

Abstract
Proteins carry out crucial tasks in organisms by exerting functions elicited from their specific three dimensional folds. Although the native structures of polypeptides fulfill many purposes, it is now recognized that most proteins can adopt an alternative assembly of beta-sheet rich amyloid. Insoluble amyloid fibrils are initially associated with multiple human ailments, but they are increasingly shown as functional players participating in various important cellular processes. In addition, amyloid deposited in patient tissues contains nonproteinaceous components, such as nucleic acids and glycosaminoglycans (GAGs). These cofactors can facilitate the formation of amyloid, resulting in the generation of different types of insoluble precipitates. By taking advantage of our understanding how proteins misfold via an intermediate stage of soluble amyloid precursor, we have devised a method to convert native proteins to amyloid fibrils in vitro. This approach allows one to prepare amyloid in large quantities, examine the properties of amyloid generated from specific proteins, and evaluate the structural changes accompanying the conversion.
Keywords: Biochemistry, Issue 82, amyloid, soluble protein oligomer, amyloid precursor, protein misfolding, amyloid fibril, protein aggregate
Introduction
Proteins are the most abundant biological macromolecules present in all types of cells. They occur in a great variety of sizes, structures, and post-translational modifications, and fulfill an enormous range of important biological functions when in their native forms. More than two dozens of aberrant polypeptides have been implicated in numerous human pathological conditions, such as Alzheimer's disease, Parkinson disease, and Type 2 Diabetes1-4. The terminal misfolded proteins accumulate as amyloid fibrils the insoluble stable aggregates that occur extracellularly or intracellularly.
Despite the implication of specific proteins in certain diseases, increasing evidence supports the notion that all polypeptides have intrinsic properties that enable amyloid transformation. A genome-wide sequence survey identified the “amylome”, by which amyloid-prone fragments constitute roughly 15% of all coding peptide segments from E. coli to humans5. Accordingly, an increasing number of functional amyloids, which participate in various important cellular processes, have been discovered in recent years. For example, bacteria assemble amyloids to form biofilm and spore structures that are critical for their survival and pathogenesis6-9. Peptide hormones form amyloid deposits during storage within mammalian secretory granules before being released, further implying that the protein amyloid form can serve beneficial biological functions6,10,11. Moreover, proteins can also assemble into amyloid fibrils to relay critical cellular signals12,13.
For many years, the most studied amyloids are prepared from individual peptides that are associated with human diseases, such as amyloid beta or prion peptide. These peptides usually lack well defined structure and spontaneously form amyloid in solution over time, a process mediated by a partially misfolded intermediate that serves as the precursor of insoluble amyloid. The precursor of amyloid is also called soluble protein oligomer, which is rare and unstable when generated from natural peptides14,15. However, as described earlier, almost any protein in principle can adopt amyloid conformation under the appropriate conditions. We recently established a method to prepare stabilized soluble oligomers of native proteins, which readily form amyloid in the presence of various cofactors16. The resulting amyloid fibrils reflect the complex nature of the terminal protein misfolding aggregates17,18. Here we use the term protein-only amyloid to refer the protein fibrils without cofactors and hybrid amyloid for the fibrils containing nonproteinaceous components.
Previously, various methods have been developed to generate amyloid in vitro by applying conditions known to favor protein misfolding, such as high temperature, hydrophobic environment, and pH variations19-23. However, they associate with various problems, such as low efficiency, reversible folding, large batch variations or slow kinetics. The approach described here is reproducible, easy to scale up, and allows one to precisely control the timing and condition of amyloid conversion.
Protocol
1. Prepare MES Buffer
Use MES buffer (0.1 M 2-(N-morpholino)ethanesulfonic acid and 0.9 M NaCl) to prepare both protein-only amyloid as well as stabilized soluble protein oligomers. Weigh 1.95 g of MES and 5.27 g of NaCl. Add H2O and adjust pH to 4.7. Make up the final volume to 100 ml. Store at 4º C for up to 6 months.
2. Prepare Protein-only Amyloid Directly from Native Proteins
Weigh protein of choice (any native protein or peptide) and reconstitute to 10 mg/ml in MES buffer.
Incubate at 65 ºC in a waterbath for 4 hr. By the end of the period, white precipitate is visible in solution.
Neutralize the sample with 10% (v/v) of Tris-HCl (1 M, pH 10.5).
Dialyze the protein in a dialysis cassette overnight to exchange to the desired buffer for further analysis.
A control sample of native protein can be prepared by following steps 2.1, 2.3, and 2.4 without heat precipitation (step 2.2).
3. Prepare Hybrid Amyloid from Stabilized Soluble Oligomers of Native Proteins
- Prepare stabilized soluble protein oligomers
- Weigh protein of choice and reconstitute to 10 mg/ml in MES buffer. Leave at room temperature for 15 min.
- Weigh 1-ethyl-3-[3-dimethylaminopropyl]carbodiimide hydrochloride (EDC) and prepare a fresh stock of 20 mg/ml in MES buffer.
- Mix protein and EDC solutions at a ratio of 2:5 (v/v), i.e. 2 mg of protein for every 10 mg of EDC.
- Incubate at room temperature for 2 hr.
- Neutralize the sample with 10% (v/v) of Tris-HCl (1 M, pH 10.5).
- Dialyze the protein in a dialysis cassette overnight to exchange to the desired buffer for further analysis.
- Store the stabilized soluble protein oligomer at 4 ºC up to 4 weeks.
- Prepare hybrid amyloids from stabilized soluble protein oligomer.
- To prepare DNA-containing amyloid, mix DNA and stabilized soluble protein oligomer at 1:1 ratio (w/w). Leave at 4 ºC for at least 2 hr. During this period, insoluble precipitate is visible in solution when the initial protein concentration is 0.5 mg/ml or higher.
- To prepare RNA-containing amyloid, mix RNA and stabilized soluble protein oligomer at 1:1 ratio (w/w). Leave at 4 ºC for at least 2 hr. During this period, insoluble precipitate is visible in solution when the initial protein concentration is 0.5 mg/ml or higher.
- To prepare heparin-containing amyloid, mix heparin and stabilized soluble protein oligomer at 1:1 ratio (w/w). Leave at 4 ºC for at least 2 hrs. During this period, insoluble precipitate is visible in solution when the initial protein concentration is 0.5 mg/ml or higher.
Representative Results
The ability to convert native proteins directly to amyloid relies on the conformational change incurred at high temperature in the MES buffer. Precipitation in the solution is a good indication of protein aggregation and possibly amyloid formation. On the other hand, to allow soluble protein oligomer to stabilize, EDC is used to crosslink the proteins to solidify the oligomeric conformation induced in MES buffer. As a result, the stabilized protein oligomers are multimeric proteins (Figure 1), distinct from monomeric native protein or heat-induced aggregates when separated by SDS-PAGE.
The stabilized soluble protein oligomers are steady in solution, thus the amyloid conversion by cofactors can be performed separately at one's convenience in a desired way to control buffer condition, relative ratio of protein to cofactor or other parameters. The process of oligomer to amyloid conversion happens rapidly, thus one should not be surprised to see insoluble precipitates formed within minutes of mixing with nucleic acids or GAGs. To ensure the quality of stabilized soluble protein oligomers, one can perform several simple tests. As characterized previously16, stabilized soluble protein oligomers can readily bind to nucleic acids, including both DNA and RNA, in a sequence-independent manner. Therefore, a gel shift assay can be performed to visualize the direct interaction between the stabilized soluble protein oligomers and DNA, which would result in the retarded migration of DNA on an agarose gel (Figure 2). Cytotoxicity is an archetypical character of soluble protein oligomers14. We have shown before that the stabilized soluble protein oligomers dose-dependently induce cell death that is detectible by FACS, trypan blue or CellTiter Blue16. As shown in Figure 3, RPMI 8226 cells are extremely susceptible to soluble protein oligomer-induced death that is measured by propidium iodide staining.
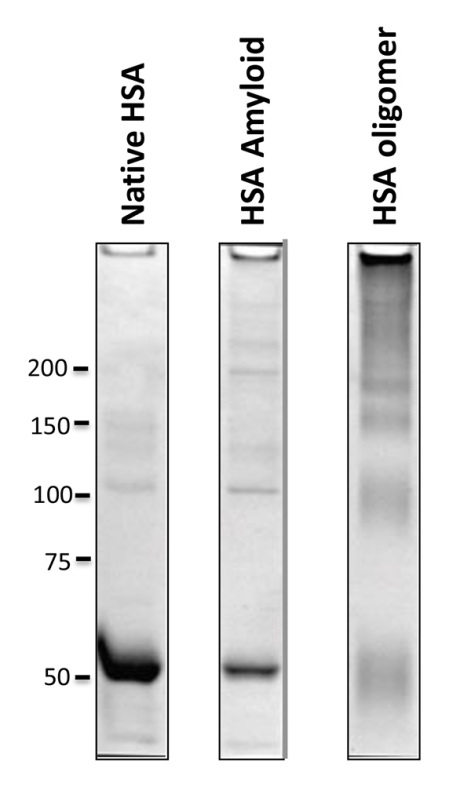 Figure 1. Molecular sizes of amyloid, amyloid precursor vs native protein. Showing human serum albumin (HSA) as an example, native HSA and HSA amyloid prepared by step 2 migrate on SDS-PAGE as monomers. Stabilized soluble HSA oligomer prepared by step 3.1 contains multiple bands that represent different oligomeric forms of HSA (right lane). Indicated on the left are protein molecular weight markers in kDa. Click here to view larger image.
Figure 1. Molecular sizes of amyloid, amyloid precursor vs native protein. Showing human serum albumin (HSA) as an example, native HSA and HSA amyloid prepared by step 2 migrate on SDS-PAGE as monomers. Stabilized soluble HSA oligomer prepared by step 3.1 contains multiple bands that represent different oligomeric forms of HSA (right lane). Indicated on the left are protein molecular weight markers in kDa. Click here to view larger image.
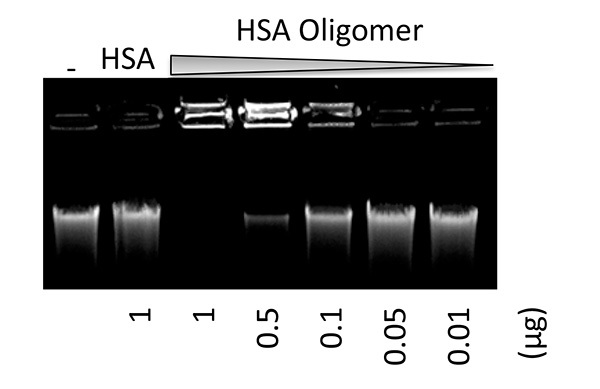 Figure 2. Binding of soluble protein oligomer with DNA. Genomic DNA (0.5 mg) from salmon sperm was mixed with different amounts of stabilized soluble HSA oligomer for 30 min. Subsequently the samples were separated on 1% agarose gel. In a control reaction, native HSA (1 mg) was mixed with DNA. The result demonstrates a general phenomenon of stabilized protein oligomer which gains ability to interact with nucleic acids, in contrast with its native form16. Click here to view larger image.
Figure 2. Binding of soluble protein oligomer with DNA. Genomic DNA (0.5 mg) from salmon sperm was mixed with different amounts of stabilized soluble HSA oligomer for 30 min. Subsequently the samples were separated on 1% agarose gel. In a control reaction, native HSA (1 mg) was mixed with DNA. The result demonstrates a general phenomenon of stabilized protein oligomer which gains ability to interact with nucleic acids, in contrast with its native form16. Click here to view larger image.
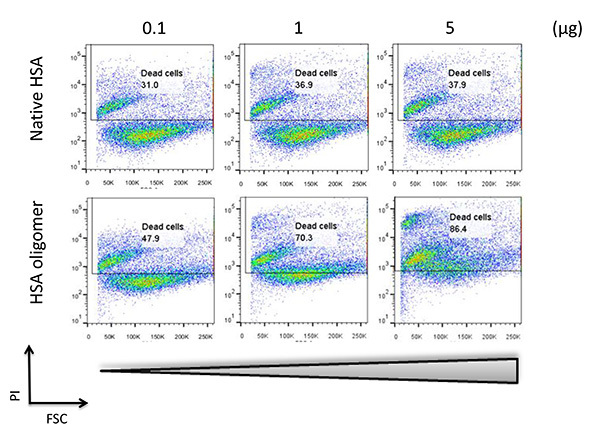 Figure 3. Cytotoxicity of soluble protein oligomer. Different amounts of native or stabilized soluble HSA oligomer were incubated with RPMI 8226 cells for 30 min. The extent of cell death was analyzed by FACS after propidium iodide (PI) staining. The percentage of dead cell population is indicated. Click here to view larger image.
Figure 3. Cytotoxicity of soluble protein oligomer. Different amounts of native or stabilized soluble HSA oligomer were incubated with RPMI 8226 cells for 30 min. The extent of cell death was analyzed by FACS after propidium iodide (PI) staining. The percentage of dead cell population is indicated. Click here to view larger image.
Discussion
The method described here offers a rapid and flexible means to prepare amyloid fibrils in vitro from virtually any protein or peptide of choice. Several different types of amyloid can be reliably prepared - protein-only fibrils or hybrid aggregates containing DNA, RNA, or glycosaminoglycans. The procedures involved are simple, straight forward, and do not require advanced technical training. The amyloid prepared by this method is quite stable and can be safely stored at 4 ºC or frozen at -80 ºC for longer period.
One of the most critical factors to ensure the successful generation of amyloid and amyloid precursor is the appropriate acidity of MES buffer. In our experience, MES buffer with a pH range of 3-5.5 affords the successful and reproducible preparation of amyloid and amyloid precursor. At pH 6.5, the efficiency to generate amyloid precursor is greatly reduced. This is consistent with the findings from other previous reports - an acetate buffer solution at pH 5.5 with trifluoroethanol was used to induce soluble oligomers of SH3 domain from bovine phosphatidyl-inositol-3'-kinase and the amino-terminal domain of the Escherichia coli HypF protein21. Human β2-microglobulin and transthyretin form amyloid fibrils efficiently under acidic conditions in vitro 22,24,25. For functional amyloidogenic protein Pme17, a critical pH regime between 5 +/- 0.5 is required for fibril formation in a specific melanosome compartment26.
Using this method, we have successfully prepared diverse amyloids from different native proteins, including human serum albumin, bovine serum albumin, chicken ovalbumin, human immunoglobulin IgG, and mouse IgG. The insoluble aggregates obtained display several characteristic properties of amyloid: they are stained positive with amyloid-specific dyes Thioflavin T, Thioflavin S, and Congo Red16,27. In addition, fibrous structures are visible when observed under electron microscopy. These observations support the concept that almost any protein can adopt either native or amyloid conformation under the appropriate conditions.
We have found that HSA oligomers can readily form amyloid the presence of sufficient amount of nucleic acids or heparin at a cofactor: protein ratio > 1:3 16. At the same concentration, heparin is slightly more potent than DNA to inhibit the cytotoxicity of prion peptide and HSA oligomers, suggesting soluble protein oligomers may differentially interact with various cofactors. Accordingly, one can modify the method by changing the ratio of soluble protein oligomers to DNA, RNA or GAGs (Protocol 3.2) to maximize the amyloid formation. Further biophysical measurement, such as X-ray diffraction or solid state FTIR, can be performed to obtain definitive structural characteristics of the aggregated material.
One of the limitations of this method is that certain quantity of native proteins or peptides are needed to prepare the soluble protein oligomers (≥1 mg). This is dictated by the high protein concentration necessary to permit the effective aggregation of partially misfolded proteins in order to form soluble protein oligomers. Previously, protein-only amyloid has been generated by treating native proteins with high temperature, hydrophobic solvent, denaturing agents, or extreme pH exposure19-22. However, soluble protein oligomers, if detectable at all, are only formed temporarily in solution with very limited abundance. To our knowledge, the protocol described here is the only established method to obtain stabilized soluble protein oligomers, which further allows one to generate different species of amyloid fibrils by mixing with various cofactors. If amyloid fibrils do not form effectively after addition of cofactors, one should examine the properties of soluble protein oligomers by analyzing their molecular sizes and ability to bind to DNA or exert cytotoxicity (as shown in Figures 1-3). If the quality is poor, one can double-check the pH of the MES buffer and also make sure that EDC is not exposed to moisture prior to reconstitution (step 3.1.2).
This protocol allows one to prepare amyloid in large quantities, examine the biochemical properties of amyloid generated from specific proteins, and evaluate the structural changes accompanying the conversion. As functional amyloids are increasingly shown to facilitate cellular signaling events12,13, ability to generate amyloid in a controlled manner would permit detailed characterization how amyloid precursors interact with other cellular factors and whether amyloid transformation plays a role in signal transduction. In addition, amyloid fibrils represent a class of danger-associated particulates that trigger inflammation via IL-1β induction28,29 . Access to homogenous amyloid will no doubt expedite further studies on the pathogenesis of diverse human diseases.
Disclosures
We have nothing to disclose.
Acknowledgments
This work is supported by grants to W.C. from National Institutes of Health Grant AI074809 and The University of Texas M. D. Anderson Cancer Center Institutional Research Grant Program.
References
- Selkoe DJ. Folding proteins in fatal ways. Nature. 2003;426:900–904. doi: 10.1038/nature02264. [DOI] [PubMed] [Google Scholar]
- Schnabel J. Protein folding: The dark side of proteins. Nature. 2010;464:828–829. doi: 10.1038/464828a. [DOI] [PubMed] [Google Scholar]
- Goldberg AL. Protein degradation and protection against misfolded or damaged proteins. Nature. 2003;426:895–899. doi: 10.1038/nature02263. [DOI] [PubMed] [Google Scholar]
- Chiti F, Dobson CM. Protein misfolding, functional amyloid, and human disease. Annu. Rev. Biochem. 2006;75:333–366. doi: 10.1146/annurev.biochem.75.101304.123901. [DOI] [PubMed] [Google Scholar]
- Goldschmidt L, Teng PK, Riek R, Eisenberg D. Identifying the amylome, proteins capable of forming amyloid-like fibrils. Proc. Natl. Acad. Sci. U.S.A. 2010;107:3487–3492. doi: 10.1073/pnas.0915166107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fowler DM, Koulov AV, Balch WE, Kelly JW. Functional amyloid - from bacteria to humans. Trends Biochem. Sci. 2007;32:217–224. doi: 10.1016/j.tibs.2007.03.003. [DOI] [PubMed] [Google Scholar]
- Chapman MR, et al. Role of Escherichia coli Curli Operons in Directing Amyloid Fiber Formation. Science. 2002;295:851–855. doi: 10.1126/science.1067484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barnhart MM, Chapman MR. Curli Biogenesis and Function. Annu. Rev. Microbiol. 2006;60:131–147. doi: 10.1146/annurev.micro.60.080805.142106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Claessen D, et al. A n olved in aerial hyphae formation in Streptomyces coelicolor by forming amyloid-like fibrils. Genes Dev. 2003;17:1714–1726. doi: 10.1101/gad.264303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maji SK, et al. Functional Amyloids As Natural Storage of Peptide Hormones in Pituitary Secretory Granules. Science. 2009;325:328–332. doi: 10.1126/science.1173155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Badtke MP, Hammer ND, Chapman MR. Functional Amyloids Signal Their Arrival. Sci. Signal. 2009;2:pe43. doi: 10.1126/scisignal.280pe43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li J, et al. The RIP1/RIP3 Necrosome Forms a Functional Amyloid Signaling Complex Required for Programmed Necrosis. Cell. 2012;150:339–350. doi: 10.1016/j.cell.2012.06.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hou F, et al. MAVS forms functional prion-like aggregates to activate and propagate antiviral innate immune response. Cell. 2011;146:448–461. doi: 10.1016/j.cell.2011.06.041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stefani M. Biochemical and biophysical features of both oligomer/fibril and cell membrane in amyloid cytotoxicity. FEBS J. 2010;277:4602–4613. doi: 10.1111/j.1742-4658.2010.07889.x. [DOI] [PubMed] [Google Scholar]
- Glabe CG. Structural classification of toxic amyloid oligomers. J. Biol. Chem. 1074;283:29639–29643. doi: 10.1074/jbc.R800016200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Di Domizio J, et al. Binding with nucleic acids or glycosaminoglycans converts soluble protein oligomers to amyloid. J. Biol. Chem. 2012;287:736–747. doi: 10.1074/jbc.M111.238477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang X, Li J-P, Lijuan Z. Heparan sulfate proteoglycans in amyloidosis. Prog. Mol. Biol. Transl. Sci. 2010;93:309–334. doi: 10.1016/S1877-1173(10)93013-5. [DOI] [PubMed] [Google Scholar]
- Jiménez JS. Protein-DNA Interaction at the Origin of Neurological Diseases: A Hypothesis. J. Alzheimer's Dis. 2010;22:375–391. doi: 10.3233/JAD-2010-100189. [DOI] [PubMed] [Google Scholar]
- Bhattacharya M, Jain N, Mukhopadhyay S. Insights into the Mechanism of Aggregation and Fibril Formation from Bovine Serum Albumin. J. Phys. Chem. B. 2011;115:4195–4205. doi: 10.1021/jp111528c. [DOI] [PubMed] [Google Scholar]
- Vetri V, et al. Bovine Serum Albumin protofibril-like aggregates formation: Solo but not simple mechanism. Arch. Biochem. Biophysics. 2011;508:13–24. doi: 10.1016/j.abb.2011.01.024. [DOI] [PubMed] [Google Scholar]
- Bucciantini M, et al. Inherent toxicity of aggregates implies a common mechanism for protein misfolding diseases. Nature. 2002;416:507–511. doi: 10.1038/416507a. [DOI] [PubMed] [Google Scholar]
- McParland VJ, Kalverda AP, Homans SW, Radford SE. Structural properties of an amyloid precursor of b2-microglobulin. Nat. Struct. Mol. Biol. 2002;9:326–331. doi: 10.1038/nsb791. [DOI] [PubMed] [Google Scholar]
- Chiti F, et al. Designing conditions for in vitro formation of amyloid protofilaments and fibrils. Proc. Natl. Acad. Sci. U.S.A. 1999;96:3590–3594. doi: 10.1073/pnas.96.7.3590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McParland VJ, et al. Partially Unfolded States of b2-Microglobulin and Amyloid Formation in vitro. Biochemistry. 2000;39:8735–8746. doi: 10.1021/bi000276j. [DOI] [PubMed] [Google Scholar]
- Liu K, Cho HS, Lashuel HA, Kelly JW, Wemmer DE. A glimpse of a possible amyloidogenic intermediate of transthyretin. Nat. Struct. Mol. Biol. 2000;7:754–757. doi: 10.1038/78980. [DOI] [PubMed] [Google Scholar]
- Pfefferkorn CM, McGlinchey RP, Lee JC. Effects of pH on aggregation kinetics of the repeat domain of a functional amyloid. Pmel17. Proc. Natl. Acad. Sci. U.S.A. 2010;107:21447–21452. doi: 10.1073/pnas.1006424107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Di Domizio J, et al. Nucleic acid-containing amyloid fibrils potently induce type I interferon and stimulate systemic autoimmunity. Proc. Natl. Acad. Sci. U.S.A. 2012;109:14550–14555. doi: 10.1073/pnas.1206923109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Halle A, Hornung V, Petzold GC, Stewart CR, Monks BG, Reinheckel T, Fitzgerald KA, Latz E, Moore KJ, Golenbock DT. The NALP3 inflammasome is involved in the innate immune response to amyloid-β. Nat. Immunol. 2008;9:857. doi: 10.1038/ni.1636. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Masters SL, O'Neill LA. Disease-associated amyloid and misfolded protein aggregates activate the inflammasome. Trends Mol. Med. 2011;17:276–282. doi: 10.1016/j.molmed.2011.01.005. [DOI] [PubMed] [Google Scholar]