Scatter plot depicting the ratio of the relative quantity (RQ) of CCL22 to CXCL10 mRNA derived from the pancreatic tissue of wild-type (n=1), RAGE-null (n=1), KC (n=6), and KCR (n=4) mice at 35 weeks of age as measured by quantitative PCR. (B) Left: Scatter plot depicting IL-6 levels detected in the serum of the peripheral blood in mice from panel A by ELISA (+/− SEM). Right: Scatter plot comparing frequencies of splenic MDSCs in mice from panel A (all strains) with <100pg/ml detectable serum IL-6 with those with >100pg/ml serum IL-6. (C) Representative flow diagrams of cells expressing CD11b, Gr1, and F4/80 in the pancreata of KC and KCR mice at 35 weeks.
We have recently reported that ablation of RAGE in the emerging tumor micro-environment attenuatets STAT-3/IL-6 pathway activation in the KC model (22). IL-6, a cytokine often associated with epithelial-derived cancers and general tissue damage, has also been implicated in MDSC recruitment and persistence through a STAT3-dependent signaling pathway (37-39). To compare serum IL-6 levels between the KC and KCR strains, peripheral blood was obtained via direct cardiac puncture of 35 week old mice and assayed for IL-6 concentration by ELISA. While serum IL-6 levels were relatively low in WT and RAGE-null mice, KC mice exhibited a significant increase in the concentration of the cytokine in their peripheral blood. This was not the case for the KCR strain, which exhibited significantly diminished levels of IL-6 (p=0.038) which were comparable with both WT and RAGE-null mice (Figure 5B left). Further illustrating the role of IL-6 in the recruitment and survival of MDSCs, mice were divided into two groups, regardless of strain: those with <100pg/ml serum IL-6 and those with >100pg/ml IL-6. The MDSC frequencies in the spleens from mice from these two groups were then assayed by flow cytometry. Mice with >100pg/ml of serum IL-6 were significantly more likely to have greater splenic MDSC accumulation (p=0.022) (Figure 5B right). Single cell suspensions derived from the pancreata of 35 week old mice from both strains were examined for MDSC frequencies as well as expression of F4/80 (Figure 5C). A failure to accumulate MDSCs in response to endogenous carcinogenesis was observed in the KCR strain. This was in contrast with the KC strain, which showed a demonstrable expansion of these cells as has been previously reported (17). Interestingly, nearly uniform F4/80 expression was observed in CD11b+ cells in the KCR strain, but not in KC mice, where the vast majority of CD11b+ cells remained negative for F4/80. Taken together, these findings suggest that RAGE has a critical role in the promotion of an immunoregulatory milieu within the emergent tumor micro-environment during pancreatic carcinogenesis. The lack of the regulatory chemokine CCL22 in relation to the immunostimulatory chemokine CXCL10, coupled with the normal serum IL-6 levels in response to the diminished incidence and timing of Kras-mediated pancreatic neoplasia could contribute to inhibited MDSC accumulation in the absence of RAGE expression.
DISCUSSION
In this study, we demonstrate that RAGE plays a critical role in promoting pancreatic neoplasia and the resultant immunosuppressive tumor micro-environment. When RAGE is ablated in mice which spontaneously form PanIN lesions due to mutant Kras expression, the emergence and progression of these malignant lesions are substantially attenuated. Concurrently, the accumulation of splenic MDSCs that occurs over time in KC mice is limited in the absence of RAGE. Our findings suggest that the integrity of RAGE is critical for pancreatic carcinogenesis and responding MDSC accumulation. Importantly, RAGE is not required for the development of MDSCs from myelopoietic progenitor cells or their specific inhibitory activity, as MDSCs are found in both RAGE-null and KCR mouse strains and are phenotypically and functionally intact. Interestingly, during pancreatic neoplasia in the absence of RAGE, a substantial majority of myeloid cells (CD11b+) exhibit a more mature phenotype manifested by the expression of the mature macrophage marker F4/80 and a loss of Gr1 expression (18, 32). When isolated and evaluated in an in vitro suppression assay, these cells had notably lost their inhibitory activity. This suggested that RAGE is a factor in either myeloid cell plasticity or determining which myeloid subsets respond to emergent pancreatic carcinogenesis. However, alternative explanations for the maturation of myeloid cells towards a more mature F4/80+ phenotype exist. Inability to recruit more suppressive cells types or apoptosis of other myeloid subsets could explain this observation. Additionally, the chemokine milieu in the pancreatic tumor micro-environment was markedly less regulatory in KCR mice illustrated by the relative abundance of CXCL10 in relation to CCL22. The levels of IL-6, which have been implicated in the STAT3-dependent recruitment and retention of MDSCs (38), were also significantly decreased in the KCR strain, and these levels directly correlated with measured MDSC frequency within the emergent pancreatic tumor micro-environment.
The proinflammatory proteins S100A8/A9 induce MDSCs by interacting with RAGE and other glycoproteins on the surface of MDSCs and promote their migration via NF-κB-dependent signaling (18). In addition, RAGE overexpressed within the tumor and stromal compartments is concurrently ligated by S100A8/A9 synthesized by MDSCs which induces a regulatory chemokine tumor gene profile and serves as a positive feed-back loop for the further recruitment of MDSCs (18, 25). Our findings suggest that RAGE ablation in the emerging tumor micro-environment leads to diminished signaling through the IL-6/STAT3 pathway. It is likely that the failure to accumulate MDSCs in KCR mice is due to a combined effect of RAGE deletion in both the tumor and bone marrow compartments in the global RAGE knockout strain. Future studies will examine targeted ablation in each exlusive compartment to further elucidate these mechanisms. Other cells of myeloid lineage (particularly dendritic cells and macrophages) have been shown by our group and others to secrete another RAGE ligand, HMGB1, in response to various maturational stimuli, therefore HMGB1 secretion by MDSCs represents an additional mechanism for MDSC recruitment to be explored (40, 41).
Our observations suggest an important mechanism driving an immunosuppressive environment during early pancreatic carcinogenesis. However, several questions remain concerning the relationship between cancer-related inflammation and the accrual of MDSCs. First it is not clear if the pancreatic carcinogenesis that promotes the accumulation of MDSCs is an epiphenomenon or if the recruitment of MDSCs contributes to carcinogenesis by limiting effective immune editing. Secondly, the failure of various immunotherapies, particularly within the context of pancreatic cancer, is attributed to the presence of host regulatory immune cells like MDSCs and Tregs (6). It is not clear if targeting MDSC function in these settings may render these therapies more effective. Our results demonstrating a critical role for RAGE in MDSC accumulation in the context of pancreatic carcinogenesis identifies a novel pathway that can be explored to address these questions possibly by assessing established strategies such as IL-2 administration in this model (42). Lastly, given the role of RAGE in promoting autophagy within tumor cells and evidence of immune cell-mediated autophagy (particularly T and NK cells), it would be interesting to explore whether interactions with MDSCs modulate these observations in KCR mice (21).
Supplementary Material
ACKNOWLEDGEMENTS
We dedicate this manuscript to the memory of Angelika Bierhaus, PhD, our friend and benefactor. She was a pioneering RAGE biologist, generous and kind in her dealings with colleagues, who provided us the mice with which we completed this work and who sadly passed away on April 15, 2012 after a long and courageous battle with cancer. Angelika had a great love of life; she was generous, kind, and warm hearted. She was always full of plans for scientific endeavors, even when her disease began to take its toll. She remained steadfast that she would not be defeated, but despite recently celebrating her 50th birthday, she was aware her remaining time was short. She dedicated herself to research, but despite her incredible strength, she was not able to overcome the disease which tragically took her life.
We would also like to acknowledge the University of Pittsburgh Cancer Institute Flow and Imaging Cytometry core facility as well as the UPCI Animal Facility.
Grant Support: NIH PO1 CA 101944-04 (Michael T. Lotze, University of Pittsburgh), 5T32 CA 082084-13 (Olivera J. Finn, University of Pittsburgh), and 3P30CA047904-22S1 (Nancy Davidson)
REFERENCES
- 1.Jemal A, Siegel R, Xu J, Ward E. Cancer Statistics, 2010. CA: A Cancer Journal for Clinicians. 2010;60:277–300. doi: 10.3322/caac.20073. [DOI] [PubMed] [Google Scholar]
- 2.Siegel R, Ward E, Brawley O, Jemal A. Cancer statistics, 2011. CA: A Cancer Journal for Clinicians. 2011;61:212–236. doi: 10.3322/caac.20121. [DOI] [PubMed] [Google Scholar]
- 3.Haugk B. Pancreatic intraepithelial neoplasia - can we detect early pancreatic cancer? Histopathology. 2010;57:503–514. doi: 10.1111/j.1365-2559.2010.03610.x. [DOI] [PubMed] [Google Scholar]
- 4.Ottenhof NA, Milne ANA, Morsink FHM, Drillenburg P, ten Kate FJW, Maitra A, Offerhaus GJ. Pancreatic Intraepithelial Neoplasia and Pancreatic Tumorigenesis: Of Mice and Men. Archives of Pathology & Laboratory Medicine. 2009;133:375–381. doi: 10.5858/133.3.375. [DOI] [PubMed] [Google Scholar]
- 5.Wachsmann MB, Pop LM, Vitetta ES. Pancreatic Ductal Adenocarcinoma: A Review of Immunologic Aspects. Journal of Investigative Medicine. 2012;60:643–663. doi: 10.231/JIM.0b013e31824a4d79. 610.231/JIM.640b013e31824a31824d31879. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Beatty GL, Chiorean EG, Fishman MP, Saboury B, Teitelbaum UR, Sun W, Huhn RD, Song W, Li D, Sharp LL, Torigian DA, O’Dwyer PJ, Vonderheide RH. CD40 Agonists Alter Tumor Stroma and Show Efficacy Against Pancreatic Carcinoma in Mice and Humans. Science. 2011;331:1612–1616. doi: 10.1126/science.1198443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Kraman M, Bambrough PJ, Arnold JN, Roberts EW, Magiera L, Jones JO, Gopinathan A, Tuveson DA, Fearon DT. Suppression of Antitumor Immunity by Stromal Cells Expressing Fibroblast Activation Protein-α. Science. 2010;330:827–830. doi: 10.1126/science.1195300. [DOI] [PubMed] [Google Scholar]
- 8.Morse MA, Hall JR, Plate JM. Countering tumor-induced immunosuppression during immunotherapy for pancreatic cancer. Expert Opinion on Biological Therapy. 2009;9:331–339. doi: 10.1517/14712590802715756. [DOI] [PubMed] [Google Scholar]
- 9.Porembka M, Mitchem J, Belt B, Hsieh C-S, Lee H-M, Herndon J, Gillanders W, Linehan D, Goedegebuure P. Pancreatic adenocarcinoma induces bone marrow mobilization of myeloid-derived suppressor cells which promote primary tumor growth. Cancer Immunology, Immunotherapy. :1–13. doi: 10.1007/s00262-011-1178-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Ostrand-Rosenberg S. Myeloid-derived suppressor cells: more mechanisms for inhibiting antitumor immunity. Cancer Immunology, Immunotherapy. 2010;59:1593–1600. doi: 10.1007/s00262-010-0855-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Gabitass R, Annels N, Stocken D, Pandha H, Middleton G. Elevated myeloid-derived suppressor cells in pancreatic, esophageal and gastric cancer are an independent prognostic factor and are associated with significant elevation of the Th2 cytokine interleukin-13. Cancer Immunology, Immunotherapy. 2011;60:1419–1430. doi: 10.1007/s00262-011-1028-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Srivastava MK, Sinha P, Clements VK, Rodriguez P, Ostrand-Rosenberg S. Myeloid-Derived Suppressor Cells Inhibit T-Cell Activation by Depleting Cystine and Cysteine. Cancer Research. 2010;70:68–77. doi: 10.1158/0008-5472.CAN-09-2587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Hanson EM, Clements VK, Sinha P, Ilkovitch D, Ostrand-Rosenberg S. Myeloid-Derived Suppressor Cells Down-Regulate L-Selectin Expression on CD4+ and CD8+ T Cells. The Journal of Immunology. 2009;183:937–944. doi: 10.4049/jimmunol.0804253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Tartour E, Pere H, Maillere B, Terme M, Merillon N, Taieb J, Sandoval F, Quintin-Colonna F, Lacerda K, Karadimou A, Badoual C, Tedgui A, Fridman W, Oudard S. Angiogenesis and immunity: a bidirectional link potentially relevant for the monitoring of antiangiogenic therapy and the development of novel therapeutic combination with immunotherapy. Cancer and Metastasis Reviews. 2011;30:83–95. doi: 10.1007/s10555-011-9281-4. [DOI] [PubMed] [Google Scholar]
- 15.Lechner M, Megiel C, Russell S, Bingham B, Arger N, Woo T, Epstein A. Functional characterization of human Cd33+ And Cd11b+ myeloid-derived suppressor cell subsets induced from peripheral blood mononuclear cells co-cultured with a diverse set of human tumor cell lines. Journal of Translational Medicine. 2011;9:90. doi: 10.1186/1479-5876-9-90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Ueha S, Shand FHW, Matsushima K. Myeloid cell population dynamics in healthy and tumor-bearing mice. International Immunopharmacology. 2011;11:783–788. doi: 10.1016/j.intimp.2011.03.003. [DOI] [PubMed] [Google Scholar]
- 17.Clark CE, Hingorani SR, Mick R, Combs C, Tuveson DA, Vonderheide RH. Dynamics of the Immune Reaction to Pancreatic Cancer from Inception to Invasion. Cancer Research. 2007;67:9518–9527. doi: 10.1158/0008-5472.CAN-07-0175. [DOI] [PubMed] [Google Scholar]
- 18.Sinha P, Okoro C, Foell D, Freeze HH, Ostrand-Rosenberg S, Srikrishna G. Proinflammatory S100 Proteins Regulate the Accumulation of Myeloid-Derived Suppressor Cells. The Journal of Immunology. 2008;181:4666–4675. doi: 10.4049/jimmunol.181.7.4666. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Turovskaya O, Foell D, Sinha P, Vogl T, Newlin R, Nayak J, Nguyen M, Olsson A, Nawroth PP, Bierhaus A, Varki N, Kronenberg M, Freeze HH, Srikrishna G. RAGE, carboxylated glycans and S100A8/A9 play essential roles in colitis-associated carcinogenesis. Carcinogenesis. 2008;29:2035–2043. doi: 10.1093/carcin/bgn188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Kang R, Tang D, Schapiro NE, Livesey KM, Farkas A, Loughran P, Bierhaus A, Lotze MT, Zeh HJ. The receptor for advanced glycation end products (RAGE) sustains autophagy and limits apoptosis, promoting pancreatic tumor cell survival. Cell Death Differ. 2010;17:666–676. doi: 10.1038/cdd.2009.149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Buchser WJ, Laskow TC, Pavlik PJ, Lin H-M, Lotze MT. Cell-mediated Autophagy Promotes Cancer Cell Survival. Cancer Research. 2012 doi: 10.1158/0008-5472.CAN-11-3396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Kang R, Loux T, Tang D, Schapiro NE, Vernon P, Livesey KM, Krasinskas A, Lotze MT, Zeh HJ. The expression of the receptor for advanced glycation endproducts (RAGE) is permissive for early pancreatic neoplasia. Proceedings of the National Academy of Sciences. 2012 doi: 10.1073/pnas.1113865109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Weiner LM, Lotze MT. Tumor-Cell Death, Autophagy, and Immunity. New England Journal of Medicine. 2012;366:1156–1158. doi: 10.1056/NEJMcibr1114526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Gebhardt C, Riehl A, Durchdewald M, Németh J, Fürstenberger G, Müller-Decker K, Enk A, Arnold B, Bierhaus A, Nawroth PP, Hess J, Angel P. RAGE signaling sustains inflammation and promotes tumor development. The Journal of Experimental Medicine. 2008;205:275–285. doi: 10.1084/jem.20070679. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Ichikawa M, Williams R, Wang L, Vogl T, Srikrishna G. S100A8/A9 Activate Key Genes and Pathways in Colon Tumor Progression. Molecular Cancer Research. 2011;9:133–148. doi: 10.1158/1541-7786.MCR-10-0394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Cheng P, Corzo CA, Luetteke N, Yu B, Nagaraj S, Bui MM, Ortiz M, Nacken W, Sorg C, Vogl T, Roth J, Gabrilovich DI. Inhibition of dendritic cell differentiation and accumulation of myeloid-derived suppressor cells in cancer is regulated by S100A9 protein. The Journal of Experimental Medicine. 2008;205:2235–2249. doi: 10.1084/jem.20080132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Liliensiek B, Weigand MA, Bierhaus A, Nicklas W, Kasper M, Hofer S, Plachky J, Grone HJ, Kurschus FC, Schmidt AM, Yan SD, Martin E, Schleicher E, Stern DM, Hammerling GG, Nawroth PP, Arnold B. Receptor for advanced glycation end products (RAGE) regulates sepsis but not the adaptive immune response. J Clin Invest. 2004;113:1641–1650. doi: 10.1172/JCI18704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Highfill SL, Rodriguez PC, Zhou Q, Goetz CA, Koehn BH, Veenstra R, Taylor PA, Panoskaltsis-Mortari A, Serody JS, Munn DH, Tolar J, Ochoa AC, Blazar BR. Bone marrow myeloid-derived suppressor cells (MDSCs) inhibit graft-versus-host disease (GVHD) via an arginase-1-dependent mechanism that is up-regulated by interleukin-13. Blood. 2010;116:5738–5747. doi: 10.1182/blood-2010-06-287839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Connolly MK, Mallen-St. Clair J, Bedrosian AS, Malhotra A, Vera V, Ibrahim J, Henning J, Pachter HL, Bar-Sagi D, Frey AB, Miller G. Distinct populations of metastases-enabling myeloid cells expand in the liver of mice harboring invasive and preinvasive intra-abdominal tumor. Journal of Leukocyte Biology. 2010;87:713–725. doi: 10.1189/jlb.0909607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Capuano G, Rigamonti N, Grioni M, Freschi M, Bellone M. Modulators of arginine metabolism support cancer immunosurveillance. BMC Immunology. 2009;10:1. doi: 10.1186/1471-2172-10-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Mundy-Bosse BL, Lesinski GB, Jaime-Ramirez AC, Benninger K, Khan M, Kuppusamy P, Guenterberg K, Kondadasula SV, Chaudhury AR, La Perle KM, Kreiner M, Young G, Guttridge DC, Carson WE. Myeloid-Derived Suppressor Cell Inhibition of the IFN Response in Tumor-Bearing Mice. Cancer Research. 2011;71:5101–5110. doi: 10.1158/0008-5472.CAN-10-2670. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Leenen PJM, de Bruijn MFTR, Voerman JSA, Campbell PA, van Ewijk W. Markers of mouse macrophage development detected by monoclonal antibodies. Journal of Immunological Methods. 1994;174:5–19. doi: 10.1016/0022-1759(94)90005-1. [DOI] [PubMed] [Google Scholar]
- 33.Mukaida N, Baba T. Chemokines in tumor development and progression. Experimental Cell Research. 2012;318:95–102. doi: 10.1016/j.yexcr.2011.10.012. [DOI] [PubMed] [Google Scholar]
- 34.Sansone P, Bromberg J. Targeting the Interleukin-6/Jak/Stat Pathway in Human Malignancies. Journal of Clinical Oncology. 2012;30:1005–1014. doi: 10.1200/JCO.2010.31.8907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Nishikawa H, Sakaguchi S. Regulatory T cells in tumor immunity. International Journal of Cancer. 2010;127:759–767. doi: 10.1002/ijc.25429. [DOI] [PubMed] [Google Scholar]
- 36.Ben-Baruch A. The multifaceted roles of chemokines in malignancy. Cancer and Metastasis Reviews. 2006;25:357–371. doi: 10.1007/s10555-006-9003-5. [DOI] [PubMed] [Google Scholar]
- 37.Wu L, Du H, Li Y, Qu P, Yan C. Signal Transducer and Activator of Transcription 3 (Stat3C) Promotes Myeloid-Derived Suppressor Cell Expansion and Immune Suppression during Lung Tumorigenesis. The American Journal of Pathology. 2011;179:2131–2141. doi: 10.1016/j.ajpath.2011.06.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Mundy-Bosse B, Young G, Bauer T, Binkley E, Bloomston M, Bill M, Bekaii-Saab T, Carson W, Lesinski G. Distinct myeloid suppressor cell subsets correlate with plasma IL-6 and IL-10 and reduced interferon-alpha signaling in CD4+ T cells from patients with GI malignancy. Cancer Immunology, Immunotherapy. 2011;60:1269–1279. doi: 10.1007/s00262-011-1029-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Chalmin F, Ladoire S, Mignot G, Vincent J, Bruchard M, Remy-Martin J-P, Boireau W, Rouleau A, Simon B, Lanneau D, De Thonel A, Multhoff G, Hamman A, Martin F, Chauffert B, Solary E, Zitvogel L, Garrido C, Ryffel B, Borg C, Apetoh L, Rébé C, Ghiringhelli F. Membrane-associated Hsp72 from tumor-derived exosomes mediates STAT3-dependent immunosuppressive function of mouse and human myeloid-derived suppressor cells. The Journal of Clinical Investigation. 2010;120:457–471. doi: 10.1172/JCI40483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Davé SH, Tilstra JS, Matsuoka K, Li F, DeMarco RA, Beer-Stolz D, Sepulveda AR, Fink MP, Lotze MT, Plevy SE. Ethyl pyruvate decreases HMGB1 release and ameliorates murine colitis. Journal of Leukocyte Biology. 2009;86:633–643. doi: 10.1189/jlb.1008662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Campana L, Bosurgi L, Bianchi ME, Manfredi AA, Rovere-Querini P. Requirement of HMGB1 for stromal cell-derived factor-1/CXCL12-dependent migration of macrophages and dendritic cells. Journal of Leukocyte Biology. 2009;86:609–615. doi: 10.1189/jlb.0908576. [DOI] [PubMed] [Google Scholar]
- 42.Liang X, de Vera ME, Buchser WJ, Romo de Vivar Chavez A, Loughran P, Beer-Stolz D, Basse P, Wang T, van Houten B, Zeh HJ, Lotze M. Inhibiting Autophagy During Interleukin 2 Immunotherapy Promotes Long Term Tumor Regression. Cancer Research. 2012 doi: 10.1158/0008-5472.CAN-12-0320. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.