

Adsorption and separation of gases is one of the primary applications of the class of materials known as metal–organic frameworks (MOFs). The role of crystallography in characterizing adsorbed gas molecules and changes in framework structure upon gas sorption is reviewed.
Keywords: metal–organic frameworks, gas sorption, framework flexibility
Abstract
Metal–organic frameworks (MOFs) are a class of porous crystalline materials of modular design. One of the primary applications of these materials is in the adsorption and separation of gases, with potential benefits to the energy, transport and medical sectors. In situ crystallography of MOFs under gas atmospheres has enabled the behaviour of the frameworks under gas loading to be investigated and has established the precise location of adsorbed gas molecules in a significant number of MOFs. This article reviews progress in such crystallographic studies, which has taken place over the past decade, but has its origins in earlier studies of zeolites, clathrates etc. The review considers studies by single-crystal or powder diffraction using either X-rays or neutrons. Features of MOFs that strongly affect gas sorption behaviour are discussed in the context of in situ crystallographic studies, specifically framework flexibility, and the presence of (organic) functional groups and unsaturated (open) metal sites within pores that can form specific interactions with gas molecules.
1. Introduction
Metal–organic frameworks (MOFs), also known as porous coordination polymers (PCPs), are a class of materials comprising metal ions or small metal clusters linked through coordination bonds via organic ligands into two-dimensional or three-dimensional periodic assemblies. This class of materials is the subject of extensive and growing worldwide research activity. The materials have been targeted for a variety of applications due to their high porosity, large surface areas, structural diversity, and both geometric and chemical tunability (Zhou et al., 2012 ▶). Prominent applications include gas storage, gaseous and other molecular separations, use as chemical sensors, and in light harvesting, biomedicine and catalysis. Each application area has been the subject of review articles (Horcajada et al., 2012 ▶; Kreno et al., 2012 ▶; Li et al., 2012 ▶; Liu et al., 2012 ▶; Sumida et al., 2012 ▶; Wang et al., 2012 ▶; Yoon et al., 2012 ▶).
The gas sorption properties of MOFs for use in storage and separation applications are considered particularly important and are frequently studied, both for new and existing MOFs (Lin et al., 2007 ▶; Collins & Zhou, 2007 ▶; Dincă & Long, 2008 ▶; Murray et al., 2009 ▶; Li et al., 2009 ▶; D’Alessandro et al., 2010 ▶; Sumida et al., 2012 ▶; Suh et al., 2012 ▶; Wu et al., 2012 ▶). Experimental studies typically involve gravimetric or volumetric adsorption measurements. These measurements allow the behaviour of the framework to be assessed and, in particular, enable both the amount of gas adsorbed at specific pressures and the overall maximum uptake achievable to be determined. Gravimetric or volumetric adsorption measurements, however, do not provide information on the location of gas molecules retained within the framework, and thereby yield only limited information on the mechanism for adsorption.
Knowledge of the sites of gas adsorption within the framework permits an understanding of the interactions occurring upon adsorption and could ultimately lead to the design of improved framework materials for gas adsorption and separation applications. Information on the binding modes of gases within MOFs has been obtained by several different methods, for example, inelastic neutron scattering (INS) for H2 adsorption in [Zn4(μ4-O)(BDC)3] (MOF-5) (Rosi et al., 2003 ▶), Raman spectroscopy for CO2 adsorption in [Zn(MeIm)2] (ZIF-8) and [Zn(SiF6)(pyz)2] (Kanoo et al., 2012 ▶; Kumari et al., 2013 ▶), IR spectroscopy studies for CO and CO2 in [Zr6(μ4-O)(μ4-OH)(BDC)6] (UiO-66) (Wiersum et al., 2011 ▶) and NO in [Fe3(μ3-O)(OH)(BDC)3] (MIL-88B(Fe)) (McKinlay et al., 2013 ▶), solid-state NMR spectroscopy for CO and CO2 adsorption in [Cu3(BTC)2] (HKUST-1) (Gul-E-Noor et al., 2013 ▶). Quantum chemical calculations have also been employed to investigate H2 adsorption (Han et al., 2009 ▶) and to characterize the mechanism involved in CO2 adsorption in the amine-functionalized MOF mmen-Mg2(dobpdc) (dobpdc4− = 4,4′-dioxido-biphenyl-3,3′-dicarboxylate; mmen = N,N′-dimethylethylenediamine; Planas et al., 2013 ▶). However, as with other areas of solid-state structural elucidation, crystallographic methods have the potential to provide the most definitive structural information.
There are many examples of crystallographic characterization of molecular guests in MOFs, in particular solvent molecules which are present in the as-synthesized materials, but such characterization is often difficult due to disorder and/or partial occupancies of the guests. Such challenges also commonly arise when determining the location of trapped gas molecules in MOFs.
Trapped gas molecules have been identified and characterized crystallographically in other porous materials prior to the development of MOFs in the mid-1990s. Work on zeolites is of particular relevance due to their similar properties to MOFs. Early examples are studies by Riley and Seff on adsorption of acetylene and carbon monoxide in various metal exchanged zeolites (Amaro & Seff, 1973 ▶; Riley & Seff, 1973 ▶, 1974 ▶; Riley et al., 1975 ▶). More recent studies of zeolites include location of noble gas atoms (Wright et al., 1984 ▶; Cho et al., 2012 ▶) and of CO2 molecules (Lozinska et al., 2012 ▶; Wong-Ng et al., 2013 ▶). All are studies by X-ray or neutron powder diffraction. Similar studies of other porous inorganic materials have also been reported, for example, N2 and H2 adsorption in a magnesium borohydride material (Filinchuk et al., 2011 ▶). Crystallographic studies on the gas hydrates have also illustrated the possibility of characterizing, by single-crystal X-ray and neutron diffraction and by powder diffraction, a variety of small gas molecules within hydrogen-bonded water clathrate cages that are templated by the included gases (Jeffrey & McMullan, 1967 ▶; Pauling & Marsh, 1952 ▶; McMullan & Kvick, 1990 ▶; Mahajan et al., 2000 ▶; Hoshikawa et al., 2006 ▶; Chakoumakos et al., 2011 ▶).
Further developments in non-ambient crystallography and, in particular, the development of crystallographic capabilities at synchrotrons (Brunelli & Fitch, 2003 ▶; Takata, 2008 ▶; Thompson et al., 2009 ▶; Jensen et al., 2010 ▶; Nowell et al., 2012 ▶; Hill, 2013 ▶) have enabled in situ gas sorption studies to be carried out on MOFs, which, due to their high porosity, often diffract quite weakly. This review focuses on such studies of MOFs that involve crystallographic location of the entrapped gas molecules. Selected examples are discussed. A more extensive list of such studies is provided in Table 1 ▶ and a list of abbreviations used in this review is included in the Appendix .
Table 1. Crystallographic studies of MOFs containing adsorbed gas molecules.
Abbreviations: NPD: neutron powder diffraction; PXRD: powder X-ray diffraction; SCND: single-crystal neutron diffraction; SCXRD: single-crystal X-ray diffraction; MEM: maximum entropy method; PDF: pair distribution function analysis; RT: room temperature. All abbreviations associated with the metal–organic framework names are provided in the Appendix .
| MOF | Formula | Gas | Anaylsis method | Temperature (K) | Gas loading | Year | Reference |
|---|---|---|---|---|---|---|---|
| [Cd(bpndc)(bpy)] | O2 | PXRD | 100 | 0.8 bar | 2008 | Tanaka et al. (2008 ▶) | |
| Ar | 110 | 3 bar | |||||
| N2 | 90 | ||||||
| COMOC-2 | [V(O)(BPDC)] | CO2 | PXRD | 233 | 0–17.5 bar | 2013 | Liu et al. (2013 ▶) |
| Co-BDP | [Co(BDP)] | N2 | PXRD | 100 | 0–15 bar | 2010 | Salles, Maurin et al. (2010 ▶) |
| [Co(HLdc)] | CO2 | PXRD | 195 | 0–1 bar | 2012 | Yang, Davies et al. (2012 ▶) | |
| CPL-1 | [Cu2(pzdc)2(pyz)] | O2 | PXRD (Rietveld/MEM method) | 300–90 | 0.8 bar | 2002 | Kitaura et al. (2002 ▶) |
| N2 | 2005 | Kitaura et al. (2005 ▶) | |||||
| Ar | |||||||
| CH4 | |||||||
| C2H2 | 393–170 | 0.1 and 1.5 bar | 2005 | Matsuda et al. (2005 ▶), Kubota et al. (2006 ▶) | |||
| H2 | 200–90 | 1.02 bar | 2005 | Kubota et al. (2005 ▶) | |||
| CPO-27-Ni (MOF-74) | [Ni2(dhtp)(OH2)2] | CO2 | PXRD | 100 | 0.2–0.5 atm | 2008 | Dietzel et al. (2008 ▶) |
| H2S | PXRD | RT | 1 atm | 2012 | Allan et al. (2012 ▶) | ||
| NO | PXRD | 298 | 1 atm | 2008 | McKinlay et al. (2008 ▶) | ||
| CPO-27-Co (MOF-74) | [Co2(dhtp)(OH2)2] | ||||||
| CPO-27-Mg (MOF-74) | [Mg2(dhtp)(OH2)2] | CO2 | NPD | 20 | 0.64 CO2/Mg | 2010 | Wu, Simmons, Srinivas et al. (2010 ▶) |
| CO2 | NPD (Rietveld/MEM method) | 20 | 0.5 and 1.75 CO2/Mg | 2011 | Queen et al. (2011 ▶) | ||
| [Cu(aip)] | CO2 | PXRD | 120 | 0–0.8 bar† | 2014 | Sato et al. (2014 ▶) | |
| PXRD (Rietveld/MEM method) | 100 | 0.5 bar | |||||
| N2 | PXRD | 120 | 0–0.8 bar† | ||||
| 77 | 0–1 bar† | ||||||
| [Cu(pyrdc)(bpp)]2 | CO2 | SCXRD | 193 | Pressure unspecified: uptake 2 CO2/Cu | 2005 | Maji et al. (2005 ▶) | |
| Cu-SIP-3 | [Cu2(OH)(C8H3O7S)] | NO | SCXRD | RT | 0.275–0.340 bar | 2010 | Allan et al. (2010 ▶) |
| DMOF | [Zn2(BDC)2(DABCO)] | CH4 | SCXRD | 90 | Pressure unspecified: uptake 3.35 CH4/Zn | 2009 | Kim et al. (2009 ▶) |
| DMOF | [Zn2(BDC)2(DABCO)]·DSB | CO2 | PXRD | 195 | 0–0.8 bar | 2011 | Yanai et al. (2011 ▶) |
| C2H2 | 0–0.6 bar | ||||||
| DMOF-(BME)2 | [Zn2(BME-BDC)2(DABCO)] | CO2 | PXRD | 195 | 0–1 bar | 2011 | Henke et al. (2011 ▶) |
| HKUST-1 | [Cu3(BTC)2] | D2 | NPD | 5 | 2–6.5 D2/Cu | 2006, 2011 | Peterson et al. (2006 ▶, 2011 ▶) |
| CD4 | NPD | 77 | 2.17–3.67 CD4/Cu | 2010 | Getzschmann et al. (2010 ▶) | ||
| CD4 | NPD | 4 | 1.1 CD4/Cu | 2010 | Wu, Simmons, Liu et al. (2010 ▶) | ||
| CO2 | NPD | 20 | 1.07–1.47 CO2/Cu | 2010 | Wu, Simmons, Srinivas et al. (2010 ▶) | ||
| Ar | NPD | 8 | 0.17 (3) Ar/Cu | 2013 | Hulvey et al. (2013 ▶) | ||
| Kr | PXRD | 140–200 | 0.075 (1)–0.374 (4) Kr/Cu (3 loadings) | 2013 | Hulvey et al. (2013 ▶) | ||
| Xe | PXRD | 240–260 | 0.072 (1)–0.315 (2) Xe/Cu (5 loadings) | 2013 | Hulvey et al. (2013 ▶) | ||
| HCu[(Cu4Cl)3(BTT)8]·3HCl | D2 | NPD | 4 | 6–30 D2/formula unit (13 Cu) | 2007 | Dincă et al. (2007 ▶) | |
| Mn3[(Mn4Cl)3(BTT)8(CH3OH)10]2 | D2 | NPD | 3.5 | 12 D2/formula unit (27 Mn) | 2006 | Dincă et al. (2006 ▶) | |
| [Mg(O2CH)2] | C2H2 | SCXRD | 90 | Pressure unspecified: uptake 0.33 C2H2/Mg (or Mn) | 2007 | Samsonenko et al. (2007 ▶) | |
| [Mn(O2CH)2] | |||||||
| MAF-2 | [Cu(etz)] | N2 | SCXRD | 93 | Pressure unspecified: uptake‡ 1.0 N2/Cu | 2008 | Zhang & Chen (2008 ▶) |
| C2H2 | 293 | 10–20 atm: uptake 1 C2H2/Cu | 2009 | Zhang & Chen (2009 ▶) | |||
| C2H2 | 123 | ≤ 0.8 bar: uptake 1 CO2 or C2H2/Cu | |||||
| CO2 | |||||||
| C2H2 | 195 | 0.05–0.8 bar: uptake 0.04–0.42 CO2 or C2H2/Cu | |||||
| CO2 | |||||||
| ZIF-8 (MAF-4) | [Zn(MeIm)2] | D2 | NPD | 3.5 | 0.5–4.67 D2/Zn | 2007 | Wu et al. (2007 ▶) |
| CD4 | NPD | 100–3.5 | 1–3 CD4/Zn | 2009 | Wu et al. (2009 ▶) | ||
| N2 | PXRD | 77 | 0.4 bar | 2011 | Fairen-Jimenez et al. (2011 ▶) | ||
| N2 | SCXRD | 423–100 | Open-flow N2 cryostat | 2012 | Zhang et al. (2012 ▶) | ||
| MAF-23 | [Zn2(BTM)2] | CO2 | SCXRD | 195 | 0–1.5 CO2/Zn | 2012 | Liao et al. (2012 ▶) |
| MAF-X7 | (Me2NH2)(Hdmf) [Co2Cl4(ppt)2] | CO2 | SCXRD | 120 | Sealed at 1 atm at 273 K | 2011 | Lin et al. (2011 ▶) |
| MCF-27 | [LiZn(BTC)] | CO2 | SCXRD | 195 | Unspecified | 2010 | Xie et al. (2010 ▶) |
| N2 | 103 | ||||||
| MFU-4l | [Zn5Cl4(BTDD)3] | Xe | PXRD (Rietveld/MEM method) | 110–150 | 0.02 bar at RT | 2012 | Soleimani-Dorcheh et al. (2012 ▶) |
| MIL-47(V) | [V(O)(BDC)] | CH4 | PXRD | 200 | 0–8.84 bar | 2010 | Rosenbach et al. (2010 ▶) |
| C3H8 | 303 | 0–8.28 bar | |||||
| CO2 | PXRD | 303 | 0–30.9 bar | 2011 | Leclerc et al. (2011 ▶) | ||
| CO2 | PXRD | 200 | 0–1.53 bar | 2010 | Salles, Jobic et al. (2010 ▶) | ||
| MIL-53(Cr) (hydrated) | [Cr(OH)(BDC)]·xH2O | CO2 | PXRD | Unspecified§ | 1–15 bar | 2006 | Llewellyn et al. (2006 ▶) |
| MIL-53(Cr) | [Cr(OH)(BDC)] | CO2 | PXRD | 293 | 0–10 bar | 2007 | Serre et al. (2007 ▶) |
| 195 | 1 bar | ||||||
| CH4 | PXRD | 303 | 0–33 bar | 2008 | Llewellyn et al. (2008 ▶) | ||
| C2H6 | 0–13.5 bar | ||||||
| C3H8 | 0–10 bar | ||||||
| C4H10 | 0–0.5 bar | ||||||
| CO2/CH4 mixture | PXRD | 303 | 0–30 bar (25:75, 50:50, 75:25) | 2009 | Hamon et al. (2009 ▶) | ||
| H2 | PXRD | 303 | 0–30 bar | 2009 | Salles et al. (2009 ▶) | ||
| MIL-53(Fe) | [Fe(OH)(BDC)] | CH4 | PXRD | 303 | 0–43 bar | 2009 | Llewellyn et al. (2009 ▶) |
| C2H6 | 0–37 bar | ||||||
| C3H8 | 0–8.6 bar | ||||||
| C4H10 | 0–2.1 bar | ||||||
| CO2 | 230 | 0–8.8 bar | 2012 | Devic et al. (2012 ▶) | |||
| [Fe(OH)(BDC-Cl)] | CH4 | PXRD | 303 | 0–38.7 bar | 2011 | Ramsahye et al. (2011 ▶) | |
| C2H6 | 0–36.3 bar | ||||||
| C3H8 | 0–8 bar | ||||||
| C4H10 | 0–2.1 bar | ||||||
| CO2 | 230 | 0–9.8 bar | 2012 | Devic et al. (2012 ▶) | |||
| [Fe(OH)(BDC-Br)] | CH4 | PXRD | 303 | 0–40.3 bar | 2011 | Ramsahye et al. (2011 ▶) | |
| C2H6 | 0–37.7 bar | ||||||
| C3H8 | 0–8.2 bar | ||||||
| C4H10 | 303–263 | 0–0.7 bar | |||||
| CO2 | 230 | 0–10.7 bar | 2012 | Devic et al. (2012 ▶) | |||
| [Fe(OH)(BDC-CH3)] | CH4 | PXRD | 303 | 0–42.0 bar | 2011 | Ramsahye et al. (2011 ▶) | |
| C2H6 | 0–38.2 bar | ||||||
| C3H8 | 0–1.2 bar | ||||||
| CO2 | 230 | 0–10 bar | 2012 | Devic et al. (2012 ▶) | |||
| [Fe(OH)(BDC-NH2)] | CO2 | PXRD | 230 | 0–11.9 bar | 2012 | ||
| [Fe(OH)(BDC-CO2H)] | CO2 | 230 | 0–11 bar | 2012 | |||
| MIL-53(Al) | [Al(OH)(BDC-NH2)] | CO2 | PXRD | 253 | 0–18 bar | 2012 | Couck et al. (2012 ▶), Serra-Crespo et al. (2012 ▶) |
| CH4 | 0–15 bar | ||||||
| [Al(OH)(BDC-F)] | CO2 | PXRD | 233–193 | 0–1.47 bar | 2013 | Biswas et al. (2013 ▶) | |
| MIL-53(Ga) | [Ga(OH)(BDC-NH2)] | CO2 | PXRD | 253 | 0–16 bar | 2012 | Serra-Crespo et al. (2012 ▶) |
| [Ga(OH)(BDC-NH2)] | CH4 | 0–11 bar | |||||
| MIL-53(In) | [In(OH)(BDC-NH2)] | CO2 | PXRD | 253 | 0–20 bar | ||
| [In(OH)(BDC-NH2)] | CH4 | 0–14 bar | |||||
| MIL-53(Sc) | [Sc(OH)(BDC)] | CO2 | PXRD | 196 | 0–0.9 bar | 2013 | Chen et al. (2013 ▶) |
| MIL-88B(Fe) | [Fe(OH)(BDC-NO2)] | NO | PXRD | RT | 1 bar | 2013 | McKinlay et al. (2013 ▶) |
| [Fe(OH)(BDC-2OH)] | NO | PXRD | RT | 1 bar | 2013 | McKinlay et al. (2013 ▶) | |
| MOF-5 | [Zn4O(BDC)3] | H2 | SCND | 300–5 | 1 atm | 2006 | Spencer et al. (2006 ▶) |
| D2 | NPD | 3.5 | 1–11.5 D2/Zn | 2005 | Yildirim & Hartman (2005 ▶) | ||
| He | PXRD | 100–500 | 1.7–150 bar | 2013 | Lock et al. (2013 ▶) | ||
| CD4 | NPD | 100–3.5 | 0.75–6 CD4/Zn | 2009 | Wu et al. (2009 ▶) | ||
| N2 | SCXRD | 293–30 | 1.25–2.5 N2 or Ar/Zn | 2005 | Rowsell, Spencer et al. (2005 ▶) | ||
| Ar | |||||||
| NOTT-202a¶ | (Me2NH2)[In(L3)] | CO2 | PXRD | 195, 273 | 0–1 bar | 2012 | Yang, Lin et al. (2012 ▶) |
| SO2 | 273 | 0–1.1 bar | 2013 | Yang et al. (2013 ▶) | |||
| NOTT-300 | [Al2(OH)2(L4)] | CO2 | PXRD | 273 | 0–1 bar | 2012 | Yang, Sun et al. (2012 ▶) |
| SO2 | |||||||
| PCN-11 | [Cu2(sbtc)] | CD4 | NPD | 4 | 2.8 CD4/Cu | 2010 | Wu, Simmons, Liu et al. (2010 ▶) |
| [Sc2(BDC)3] | CO2 | SCXRD | 235 | 1 bar | 2009 | Miller et al. (2009 ▶) | |
| H2 | 80 | 0.25 bar | |||||
| CH4 | 230 | 9 bar | |||||
| C2H6 | 230 | 5 bar | |||||
| SNU-110 | [Zn2(mpm-PBODB)2(bpy)] | CO2 | PXRD | 248 | 1 atm (CO2 stream) | 2012 | Hong & Suh (2012 ▶) |
| YO-MOF | [Zn2(L1)(L2)] | CO2 | PXRD–PDF analysis | 260–RT | 1 atm | 2010 | Mulfort et al. (2010 ▶) |
| Y(BTC) | D2 | NPD | 4 | 0.64 (5)–5.53 (3) D2/Y (6 loadings) | 2008 | Luo et al. (2008 ▶) | |
| [Zn2(Atz)2(ox)] | CO2 | SCXRD | 123–293 | 0.65 CO2/Zn | 2010 | Vaidhyanathan et al. (2010 ▶) | |
| [Zn2(btdc)2(bpy)] (threefold interpenetrated) | CO2 | PXRD | 195 | 0–1 bar† | 2010 | Bureekaew et al. (2010 ▶) | |
| [Zn2(btdc)2(bpy)] (twofold interpenetrated) | |||||||
| [Zn2(sdb)2(bpy)] | CO2 | PXRD | 195–295 | 0.1–0.9 bar | 2013 | Hijikata et al. (2013 ▶) | |
| [Zn(TCNQ-TCNQ)(bpy)] | O2 | PXRD | Unspecified | 7.5 O2/Zn | 2010 | Shimomura et al. (2010 ▶) | |
| NO | 9 NO/Zn | ||||||
Simultaneous measurement of adsorption isotherm and X-ray powder pattern.
The same study is cited by the same authors in a later report (Zhang & Chen, 2009 ▶), but listed as having an uptake of 0.5 N2/Cu (i.e. formula MAF-2·0.5N2).
Accompanying adsorption isotherms over similar pressure range are conducted at 304 K.
NOTT-202a is the desolvated form of NOTT-202, which has the formula (Me2NH2)1.75[In(L3)]1.75·12DMF·10H2O (Yang, Lin et al., 2012 ▶).
2. Early examples
The possibility of gas sorption in MOFs was recognized very early in the development of the field (Kondo et al., 1997 ▶; Li et al., 1998 ▶, 1999 ▶; Chui et al., 1999 ▶; Eddaoudi et al., 2000 ▶, 2002 ▶; Fletcher et al., 2001 ▶; Barthelet et al., 2002 ▶). The first in situ studies by crystallographic methods of gas sorption in MOFs followed soon after. In 2002, Kitagawa, Takata and coworkers used synchrotron X-ray powder diffraction to examine physisorbed O2 in the channels of [Cu(pzdc)(pyz)], CPL-1 (pzdc = 2,3-pyrazinedicarboxylate; pyz = pyrazine; Kitaura et al., 2002 ▶). The study used the maximum entropy method (MEM)/Rietveld methodology developed by Takata (Takata et al., 1995 ▶, 2001 ▶; Tanaka et al. 2002 ▶) to produce a precise electron-density map from high-resolution X-ray powder diffraction (PXRD) patterns (Takata, 2008 ▶) collected under an O2 atmosphere. The data were recorded using an O2 pressure of 80 kPa (0.8 bar) and temperatures from 90 to 300 K at the SPring-8 synchrotron. Initial removal of guest water molecules was carried out by heating under reduced pressure with no change in the framework structure. The electron density map generated from the MEM analysis was then used to identify the O2 sites within the pores (at 90 K; Fig. 1 ▶). Final Rietveld refinement yielded indices of fit R wp = 0.021 and R I = 0.039. The oxygen sites were observed as peanut-shaped electron densities in the middle of the channels, accounting for 15.8 (1) electrons based on the MEM analysis and suggesting diatomic oxygen. A ratio of one O2 molecule per Cu atom was observed, corresponding well to the adsorption isotherms recorded at 77 K, which suggested a saturation of one O2 per Cu atom. The ordering of the O2 molecules indicated that they more closely resembled O2 in the solid state rather than the liquid or gaseous state despite the experimental conditions (T > b.p. of O2), suggesting a significant confinement effect. Furthermore, the intermolecular distance between pairs of O2 molecules [3.28 (4) Å] was observed to be lower than the minimum of the Lennard–Jones potential (3.9 Å) and suggests the formation of van der Waals dimers (Fig. 2 ▶). The dimers align along the a-axis to form a one-dimensional ladder structure, leading to antiferromagnetic coupling as confirmed by magnetic susceptibility measurements.
Figure 1.

MEM electron densities of (a) anhydrous CPL-1 without O2 molecules at 120 K and (b) CPL-1 with adsorbed O2 at 90 K as an equal-density contour surface. The equicontour level is 1.0 e Å−3. (Reproduced from Kitaura et al., 2002 ▶, with permission from the AAAS.)
Figure 2.

Crystal structure of CPL-1 with adsorbed O2 at 90 K. View down the a-axis (top). View down the c-axis (bottom). (Reproduced from Kitaura et al., 2002 ▶, with permission from the AAAS.)
Further crystallographic studies have been conducted of CPL-1 under atmospheres of a number of gases, including N2, Ar, CH4, H2 and C2H2 (Kitaura et al., 2005 ▶; Kubota et al., 2005 ▶, 2006 ▶, 2007 ▶; Matsuda et al., 2005 ▶). The nitrogen uptake showed similar results to oxygen, forming van der Waals dimers in one-dimensional arrays, but the accessible pore surface for both the Ar and CH4-filled frameworks was seen to be significantly different to those of the oxygen and nitrogen-containing versions. This was suggested to result from framework flexibility and considered to be an effect of induced fitting via molecule-to-pore surface interactions. The confinement of acetylene (C2H2) within CPL-1 was even more interesting. The structure at different acetylene loadings was shown to have an intermediate and a saturated phase, which exhibit different acetylene-to-framework interactions (Fig. 3 ▶; Matsuda et al., 2005 ▶). At lower loadings, a meta-stable phase formed with an interaction between the acetylene and the two metal-coordinated carboxylate O atoms, but under saturated conditions a slight rotation of the acetylene molecules occurs, which aligns them with two uncoordinated O atoms of the carboxylates to form stronger C—H⋯O hydrogen bonds. Subsequently, the pyrazine rings of the framework are then seen to rotate. Contractions are observed in the unit cell on going from the intermediate phase to the acetylene-saturated phase, which contains acetylene at storage densities 200 times the compression limit of free acetylene. These specific guest-framework interactions provide a clear rationale for the greatly enhanced adsorption of C2H2 over CO2, particularly at low pressures. Analogous studies on hydrogen adsorption also suggest interactions with carboxylate O atoms. Although H atoms are not clearly resolved, an occupancy of 0.3H2 per site is recovered from the MEM/Rietveld analysis (Kubota et al., 2005 ▶), consistent with an estimate of adsorbed H2.
Figure 3.

Crystal structure of CPL-1 viewed along the a-axis, in its evacuated form, and with channels partially filled (intermediate phase) and filled (full adsorbed phase), showing changes in acetylene-to-framework C—H⋯O hydrogen bonding on increasing gas loading. (Reproduced from Takata, 2008 ▶, with permission from the IUCr.)
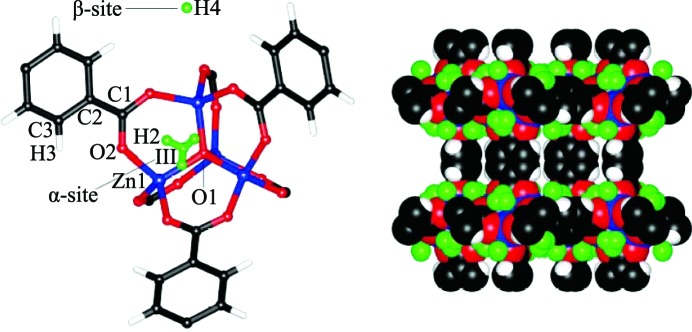
In 2005, Yaghi, Howard and coworkers reported single-crystal X-ray diffraction studies of adsorption experiments of Ar and N2 in MOF-5 (Rowsell, Spencer et al., 2005 ▶), a primitive cubic framework, comprising Zn4(μ4-O)6+ clusters linked via terephthalate ligands. Single-crystal X-ray diffraction had been previously used to determine the positions of gas molecules, specifically CO2, in a dynamically porous one-dimensional coordination polymer under a sealed gaseous atmosphere (Takamizawa et al., 2003 ▶), but not on a three-dimensional framework with such large voids and with the potential for many crystallographically independent adsorption sites. The study indicated eight different adsorption sites: five close to the framework and an additional three forming a second layer within the pores of the MOF (Fig. 4 ▶). All the sites exhibited partial occupancies and seemed to be intrinsic to the framework, with the same sites being observed in both the N2 and Ar adsorption cases, albeit with different relative populations. Diffraction studies were conducted on a laboratory diffractometer at temperatures from 293 to 30 K. By controlling the temperature and looking at the relative occupancies the authors were able to rank the preferential adsorption sites. Gas molecules could not be located at 293 K, but electron density attributable to the gas molecules was clearly evident at 30 K, which is below the freezing point of the gases. The most populated site for both gases (site α) was very close to the metal cluster, with interactions involving three carboxylate groups and three Zn atoms. Other binding sites, such as interactions with the edge of the aromatic ring (sites δ and ∊; see d and e in Fig. 4 ▶), were previously unobserved in gas sorption studies in the presence of aromatic moieties.
Figure 4.

Eight symmetry-independent adsorption sites in MOF-5, each partially occupied by Ar atoms, as identified by single-crystal X-ray diffraction at 30 K. Sites α–∊ shown in (a)–(e) are in close proximity to framework atoms. Nitrogen molecules only populate sites α, γ and ∊ at 30 K, but instead populate sites β and δ alongside α at higher temperatures. (Reproduced from Rowsell, Spencer et al., 2005 ▶, with permission from the AAAS.)
Over the last decade, crystallographic characterization of gas-containing MOFs has progressed and a number of different studies have been reported. This review will focus on selected examples to illustrate different aspects of the field. A more complete list of studies is provided in Table 1 ▶. We will begin with a survey of some examples that illustrate the different crystallographic techniques used. The review will then consider situations where the unique properties of the framework, such as its flexibility, or the presence of functional groups or open metal sites have been shown to influence the location of the gas sorption sites.
3. X-ray versus neutron diffraction
Single-crystal X-ray diffraction has been the main method for definitive structural characterization over the last century and has recently been used effectively to locate gas molecules entrapped in the pores of MOFs in several studies similar to those by Yaghi, Howard and co-workers (see above). These in situ diffraction experiments typically adopt one of two approaches, in both of which the crystal is situated inside a glass capillary. A simpler experimental design involves the capillary being filled with the desired gas to a specified pressure and sealed to maintain the atmosphere, enabling measurement at a single pressure. A change in temperature can be used to change the relative pressure (p/p 0). Alternatively, and now more commonly, the capillary is connected via tubing to a gas manifold that enables evacuation and then dosing the capillary with gas to a desired pressure. The latter arrangement has the advantage of enabling a sequence of measurements to be made at different pressures at the same temperature (as well as varying temperature), and for measurements involving several gases to be made on the same crystal. Such experimental set-ups, known as static pressure cells, are now found at a number of beamlines at synchrotron facilities worldwide. A further alternative is a flow cell, in which the gas is continuously flowed over the crystalline sample within an environmental cell during the diffraction experiment.
An illustrative example of a static cell study is that by Miller et al. carried out at the Swiss–Norwegian beamline (BM01) at ESRF (Miller et al., 2009 ▶). The study examined gas adsorption sites in a Sc(BDC) MOF (BDC = 1,4-benzenedicarboxylate or terephthalate), which is a small-pore MOF with one-dimensional channels and a poor affinity for water. Single crystals were mounted on a glass fibre and glued inside a 0.3 mm quartz capillary. The capillary was evacuated and the diffraction pattern checked under vacuum, before separate introduction of a sequence of gases. The gases studied were CO2, H2, CH4 and C2H6, each at a single gas pressure and temperature selected based on previously measured adsorption isotherms and chosen to ensure sufficient gas uptake to enable crystallographic detection of the gas molecules. The diffraction experiment was performed at 0.25 bar and 80 K for H2, 1 bar and 235 K for CO2, and at 230 K for CH4 (9 bar) and C2H6 (5 bar). The CO2 molecules and the C atoms of adsorbed CH4 and C2H6 molecules could be modelled in the pores of the MOF. The hydrocarbon gas uptake resulted in no change to the framework structure or the space group of the crystals (Fig. 5 ▶). Introduction of CO2 (Fig. 6 ▶) or H2, by contrast, resulted in a lowering of the symmetry from orthorhombic Fddd to monoclinic C2/c. This change arises due to rotation of the terephthalate groups upon adsorption of CO2 or H2, leading to adjacent channels becoming inequivalent. In the case of CO2, weak C—H⋯O interactions (H⋯O 2.87–2.98 Å) involving phenyl H atoms are observed. Calorimetric studies indicate that binding to CO2 is the strongest of the four gases, but still of modest strength (ΔH ads = −20 kJ mol−1 at room temperature). Thus, the authors note that it is the small size of the channels that enables crystallographic location of the physisorbed gas molecules even at 230 K.
Figure 5.

Location of (a) adsorbed CH4 (9 bar) and (b) adsorbed C2H6 (5 bar) in the channels of the MOF [Sc(BDC)]. Disorder of CH4 molecules is shown. One of three locations in the disordered model for C2H6 is shown. H atoms are not shown. (Reproduced from Miller et al., 2009 ▶, with permission from the American Chemical Society.)
Figure 6.

One of two inequivalent channels in [Sc(BDC)] following adsorption of CO2 (1 bar). CO2 molecules are disordered over the two sites (as shown) in this channel. (Reproduced from Miller et al., 2009 ▶, with permission from the American Chemical Society.)
Although X-ray crystallographic studies of gas adsorption in MOFs have primarily involved the use of synchrotron facilities, laboratory diffractometers have also been used (Takamizawa et al., 2003 ▶; Rowsell, Spencer et al., 2005 ▶). One such example is the work by Zhang and Chen on the absorption of N2, CO2 and C2H2 in a metal azolate framework, [Cu(etz)]n (MAF-2, Hetz = 3,5-diethyl-1,2,4-triazole; Zhang & Chen, 2009 ▶). Single crystals of MAF-2 were sealed inside glass capillaries together with the desired gas (C2H2 or CO2). X-ray data were collected at temperatures of 123 and 293 K for crystals with maximum loading (MAF-2·C2H2 and MAF-2·CO2). The position and anisotropic displacement parameters of the entrapped acetylene molecules could be refined without restraints, using the 123 K data, consistent with strong localization of the acetylene molecules. In contrast, entrapped CO2 molecules could only be modelled using crystallographic restraints. The acetylene-containing crystal structure could also be modelled at room temperature, whereas at this temperature CO2 within the pores could not. The crystallographic results are consistent with gravimetric gas adsorption measurements, which suggested a preferential uptake of C2H2 over CO2 (Zhang & Chen, 2009 ▶). Although the space group was maintained upon adsorption of the two gases, the crystallographic models showed slight deformations of the framework structure even at very low loadings of gas.
Accurate location of H atoms of the guest molecules is typically not possible using X-ray diffraction, since scattering intensity is related to electron density. The situation is particularly problematic when considering the location of H2 molecules in the pores of MOFs. Most studies involving H2 molecule location within MOFs therefore typically use neutron diffraction instead, for which the scattering length for hydrogen is more comparable to that of other elements. Two such studies using different neutron diffraction techniques to identify the hydrogen adsorption sites in MOF-5 were reported in 2005–2006 (Yildirim & Hartman, 2005 ▶; Spencer et al., 2006 ▶). Yaghi, Howard and co-workers extended their earlier single-crystal X-ray diffraction studies by using single-crystal neutron diffraction and Yildirim and Hartman used neutron powder diffraction (NPD). The single-crystal diffraction data were recorded at various temperatures and identified two different sites for hydrogen adsorption (Fig. 7 ▶). These directly correspond, respectively, to the α and β sites from the previous X-ray study using argon and nitrogen (Figs. 4 ▶ a and b). Location of H2 at the α site could be modelled at 50 K or below, including modelling of atomic positions, whereas population of the β site could only be identified at 5 K, but with individual atoms unable to be modelled. This indicates that the molecules are not highly localized, particularly at higher temperatures. The work only partially agreed with previous inelastic neutron scattering (INS) data (Rosi et al., 2003 ▶; Rowsell, Eckert & Yaghi, 2005 ▶), which suggest that the hydrogen did indeed interact with the α site, but also with the γ site identified in the previous X-ray study of Ar and N2 uptake (Rowsell, Spencer et al., 2005 ▶), although not with the β site identified in the neutron diffraction study (Spencer et al., 2006 ▶).
Figure 7.
The two H2 sites (α and β) identified by single-crystal neutron diffraction in MOF-5, shown for a single framework node (left) and for a section of the framework (right). (Reproduced from Spencer et al., 2006 ▶, http://dx.doi.org/10.1039/B511941C, with permission from the Royal Society of Chemistry ).
The use of hydrogen in neutron diffraction experiments can present difficulties due to its large incoherent scattering cross-section, a problem that is more challenging for powder diffraction than single-crystal diffraction experiments. Such a problem does not arise for deuterium, which exhibits much smaller incoherent scattering, as well as a larger (coherent) scattering length. Therefore, replacement of hydrogen by deuterium, where possible, in the materials studied is a common experimental approach in neutron diffraction. Considering gas sorption by MOFs, this can involve MOFs comprising perdeuterated ligands or the use of D2 instead of H2 (or, more generally, the use of deuterated gases; Peterson et al., 2006 ▶; Wu, Simmons, Liu et al., 2010 ▶). The NPD study by Yildirim and Hartman was conducted at 3.5 K using a perdeuterated MOF-5 sample loaded with D2 and the structure refined by Rietveld methods (Yildirim & Hartman, 2005 ▶). Analyses were carried out for several gas loadings ranging from 4D2 molecules per Zn centre to 46D2 per Zn. Four different sites were identified for the location of D2 molecules. These sites correspond to sites α–δ from the single-crystal X-ray study (of Ar and N2) and the first two relate directly to the two sites identified in the single-crystal neutron study. One of the other two sites (γ) therefore also matches the secondary site suggested by the INS study (Rowsell, Eckert & Yaghi, 2005 ▶). It is not clear if the increased number of binding sites observed in this study is due to the lower temperature or the deuteration (of framework and gas). Although there are some differences, this series of studies by different groups has reached similar conclusions regarding the preferred binding sites.
In further studies by Yildrium and co-workers, NPD data were recorded at 3.5 K with various D2:Zn ratios for the MOF [Zn(mIm)2]n (ZIF-8; ZIF = zeolitic imidazolate framework; HmIm = 2-methylimidazole). Six different D2 sites were identified after Rietveld refinement of the structural model (Wu et al., 2007 ▶). The principal site, designated based on the relative D2 occupancies, involves interaction with the imidazolate linker and contrasts with the previous examples discussed where the main binding mode was to the metal cluster.
In situ NPD has also been used in studies of adsorption of gases other than hydrogen. Wu et al. used Rietveld refinements to determine CD4 locations ZIF-8 and MOF-5 (Wu et al., 2009 ▶). Upon low loadings of gas molecules both frameworks were shown to adsorb methane at the primary binding sites discussed in the previous studies (two sites for ZIF-8 and one site for MOF-5). Population of these binding sites resulted in no change in the crystalline phase of the framework upon cooling to 3.5 K and gave defined CD4 orientations which fitted with the symmetry of the space group (Fig. 8 ▶). Higher loadings at 80 K did not show any additional well defined CD4 molecules, but the primary binding sites remained occupied. Cooling these samples below 60 K resulted in a reversible change in crystalline phase and the crystal structures had to be solved in a lower symmetry supercell. This was attributed to slight deformations in the framework caused by intermolecular repulsions from the confined methane (Wu et al., 2009 ▶).
Figure 8.
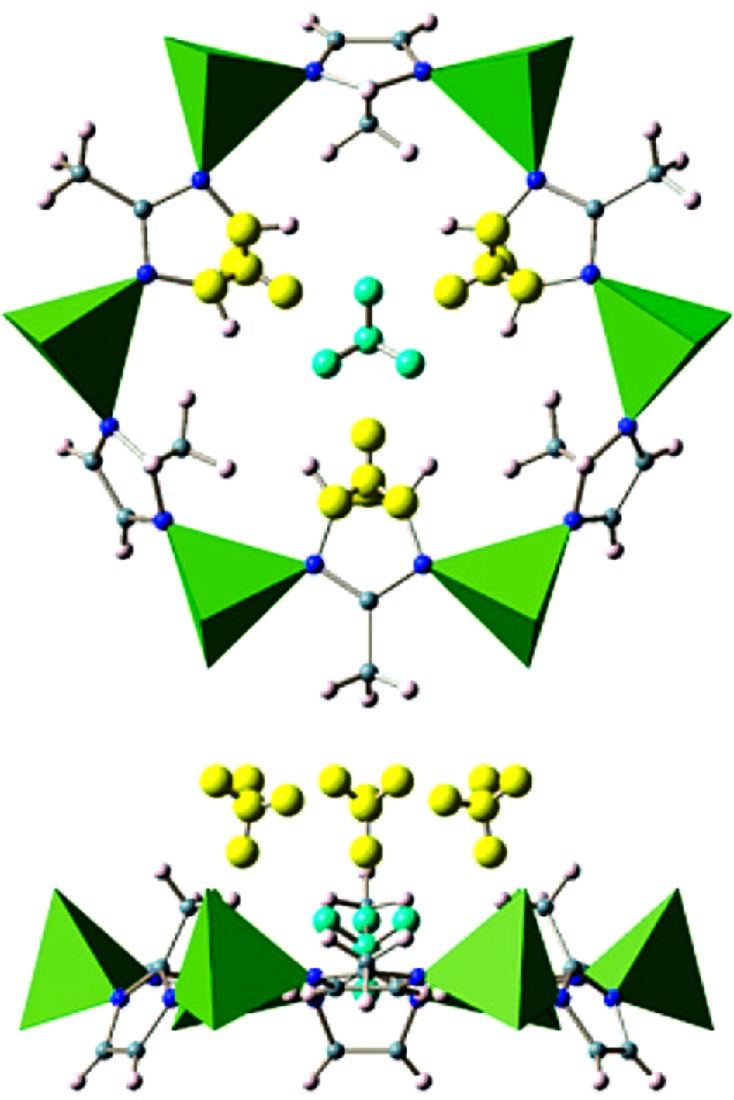
Primary (yellow) and secondary (cyan) sites for methane (CD4) in ZIF-8 shown in two perpendicular views of the hexagonal pore. (Reproduced from Wu et al., 2009 ▶, with permission from the American Chemical Society).
There are several reports in which useful information about the behaviour of MOFs has been found from the in situ powder diffraction studies of gas-loaded samples, without the use of Rietveld refinements to determine the location of the gas molecules. These include the use of in situ X-ray powder diffraction to examine phase transitions in [Zn(TCNQ-TCNQ)(bpy)] occurring upon the selective adsorption of O2 and NO gas (Shimomura et al., 2010 ▶), investigation of structural changes in DMOF, [Zn2(BDC)2(DABCO)], upon co-adsorption of CO2 and fluorescent guest molecules (Yanai et al., 2011 ▶), and studies of framework flexibility and reversibility upon CO2 adsorption above and below its triple point (Yang, Lin et al., 2012 ▶).
4. Flexible MOFs
Guest-responsive behaviour in some MOFs, for example the ability to change pore size or framework structure upon the introduction of various gases or other guest molecules, may potentially lead to important applications in gas sorption and separation. The effects are often apparent from the shape of adsorption isotherms, but can also be followed by crystallographic methods. Such processes usually involve a change of the unit-cell dimensions which is identifiable by the shifts of the Bragg peaks in the powder diffraction pattern. Changes in the framework structure can be reversible upon desorption of the gas and such behaviour is often termed ‘breathing’.
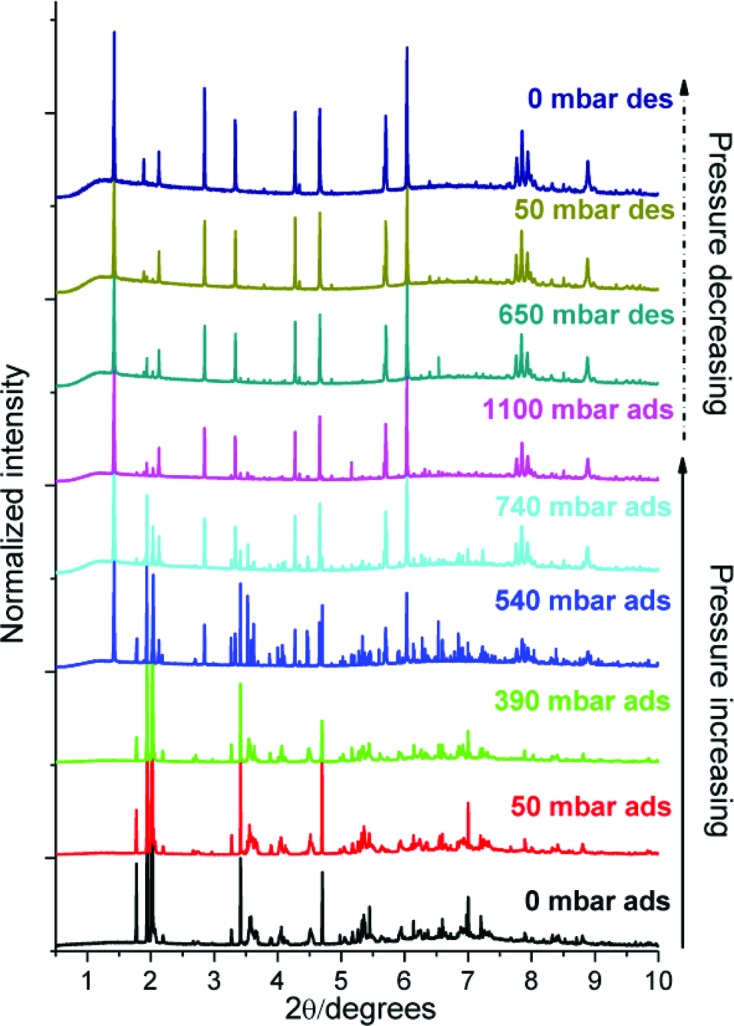
The most-studied MOF family that exhibits ‘breathing’ behaviour is probably the MIL-53(M) series. These isostructural MOFs involve trivalent metal ions (M 3+) coordinated to terephthalate (BDC) linkers and adopt large or narrow pore structures depending on the absorption of particular guests (Hamon et al., 2009 ▶). In 2006 Llewellyn et al. reported on the framework breathing of hydrated MIL-53(Cr) and MIL-53(Al) on the introduction of CO2 gas in a study by in situ powder X-ray diffraction (Llewellyn et al., 2006 ▶). In a more detailed follow-up study in 2007, Serre and Férey examined anhydrous MIL-53(Cr) upon addition of CO2 between 0 and 10 bar, and studied the response of the framework in situ by synchrotron XRPD (Fig. 9 ▶). On initial adsorption (up to 4 bar) the framework adopts the low pressure (narrow pore) form (Serre et al., 2007 ▶). Upon higher loadings of gas (above 5 bar) the framework expands to give the high-pressure (large pore) form. Upon slowly removing the CO2 pressure the large pore phase is retained down to 2 bar, before fully converting to the narrow pore version again, showing a hysteresis that correlates with the corresponding adsorption isotherms. The framework also showed a reversible cycling effect upon removal and re-dosing of CO2, which could be important for future gas storage/separation applications.
Figure 9.

‘Breathing’ effect of MIL-53(Cr) at different pressures of CO2, demonstrated by in situ X-ray powder diffraction and the corresponding structural changes (LP = low pressure; HP = high pressure). (Reproduced from Serre et al., 2007 ▶, with permission from Wiley.)
The study also shows that, upon degassing, the MIL-53(Cr) framework exists in the large pore form, and the addition of a small amount of CO2 causes the framework to close due to host–guest interactions. To study this behaviour, Rietveld refinements on a powder pattern measured at below 1 bar CO2 were used in conjunction with periodic DFT calculations and in situ IR spectroscopy. An electron donor–acceptor interaction between the hydroxyl O atom of the framework and the adsorbed CO2 is indicated to be responsible for the breathing effect (Serre et al., 2007 ▶).
The differences between the chromium and iron analogues of the MOF, MIL-53(Cr) and MIL-53(Fe), under the absorption of light gaseous hydrocarbons have also been explored by powder X-ray diffraction (Llewellyn et al., 2008 ▶, 2009 ▶; Rosenbach et al., 2010 ▶). MIL-53(Fe) showed much more complex behaviour than its sister compounds with a multi-step breathing process (Fig. 10 ▶). A total of four different pore-opening stages were reported upon increasing gas pressure, starting with a very narrow pore (vnp) phase (C2/c), then moving to an intermediate phase ( ), next to the narrow pore phase analogous to the Cr and Al frameworks (C2/c) and finally the large pore form (Imcm) at the highest pressure. In addition, the relative pressures required to convert from one phase to another were shown to greatly vary based on the hydrocarbon chain length. Introduction of CH4 caused the structure to change to the intermediate phase at around 12 bar pressure, but then gave no further structural changes, whereas uptake of C4H10 caused the framework to go through all four phases and be fully converted to the large pore form before reaching 2 bar. C2H6 and C3H8 showed intermediate activity, confirming a trend in behaviour of the framework that correlates with size of the alkane gas molecules (Llewellyn et al., 2009 ▶; Fig. 11 ▶). A similar trend is observed in the pressure at which the narrow-to-large pore transitions occur in MIL-53(Cr) (Llewellyn et al., 2008 ▶). MIL-53(Fe) was also shown to exist in the closed (very narrow pore) form when evacuated in contrast to the Cr (and Al) analogue which require the absorption of a small amount of guest for it to close (Llewellyn et al., 2009 ▶).
), next to the narrow pore phase analogous to the Cr and Al frameworks (C2/c) and finally the large pore form (Imcm) at the highest pressure. In addition, the relative pressures required to convert from one phase to another were shown to greatly vary based on the hydrocarbon chain length. Introduction of CH4 caused the structure to change to the intermediate phase at around 12 bar pressure, but then gave no further structural changes, whereas uptake of C4H10 caused the framework to go through all four phases and be fully converted to the large pore form before reaching 2 bar. C2H6 and C3H8 showed intermediate activity, confirming a trend in behaviour of the framework that correlates with size of the alkane gas molecules (Llewellyn et al., 2009 ▶; Fig. 11 ▶). A similar trend is observed in the pressure at which the narrow-to-large pore transitions occur in MIL-53(Cr) (Llewellyn et al., 2008 ▶). MIL-53(Fe) was also shown to exist in the closed (very narrow pore) form when evacuated in contrast to the Cr (and Al) analogue which require the absorption of a small amount of guest for it to close (Llewellyn et al., 2009 ▶).
Figure 10.

Structural evolution of the various MIL-53 series analogues with increasing amount of alkane guest. Top MIL-53(Fe), bottom (MIL-53(Cr,Al). (Reproduced from Llewellyn et al., 2009 ▶, with permission from the American Chemical Society.)
Figure 11.

In situ XRPD studies of hydrocarbon adsorption in MIL-53(Fe). Phase changes upon increasing pressure are indicated by colour changes in patterns (blue = vnp; green = intermediate; red = np; black = lp). (Reproduced from Llewellyn et al., 2009 ▶, with permission from the American Chemical Society.)
The effect of mixed gases on the breathing behaviour of MIL-53(Cr) has also been crystallographically explored using co-adsorption of CO2 and CH4. It was found that mixtures with equimolar or high levels of CO2 followed the normal breathing behaviour, where the pores initially closed at low gas pressures and then reopened at higher pressures. However, mixtures high in CH4 did not show the breathing behaviour and constantly remained in the large pore form, similar to that observed for adsorption of pure methane. Control of the breathing behaviour, therefore, was attributed to the partial pressure of CO2, with the narrow pore form thought to exclude CH4 and contain mainly CO2. This could potentially offer high selectivity for separation of these two gases (Hamon et al., 2009 ▶).
The changes that occur when terephthalate ligands containing substituents, including Br, Cl, CH3, NH2, CO2H and F, are used in the MIL-53 framework structure have also been explored in several studies (Ramsahye et al., 2011 ▶; Couck et al., 2012 ▶; Devic et al., 2012 ▶; Biswas et al., 2013 ▶) as well as the effect of other metals (Ga and In; Serra-Crespo et al., 2012 ▶). The substituents investigated showed significant differences in the breathing behaviour under CO2 and hydrocarbon introduction, including strong intra-framework interactions that hold the framework constantly in the very narrow pore form, as well as changes in the phase transition behaviour, such as skipping the intermediate pore form upon gas uptake.
Similar breathing effects, in which transitions between narrow and large pores take place, have been studied crystallographically for a number of other MOFs, including: CO2 adsorption in MIL-47(V), where a breathing transition is observed along with a change from a monoclinic to orthorhombic unit cell (Leclerc et al., 2011 ▶), N2 uptake in [Co(BDP)] (BDP = benzene-1,4-dipyrazolate) showing a dry (desolvated) phase, three distinct intermediate forms and a filled phase (Salles, Maurin et al., 2010 ▶), CO2 adsorption in [Zn(BME-BDC)2(dabco)] (BME-BDC = 2,5-bis(2-methoxyethoxy)-1,4-benzenedicarboxylate) exhibiting narrow, intermediate and open pore forms (Henke et al., 2011 ▶) and VO(BPDC) (BPDC = biphenyldicarboxylate) which exhibits a large and narrow pore form, but for which periodic DFT-D calculations predict an additional overstretched narrow pore form (Liu et al., 2013 ▶).
In situ X-ray powder diffraction studies on [Co(HLdc)], [Cd(bpndc)(bpy)] and ZIF-8 have also shown evidence of gate-opening effects due to phase transitions in these flexible frameworks (Yang, Davies et al., 2012 ▶; Tanaka et al., 2008 ▶; Fairen-Jimenez et al., 2011 ▶). The work on ZIF-8 (aka MAF-4) provides a good example of the relevance of such studies. The framework contains large cavities accessed through narrow windows which should provide a molecular sieving effect, denying access to larger gas molecules but allowing H2 in, yet experimental evidence demonstrates absorption of both CH4 and N2 (Fairen-Jimenez et al., 2011 ▶). To address this problem Fairen-Jimenez et al. used in situ X-ray powder diffraction studies to show that the behaviour of the framework upon introduction of the gas was analogous to its behaviour under high physical pressures (i.e. 14.7 kbar in a diamond–anvil cell). The result was an enlargement of window size due to a swing effect involving the imidazolate rings, which allows the gas molecules to diffuse through the framework (Fairen-Jimenez et al., 2011 ▶). This work was followed up by Zhang et al. (2012 ▶) who studied the effect by a combination of single-crystal X-ray diffraction and Raman spectroscopy. Although they showed the same two high- and low-pressure phases, they also found differences in the determined positions of the nitrogen molecules (Zhang et al., 2012 ▶). Additionally they suggested the possibility of an intermediate phase, which comprised a solid solution of the geometries in the two identified phases.
Similar work by Schröder, Yang and co-workers on adsorption of SO2 in NOTT-202a (Me2NH2[In(L3)]) [H4L3 = biphenyl-3,3′,5,5′-tetra-(phenyl-4-carboxylic acid)] developed an interesting idea for accessing new MOF topologies. Adsorption of SO2 by the framework resulted in a phase transition which could be monitored by powder diffraction (Fig. 12 ▶). The introduction of the gas was not observed to break any bonds but just to cause a structural re-ordering that was maintained after removal of the gas (irreversible process). The unit cell of the new MOF (NOTT-202b) is related to the original structure and a model for the new architecture was obtained by analysis of the changes in symmetry. Unfortunately due to the possible disorder within the structure and insufficient data quality, Rietveld refinements were not successful, but the calculated powder pattern based upon the proposed structural model for NOTT-202b closely resembles the experimental pattern. The new MOF does not appear to be accessible by conventional synthetic means and therefore there may be a potential for using SO2 in a catalytic manner for MOF framework transformation (Yang et al., 2013 ▶). Similarly, CO2 adsorption, coupled with the loss of coordinated solvent, has been implicated in the transformation of a three-dimensional MOF framework [Zn4(L5)2(DMF)3(OH2)3]·4H2O [H4L5 = 1,4-bis(3,5-dicarboxyphen-1-oxy)but-2-ene] to a two-dimensional framework [Zn2(L5)(OH2)2]·2H2O (Hawxwell et al., 2007 ▶). The transformation involves a change in conformation of a flexible tetracarboxylate ligand (L5) from twisted to planar conformation. The structure of the resulting new MOF was determined ab initio by X-ray powder diffraction.
Figure 12.
Powder diffraction patterns showing the irreversible structure changes that occur upon increased SO2 loading of NOTT-202a. (Reproduced from Yang et al., 2013 ▶, http://dx.doi.org/10.1021/ja401061m, with permission from the Royal Society of Chemistry.)
5. Functional groups
Most MOFs employ linker ligands that contain only functional groups that bond to the metal ions in the framework. Such functional groups are usually carboxylate or azolate groups. Thus, the interior surfaces of most MOFs permit only relatively weak interactions with adsorbed guest molecules, including gases. A growing number of MOFs contain additional functional groups, either as part of secondary building units comprised of small metal-oxo/hydroxo clusters (i.e. as μ-OH or μ3-OH groups) or, increasingly, as substituents on the hydrocarbon backbone of the linker ligands. These functional groups provide the possibility of stronger and more directional host–guest interactions. Recent studies of gas adsorption in such functionalized MOFs have shown promise in either increased gas uptake over particular pressure ranges or increased selectivity for one gas over another due to favourable interactions between the framework and the gas molecules (Bourrelly et al., 2005 ▶; Arstad et al., 2008 ▶; Bae et al., 2009 ▶; Couck et al., 2009 ▶; Demessence et al., 2009 ▶; Neofotistou et al., 2009 ▶; Choi et al., 2012 ▶). Crystallographic studies can reveal the role of the available functional groups within MOF pores in forming interactions with adsorbed gas molecules. This in turn may lead to the design of new MOFs with improved gas uptake characteristics.
Shimizu and co-workers used in situ single-crystal X-ray diffraction to locate CO2 molecules inside [Zn2(Atz)2(ox)], a Zn-based MOF constructed using amino-functionalized 1,2,4-triazoles (Atz) and oxalate ligands (ox) (Vaidhyanathan et al., 2009 ▶, 2010 ▶). The crystal structure was determined at four temperatures from 123 to 293 K. The best refinement (R = 0.027) was obtained at 173 K and indicated a loading of 1.3 CO2 per MOF formula unit, i.e. [Zn2(Atz)2(ox)](CO2)1.30, consistent with gravimetric adsorption measurements. Two independent CO2 molecules were located crystallographically inside the pore, with occupancies of 0.8 and 0.5 (Fig. 13 ▶). The molecule in site (I) forms an Nδ−⋯Cδ+ interaction between the N atom of the amino group and the C atom of a CO2 at a distance of 3.151 (8) Å (cf. ΣvdW = 3.25 Å). The H atoms on the amine were located crystallographically, and noted to be bent out of the plane of the triazole, confirming that the amino lone pair was not delocalized into the ring and that therefore it could be responsible for the CO2 binding. Further, weak (long and highly non-linear) hydrogen bonds were identified between the amino groups and CO2 O atoms, as well as a characteristic cooperative T-shaped Oδ−⋯Cδ+ interaction between the two independent CO2 molecules.
Figure 13.

Representation of the different binding modes for CO2 molecules in [Zn2(Atz)2(ox)]·(CO2)1.30 at 173 K (Reproduced from Vaidhyanathan et al., 2010 ▶, with permission from the AAAS.)
Similar single-crystal diffraction studies on triazole-containing MOFs MAF-X7 and MAF-23 under CO2 loadings have also been carried out by Zhang and co-workers. The work on MAF-X7 showed one CO2 molecule within the framework that had a contact between the central carbon of the CO2 and the triazolate 4-nitrogen [3.26 (4) Å] at a separation close to the sum of their van der Waals radii. Additional, separate weak (long) C—H⋯O hydrogen bonds are noted involving the (DMF)H+ cation and a ring C—H group, each with CO2 O atoms (Lin et al., 2011 ▶). In MAF-23 two independent CO2 sites were observed, both held in place by claw-like interactions from N atoms on two different triazolate rings. Most of the interaction distances were smaller than the sum of van der Waals radii and the site with a narrower claw angle showed a high CO2 occupancy (Liao et al., 2012 ▶).
The N atoms of the amine or imine groups are not the only functional groups capable of affecting the adsorption of gases. Studies by Yang and Schröder using a combination of powder diffraction and inelastic neutron scattering has shown that the primary binding site for CO2 in NOTT-300 is a pocket containing a hydrogen bond from a μ2-OH group with an additional weak cooperative hydrogen bond from an aromatic C—H group (Yang, Sun et al., 2012 ▶). The study also found a second CO2 molecule around the centre of the pore which interacted with the first CO2 (oxygen to carbon). Gravimetric measurements also suggested a high uptake of SO2 and crystallographic studies confirmed that the molecules sit in the same sites.
6. Open metal coordination sites
Similar to the effect of unsaturated functional groups (see §5), MOFs with open metal coordination sites provide opportunities for direct interactions with gas molecules which may determine the location and even dominate the binding energy of the gas molecules. Several different frameworks with open metal coordination sites have been studied crystallographically under different gas environments. Work by Long and co-workers using neutron powder diffraction reported on the D2 binding sites in a MOF containing exposed Mn2+ ions (Dincă et al., 2006 ▶). The work suggested two primary sites around the basic framework unit, with two further sites occupied at higher gas pressures. The first site reported was only 2.27 Å away from the exposed Mn2+ ion, suggesting a strong interaction from the ion. Despite this interaction, the occupancy of the site was only 29% due to competitive binding of methanol. Further work involved replacing the Mn2+ centres with Cu2+, which had a longer interaction with D2 of 2.47 Å, but resulted in increased occupancy of 93% due to more facile loss of the methanol (Dincă et al., 2007 ▶). A contemporaneous study by Peterson et al. (2006 ▶) reported on six different D2 sites within the well studied MOF [Cu3(BTC)2] (HKUST-1) (Fig. 14 ▶). By refining the occupancies of the sites at different D2 loadings ranging from 0.5 D2 per Cu to 4 D2 per Cu they showed a progressive filling of the different adsorption sites within the framework. The main site was observed to be occupying the uncoordinated axial sites of the Cu paddlewheels at a distance of 2.39 (1) Å [cf. Cu—O 2.17 (1) Å in hydrated material]. Only at the highest loading were all six sites occupied, some still partially. Further work by the group in 2011 showed an additional three metastable sites at higher loadings of D2, up to 6.5 D2 per Cu (Peterson et al., 2011 ▶), and illustrated the redistribution between sites that occurs on increasing gas loading. All experiments were carried out at 5 K after first loading the gas at higher temperatures.
Figure 14.
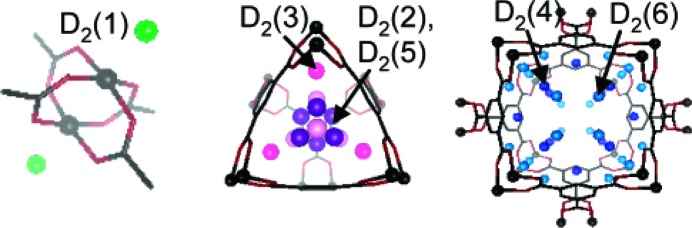
Six D2 adsorption sites identified in HKUST-1: the axial coordination site (left); view along the [111] direction showing sites close to the aromatic rings (sites 2 and 5) and carboxylate groups (site 3; middle); and view down the channels along the [100] direction (right). (Adapted from Peterson et al., 2006 ▶, with permission from the American Chemical Society.)
The importance of open metal sites in HKUST-1 and also the limit of their involvement in interaction with gas molecules is exemplified in the neutron powder diffraction study of CD4 gas uptake reported by Kaskel and coworkers (Getzschmann et al., 2010 ▶; Wu, Simmons, Liu et al., 2010 ▶) and studies of noble gas uptake using powder diffraction with X-rays (Xe, Kr) and neutrons (Ar) by Forster and coworkers (Hulvey et al., 2013 ▶). The crystallographic study by Kaskel identifies eight CD4 sites, four primary and four secondary sites, although not all can be occupied simultaneously due to the close proximity of some sites. Rietveld refinements for the evacuated MOF and at the two highest gas loadings (2.17 CD4/Cu and 3.67 CD4/Cu) show a reduction in Cu⋯Cu separation within the paddlewheel compared with the parent material with an axially coordinated water molecule. There is also a very small reduction in unit-cell volume upon gas uptake. The crystallographic study is complemented by a gravimetric gas adsorption study and a Grand Canonical Monte Carlo (GCMC) simulation of the adsorption at pressures and temperatures to match the experimental studies. The GCMC calculations provide a semi-quantitative model for the adsorption isotherms and identify methane sites that match most of those located crystallographically. However, the NPD study notably identifies a highly populated site that requires interaction with the open Cu sites (Cu⋯C 3.075 Å). The GCMC calculations are not parameterized to take into account this interaction with the metal centre and therefore do not identify this site. Zhou and Yildirim have studied the CO2 adsorption sites within HKUST-1 (Wu, Simmons, Srinivas et al., 2010 ▶). The XRPD study showed two primary adsorption sites, one around the axial Cu coordination site and the other termed the ‘small cage window site’. The metal coordination site relates to the highest occupancy D2 site reported by Peterson et al. for HKUST-1 and the small cage window site is close to the corresponding second, third and sixth occupied sites (Peterson et al., 2011 ▶). The two sites also resemble those observed for CD4 uptake in HKUST-1 in a similar study by Zhou and coworkers, which also examined CD4 uptake in other open-site Cu-MOFs experimentally and computationally (Wu, Simmons, Liu et al., 2010 ▶). In contrast to these studies, investigation of noble gas uptake by Forster provided no evidence for interaction of the noble gas atoms with the open CuII sites (Hulvey et al., 2013 ▶).
Very recently Matsuda, Kitagawa and coworkers have demonstrated the ability of a two-dimensional layered MOF, [Cu(aip)(OH2)]·n(solvent) (H2aip = 5-azidoisophthalic acid), which is based on the common M 2(O2CR)4 paddlewheel motif, to undergo a transformation upon desolvation in which paddlewheel units from neighbouring layers become covalently linked into columns via bridging Cu—O bonds. This is a less extreme form of the reversible transformations between paddlewheels and metal carboxylate columns recently reported by us (Smart et al., 2013 ▶) and by Bradshaw, Rossesinsky and coworkers (Stylianou et al., 2012 ▶). The structural transformation is not only reversible upon resolvation, but provides a potential means for separation of CO and N2 gases. Study of the collapsed desolvated form [Cu(aip)] via simultaneous measurement of adsorption isotherms and X-ray powder diffraction at 120 K showed that initial adsorption of CO and N2 is analogous, involving filling of the larger of two channels, which accommodates 0.76 CO molecules (0.85 N2 molecules) per Cu. On increasing the pressure, there is a step change in the CO isotherm, but no such change in the N2 isotherm. Powder diffraction reveals a return of the framework containing CO to the two-dimensional layered arrangement of its solvated form, but in which CO molecules are axially coordinated at the CuII sites as well as filling both large and small channels (2.10 CO per Cu). Studies using CO/N2 gas mixtures demonstrate significant enrichment in CO upon adsorption by [Cu(aip)]. Although quite weakly coordinating, the results suggest that stronger coordination by CO than by N2 enables the structural transformation and therefore increased uptake of CO. Only upon expansion of the framework, following CO coordination, does the small channel become accessible, and in gas mixtures this is filled by CO rather than by N2, thereby accentuating the enrichment process. In a comparative study, the authors show that HKUST-1 exhibits no difference between adsorption isotherms of CO and N2, despite its well established ability to coordinate other adsorbed gas molecules at the open CuII sites (see above).
The channel-MOF CPO-27-Ni (Ni-MOF-74; Fig. 15 ▶) presents open Ni metal sites in the channel walls following desolvation. In 2008, both Morris and Blom reported in situ X-ray powder diffraction studies under loadings of NO (McKinlay et al., 2008 ▶) and CO2 gas (Dietzel et al., 2008 ▶), respectively. Both studies showed that the gas molecules were bound to the unsaturated Ni site with bond lengths shorter than the sum of the van der Waals radii. The NO gas bonded through the nitrogen and the carbon dioxide was bound end-on through an O atom but with a bent molecular arrangement (162°). In the same study Morris reported similar results for the cobalt analogue of the framework, which suggests that the effect is not specific to the metal and perhaps other less toxic metals could be targeted for applications such as in vitro release of NO. Along these lines it was suggested that the zinc analogue would be suitable, but unfortunately it proved considerably harder to activate for NO absorption (McKinlay et al., 2008 ▶). A later study on CPO-27-Ni also showed applicability of the MOF to in vitro delivery of H2S, which similarly to the NO and CO2 studies was bound to the nickel centres, and was located using X-ray powder diffraction (Allan et al., 2012 ▶).
Figure 15.

Crystal structure of CPO-27-Ni with adsorbed CO2 next to the metal atom. (a) Hexagonal channels viewed along the [001] direction. (b) Coordination environment of the CO2 molecules. (Reproduced from Dietzel et al., 2008 ▶, http://dx.doi.org/10.1039/B810574J, with permission from the Royal Society of Chemistry.)
The work on the CPO-27/MOF-74 series was further explored by Zhou and Yildirim who used neutron powder diffraction to investigate CO2 adsorption in the Mg analogue (Wu, Simmons, Srinivas et al., 2010 ▶). The gas was adsorbed into the empty framework at 240 K and the sample cooled to 196 K, whereupon all gas had been absorbed. Further cooling to 20 K was undertaken prior to NPD data collection. The crystal structure model from Rietveld fitting (R wp = 0.024) showed an end-on CO2 bound to the metal with a bent geometry (O=C=O ≃ 160.5°), which the authors attribute to effects of (unmodelled) disorder, since calculations suggest a significant energy penalty for such a large deviation from linearity. Indeed, a subsequent study by Brown and Yaghi, using NPD and MEM, suggested a larger O=C=O angle of 170° (Queen et al., 2011 ▶). This study also found evidence of a second adsorption site within the pores that involved interaction of CO2 molecules with carbon and oxygen framework atoms.
Overall it can be seen that the open metal sites tend to have a significant effect on the location of a variety of different gases.
7. Gas sorption in molecular crystalline materials
Although not formally the focus of this review it is pertinent to place the work on MOFs in a broader context and refer the reader to some examples of the growing number of crystallographic studies of gas and vapour sorption involving molecular crystals and coordination polymers. Such materials include flexible one-dimensional coordination polymers that adsorb a wide variety of gases (CO2, CH4, O2, H2, Ar, Kr, Xe) into small spaces between polymer strands (Takamizawa et al., 2003 ▶, 2004 ▶, 2005 ▶; Takamizawa, Nakata & Akatsuka, 2006 ▶; Takamizawa, Kojima & Akatsuka, 2006 ▶; Takamizawa, Nakata, Akatsuka, Kachi-Terajima & Miyake, 2008 ▶, 2010 ▶; Takamizawa & Nakata, 2005 ▶; Ueda et al., 2007 ▶; Kosaka, Yamagishi, Hori et al., 2013 ▶; Kosaka, Yamagishi, Yoshida et al., 2013 ▶;) or within cages (Coronado et al., 2013 ▶) and both metal complexes and one-dimensional coordination polymers that adsorb gases or vapours via metal coordination: SO2 (Albrecht et al., 2000 ▶); HCl/HBr (Mínguez Espallargas et al., 2006 ▶, 2007 ▶, 2010 ▶, 2011 ▶; Adams et al., 2007 ▶, 2010 ▶; Vitórica-Yrezábal et al., 2011 ▶; Coronado et al., 2012 ▶); N2/O2/H2/CO/C2H4/NH3 (Huang et al., 2010 ▶); alcohol (ROH) vapours (Vitórica-Yrezábal et al., 2013 ▶; Libri et al., 2008 ▶). Organic molecular crystals which adsorb gases have been subject to crystallographic characterization for CO2 (Jacobs et al., 2012 ▶, 2014 ▶) and Xe (Taratula et al., 2010 ▶).
8. Conclusions
Crystallographic studies in which adsorbed gas molecules are allocated within the pores of MOFs (or other porous materials) present significant experimental and structure refinement challenges. However, such studies have been successfully conducted on a range of metal–organic frameworks using both single-crystal and powder diffraction, and employing both X-rays and neutrons (Table 1 ▶). These studies have enabled important structural information on the position of gas molecules contained within these porous materials to be determined and the nature of the interactions involved in holding these molecules in place to be investigated. This knowledge can now be applied to help design the next generation of porous materials. As diffraction capabilities continue to advance, it is anticipated that crystallographic characterization of gas molecules adsorbed within MOFs and related porous materials will become more routinely undertaken. Such studies will continue to make important contributions not only to the development of MOFs and related materials, but in driving crystallography towards new frontiers. The latter is a most apt consideration at this time of the centenary of the field of crystallography.
Acknowledgments
We are grateful to EPSRC for funding (E-Futures Doctoral Training Centre grant EP/G037477/1 for EJC; grant EP/F02195X/1 and a Doctoral Prize fellowship for IJVY). We are grateful for support for our own work on solid–gas reactions and gas adsorption over a number of years from colleagues at synchrotron beamlines at ESRF, Diamond Light Source and Daresbury SRS (ESRF, ID31: Andy Fitch, Michaela Brunelli, Caroline Curfs, Adrian Hill; Diamond, I11: Chiu Tang and Stephen Thompson; Diamond I19: Dave Allan and Mark Warren; Daresbury SRS, 9.8: John Warren).
Appendix A. Abbreviations
H2aip: 5-azidoisophthalic acid
Atz: 3-amino-1,2,4-triazole
BDC: 1,4-benzenedicarboxylate
BDP: 1,4-benzenedipyrazolate
BME-BDC: 2,5-bis(2-methoxyethoxy)-1,4-benzenedicarboxylate
BPDC: biphenyl-4,4′-dicarboxylate
H2bpndc: benzophenone-4,4′-dicarboxylic acid
bpy: 4,4′-bipyridyl
BTC: 1,3,5-benzenetricarboxylate
H2btdc: 2,2′-bithiophene-5,5′-dicarboxylic acid
H2BTDD: bis(1H-1,2,3-triazolo[4,5-b],[4′,5′-i])dibenzo[1,4]dioxin
H3BTT: 1,3,5-tris(tetrazol-5-yl)benzene
DABCO: 1,4-diazabicyclo[2.2.2]octane
DMF: dimethylformamide
DSB: distyrylbenzene
Hetz: 3,5-diethyl-1,2,4-triazole
H4L1: 4,4′,4′′,4′′′-benzene-1,2,4,5-tetrayl-tetrabenzoic acid
L2: N,N′-di-(4-pyridyl)-1,4,5,8-naphthalenetetracarboxydiimide
H4L3 (NOTT-202a): biphenyl-3,3′,5,5′-tetra-(phenyl-4-carboxylic acid)
H4L4 (NOTT-300): biphenyl-3,3′,5,5′-tetracarboxylic acid
H4L5: 1,4-bis(3,5-dicarboxyphen-1-oxy)but-2-ene
H3Ldc: 5-{4-[3-carboxy-2,6-bis(pyridin-4-yl)pyridin-4-yl]phenyl}benzene-1,3-dicarboxylic acid
MeIm: 2-methylimidazolate
H2mpm-PBODB: 3,3′-(1,4-phenylenebis(oxy))dibenzoic acid
ox: oxalate
H2ppt: 3-(2-phenol)-5-(4-pyridyl)-1,2,4-triazole
pyz: pyrazine
pzdc: 2,3-pyrazinedicarboxylate
—R: (—CH3) [with additional group (methyl) instead of a H atom]
H2sdb: 4,4-sulfonyldibenzoic acid
H2sbtc: trans-stilbene-3,3′,5,5′-tetracarboxylic acid
TCNQ: 7,7,8,8-tetracyano-p-quino-dimethane
References
- Adams, C. J., Colquhoun, H. M., Crawford, P. C., Lusi, M. & Orpen, A. G. (2007). Angew. Chem. Int. Ed. 46, 1124–1128. [DOI] [PubMed]
- Adams, C. J., Haddow, M. F., Lusi, M. & Orpen, A. G. (2010). Proc. Nat. Acad. Sci. USA, 107, 16033–16038. [DOI] [PMC free article] [PubMed]
- Albrecht, M., Lutz, M., Spek, A. L. & van Koten, G. (2000). Nature, 406, 970–974. [DOI] [PubMed]
- Allan, P. K., Wheatley, P. S., Aldous, D., Mohideen, M. I., Tang, C., Hriljac, J. A., Megson, I. L., Chapman, K. W., De Weireld, G., Vaesen, S. & Morris, R. E. (2012). Dalton Trans. 41, 4060–4066. [DOI] [PubMed]
- Allan, P. K., Xiao, B., Teat, S. J., Knight, J. W. & Morris, R. E. (2010). J. Am. Chem. Soc. 132, 3605–3611. [DOI] [PubMed]
- Amaro, A. A. & Seff, K. (1973). J. Phys. Chem. 77, 906–910.
- Arstad, B., Fjellvåg, H., Kongshaug, K. O., Swang, O. & Blom, R. (2008). Adsorption, 14, 755–762.
- Bae, Y., Farha, O. K., Hupp, J. T. & Snurr, R. Q. (2009). J. Mater. Chem. 19, 2131.
- Barthelet, K., Marrot, J., Riou, D. & Férey, G. (2002). Angew. Chem. Int. Ed. 41, 281–284. [DOI] [PubMed]
- Biswas, S., Rémy, T., Couck, S., Denysenko, D., Rampelberg, G., Denayer, J. F., Volkmer, D., Detavernier, C. & Van Der Voort, P. (2013). Phys. Chem. Chem. Phys. 15, 3552–3561. [DOI] [PubMed]
- Bourrelly, S., Llewellyn, P. L., Serre, C., Millange, F., Loiseau, T. & Férey, G. (2005). J. Am. Chem. Soc. 127, 13519–13521. [DOI] [PubMed]
- Brunelli, M. & Fitch, A. N. (2003). J. Synchrotron Rad. 10, 337–339. [DOI] [PubMed]
- Bureekaew, S., Sato, H., Matsuda, R., Kubota, Y., Hirose, R., Kim, J., Kato, K., Takata, M. & Kitagawa, S. (2010). Angew. Chem. Int. Ed. 49, 7660–7664. [DOI] [PubMed]
- Chakoumakos, B. C., Rawn, C. J., Rondinone, A. J., Stern, L. A., Circone, S., Kirby, S. H., Ishii, Y., Jones, C. Y. & Toby, B. H. (2011). Can. J. Phys. 81, 183–189.
- Chen, L., Mowat, J. P., Fairen-Jimenez, D., Morrison, C. A., Thompson, S. P., Wright, P. A. & Düren, T. (2013). J. Am. Chem. Soc. 135, 15763–15773. [DOI] [PubMed]
- Cho, H. S., Miyasaka, K., Kim, H., Kubota, Y., Takata, M., Kitagawa, S., Ryoo, R. & Terasaki, O. (2012). J. Phys. Chem. C, 116, 25300–25308.
- Choi, S., Watanabe, T., Bae, T., Sholl, D. S. & Jones, C. W. (2012). J. Phys. Chem. Lett, 3, 1136–1141. [DOI] [PubMed]
- Chui, S. S., Lo, S. M., Charmant, J. P., Orpen, A. G. & Williams, I. D. (1999). Science, 283, 1148–1150. [DOI] [PubMed]
- Collins, D. J. & Zhou, H. (2007). J. Mater. Chem. 17, 3154–3160.
- Coronado, E., Giménez-Marqués, M., Mínguez Espallargas, G. & Brammer, L. (2012). Nat. Commun. 3, 828. [DOI] [PubMed]
- Coronado, E., Giménez-Marqués, M., Mínguez Espallargas, G., Rey, F. & Vitórica-Yrezábal, I. J. (2013). J. Am. Chem. Soc. 135, 15986–15989. [DOI] [PubMed]
- Couck, S., Denayer, J. F., Baron, G. V., Rémy, T., Gascon, J. & Kapteijn, F. (2009). J. Am. Chem. Soc. 131, 6326–6327. [DOI] [PubMed]
- Couck, S., Gobechiya, E., Kirschhock, C. E., Serra-Crespo, P., Juan-Alcañiz, J., Martinez Joaristi, A., Stavitski, E., Gascon, J., Kapteijn, F., Baron, G. V. & Denayer, J. F. (2012). ChemSusChem, 5, 740–750. [DOI] [PubMed]
- D’Alessandro, D., Smit, B. & Long, J. (2010). Angew. Chem. Int. Ed. 49, 6058–6082. [DOI] [PubMed]
- Demessence, A., D’Alessandro, D. M., Foo, M. L. & Long, J. R. (2009). J. Am. Chem. Soc. 131, 8784–8786. [DOI] [PubMed]
- Devic, T., Salles, F., Bourrelly, S., Moulin, B., Maurin, G., Horcajada, P., Serre, C., Vimont, A., Lavalley, J., Leclerc, H., Clet, G., Daturi, M., Llewellyn, P. L., Filinchuk, Y. & Férey, G. (2012). J. Mater. Chem. 22, 10266.
- Dietzel, P. D. C., Johnsen, R. E., Fjellvåg, H., Bordiga, S., Groppo, E., Chavan, S. & Blom, R. (2008). Chem. Commun. pp. 5125–5127. [DOI] [PubMed]
- Dincă, M., Dailly, A., Liu, Y., Brown, C. M., Neumann, D. A. & Long, J. R. (2006). J. Am. Chem. Soc. 128, 16876–16883. [DOI] [PubMed]
- Dincă, M., Han, W., Liu, Y., Dailly, A., Brown, C. M. & Long, J. R. (2007). Angew. Chem. Int. Ed. 46, 1419–1422. [DOI] [PubMed]
- Dincă, M. & Long, J. R. (2008). Angew. Chem. Int. Ed. 47, 6766–6779. [DOI] [PubMed]
- Eddaoudi, M., Kim, J., Rosi, N., Vodak, D., Wachter, J., O’Keeffe, M. & Yaghi, O. M. (2002). Science, 295, 469–472. [DOI] [PubMed]
- Eddaoudi, M., Li, H. & Yaghi, O. M. (2000). J. Am. Chem. Soc. 122, 1391–1397.
- Fairen-Jimenez, D., Moggach, S. A., Wharmby, M. T., Wright, P. A., Parsons, S. & Düren, T. (2011). J. Am. Chem. Soc. 133, 8900–8902. [DOI] [PubMed]
- Filinchuk, Y., Richter, B., Jensen, T. R., Dmitriev, V., Chernyshov, D. & Hagemann, H. (2011). Angew. Chem. Int. Ed. 50, 11162–11166. [DOI] [PubMed]
- Fletcher, A. J., Cussen, E. J., Prior, T. J., Rosseinsky, M. J., Kepert, C. J. & Thomas, K. M. (2001). J. Am. Chem. Soc. 123, 10001–10011. [DOI] [PubMed]
- Getzschmann, J., Senkovska, I., Wallacher, D., Tovar, M., Fairen-Jimenez, D., Düren, T., van Baten, J. M., Krishna, R. & Kaskel, S. (2010). Microporous Mesoporous Mater. 136, 50–58.
- Gul-E-Noor, F., Mendt, M., Michel, D., Pöppl, A., Krautscheid, H., Haase, J. & Bertmer, M. (2013). J. Phys. Chem. C, 117, 7703–7712.
- Hamon, L., Llewellyn, P. L., Devic, T., Ghoufi, A., Clet, G., Guillerm, V., Pirngruber, G. D., Maurin, G., Serre, C., Driver, G., van Beek, W., Jolimaître, E., Vimont, A., Daturi, M. & Férey, G. (2009). J. Am. Chem. Soc. 131, 17490–17499. [DOI] [PubMed]
- Han, S. S., Mendoza-Cortés, J. L. & Goddard, W. A. (2009). Chem. Soc. Rev. 38, 1460–1476. [DOI] [PubMed]
- Hawxwell, S. M., Mínguez Espallargas, G., Bradshaw, D., Rosseinsky, M. J., Prior, T. J., Florence, A. J., van de Streek, J. & Brammer, L. (2007). Chem. Commun. pp. 1532–1534. [DOI] [PubMed]
- Henke, S., Florian Wieland, D. C., Meilikhov, M., Paulus, M., Sternemann, C., Yusenko, K. & Fischer, R. A. (2011). CrystEngComm, 13, 6399–6404.
- Hijikata, Y., Horike, S., Sugimoto, M., Inukai, M., Fukushima, T. & Kitagawa, S. (2013). Inorg. Chem. 52, 3634–3642. [DOI] [PubMed]
- Hill, A. H. (2013). J. Appl. Cryst. 46, 570–572.
- Hong, D. H. & Suh, M. P. (2012). Chem. Commun. 48, 9168–9170. [DOI] [PubMed]
- Horcajada, P., Gref, R., Baati, T., Allan, P. K., Maurin, G., Couvreur, P., Férey, G., Morris, R. E. & Serre, C. (2012). Chem. Rev. 112, 1232–1268. [DOI] [PubMed]
- Hoshikawa, A., Igawa, N., Yamauchi, H. & Ishii, Y. (2006). J. Chem. Phys. 125, 034505. [DOI] [PubMed]
- Huang, Z., White, P. S. & Brookhart, M. (2010). Nature, 465, 598–601. [DOI] [PubMed]
- Hulvey, Z., Lawler, K. V., Qiao, Z., Zhou, J., Fairen-Jimenez, D., Snurr, R. Q., Ushakov, S. V., Navrotsky, A., Brown, C. M. & Forster, P. M. (2013). J. Phys. Chem. C, 117, 20116–20126.
- Jacobs, T., Lloyd, G. O., Gertenbach, J., Müller-Nedebock, K. K., Esterhuysen, C. & Barbour, L. J. (2012). Angew. Chem. Int. Ed. 51, 4913–4916. [DOI] [PubMed]
- Jacobs, T., Smith, V. J., Thomas, L. H. & Barbour, L. J. (2014). Chem. Commun. 50, 85–87. [DOI] [PubMed]
- Jeffrey, G. A. & McMullan, R. K. (1967). Prog. Inorg. Chem. 8, 43–108.
- Jensen, T. R., Nielsen, T. K., Filinchuk, Y., Jørgensen, J.-E., Cerenius, Y., Gray, E. M. & Webb, C. J. (2010). J. Appl. Cryst. 43, 1456–1463. [DOI] [PMC free article] [PubMed]
- Kanoo, P., Reddy, S. K., Kumari, G., Haldar, R., Narayana, C., Balasubramanian, S. & Maji, T. K. (2012). Chem. Commun. 48, 8487. [DOI] [PubMed]
- Kim, H., Samsonenko, D. G., Das, S., Kim, G. H., Lee, H. S., Dybtsev, D. N., Berdonosova, E. A. & Kim, K. (2009). Chem. Asian J. 4, 886–891. [DOI] [PubMed]
- Kitaura, R., Kitagawa, S., Kubota, Y., Kobayashi, T. C., Kindo, K., Mita, Y., Matsuo, A., Kobayashi, M., Chang, H. C., Ozawa, T. C., Suzuki, M., Sakata, M. & Takata, M. (2002). Science, 298, 2358–2361. [DOI] [PubMed]
- Kitaura, R., Matsuda, R., Kubota, Y., Kitagawa, S., Takata, M., Kobayashi, T. C. & Suzuki, M. (2005). J. Phys. Chem. B, 109, 23378–23385. [DOI] [PubMed]
- Kondo, M., Yoshitomi, T., Matsuzaka, H., Kitagawa, S. & Seki, K. (1997). Angew. Chem. Int. Ed. 36, 1725–1727.
- Kosaka, W., Yamagishi, K., Hori, A., Sato, H., Matsuda, R., Kitagawa, S., Takata, M. & Miyasaka, H. (2013). J. Am. Chem. Soc. 135, 18469–18480. [DOI] [PubMed]
- Kosaka, W., Yamagishi, K., Yoshida, H., Matsuda, R., Kitagawa, S., Takata, M. & Miyasaka, H. (2013). Chem. Commun. 49, 1594–1596. [DOI] [PubMed]
- Kreno, L. E., Leong, K., Farha, O. K., Allendorf, M., Van Duyne, R. P. & Hupp, J. T. (2012). Chem. Rev. 112, 1105–1125. [DOI] [PubMed]
- Kubota, Y., Takata, M., Kobayashi, T. & Kitagawa, S. (2007). Coord. Chem. Rev. 251, 2510–2521.
- Kubota, Y., Takata, M., Matsuda, R., Kitaura, R., Kitagawa, S., Kato, K., Sakata, M. & Kobayashi, T. C. (2005). Angew. Chem. Int. Ed. 44, 920–923. [DOI] [PubMed]
- Kubota, Y., Takata, M., Matsuda, R., Kitaura, R., Kitagawa, S. & Kobayashi, T. C. (2006). Angew. Chem. Int. Ed. 45, 4932–4936. [DOI] [PubMed]
- Kumari, G., Jayaramulu, K., Maji, T. K. & Narayana, C. (2013). J. Phys. Chem. A, 117, 11006–11012. [DOI] [PubMed]
- Leclerc, H., Devic, T., Devautour-Vinot, S., Bazin, P., Audebrand, N., Férey, G., Daturi, M., Vimont, A. & Clet, G. (2011). J. Phys. Chem. C, 115, 19828–19840.
- Li, H., Eddaoudi, M., Groy, T. L. & Yaghi, O. M. (1998). J. Am. Chem. Soc. 120, 8571–8572.
- Li, H., Eddaoudi, M., O’Keeffe, M. & Yaghi, O. M. (1999). Nature, 402, 276–279.
- Li, J. R., Kuppler, R. J. & Zhou, H. C. (2009). Chem. Soc. Rev. 38, 1477–1504. [DOI] [PubMed]
- Li, J. R., Sculley, J. & Zhou, H. C. (2012). Chem. Rev. 112, 869–932. [DOI] [PubMed]
- Liao, P. Q., Zhou, D. D., Zhu, A. X., Jiang, L., Lin, R. B., Zhang, J. P. & Chen, X. M. (2012). J. Am. Chem. Soc. 134, 17380–17383. [DOI] [PubMed]
- Libri, S., Mahler, M., Mínguez Espallargas, G., Singh, D. C. N. G., Soleimannejad, J., Adams, H., Burgard, M. D., Rath, N. P., Brunelli, M. & Brammer, L. (2008). Angew. Chem. Int. Ed. 47, 1693–1697. [DOI] [PubMed]
- Lin, X., Jia, J., Hubberstey, P., Schröder, M. & Champness, N. R. (2007). CrystEngComm, 9, 438–448.
- Lin, J., Xue, W., Zhang, J. & Chen, X. (2011). Chem. Commun. 47, 926–928. [DOI] [PubMed]
- Liu, J., Thallapally, P. K., McGrail, B. P., Brown, D. R. & Liu, J. (2012). Chem. Soc. Rev. 41, 2308–2322. [DOI] [PubMed]
- Liu, Y. Y., Couck, S., Vandichel, M., Grzywa, M., Leus, K., Biswas, S., Volkmer, D., Gascon, J., Kapteijn, F., Denayer, J. F., Waroquier, M., Van Speybroeck, V. & Van Der Voort, P. (2013). Inorg. Chem. 52, 113–120. [DOI] [PubMed]
- Llewellyn, P. L., Bourrelly, S., Serre, C., Filinchuk, Y. & Férey, G. (2006). Angew. Chem. Int. Ed. 45, 7751–7754. [DOI] [PubMed]
- Llewellyn, P. L., Horcajada, P., Maurin, G., Devic, T., Rosenbach, N., Bourrelly, S., Serre, C., Vincent, D., Loera-Serna, S., Filinchuk, Y. & Férey, G. (2009). J. Am. Chem. Soc. 131, 13002–13008. [DOI] [PubMed]
- Llewellyn, P. L., Maurin, G., Devic, T., Loera-Serna, S., Rosenbach, N., Serre, C., Bourrelly, S., Horcajada, P., Filinchuk, Y. & Férey, G. (2008). J. Am. Chem. Soc. 130, 12808–12814. [DOI] [PubMed]
- Lock, N., Christensen, M., Kepert, C. J. & Iversen, B. B. (2013). Chem. Commun. 49, 789–791. [DOI] [PubMed]
- Lozinska, M. M., Mangano, E., Mowat, J. P., Shepherd, A. M., Howe, R. F., Thompson, S. P., Parker, J. E., Brandani, S. & Wright, P. A. (2012). J. Am. Chem. Soc. 134, 17628–17642. [DOI] [PubMed]
- Luo, J., Xu, H., Liu, Y., Zhao, Y., Daemen, L. L., Brown, C., Timofeeva, T. V., Ma, S. & Zhou, H. C. (2008). J. Am. Chem. Soc. 130, 9626–9627. [DOI] [PubMed]
- Mahajan, D., Koetzle, T. F., Klooster, W. T., Brammer, L., McMullan, R. K. & Goland, A. N. (2000). Ann. New York Acad. Sci. 912, 940–949.
- Maji, T. K., Mostafa, G., Matsuda, R. & Kitagawa, S. (2005). J. Am. Chem. Soc. 127, 17152–17153. [DOI] [PubMed]
- Matsuda, R., Kitaura, R., Kitagawa, S., Kubota, Y., Belosludov, R. V., Kobayashi, T. C., Sakamoto, H., Chiba, T., Takata, M., Kawazoe, Y. & Mita, Y. (2005). Nature, 436, 238–241. [DOI] [PubMed]
- McKinlay, A. C., Eubank, J. F., Wuttke, S., Xiao, B., Wheatley, P. S., Bazin, P., Lavalley, J.-C., Daturi, M., Vimont, A., De Weireld, G., Horcajada, P., Serre, C. & Morris, R. E. (2013). Chem. Mater. 25, 1592–1599.
- McKinlay, A. C., Xiao, B., Wragg, D. S., Wheatley, P. S., Megson, I. L. & Morris, R. E. (2008). J. Am. Chem. Soc. 130, 10440–10444. [DOI] [PubMed]
- McMullan, R. K. & Kvick, Å. (1990). Acta Cryst. B46, 390–399.
- Miller, S. R., Wright, P. A., Devic, T., Serre, C., Férey, G., Llewellyn, P. L., Denoyel, R., Gaberova, L. & Filinchuk, Y. (2009). Langmuir, 25, 3618–3626. [DOI] [PubMed]
- Mínguez Espallargas, G., Brammer, L., van de Streek, J., Shankland, K., Florence, A. J. & Adams, H. (2006). J. Am. Chem. Soc. 128, 9584–9585. [DOI] [PubMed]
- Mínguez Espallargas, G., Florence, A. J., van de Streek, J. & Brammer, L. (2011). CrystEngComm, 13, 4400–4404.
- Mínguez Espallargas, G., Hippler, M., Florence, A. J., Fernandes, P., van de Streek, J., Brunelli, M., David, W. I., Shankland, K. & Brammer, L. (2007). J. Am. Chem. Soc. 129, 15606–15614. [DOI] [PubMed]
- Mínguez Espallargas, G., van de Streek, J., Fernandes, P., Florence, A. J., Brunelli, M., Shankland, K. & Brammer, L. (2010). Angew. Chem. Int. Ed. 49, 8892–8896. [DOI] [PubMed]
- Mulfort, K. L., Farha, O. K., Malliakas, C. D., Kanatzidis, M. G. & Hupp, J. T. (2010). Chem. Eur. J. 16, 276–281. [DOI] [PubMed]
- Murray, L. J., Dincă, M. & Long, J. R. (2009). Chem. Soc. Rev. 38, 1294–1314. [DOI] [PubMed]
- Neofotistou, E., Malliakas, C. D. & Trikalitis, P. N. (2009). Chem. Eur. J. 15, 4523–4527. [DOI] [PubMed]
- Nowell, H., Barnett, S. A., Christensen, K. E., Teat, S. J. & Allan, D. R. (2012). J. Synchrotron Rad. 19, 435–441. [DOI] [PubMed]
- Pauling, L. & Marsh, R. E. (1952). Proc. Nat. Acad. Sci. USA, 38, 112–118. [DOI] [PMC free article] [PubMed]
- Peterson, V. K., Brown, C. M., Liu, Y. & Kepert, C. J. (2011). J. Phys. Chem. C, 115, 8851–8857.
- Peterson, V. K., Liu, Y., Brown, C. M. & Kepert, C. J. (2006). J. Am. Chem. Soc. 128, 15578–15579. [DOI] [PubMed]
- Planas, N., Dzubak, A. L., Poloni, R., Lin, L. C., McManus, A., McDonald, T. M., Neaton, J. B., Long, J. R., Smit, B. & Gagliardi, L. (2013). J. Am. Chem. Soc. 135, 7402–7405. [DOI] [PubMed]
- Queen, W. L., Brown, C. M., Britt, D. K., Zajdel, P., Hudson, M. R. & Yaghi, O. M. (2011). J. Phys. Chem. C, 115, 24915–24919.
- Ramsahye, N. A., Trung, T. K., Bourrelly, S., Yang, Q., Devic, T., Maurin, G., Horcajada, P., Llewellyn, P. L., Yot, P., Serre, C., Filinchuck, Y., Fajula, F., Férey, G. & Trens, P. (2011). J. Phys. Chem. C, 115, 18683–18695.
- Riley, P. E., Kunz, K. B. & Seff, K. (1975). J. Am. Chem. Soc. 97, 537–542.
- Riley, P. E. & Seff, K. (1973). J. Am. Chem. Soc. 95, 8180–8182.
- Riley, P. E. & Seff, K. (1974). Inorg. Chem. 13, 1355–1360.
- Rosenbach, N., Ghoufi, A., Déroche, I., Llewellyn, P. L., Devic, T., Bourrelly, S., Serre, C., Férey, G. & Maurin, G. (2010). Phys. Chem. Chem. Phys. 12, 6428–6437. [DOI] [PubMed]
- Rosi, N. L., Eckert, J., Eddaoudi, M., Vodak, D. T., Kim, J., O’Keeffe, M. & Yaghi, O. M. (2003). Science, 300, 1127–1129. [DOI] [PubMed]
- Rowsell, J. L. C., Eckert, J. & Yaghi, O. M. (2005). J. Am. Chem. Soc. 127, 14904–14910. [DOI] [PubMed]
- Rowsell, J. L. C., Spencer, E. C., Eckert, J., Howard, J. A. K. & Yaghi, O. M. (2005). Science, 309, 1350–1354. [DOI] [PubMed]
- Salles, F., Jobic, H., Devic, T., Llewellyn, P. L., Serre, C., Férey, G. & Maurin, G. (2010). ACS Nano 4, 143–152. [DOI] [PubMed]
- Salles, F., Kolokolov, D. I., Jobic, H., Maurin, G., Llewellyn, P. L., Devic, T., Serre, C. & Férey, G. (2009). J. Phys. Chem. C, 113, 7802–7812.
- Salles, F., Maurin, G., Serre, C., Llewellyn, P. L., Knöfel, C., Choi, H. J., Filinchuk, Y., Oliviero, L., Vimont, A., Long, J. R. & Férey, G. (2010). J. Am. Chem. Soc. 132, 13782–13788. [DOI] [PubMed]
- Samsonenko, D. G., Kim, H., Sun, Y., Kim, G. H., Lee, H. S. & Kim, K. (2007). Chem. Asian J. 2, 484–488. [DOI] [PubMed]
- Sato, H., Kosaka, W., Matsuda, R., Hori, A., Hijikata, Y., Belosludov, R. V., Sakaki, S., Takata, M. & Kitagawa, S. (2014). Science, 343, 167–170. [DOI] [PubMed]
- Serra-Crespo, P., Gobechiya, E., Ramos-Fernandez, E. V., Juan-Alcañiz, J., Martinez-Joaristi, A., Stavitski, E., Kirschhock, C. E., Martens, J. A., Kapteijn, F. & Gascon, J. (2012). Langmuir, 28, 12916–12922. [DOI] [PubMed]
- Serre, C., Bourrelly, S., Vimont, A., Ramsahye, N. A., Maurin, G., Llewellyn, P., Daturi, M., Filinchuk, Y., Leynaud, O., Barnes, P. & Férey, G. (2007). Adv. Mater. 19, 2246–2251.
- Shimomura, S., Higuchi, M., Matsuda, R., Yoneda, K., Hijikata, Y., Kubota, Y., Mita, Y., Kim, J., Takata, M. & Kitagawa, S. (2010). Nat. Chem. 2, 633–637. [DOI] [PubMed]
- Smart, P., Mason, C. A., Loader, J. R., Meijer, A. J. H. M., Florence, A. J., Shankland, K., Fletcher, A. J., Thompson, S. P., Brunelli, M., Hill, A. H. & Brammer, L. (2013). Chem. Eur. J. 19, 3552–3557. [DOI] [PubMed]
- Soleimani-Dorcheh, A., Dinnebier, R. E., Kuc, A., Magdysyuk, O., Adams, F., Denysenko, D., Heine, T., Volkmer, D., Donner, W. & Hirscher, M. (2012). Phys. Chem. Chem. Phys. 14, 12892–12897. [DOI] [PubMed]
- Spencer, E. C., Howard, J. A. K., McIntyre, G. J., Rowsell, J. L. C. & Yaghi, O. M. (2006). Chem. Commun. pp. 278–280. [DOI] [PubMed]
- Stylianou, K. C., Rabone, J., Chong, S. Y., Heck, R., Armstrong, J., Wiper, P. V., Jelfs, K. E., Zlatogorsky, S., Bacsa, J., McLennan, A. G., Ireland, C. P., Khimyak, Y. Z., Thomas, K. M., Bradshaw, D. & Rosseinsky, M. J. (2012). J. Am. Chem. Soc. 134, 20466–20478. [DOI] [PubMed]
- Suh, M. P., Park, H. J., Prasad, T. K. & Lim, D. W. (2012). Chem. Rev. 112, 782–835. [DOI] [PubMed]
- Sumida, K., Rogow, D. L., Mason, J. A., McDonald, T. M., Bloch, E. D., Herm, Z. R., Bae, T. H. & Long, J. R. (2012). Chem. Rev. 112, 724–781. [DOI] [PubMed]
- Takamizawa, S., Kojima, K. & Akatsuka, T. (2006). Inorg. Chem. 45, 4580–4582. [DOI] [PubMed]
- Takamizawa, S. & Nakata, E. (2005). CrystEngComm, 7, 476–479.
- Takamizawa, S., Nakata, E. & Akatsuka, T. (2006). Angew. Chem. Int. Ed. 45, 2216–2221. [DOI] [PubMed]
- Takamizawa, S., Nakata, E., Akatsuka, T., Kachi-Terajima, C. & Miyake, R. (2008). J. Am. Chem. Soc. 130, 17882–17892. [DOI] [PubMed]
- Takamizawa, S., Nataka, E., Akatsuka, T., Miyake, R., Kakizaki, Y., Takeuchi, H., Maruta, G. & Takeda, S. (2010). J. Am. Chem. Soc. 132, 3783–3792. [DOI] [PubMed]
- Takamizawa, S., Nakata, E. & Saito, T. (2004). Angew. Chem. Int. Ed. 43, 1368–1371. [DOI] [PubMed]
- Takamizawa, S., Nakata, E., Saito, T. & Akatsuka, T. (2005). Inorg. Chem. 44, 1362–1366. [DOI] [PubMed]
- Takamizawa, S., Nakata, E., Yokoyama, H., Mochizuki, K. & Mori, W. (2003). Angew. Chem. Int. Ed. 42, 4331–4334. [DOI] [PubMed]
- Takata, M. (2008). Acta Cryst. A64, 232–245. [DOI] [PubMed]
- Takata, M., Nishibori, E. & Sakata, M. (2001). Z. Kristallogr. 216, 71–86.
- Takata, M., Umeda, B., Nishibori, E., Sakata, M., Saitot, Y., Ohno, M. & Shinohara, H. (1995). Nature, 377, 46–49.
- Tanaka, D., Nakagawa, K., Higuchi, M., Horike, S., Kubota, Y., Kobayashi, T., Takata, M. & Kitagawa, S. (2008). Angew. Chem. Int. Ed. 47, 3914–3918. [DOI] [PubMed]
- Tanaka, H., Takata, M., Nishibori, E., Kato, K., Iishi, T. & Sakata, M. (2002). J. Appl. Cryst. 35, 282–286.
- Taratula, O., Hill, P. A., Khan, N. S., Carroll, P. J. & Dmochowski, I. J. (2010). Nat. Commun. 1, article 148. [DOI] [PMC free article] [PubMed]
- Thompson, S. P., Parker, J. E., Potter, J., Hill, T. P., Birt, A., Cobb, T. M., Yuan, F. & Tang, C. C. (2009). Rev. Sci. Instrum. 80, 075107. [DOI] [PubMed]
- Ueda, T., Kurokawa, K., Eguchi, T., Kachi-Terajima, C. & Takamizawa, S. (2007). J. Phys. Chem. C, 111, 1524–1534.
- Vaidhyanathan, R., Iremonger, S. S., Dawson, K. W. & Shimizu, G. K. H. (2009). Chem. Commun. 35, 5230–5232. [DOI] [PubMed]
- Vaidhyanathan, R., Iremonger, S. S., Shimizu, G. K., Boyd, P. G., Alavi, S. & Woo, T. K. (2010). Science, 330, 650–653. [DOI] [PubMed]
- Vitórica-Yrezábal, I. J., Mínguez Espallargas, G., Soleimannejad, J., Florence, A. J., Fletcher, A. J. & Brammer, L. (2013). Chem. Sci. 4, 696–708.
- Vitórica-Yrezábal, I. J., Sullivan, R. A., Purver, S. L., Curfs, C., Tang, C. C. & Brammer, L. (2011). CrystEngComm, 13, 3189–3196.
- Wang, J., Wang, C. & Lin, W. (2012). ACS Catal. 2, 2630–2640.
- Wiersum, A. D., Soubeyrand-Lenoir, E., Yang, Q., Moulin, B., Guillerm, V., Yahia, M. B., Bourrelly, S., Vimont, A., Miller, S., Vagner, C., Daturi, M., Clet, G., Serre, C., Maurin, G. & Llewellyn, P. L. (2011). Chem. Asian J. 6, 3270–3280. [DOI] [PubMed]
- Wong-Ng, W., Kaduk, J. A., Huang, Q., Espinal, L., Li, L. & Burress, J. (2013). Microporous Mesoporous Mater. 172, 95–104.
- Wright, P. A., Thomas, J. M., Ramdas, S. & Cheetham, A. K. (1984). J. Chem. Soc. Chem. Commun. pp. 1338–1339.
- Wu, H., Gong, Q., Olson, D. H. & Li, J. (2012). Chem. Rev. 112, 836–868. [DOI] [PubMed]
- Wu, H., Simmons, J. M., Liu, Y., Brown, C. M., Wang, X.-S., Ma, S., Peterson, V. K., Southon, P. D., Kepert, C. J., Zhou, H.-C., Yildirim, T. & Zhou, W. (2010). Chem. Eur. J. 16, 5205–5214. [DOI] [PubMed]
- Wu, H., Simmons, J. M., Srinivas, G., Zhou, W. & Yildirim, T. (2010). J. Phys. Chem. Lett, 1, 1946–1951.
- Wu, H., Zhou, W. & Yildirim, T. (2007). J. Am. Chem. Soc. 129, 5314–5315. [DOI] [PubMed]
- Wu, H., Zhou, W. & Yildirim, T. (2009). J. Phys. Chem. C, 113, 3029–3035.
- Xie, L.-H., Lin, J.-B., Liu, X.-M., Wang, Y., Zhang, W.-X., Zhang, J.-P. & Chen, X.-M. (2010). Inorg. Chem. 49, 1158–1165. [DOI] [PubMed]
- Yanai, N., Kitayama, K., Hijikata, Y., Sato, H., Matsuda, R., Kubota, Y., Takata, M., Mizuno, M., Uemura, T. & Kitagawa, S. (2011). Nat. Mater. 10, 787–793. [DOI] [PubMed]
- Yang, W., Davies, A. J., Lin, X., Suyetin, M., Matsuda, R., Blake, A. J., Wilson, C., Lewis, W., Parker, J. E., Tang, C. C., George, M. W., Hubberstey, P., Kitagawa, S., Sakamoto, H., Bichoutskaia, E., Champness, N. R., Yany, S. & Schröder, M. (2012). Chem. Sci. 3, 2993–2999.
- Yang, S., Lin, X., Lewis, W., Suyetin, M., Bichoutskaia, E., Parker, J. E., Tang, C. C., Allan, D. R., Rizkallah, P. J., Hubberstey, P., Champness, N. R., Thomas, K. M., Blake, A. J. & Schröder, M. (2012). Nat. Mater. 11, 710–716. [DOI] [PubMed]
- Yang, S., Liu, L., Sun, J., Thomas, K. M., Davies, A. J., George, M. W., Blake, A. J., Hill, A. H., Fitch, A. N., Tang, C. C. & Schröder, M. (2013). J. Am. Chem. Soc. 135, 4954–4957. [DOI] [PubMed]
- Yang, S., Sun, J., Ramirez-Cuesta, A. J., Callear, S. K., David, W. I. F., Anderson, D. P., Newby, R., Blake, A. J., Parker, J. E., Tang, C. C. & Schröder, M. (2012). Nat. Chem. 4, 887–894. [DOI] [PubMed]
- Yildirim, T. & Hartman, M. R. (2005). Phys. Rev. Lett. 95, 215504. [DOI] [PubMed]
- Yoon, M., Srirambalaji, R. & Kim, K. (2012). Chem. Rev. 112, 1196–1231. [DOI] [PubMed]
- Zhang, J.-P. & Chen, X.-M. (2008). J. Am. Chem. Soc. 130, 6010–6017. [DOI] [PubMed]
- Zhang, J.-P. & Chen, X.-M. (2009). J. Am. Chem. Soc. 131, 5516–5521. [DOI] [PubMed]
- Zhang, J., Zhu, A. & Chen, X. (2012). Chem. Commun. 48, 11395. [DOI] [PubMed]
- Zhou, H. C., Long, J. R. & Yaghi, O. M. (2012). Chem. Rev. 112, 673–674. [DOI] [PubMed]