

Abstract
Background
The proto-oncogene Casitas b-lineage lymphoma (c-Cbl) is an adaptor protein with an intrinsic E3 ubiquitin ligase activity that targets receptor and non-receptor tyrosine kinases, resulting in their ubiquitination and down-regulation. However, the function of c-Cbl in the control of cardiac function is currently unknown. In this study, we examined the role of c-Cbl in myocyte death and cardiac function after myocardial ischemia.
Methods and Results
We show increased c-Cbl expression in human ischemic and dilated cardiomyopathy hearts and in response to pathological stress stimuli in mice. c-Cbl deficient mice demonstrated a more robust functional recovery after myocardial ischemia reperfusion injury, as well as significantly reduced myocyte apoptosis and improved cardiac function. Ubiquitination and downregulation of key survival c-Cbl targets, epidermal growth factor receptors and focal adhesion kinase, were significantly reduced in c-Cbl knockout mice. Inhibition of c-Cbl expression or its ubiquitin ligase activity in cardiac myocytes offered protection against H2O2 stress. Interestingly, c-Cbl deletion reduced the risk of death and increased cardiac functional recovery after chronic myocardial ischemia. This beneficial effect of c-Cbl deletion was associated with enhanced neoangiogenesis and increased expression of vascular endothelial growth factor (VEGF)-a and VEGF receptor type 2 in the infarcted region.
Conclusions
c-Cbl activation promotes myocyte apoptosis, inhibits angiogenesis and causes adverse cardiac remodeling after myocardial infarction. These findings point to c-Cbl as a potential therapeutic target for the maintenance of cardiac function and remodeling after myocardial ischemia.
Keywords: Myocardial ischemia, ubiquitin ligase, cardioprotection, angiogenesis, apoptosis
Introduction
Myocardial infarction (MI) due to coronary artery occlusion is a leading cause of death worldwide.1 The loss of blood flow after MI results in loss of cardiac myocytes in the ischemic zone, which diminish cardiac contractility and impede angiogenesis and repair. Although much is known about the pathways that promote the pathological remodeling responses, mechanisms that promote myocyte loss and impair cardiac function have not been as clearly defined. The ubiquitin proteasome system (UPS) is largely responsible for the degradation of misfolded and damaged proteins as well as proteins involved in the control of components of the contractile apparatus and hypertrophic gene expression, thereby regulating cardiac hypertrophy and remodeling.2 Activation of the UPS in heart reduces muscle mass as occurs during muscle atrophy,3,4 promotes myocyte hypertrophy and impairs cardiac remodeling.5
The formation of ubiquitin-protein conjugates involves three components that participate in a cascade of ubiquitin transfer reactions: a ubiquitin-activation enzyme (E1), a ubiquitin-conjugating enzyme (E2) and a ubiquitin ligase (E3) that acts at the last step of the cascade and presents the greatest tissue and substrate specificity.6,7 Once a protein has been marked with polyubiquitin conjugates, it is destined for degradation by the multicatalytic protease, the 26S proteasome complex.8 The proto-oncogene c-Cbl contains a highly conserved helical region and an adjacent RING finger domain that together bind an E2; as such c-Cbl acts as a RING type ubiquitin ligase (E3) and negatively regulates receptor and non-receptor tyrosine kinases by promoting their ubiquitination and lysosomal/proteasomal degradation.9 In addition to a tyrosine kinase-binding domain for binding to activated tyrosine kinases, c-Cbl also contains multiple protein interaction motifs, a tyrosine kinase binding domain (TKB) that encompasses a 4-helical bundle, an EF hand and a a Src homology domain 2 (SH2), and a proline-rich motif.10 These domains/motifs allow the interaction of c-Cbl with multiple signal transducers, including the p85 subunit of PI3 kinase, the Crk and Grb2 adaptor proteins, and Src family kinases suggesting that c-Cbl acts as a docking protein to integrate signaling pathways.9 Although c-Cbl phosphorylation in response to a variety of cell surface receptors has been documented, the precise biological function of c-Cbl is unclear as both negative and positive roles for c-Cbl have been proposed.9 In the heart, c-Cbl expression increases in response to right ventricular pressure overload stimuli11 and one study linked c-Cbl to reduced insulin responsiveness in adult rat cardiomyocytes in vitro.12 However, specific functions of c-Cbl in myocyte growth and cardiac function are still not well understood and no loss-of-function cardiac phenotypes of c-Cbl gene have yet been described.
We recently showed that activation of c-Cbl ubiquitin ligase by neutrophil derived serine proteases leads to focal adhesion protein turnover and myofibril degeneration.13 In the present study, we show that c-Cbl expression increases in human cardiomyopathies and in response to myocardial stress in mice. Mice lacking c-Cbl display normal cardiac function but exhibit less myocyte loss and cardiac dysfunction in response to ischemia reperfusion (IR) injury. c-Cbl deficient mice are also protected against pathological remodeling of the heart following chronic myocardial infarction (MI). This protection appears to be mediated, at least in part, to neoangiogenesis in the infarcted region. We conclude that c-Cbl negatively regulates ventricular myocyte survival and angiogenesis after myocardial ischemia injury.
Materials & Methods
Subjects and tissue preparation
Human left ventricular (LV) myocardial tissue was obtained at the time of cardiac transplantation from patients with end-stage heart failure ischemic cardiomyopathy (ICM, n=5) and dilated cardiomyopathy (DCM, n=5). Left ventricular tissue was also obtained from non-failing hearts (n=5) of brain-dead organ donors that could not be used for transplantation. Relevant clinical data were collected from all subjects providing heart tissue (see Supplemental Table S1). Our protocol was approved by Temple University Institutional Review Board, according to the guidelines noted in our Instructions to Authors. All hearts were arrested in situ with cold, blood-containing, cardioplegia solution and promptly transported to the laboratory in Krebs-Henseleit buffer (KHB) solution as previously described.14 LV tissue slices were immediately snap frozen in liquid nitrogen and stored at -80°C or fixed in formalin.
Myocardial ischemia and IR injury procedure
All mice were maintained and studied using protocols approved by the Animal Care and Use Committee of Temple University. c-Cbl knockout (KO) and Cblflox/flox mice (provided by Dr. Hua Gu, IRCM) and were genotyped as described previously.15,16 Generation of α-myosin heavy chain (α-MHC)- MerCreMer/c-Cblflox/flox mice is described in the Methods section of the online-only Data Supplement (Supplemental Figure S1). For easier reading, we refer these mice as CM-Cbl KO mice. CM-Cbl KO mice were paired with age- and sex-matched c-Cblflox/flox littermates for all experiments. 10-12 weeks old wild type (WT) control, c-Cbl KO, CM-Cbl KO and c-Cblflox/flox mice were anesthetized with a mixture of ketamine (100 mg/kg) and xylazine (10 mg/kg) to perform a left thoracotomy under mechanical ventilation. The body temperatures of the mice were maintained by a heated surgical platform and body temperature was monitored using a rectal sensor during the surgical procedure. A 6–0 suture with a slipknot was tied around the left anterior descending (LAD) coronary artery to produce ischemia. Consistent elevation of the ST segment was observed in lead II tracings following occlusion of the LAD coronary vessel. One group of mice was revived for a 30 minutes ischemia period after which the knot was released and reperfusion in the heart occurred. The chest wall was closed with 8-0 silk and then the animal was removed from the ventilator and kept warm in the cage maintained at 37°C overnight. A sham procedure constituted the surgical incision without LAD ligation. Hearts were harvested after 2, 7 or 30 days (for animals subjected to myocardial ischemia) or after 24 hours of reperfusion (for animal subjected to IR injury). All mice were randomized to the experimental protocol described above.
Data Analysis
Summary data are presented as mean±SEM. For comparisons of >2 groups, one-way ANOVA or, more generally, the generalized linear regression approach was employed for normal distributions and the Kruskal Wallis test for non-normal or small sample situations. Two-group comparisons were analyzed by the two-sample t test or nonparametric Wilcoxon rank test, whenever appropriate (e.g., when the sample size was small and/or the distribution was not normal). Bonferroni post-hoc test adjustments were employed for multiple pairwise group comparisons after the overall F or Kruskal Wallis test showed a statistical significance. The exact testing was used when the sample size was small (e.g., when all group sizes <10). The survival time was analyzed using the Kaplan-Meier product-limit approach and compared by the log-rank test. To make the plot and interpretation of the fold increase over WT sham easier to understand, we scaled the data value from each animal in each of the four groups, including individual sham values, using the mean of the WT sham group. All in vitro experiments were performed at least three times from three different cultures and the data values were scaled to controls. A value of P<0.05 was considered statistically significant.
An expanded Materials and Methods section is in the online-only Data Supplement.
Results
c-Cbl expression is developmentally downregulated
To determine the temporal pattern of c-Cbl expression during normal cardiac development in the mouse, we analyzed total protein extracts from series of fetal and postnatal time points. c-Cbl protein was highly expressed in fetal hearts at 12.5 day (d) and this expression decreased gradually throughout fetal life (-17 ±3% at E19.5 compared to E12.5) (Figure 1A). After birth, c-Cbl expression decreased significantly at 3d and 7d post-natal (-30 ±2% at 1d, -37 ±2% at 3d, and −46 ±3% at 7d compared to E12.5) and reached low but detectable levels in adult hearts (-80 ±5% compared to E12.5). This decrease in c-Cbl expression in adult hearts was associated with a decrease in c-Cbl expression in adult isolated myocytes (Figure 1B).
Figure 1.
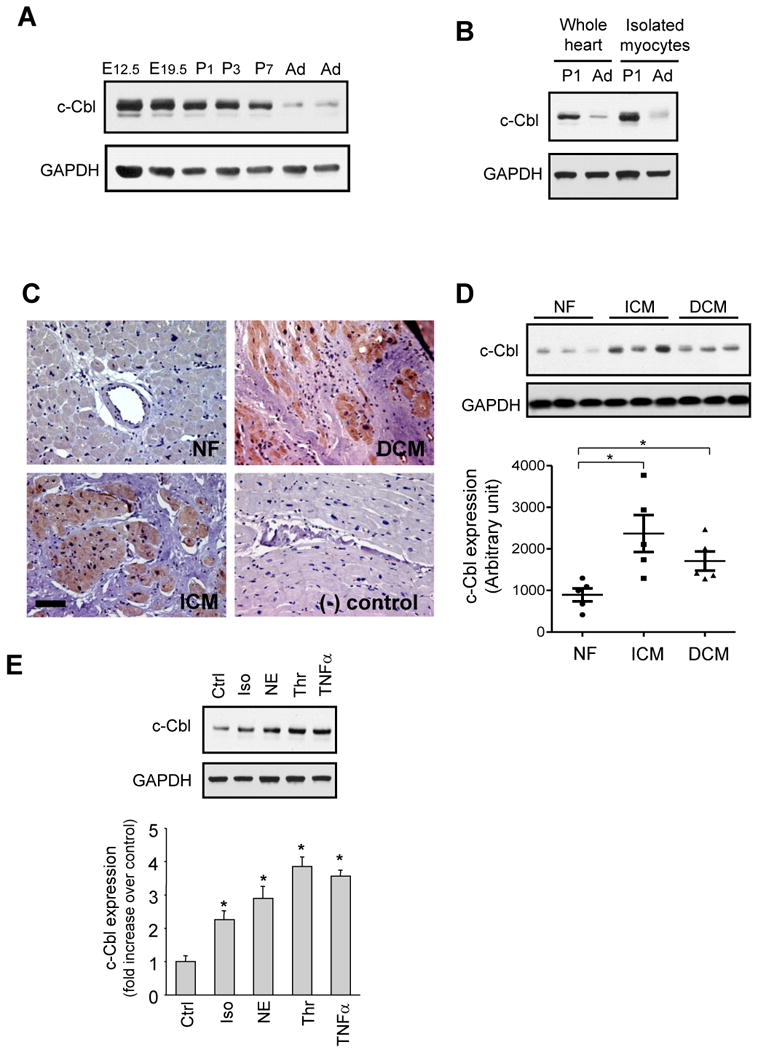
c-Cbl expression is upregulated in human cardiomyopathies. (A and B) Representative immunoblots of cardiac lysates from indicated embryonic (E), postnatal day (P), or adult (Ad) Sprague Dawley rats (A) or cardiomyocytes isolated from 1 day post-natal or adult rat hearts (B). GAPDH was shown as a loading control. Western blots are representative of 3 separate experiments. (C) Representative immunostainings of paraffin-embedded non failing (NF), ischemic (ICM) and dilated cardiomyopathy (DCM) human heart sections stained for c-Cbl and counterstained with hematoxylin. Scale Bar, 40 µm. *P<0.05 vs. NF controls. (D) Immunoblot analysis of whole lysates from NF (n=5), ICM (n=5), and DCM (n=5) human hearts. (E) Lysates from NRCMs untreated or treated with isoproterenol (Iso, 10 µM), norepinephrine (NE, 10 µM), thrombin (Thr, 1 U/ml), or TNFα (100 ng/ml) for 48 hours. Top, c-Cbl immunoblot with GAPDH taken as a loading control. Bottom, Quantification of experiments expressed as mean ± S.E from three separate cultures. *P<0.05 vs. control.
c-Cbl expression is upregulated in failing human myocardium
Cardiac explants from human donors (clinical characteristics of heart donors are summarized in Supplemental Table 1) were assessed for c-Cbl expression using Western blot and immunohistochemistry. Figure 1C shows increased c-Cbl immunostaining in both ischemic (ICM) and dilated cardiomyocpathies (DCM) compared to non-failing hearts. This c-Cbl accumulation was mainly detected in myocytes and no appreciable labeling was observed at the level of fibroblasts in the fibrotic zone, smooth muscle cells of medial arteries, or endothelial cells. Some c-Cbl immunostaining was detected at the medial layer of small arterioles (data not shown). c-Cbl accumulation in failing human heart samples was 2.3-fold and 1.5-fold higher in the ICM and DCM human hearts, respectively, compared to signals in non-failing hearts (Figure 1D).
To explore the mechanism of induction of c-Cbl, we examined the effect of stress stimuli on c-Cbl expression in neonatal rat cardiomyocytes (NRCMs). Myocytes challenged with hypertrophic agonists norepinephrine (primarily an α-adrenergic receptor agonist), isoproterenol (a non-selective β-adrenergic receptor agonist), thrombin, or TNF-α for 48 hours showed an increase in c-Cbl accumulation compared to controls (Figure 1E). These data together show the effect of hypertrophic agonists and inflammatory cytokines in modulating c-Cbl expression in cardiomyocytes.
c-Cbl ablation protects the heart from ischemia/reperfusion injury
To explore the functional effects associated with c-Cbl induction in the failing hearts, c-Cbl KO mice and their WT controls were subjected to transient LAD ligation for 30 minutes followed by reperfusion for 24 hours. c-Cbl KO mice did not differ from WT mice with regard to their baseline heart weight-to-body weight ratio, heart rate (WT 457±10 beats/minute; c-Cbl KO 471±24 beats/minute), LV end systolic pressure (WT 89±9 mm Hg; c-Cbl KO 86±5 mm Hg), LV end diastolic pressure (WT 5.2 ±0.8 mmHg; c-Cbl KO 6.3 ±0.9 mm Hg), and maximal dP/dt (WT 8517 ±780 mmHg.minutes–1; c-Cbl KO 7517 ±395 mmHg.minutes–1); hemodynamic data were obtained in n=8 WT mice and n=8 c-Cbl KO mice. Interestingly, c-Cbl KO mice subjected to IR injury showed a significantly less impairment of cardiac function as assessed by LV ejection fraction and fractional shortening percentage compared to WT mice (Figures 2A and 2B and Table 1). Ablation of c-Cbl also reduced the percentage of infarcted area normalized to area at risk (AAR) (22 ± 3%), compared with WT mice (34 ± 5%) (Figures 2C and 2D). Apoptotic cell death in the AAR, as evaluated by terminal deoxynucleotidyl transferase dUTP nick end labeling staining, was significantly lower in c-Cbl KO mice compared with that in WT mice (Figures 2E and 2F). Under baseline conditions, the proportion of TUNEL-positive cardiomyocytes was very low (<0.1%) in c-Cbl KO and WT mice. Consistent with TUNEL staining, caspase-3 activity was also lower in c-Cbl KO infracted hearts compared to WT, thus confirming a lesser propensity for myocyte death by apoptosis in c-Cbl KO mice subjected to IR (Figure 2G). Circulating plasma levels of the cardiac-specific isoform of troponin-I (cTnI) were evaluated as an additional marker of myocardial injury (Figure 2H). Serum samples from c-Cbl KO mice subjected to IR injury were found to contain lower levels of cTnI compared to WT mice (43.2 ±2.2 in WT vs. 28.1 ± 3.5 ng/ml per mg protein in c-Cbl KO), thus confirming the cytoprotective effect of c-Cbl deletion.
Figure 2.
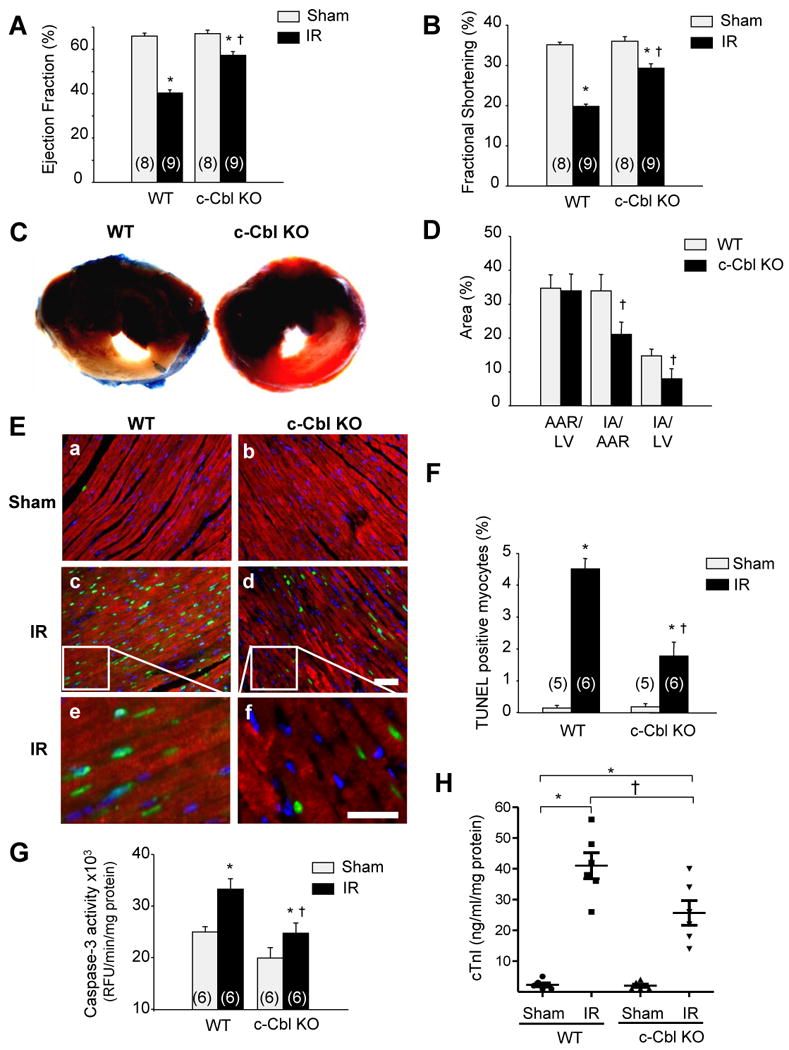
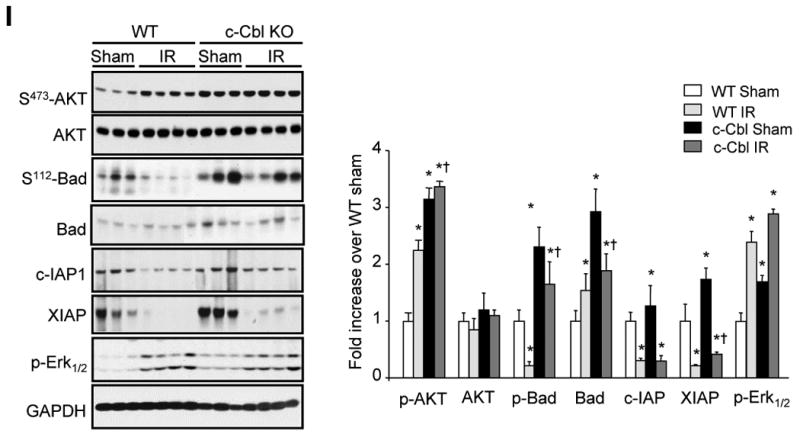
c-Cbl ablation protects against IR injury. The left anterior descending artery was ligated for 30 minutes to induce ischemia and it subsequently was reperfused for 24 hours. (A-B) Echocardiography measurement of ejection fraction (A) and fractional shortening (B) in WT and c-Cbl KO animals (n=8 for sham groups, n=9 for IR groups). (C) Representative cross-sections were stained with triphenyl tetrazolium chloride and Evans blue to determine the extent of injury. (D) Quantification of infarct area (IA) vs. area at risk (AAR) after IR injury in the indicated groups (n=6 per group). (E) LV tissue sections were assessed for apoptosis using TUNEL assay (green), tropomyosin (red), and DAPI (4′,6-diamidino-2-phenylindole) (blue) staining. Scale bars: 40 µm (a-d) or 20 µm (e, f). (F) The number of TUNEL-positive myocytes in the ischemic area was expressed as a percentage of total nuclei detected by DAPI staining. (G) Quantification of caspase-3 activity in LV using caspase-3 specific fluorogenic substrate. (H) Serum levels of cardiac troponin I after IR injury in WT and c-Cbl KO mice (n=6 each group). (I) Representative immunoblots of LV lysates from WT or c-Cbl KO animals. GAPDH was included as a loading control. Left, Representative autoradiogram (with each lane from a single gel exposed for the same duration). Right, Fold induction (n=6 each group). *P<0.05 vs. WT shams, †P<0.05 vs. WT IR.
Table 1.
Echocardiographic measurements in WT and c-Cbl KO mice subjected or not to IR injury.
| Sham | IR | |||
|---|---|---|---|---|
|
| ||||
| WT (n=8) |
c-Cbl KO (n=9) |
WT (n=8) |
c-Cbl KO (n=9) |
|
| HR (bpm) | 480 ±10 | 471 ±24 | 457 ±12 | 497 ±37 |
| HW (mg) | 126 ±1 | 117 ±10 | 135 ±4 | 129 ±10 |
| BW (g) | 24.5 ±0.9 | 21.6 ±0.9 | 24.2 ±0.6 | 22.5 ±0.9 |
| HW/BW (mg/g) | 5.3 ±0.1 | 5.5 ±0.3 | 5.6 ±0.2 | 5.8 ±0.3 |
| LVEDD (mm) | 3.34 ±0.11 | 3.15 ±0.15 | 3.66 ±0.09 | 3.29 ±0.09 |
| LVESD (mm) | 2.19 ±0.1 | 2.00 ±0.12 | 2.85 ±0.04* | 2.32 ±0.07*† |
| LVPWTd (mm) | 0.98 ±0.01 | 0.94 ±0.01 | 0.96 ±0.02 | 0.92 ±0.03 |
| LVPWTs (mm) | 1.45 ±0.04 | 1.33 ±0.03 | 1.42 ±0.08 | 1.30 ±0.11 |
HR indicates heart rate; HW, heart weight; BW, body weight; LVEDD, LV end diastolic dimension; LVESD, LV end systolic dimension; LVPWTd, LV posterior wall thickness diastole; and LVPWTs, LV posterior wall thickness systole.
P<0.05 vs. WT shams,
P<0.05 vs. WT IR
c-Cbl deletion exhibits protective signaling
Activation of c-Cbl enables it to act as a multivalent adaptor for a plethora of SH2 or SH3 domain-containing signaling proteins to positively or negatively regulate signaling pathways and cell growth.9 Therefore, we next examined the molecular mechanisms responsible for c-Cbl-mediated cardioprotection. Relative to WT samples, c-Cbl KO heart samples showed increases in the levels of phospho-Akt(S473) (3.2 ±0.2-fold), phospho-Bad(S112) (3.3 ±0.4-fold), Bad (2.9 ±0.5-fold), XIAP (1.8 ±0.2-fold), and phospho-Erk1/2 (1.7 ±0.2-fold) (Figure 2I). However, we observed no significant increase in the expression of total-Akt or c-IAP1 in c-Cbl KO as compared to WT control heart samples. We also examined these signaling molecules after 1-day following IR injury; relative to sham-operated controls, WT mice after IR injury showed significant increases in phospho-Akt(S473) (2.2 ±0.2-fold) and phospho-Erk1/2 (2.4 ±0.2-fold), along with a significant decrease in the expression of phospho-Bad(S112) (-0.78 ±0.07-fold), c-IAP-1 (-0.70 ±0.1-fold), and XIAP (-0.80 ±0.14-fold). In comparison, c-Cbl-KO mice subjected to IR injury showed a stronger increase in phospho-Bad(S112), c-IAP, and XIAP expression compared to WT subjected to IR injury. No significant effect on total-Akt expression or phosphorylation between WT and c-Cbl KO mice subjected to IR injury was detected. Thus, as compared to WT hearts, c-Cbl-KO hearts show significantly more elevation in the levels of anti-apoptotic signaling molecules.
Cardiac-specific deletion of c-Cbl is cardioprotective
In light of the cardioprotective effect of global c-Cbl deletion, we generated mice homozygous for a loxP-flanked c-Cbl allele and positive for tamoxifen-inducible Cre recombinase driven by the cardiomyocyte-specific α-myosin heavy chain (MHC) promoter (α-MHC-MerCreMer/c-Cblflox/flox). Tamoxifen injections at the age of 8 weeks for 5 days induced Cre-mediated recombination after 4 weeks, and Western analysis of protein lysates confirmed that c-Cbl protein was reduced by 90% in cardiac tissue (Supplemental Figure S1. To assess the potential consequences of cardiac c-Cbl loss-of-function on IR injury, we subjected CM-Cbl KO and c-Cblflox/flox to IR for 24 hours. Echocardiography before surgery showed no difference in cardiac function and geometry between the groups (Supplemental Table 2). 24 hours after surgery, CM-Cbl KO showed significantly improved ejection fraction and fractional shortening compared to c-Cblflox/flox mice (Supplemental Figures S2A and S2B). This was associated with reduced LV internal diameter (Supplemental Figures S2C and S2D). Cardiac myocyte specific deletion of c-Cbl also reduced the percentage of infarcted area normalized to area at risk (AAR) (25 ± 4%, n=6), compared with c-Cblflox/flox hearts mice (38 ± 3%, n=5)(Supplemental Figure S2E), reduced occurrence of TUNEL-positive cells in the infarcted heart (Supplemental Figures S3A-3B), and attenuated caspase-3 activity in infracted hearts compared to c-Cblflox/flox mice (Supplemental Figure S3C). These data collectively demonstrate that deletion of c-Cbl in cardiomyocytes offers cardioprotection in response to IR injury.
c-Cbl deletion prevents EGFR and FAK protein ubiquitination and degradation induced after IR injury
c-Cbl proteins contain a RING domain through which they interact with E2 ubiquitin-conjugating enzymes and which is required for their ubiquitin ligase activity.9 Immunoprecipitation analysis coupled with in vitro ubiquitination assay revealed that c-Cbl ubiquitin ligase activity was increased in WT heart samples subjected to IR injury compared to WT controls (Figure 3A). c-Cbl tyrosine phosphorylation at Y774 residue, a site involved in c-Cbl interaction with PI3 kinase, was also increased after IR. No change in c-Cbl expression was detectable after IR injury.
Figure 3.
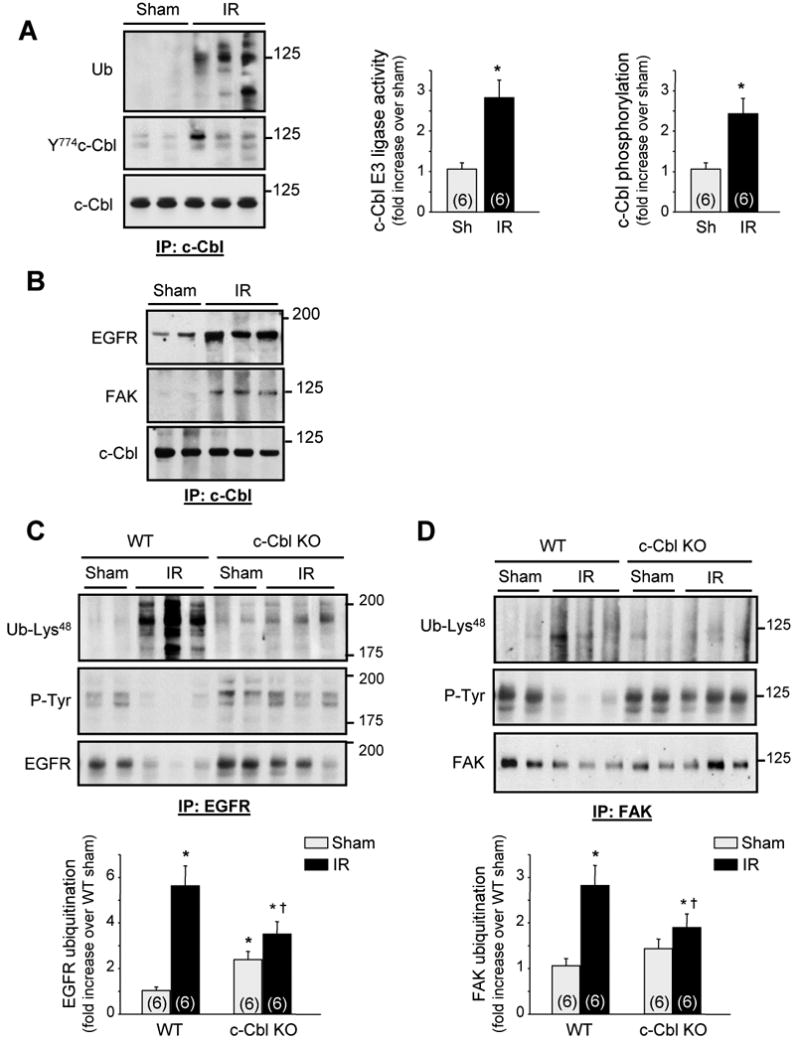
c-Cbl ablation protects against EGFR and FAK ubiquitination and downregulation induced after IR injury. The left anterior descending artery was ligated for 30 minutes to induce ischemia and it subsequently was reperfused for 24 hours. (A-B) c-Cbl immunoprecipitates were assayed for auto-ubiquitination assay and immunoblot analysis. (C-D) EGFR or FAK immunoprecipitates from LV lysates of shams and mice subjected to IR for 24 hours were immunoblotted with ubiquitin-Lys48, phosphotyrosine (P-Tyr), EGFR, or FAK antibodies. Top, Representative autoradiogram (with each lane from a single gel exposed for the same duration). Bottom, Fold induction (n=6 each group). *P<0.05 compared to WT shams, †P<0.05 compared to WT IR.
To explore the significance of c-Cbl activation in vivo, we assessed next whether c-Cbl mediates ubiquitination of epidermal growth factor receptor (EGFR) and focal adhesion kinase (FAK), two key signaling targets of c-Cbl that control myocyte hypertrophy and survival.17,18 We found that IR injury induced an increase in c-Cbl interaction with EGFR and FAK (Figure 3B), along with an increase in their ubiquitination at Lysine 48 residue, a ubiquitin chain structure that is usually required for substrate targeting to the 26S proteasome for degradation (Figures 3C and 3D).19 Notably, EGFR and FAK tyrosine phosphorylation and expression levels decreased significantly in WT mice subjected to IR injury compared to shams, suggesting a link between EGFR/FAK ubiquitination and degradation. c-Cbl deletion attenuated EGFR and FAK ubiquitination and degradation induced by IR injury compared to WT. No significant difference in EGFR or FAK protein accumulation was detected between c-Cbl KO and WT shams, although a slight increase in basal ubiquitination of EGFR was detected in c-Cbl KO compared to WT sham hearts. This effect of c-Cbl deletion on basal EGFR ubiquitination could result from decreased EGFR degradation, increased EGFR association with c-Cbl homolog Cbl-b,20 or both. Collectively, these data show that c-Cbl activation after IR mediates ubiquitination and degradation of signaling molecules involved in myocyte survival.
c-Cbl knockdown induces protection against oxidative stress-induced myocyte death
To directly demonstrate that the cardioprotective effect of c-Cbl deletion after IR results from endogenous expression of c-Cbl in cardiomyocytes, we investigated the role of c-Cbl in oxidative stress-induced cardiac myocyte death. Treatment of neonatal rat cardiomyocytes (NRCMs) with H2O2 induced an increase in c-Cbl ubiquitin ligase activity as assessed by in vitro auto-ubiquitination assay (Figure 4A). This increase was rapid, was sustained for over 60 minutes after H2O2 treatment and was potent compared to cells treated with EGF for 5 minutes. H2O2 also caused a transient increase in c-Cbl Y774 phosphorylation with maximum phosphorylation observed at 30 minutes after H2O2 treatment. Concomitant with increased c-Cbl auto-ubiquitination, H2O2 treatment increased c-Cbl interaction with EGFR along with increased EGFR ubiquitination (Figure 4B), suggesting that c-Cbl mediates EGFR ubiquitination. Transduction of adenovirus carrying shRNA c-Cbl (Ad-shCbl), which reduced c-Cbl expression by ∼75-85% compared to cells infected with shRNA control (Ad-shCtrl), significantly decreased EGFR ubiquitination and p38 MAPK and AKT phosphorylation induced by H2O2 or EGF treatment for 10 minutes (Figure 4C and Supplemental Figure S4). Expression of Ad-shCbl enhanced the basal phosphorylation of pro-survival signaling molecules, ERK1/2 and AKT, compared to Ad-shCtrl infected cells. Furthermore, H2O2 treatment did not further increase or induce ERK1/2 or JNK phosphorylation, respectively. Ad-shCbl infected cells also showed a significantly attenuated phosphorylation of these two kinases in response to EGF treatment compared to Ad-shCtrl infected cells. These data show that inhibition of c-Cbl expression enhances pro-survival signaling and suggest that c-Cbl-dependent and independent pathways are involved in H2O2-induced downstream signaling.
Figure 4.
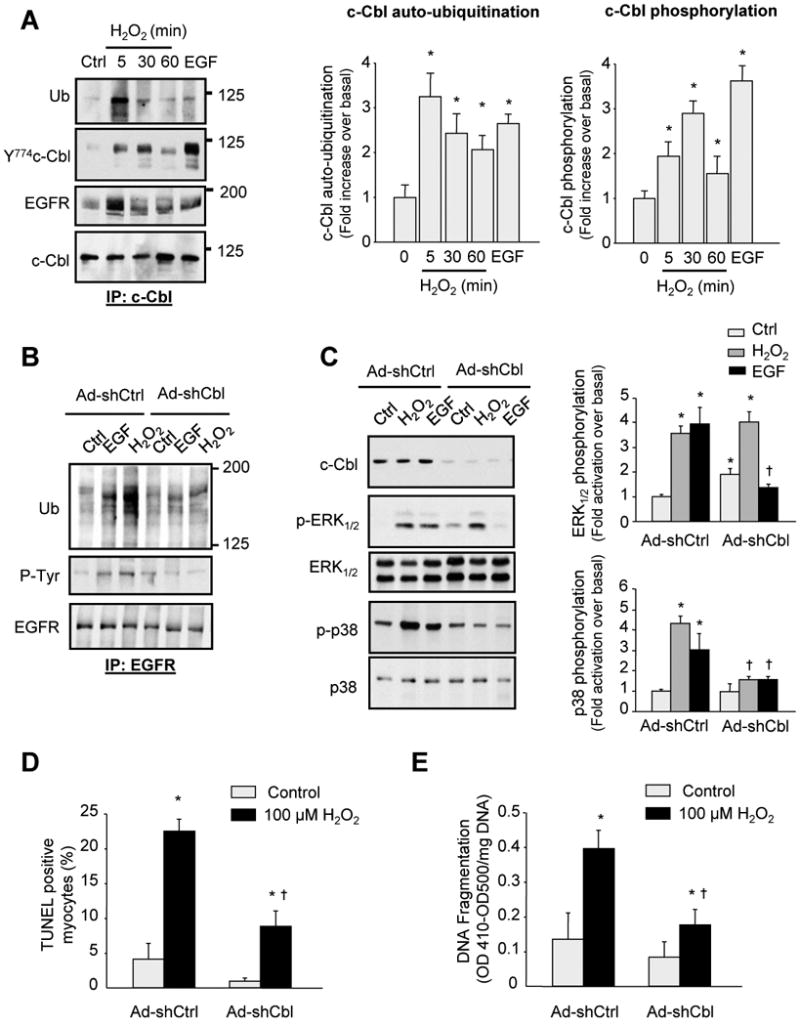
c-Cbl deletion protects myocytes from H2O2-induced apoptosis. (A) Neonatal rat cardiac myocytes treated with H2O2 (100 µmol/L) for the indicated time were evaluated for auto-ubiquitination assay and immunoblot analysis. The result of densitometric analyses is shown. (B-C) Cardiac myocytes were transduced with adenoviruses expressing shRNA Cbl (Ad-shCbl, 10 pfu/cell) or shRNA control (Ad-shCtrl, 10 pfu/cell) for 48 hours and then untreated or treated with 100 ng/ml EGF or 100 µM H2O2 for 10 minutes. Cell lysates were assayed for EGFR immunoprecipitation assays (B) or immunoblot analysis (C). (D-E) Myocyte apoptosis induced by H2O2 as assessed by the percentage of TUNEL-positive myocytes in culture (D) or DNA fragmentation ELISA assay (E). Results are expressed as (OD410-OD500)/mg DNA (D) for triplicate determinations from a single experiment (mean ± S.E). All experiments were performed at least three times from three different cultures and the data values were scaled to untreated controls. *P<0.05 vs. control; †P<0.05 vs. treated myocytes.
To further demonstrate the role of c-Cbl ligase activity on EGFR ubiquitination and EGFR downstream signaling activation, we infected myocytes with recombinant adenoviruses expressing Lac.Z or 70Z-Cbl, a c-Cbl mutant in which 17 amino acid deletion in the helix linker and RING finger domain abolishes the ubiquitin ligase activity resulting in a dominant-negative mutant.21 Overexpression of 70Z-Cbl significantly attenuated EGFR ubiquitination and p38 MAPK phosphorylation induced by H2O2 or EGF treatment compared to Lac.Z infected cells (Supplemental Figure S5).
To assess whether c-Cbl activation is involved in H2O2-induced myocyte apoptosis, myocytes infected with recombinant adenovirus expressing shRNA c-Cbl, shRNA control, Lac.Z, or 70Z-Cbl were evaluated for apoptotic signaling. Overexpression of c-Cbl shRNA or 70Z-Cbl significantly attenuated myocyte apoptosis induced by H2O2 treatment for 16 hours, as demonstrated by a decrease in the percentage of TUNEL positive myocytes and DNA fragmentation (Figures 4D and 4E and Supplemental Figure S5). No effect of Ad-shCbl or 70Z-Cbl expression on myocyte apoptosis was observed in control myocytes. These results suggest that inhibition of endogenous c-Cbl during oxidative stress is protective and that the protective effect of c-Cbl is cardiomyocytes cell-autonomous.
Mice lacking c-Cbl are protected against pathological remodeling induced after myocardial ischemia
To explore the potential influence of c-Cbl on cardiac remodeling responses following myocardial ischemia, we examined c-Cbl localization and expression 2, 7 and 30 days after MI. Figure 5A shows increased c-Cbl immunostaining in border-zone cardiomyocytes and infiltrating inflammatory cells at 7d and 30d after MI. c-Cbl was not immunodetected in fibroblasts or myofibroblasts of the infarcted area. Quantification of c-Cbl accumulation in shams and the border zone of the infarct showed 2.7 ±0.6, 3.5 ±0.2, and 3.7 ±0.3-fold increase over sham control at 2, 7, and 30d after MI, respectively (Figure 5B).
Figure 5.
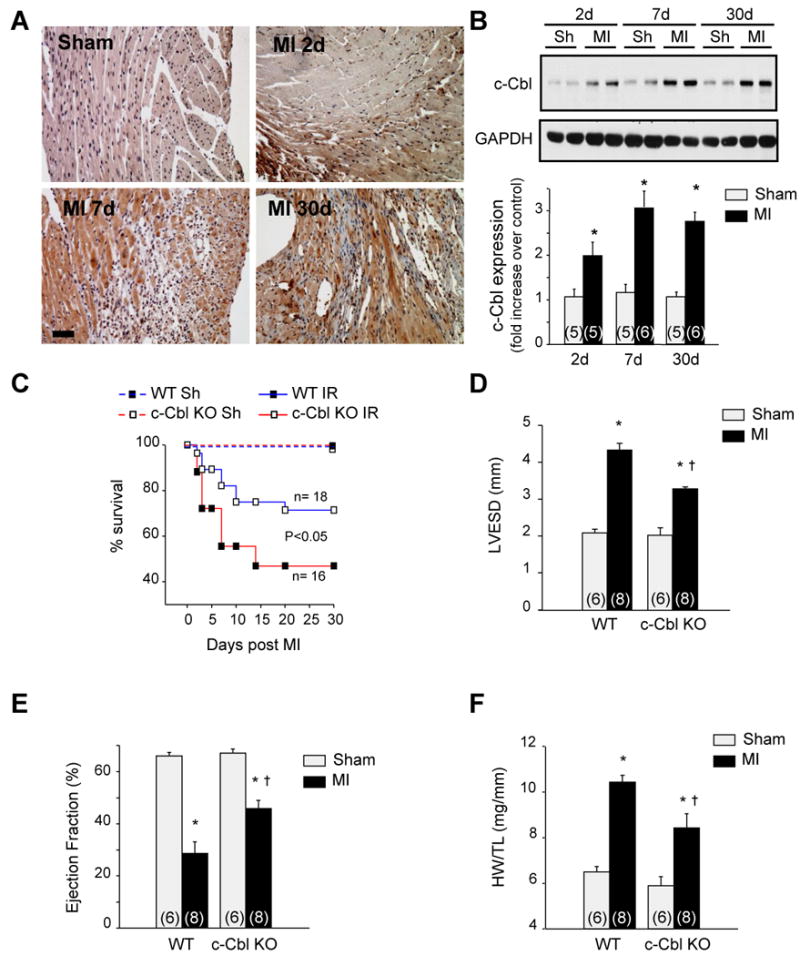

c-Cbl KO mice are protected against post-MI cardiac remodeling. (A) Representative micrographs of paraffin-embedded sections from mouse hearts subjected to permanent left coronary artery ligation for 2, 7, and 30 days. Scale bar: 40 µm. (B) Top: Representative immunoblot of LV heart lysates for c-Cbl. Bottom: Fold induction of c-Cbl accumulation (n=5 for sham groups; n=6 for MI groups). (C) Comparison of post-MI mortality between WT (n=18) and c-Cbl KO (n=16) mice. c-Cbl KO mice showed an increase in survival after MI compared to WT (p<0.05). (D-E) Cardiac function was measured by echocardiography 4 weeks after MI. The data demonstrate cardiac dilation and loss of contractile function in WT mice as indicated by left ventricular end-systolic dimension (LVESD) (D) and ejection fraction (E), whereas the loss of cardiac function was substantially attenuated in c-Cbl KO mice (n=6 for sham groups; n=8 for MI groups). (F and G) c-Cbl deletion attenuated MI-induced increase in the ratio of the heart weight (HW) to tibia length (TL) (F) and the ratio of lung weight (LW) to TL (G). (H) The infarct size 4 weeks post-MI expressed as a fraction of the total cross-sectional circumference of the LV indicates that the infarct size in c-Cbl KO mice is significantly smaller than the infarct size in WT. *P<0.05 compared with WT sham group. †P<0.05 compared with the WT MI group.
Survival up to 4 weeks after MI was comparable in WT and c-Cbl KO shams (97% versus 98%, respectively). However, c-Cbl deficient mice displayed remarkably improved post-infarction survival compared to WT (85% in c-Cbl KO mice compared to 55% in WT mice, p<0.05) especially in the first week after the insult (Figure 5C). Echocardiography 1 and 4 weeks after MI surgery indicated that left ventricular (LV) end-diastolic dimension, LV end-systolic dimension and LV ejection fraction improved significantly in Cbl KO compared to WT mice, whereas there were no differences in the sham-operated animals (Figures 5D and 5E and Supplemental Tables 3 and 4). Morphometric analysis at 4 weeks post-MI indicated the presence of cardiac hypertrophy and concomitant signs of cardiac failure, such as an increase in lung weight, in WT mice, whereas this pathological remodeling response was lacking in the c-Cbl KO mice (Figures 5F and 5G). Myocyte cross-sectional area, an index of myocyte size, mirrored the changes in cardiac hypertrophy and was significantly attenuated in c-Cbl KO compared to WT infarcted hearts (Supplemental Figure S6). Infarct size measurements as percentage of LV circumference indicated 45 ±5% of the LV to be affected by the infarct in WT, whereas the infarct accounted for 29 ±4% of the LV in c-Cbl KO mice (Figure 5H). Examination of cell death in the border zone indicated less TUNEL-positive cells in the c-Cbl KO mice compared to WT animals subjected to MI (data not shown). Thus, the integrity of the infarcted area was better maintained after MI in c-Cbl KO mice and likely prevented the heart from dilating to the same extent as in the WT mice, which results in a better maintenance of cardiac function post-MI.
Upregulation of VEGFa/b and VEGFr2 in the infarcted region of c-Cbl KO mice
One of the beneficial effects of c-Cbl deficiency on the cardiovascular system may be through an improvement in myocardial perfusion by the formation of new capillaries and by the enlargement of preexisting collateral vessels. One key regulator of this pro-angiogenic response is VEGFa, which encodes an extracellular ligand for its corresponding endothelial receptors VEGF receptor (VEGFr)1 and VEGFr2.22,23 VEGFa, VEGFb and their receptors VEGFr2 were significantly upregulated in c-Cbl KO compared to WT hearts in both sham and MI groups (Figures 6A and 6B). This observation is consistent with the fact that VEGFa induces expression of its receptors to create a positive feed-forward loop allowing endothelial cells to become responsive to and activated by VEGF.24 Immunohistochemistry 7 days after MI indicated that VEGFr2 expression was significantly induced in the infarct area and the border zone, which was even more pronounced in the c-Cbl KO (Figure 6C). Erk1/2 and AKT phosphorylation was also upregulated in c-Cbl KO compared to WT hearts in shams (Supplemental Figures 7A and 7B). However, no change in Erk1/2 and AKT phosphorylation levels was observed between c-Cbl KO and WT after MI. Collectively, these data indicate that removal of c-Cbl increases the expression of VEGFa and its downstream receptor, which likely promotes cardioprotection after ischemic damage.
Figure 6.
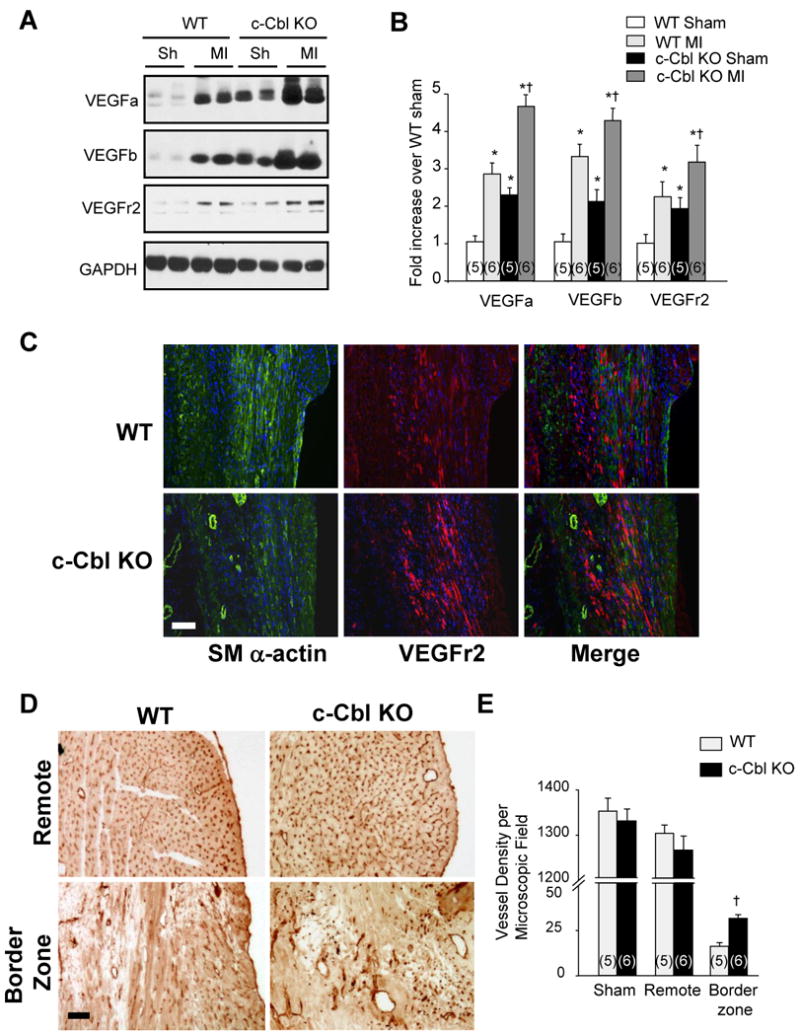
c-Cbl deletion enhances angiogenic response in infarcted region after MI. (A) Immunoblot analysis for VEGF and VEGFr2 indicates an increased angiogenic response in the infarcted region of the c-Cbl KO mice vs. the WT 7 days post-MI. (B) Data are represented as fold change compared to WT animals (n=5 for sham groups, n=6 for MI groups). (C) Immunohistochemistry analysis reveals an increase in VEGFr2 expression and smooth muscle α-actin (SM α-actin) positive blood vessels at the border zone of the infarcted region 7 days post-MI, which is more pronounced in the Cbl KO mice. Scale bar: 40 μm. (D) Representative sections stained for CD31 labeling show an equal distribution of capillaries surrounding cardiomyocytes in the non-infarcted, remote areas, whereas the ischemic region shows irregular patterning of vasculature with additional and enlarged vessels in the border zone and the infarcted region of the Cbl KO mice compared to the WT 30 days post-MI. Scale bars: 40 μm. (E) Semi-quantitative analysis of capillary density in the myocardium indicates an increase in vessel density in the border zone of c-Cbl KO mice compared to WT. The capillary count of these sections is expressed as number of vessels per microscopic field (n=5 for sham groups, n=6 for MI groups). *P<0.05 compared with the WT sham group, †P<0.05 compared with the WT MI group.
We next visualized vasculature using anti-CD31 as an endothelial surface marker and smooth muscle α-actin as a marker for smooth muscle cells. There was a regular distribution of capillaries around cardiomyocytes in both sham-operated groups, with no detectable differences is vessel density (Figures 6D and 6E). Four weeks after MI, the border zone of the infarcted area contained regions of low vascularity in the WT animals, with a more pronounced reduction in the infarct. However, both the border zone and the infarcted region of the c-Cbl KO appeared to be highly vascularized, with enlarged thin-walled vessels (Figures 6C and 6D). The number of capillaries counted in the remote region showed no significant difference between WT and c-Cbl KO mice post-MI, whereas this number was significantly increased in the border zone of the c-Cbl KO mice (Figures 6E). These findings suggest that the cardioprotection observed in Cbl KO mice is attributable, at least in part, to an increase in vessel formation post-MI, which might involve VEGFa and its receptor VEGFr2.23
Discussion
In this study, we identified a novel role for the E3 ubiquitin ligase and adaptor molecule c-Cbl in mediating early stress responses in the heart. c-Cbl expression was strongly induced in human failing hearts and its inhibition provided protection against IR injury, evidenced by decreased cardiac apoptosis and improved LV function. c-Cbl directly ubiquitinates EGFR and FAK such that decreased EGFR and FAK ubiquitination in c-Cbl KO mice reduced their degradation and attenuated cardiomyocyte death induced after IR. More interestingly, c-Cbl deletion attenuated LV hypertrophy, enhanced angiogenesis and improved LV contractility after chronic ischemic insult. This seems to be related, in part, to increased VEGFa and VEGFr2 expression. These data highlight c-Cbl as one of the important signaling molecules that is specifically involved in pathological cardiac remodeling.
Using c-Cbl KO mice, we provide the first evidence, to our knowledge, that this multifaceted protein is involved in the early responses induced by IR injury. Adult c-Cbl-deficient mice have virtually normal hearts under non-stressed, baseline conditions. Moreover, baseline heart rate, blood pressure, and maximal dP/dt were comparable in c-Cbl-deficient and WT mice. However, c-Cbl-deficient mice had smaller infarct sizes, reduced cardiomyocyte apoptosis in the infarct border zone after IR injury, and preserved cardiac contractile function compared with WT controls, indicating that endogenous c-Cbl mediates myocardial tissue damage in vivo. The cardioprotective effects of c-Cbl deletion correlate with the induction of XIAP expression as well as the phosphorylation of Bad, two molecules known to prevent myocyte death.25 We also found high basal levels of Akt and Erk1/2 phosphorylation in c-Cbl deficient mice compared to WT controls, suggesting cardioprotective effects of increased Akt and Erk1/2 phosphorylation in these mice. Interestingly these beneficial effects of c-Cbl deletion on myocyte apoptosis and cardiac function induced after IR injury occurred independently of c-Cbl effects on immune system, as shown by the lack of significant changes in leukocyte infiltration and activation between c-Cbl KO and WT mice (Supplemental Figure S8). Rather, deletion of c-Cbl in cardiac myocytes conferred this protection against IR injury. Using CM-Cbl KO mice, we demonstrated that endogenous deletion of c-Cbl in cardiac myocytes offered cardioprotection against IR injury that mirrored the effect of global c-Cbl deletion. CM-Cbl KO mice showed less myocyte apoptosis, reduced infarct size and improved cardiac function in response to IR injury. Furthermore, inhibition of c-Cbl expression or of its E3 ubiquitin ligase activity markedly attenuated myocyte apoptosis induced in response to oxidative stress, suggesting that endogenous expression of c-Cbl in myocytes plays an important role in mediating the deleterious effect of IR injury.
c-Cbl activation occurs in response to a variety of stimuli, including activated EGFR, cytokines and hormones.9 We describe here that c-Cbl ubiquitin ligase activity and its tyrosine phosphorylation were increased in hearts of mice subjected to IR injury and in cultured myocytes after H2O2 stimulation. Furthermore, inhibition of c-Cbl expression by shRNA or inhibition of its ubiquitin ligase activity significantly reduced myocyte apoptosis in response to H2O2. Given that H2O2 is a potent inducer of myocyte death that is released as a result of IR injury, it may account for the c-Cbl activation and myocyte death in the early IR injury. However, stimulation from other factors released after IR injury could not be excluded. In this regard, we showed recently that c-Cbl ubiquitin ligase activity is involved in mediating focal adhesion and myofibril protein degradation and myocyte death induced by neutrophil derived serine proteases.13 Together, these findings show that activation of c-Cbl following IR increases myocyte apoptosis and that a similar mechanism may be critical in other settings of cardiac stress.
An important finding of the present study is the identification of the molecular mechanisms by which the c-Cbl exerts its cardioprotective effect. c-Cbl RING finger has intrinsic E3 ligase activity and can recruit E2s for the transfer of ubiquitin to substrates.9 Activation of c-Cbl ligase activity has been shown to target several activated receptor and non-receptor tyrosine kinases and mediates their downregulation, thus providing a means by which signaling processes can be negatively regulated.9,26 In this study we found that c-Cbl activation after IR injury correlated with c-Cbl interaction with EGFR and FAK, two known targets of c-Cbl.9 Interestingly, EGFR and FAK ubiquitination and degradation were significantly decreased in c-Cbl KO mice, suggesting that c-Cbl activation presumably mediates EGFR and FAK ubiquitination and degradation induced by IR injury. To our knowledge, this is the first demonstration that EGFR and FAK are being ubiquitinated and downregulated in vivo. Similar decrease in EGFR and FAK expression was observed in human failing hearts, but the mechanisms of such downregulation have never been elucidated.27,28 Because selective downregulation of EGFR and focal adhesion proteins appears to be critical for reduction in myocyte survival,17,18 they might be directly linked to reduced myocyte loss and enhanced cardiac contractility observed after IR injury in c-Cbl KO mice. These data extend our pervious findings showing the role of c-Cbl ubiquitin ligase activity in FAK and myofibril protein degradation and myocyte death in response to neutrophil derived serine proteases.13 While our data that deletion of c-Cbl or inhibition of its ubiquitin ligase activity results in reduced EGFR ubiquitination and attenuated myocyte apoptosis in response to H2O2 clearly support the established model that c-Cbl is a negative regulator of tyrosine kinase signaling, the implication of the PI3 kinase interacting domain of c-Cbl (located at Y731,Y770,Y774)9 remains to be elucidated. In this regard, c-Cbl has been suggested to function as a positive regulator of signaling pathways through its adaptor function to recruit PI3 kinase and other SH2 domain-containing molecules in some systems.9,26 It is noteworthy that hearts from c-Cbl KO mice showed high basal levels of Akt and Erk1/2 phosphorylation compared to WT control mice. However, this basal increase in Akt and Erk1/2 phosphorylation did not affect heart weight in c-Cbl KO mice. The explanation of this lack of hypertrophy in c-Cbl KO mice needs further investigation. However it has been reported that c-Cbl KO mice exhibit elevated energy expenditure and improved insulin action29 which could, in an organ with high energy demands like the heart, play a role in maintaining cardiac structure and function and prevent cardiac hypertrophy.
In addition to its role in protecting myocyte death, our results show that c-Cbl deficiency also affected the levels of VEGFa and its receptor VEGFr2 in response to chronic myocardial ischemia. As the angiogenic activity of VEGF is dependent on its expression levels,30 this increase in total VEGF levels would be expected to result in vascular development/angiogenesis. However, c-Cbl KO mice showed no obvious defects in vascular development in shams despite increased levels of VEGFa and VEGFr2. Early studies have demonstrated that loss of c-Cbl alone is dispensable for normal embryonic development15 whereas loss of both c-Cbl and Cbl-b was embryonic lethal.16 Therefore, it is tempting to speculate that c-Cbl activity is not stringently required during development, and the function of Cbl family proteins may be compensatory in their ability to regulate the activity of target proteins. These data are consistent with previous studies showing normal embryonic and postnatal angiogenesis but impaired ischemic-induced angiogenesis in several gene KO mouse models,31,32 suggesting that c-Cbl selectively plays a role in pathological angiogenesis. Moreover, c-Cbl inactivation has been shown to enhance endothelial cell proliferation and tube formation in response to VEGF in vitro, and results in increased tumor angiogenesis and retinal neovascularization in vivo.33,34 This latter effect of c-Cbl is mediated by a direct regulation of VEGFr2 expression through its phosphorylation on Y1052 and Y1057 residues, as well as indirectly through PLCγ1 activation.33,34 In addition, it is notable that the enhancement of overall vascularization may not exclusively come from neoangiogenesis but also from protecting pre-existing vascular cells. In fact, c-Cbl deletion protects cells from undergoing apoptosis after MI. Irrespective of whether c-Cbl removal enhances cardiac protection through upregulation of angiogenesis, through an increase in protection of pre-existing vascular cells, or a combination of both, our findings show that c-Cbl is a negative regulator for angiogenesis by functioning as a molecular switch to fine-tune angiogenic events during pathological conditions such as MI.
The role of muscle-specific protein ubiquitin ligases as molecules that affect the severity of cardiac diseases is complex and often ambiguous. Activation of E3 ligases CHIP and MDM2 protected myocardium from IR injury,35,36 while activation of Atrogin1 and MuRF1 attenuated cardiac hypertrophy.37,38 The current study adds c-Cbl to the list of E3 ubiquitin ligases that play a role in cardiac protection following myocardial ischemia. Expression of c-Cbl was increased in human hearts with ICM and DCM and its activation provided a means by which signaling emanating from EGFR and FAK can be negatively regulated. Inactivation of ubiquitin ligase activity of c-Cbl or inhibition of its expression may lead to enhanced and prolonged signaling, a function which can explain the increased survival signaling in myocytes expressing ligase deficient mutant of c-Cbl or in c-Cbl KO hearts, respectively. These data, along with the recent findings linking mutation in c-Cbl RING finger domain to Noonan Syndrome, one of the most common genetic syndromes associated with congenital heart disease in human,39 suggest that c-Cbl inhibition may constitute a novel therapy offering cardioprotection.
In conclusion, the results of this study suggest that c-Cbl deletion protects the heart against IR injury by decreasing/preventing ubiquitination and subsequent degradation of EGFR and FAK and myocyte death in response to injury. These findings provide evidence for an important role of c-Cbl in direct modulation of EGFR and focal adhesion protein turnover in the heart. Given that EGFR and FAK signaling downregulation also underlie cardiac hypertrophy and other form of heart disease, it is likely that c-Cbl plays a role in a variety of stress settings. Thus, modulating c-Cbl activation to attenuate myocyte death and to enhance angiogenesis may have potential implications in the treatment of cardiac disease.
Supplementary Material
Acknowledgments
Funding Sources: This work was supported by the National Institute of Health (HL360338 and HL 360343 to A.S. and CA105489, CA99163, CA87986 and CA116552 to H.B). K Rafiq is a recipient of an American Heart Association award (240052).
Footnotes
Conflict of Interest Disclosures: None.
References
- 1.Members WG, Roger VL, Go AS, Lloyd-Jones DM, Benjamin EJ, Berry JD, Borden WB, Bravata DM, Dai S, Ford ES, Fox CS, Fullerton HJ, Gillespie C, Hailpern SM, Heit JA, Howard VJ, Kissela BM, Kittner SJ, Lackland DT, Lichtman JH, Lisabeth LD, Makuc DM, Marcus GM, Marelli A, Matchar DB, Moy CS, Mozaffarian D, Mussolino ME, Nichol G, Paynter NP, Soliman EZ, Sorlie PD, Sotoodehnia N, Turan TN, Virani SS, Wong ND, Woo D, Turner MB. Heart Disease and Stroke Statistics ”2012 Update”. Circulation. 2012;125:e2–e220. doi: 10.1161/CIR.0b013e31823ac046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Wang X, Robbins J. Heart failure and protein quality control. Circ Res. 2006;99:1315–1328. doi: 10.1161/01.RES.0000252342.61447.a2. [DOI] [PubMed] [Google Scholar]
- 3.Razeghi P, Sharma S, Ying J, Li YP, Stepkowski S, Reid MB, Taegtmeyer H. Atrophic remodeling of the heart in vivo simultaneously activates pathways of protein synthesis and degradation. Circulation. 2003;108:2536–2541. doi: 10.1161/01.CIR.0000096481.45105.13. [DOI] [PubMed] [Google Scholar]
- 4.Glass DJ. Signalling pathways that mediate skeletal muscle hypertrophy and atrophy. Nat Cell Biol. 2003;5:87–90. doi: 10.1038/ncb0203-87. [DOI] [PubMed] [Google Scholar]
- 5.Depre C, Wang Q, Yan L, Hedhli N, Peter P, Chen L, Hong C, Hittinger L, Ghaleh B, Sadoshima J, Vatner DE, Vatner SF, Madura K. Activation of the cardiac proteasome during pressure overload promotes ventricular hypertrophy. Circulation. 2006;114:1821–1828. doi: 10.1161/CIRCULATIONAHA.106.637827. [DOI] [PubMed] [Google Scholar]
- 6.Ciechanover A, Orian A, Schwartz AL. The ubiquitin-mediated proteolytic pathway: mode of action and clinical implications. J Cell Biochem Supplement. 2000;34:40–51. doi: 10.1002/(sici)1097-4644(2000)77:34+<40::aid-jcb9>3.0.co;2-6. [DOI] [PubMed] [Google Scholar]
- 7.Friedman J, Xue D. To live or die by the sword: The regulation of apoptosis by the proteasome. Develop Cell. 2004;6:460–461. doi: 10.1016/s1534-5807(04)00104-2. [DOI] [PubMed] [Google Scholar]
- 8.Coux O, Tanaka K, Goldberg AL. Structure and functions of the 20S and 26S proteasomes. Ann Rev Biochem. 1996;65:801–847. doi: 10.1146/annurev.bi.65.070196.004101. [DOI] [PubMed] [Google Scholar]
- 9.Schmidt MH, Dikic I. The Cbl interactome and its functions. Nat Rev Mol Cell Biol. 2005;6:907–918. doi: 10.1038/nrm1762. [DOI] [PubMed] [Google Scholar]
- 10.Meng W, Sawasdikosol S, Burakoff SJ, Eck MJ. Structure of the amino-terminal domain of Cbl complexed to its binding site on ZAP-70 kinase. Nature. 1999;398:84–90. doi: 10.1038/18050. [DOI] [PubMed] [Google Scholar]
- 11.Balasubramanian S, Mani S, Shiraishi H, Johnston RK, Yamane K, Willey CD, Cooper G, IV, Tuxworth WJ, Kuppuswamy D. Enhanced ubiquitination of cytoskeletal proteins in pressure overloaded myocardium is accompanied by changes in specific E3 ligases. J Mol Cell Cardiol. 2006;41:669–679. doi: 10.1016/j.yjmcc.2006.04.022. [DOI] [PubMed] [Google Scholar]
- 12.Rosenblatt-Velin N, Lerch R, Papageorgiou I, Montessuit C. Insulin resistance in adult cardiomyocytes undergoing dedifferentiation: role of GLUT4 expression and translocation. FASEB J. 2004;18:872–874. doi: 10.1096/fj.03-1095fje. [DOI] [PubMed] [Google Scholar]
- 13.Rafiq K, Guo J, Vlasenko L, Guo X, Kolpakov MA, Sanjay A, Houser SR, Sabri A. C-CBL ubiquitin ligase regulates focal adhesion protein turnover and myofibril degeneration induced by neutrophil protease cathepsin G. J Biol Chem. 2012;287:5327–5339. doi: 10.1074/jbc.M111.307009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Chaudhary KW, Rossman EI, Piacentino V, III, Kenessey A, Weber C, Gaughan JP, Ojamaa K, Klein I, Bers DM, Houser SR, Margulies KB. Altered myocardial Ca2+ cycling after left ventricular assist device support in the failing human heart. J Amer Coll Cardiol. 2004;44:837–845. doi: 10.1016/j.jacc.2004.05.049. [DOI] [PubMed] [Google Scholar]
- 15.Murphy MA, Schnall RG, Venter DJ, Barnett L, Bertoncello I, Thien CBF, Langdon WY, Bowtell DDL. Tissue hyperplasia and enhanced T-cell signalling via ZAP-70 in c-Cbl-deficient mice. Mol Cell Biol. 1998;18:4872–4882. doi: 10.1128/mcb.18.8.4872. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Naramura M, Jang IK, Kole H, Huang F, Haines D, Gu H. c-Cbl and Cbl-b regulate T cell responsiveness by promoting ligand-induced TCR down-modulation. Nature Immunol. 2002;3:1192–1199. doi: 10.1038/ni855. [DOI] [PubMed] [Google Scholar]
- 17.Pentassuglia L, Sawyer DB. The role of Neuregulin-1b/ErbB signaling in the heart. Exp Cell Res. 2009;315:627–637. doi: 10.1016/j.yexcr.2008.08.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Franchini KG. Focal adhesion kinase-The basis of local hypertrophic signaling domains. J Mol Cell Cardiol. 2012;52:485–492. doi: 10.1016/j.yjmcc.2011.06.021. [DOI] [PubMed] [Google Scholar]
- 19.Chau V, Tobias J, Bachmair A, Marriott D, Ecker D, Gonda D, Varshavsky A. A multiubiquitin chain is confined to specific lysine in a targeted short-lived protein. Science. 1989;243:1576–83. doi: 10.1126/science.2538923. [DOI] [PubMed] [Google Scholar]
- 20.Pennock S, Wang Z. A tale of two Cbls: Interplay of c-Cbl and Cbl-b in epidermal growth factor receptor downregulation. Mol Cell Biol. 2008;28:3020–3037. doi: 10.1128/MCB.01809-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Joazeiro CAnP, Wing SS, Huang Hk, Leverson JD, Hunter T, Liu YC. The tyrosine kinase negative regulator c-Cbl as a RING-Type, E2-dependent ubiquitin-protein ligase. Science. 1999;286:309–312. doi: 10.1126/science.286.5438.309. [DOI] [PubMed] [Google Scholar]
- 22.Ferrara N, Gerber HP, LeCouter J. The biology of VEGF and its receptors. Nat med. 2003;9:669–676. doi: 10.1038/nm0603-669. [DOI] [PubMed] [Google Scholar]
- 23.Dor Y, Djonov V, Abramovitch R, Itin A, Fishman GI, Carmeliet P, Goelman G, Keshet E. Conditional switching of VEGF provides new insights into adult neovascularization and pro-angiogenic therapy. Embo J. 2002;21:1939–1947. doi: 10.1093/emboj/21.8.1939. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Herve MAJ, Meduri G, Petit FG, Domet TS, Lazennec G, Mourah S, Perrot-Applanat M. Regulation of the vascular endothelial growth factor (VEGF) receptor Flk-1/KDR by estradiol through VEGF in uterus. J endocrinol. 2006;188:91–99. doi: 10.1677/joe.1.06184. [DOI] [PubMed] [Google Scholar]
- 25.Dorn GW., II Apoptotic and non-apoptotic programmed cardiomyocyte death in ventricular remodeling. Cardiovasc Res. 2009;81:465–473. doi: 10.1093/cvr/cvn243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Thien CBF, Langdon WY. Cbl: many adaptations to regulate protein tyrosine kinases. Nat Rev Mol Cell Biol. 2001;2:294–307. doi: 10.1038/35067100. [DOI] [PubMed] [Google Scholar]
- 27.Pfister R, Acksteiner C, Baumgarth J, Burst V, Geissler HJ, Margulies KB, Houser S, Bloch W, Flesch M. Loss of ß1D-integrin function in human ischemic cardiomyopathy. Basic Res Cardiol. 2007;102:257–264. doi: 10.1007/s00395-006-0640-1. [DOI] [PubMed] [Google Scholar]
- 28.Rohrbach S, Niemann B, Silber RE, Holtz J. Bas Res Cardiol. Springer; Berlin/Heidelberg: 2005. Neuregulin receptors erbB2 and erbB4 in failing human myocardium; pp. 240–249. [DOI] [PubMed] [Google Scholar]
- 29.Molero JC, Jensen TE, Withers PC, Couzens M, Herzog H, Thien CBF, Langdon WY, Walder K, Murphy MA, Bowtell DDL, James DE, Cooney GJ. c-Cbl deficient mice have reduced adiposity, higher energy expenditure, and improved peripheral insulin action. J Clin Invest. 2004;114:1326–1333. doi: 10.1172/JCI21480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Carmeliet P, Ferreira V, Breier G, Pollefeyt S, Kieckens L, Gertsenstein M, Fahrig M, Vandenhoeck A, Harpal K, Eberhardt C, Declercq C, Pawling J, Moons L, Collen D, Risau W, Nagy A. Abnormal blood vessel development and lethality in embryos lacking a single VEGF allele. Nature. 1996;380:435–439. doi: 10.1038/380435a0. [DOI] [PubMed] [Google Scholar]
- 31.Carmeliet P, Moons L, Luttun A, Vincenti V, Compernolle V, De Mol M, Wu Y, Bono F, Devy L, Beck H, Scholz D, Acker T, DiPalma T, Dewerchin M, Noel A, Stalmans I, Barra A, Blacher S, Vandendriessche T, Ponten A, Eriksson U, Plate KH, Foidart JM, Schaper W, Charnock-Jones DS, Hicklin DJ, Herbert JM, Collen D, Persico MG. Synergism between vascular endothelial growth factor and placental growth factor contributes to angiogenesis and plasma extravasation in pathological conditions. Nature Med. 2001;7:575–583. doi: 10.1038/87904. [DOI] [PubMed] [Google Scholar]
- 32.Chen J, Somanath PR, Razorenova O, Chen WS, Hay N, Bornstein P, Byzova TV. Akt1 regulates pathological angiogenesis, vascular maturation and permeability in vivo. Nature Med. 2005;11:1188–1196. doi: 10.1038/nm1307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Meyer RD, Husain D, Rahimi N. c-Cbl inhibits angiogenesis and tumor growth by suppressing activation of PLC[gamma]1. Oncogene. 2011;30:2198–2206. doi: 10.1038/onc.2010.597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Singh AJ, Meyer RD, Navruzbekov G, Shelke R, Duan L, Band H, Leeman SE, Rahimi N. A cri c-Cbl in VEGFR-2-mediated PLCγ1 activation and angiogenesis. Proc Nat Acad Sci USA. 2007;104:5413–5418. doi: 10.1073/pnas.0700809104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Zhang C, Xu Z, He XR, Michael LH, Patterson C. CHIP, a cochaperone/ubiquitin ligase that regulates protein quality control, is required for maximal cardioprotection after myocardial infarction in mice. Am J Physiol Heart Circ Physiol. 2005;288:H2836–2842. doi: 10.1152/ajpheart.01122.2004. [DOI] [PubMed] [Google Scholar]
- 36.Toth A, Nickson P, Qin LL, Erhardt P. Differential regulation of cardiomyocyte survival and hypertrophy by MDM2, an E3 ubiquitin ligase. J Biol Chem. 2006;281:3679–3689. doi: 10.1074/jbc.M509630200. [DOI] [PubMed] [Google Scholar]
- 37.Arya R, Kedar V, Hwang JR, McDonough H, Li HH, Taylor J, Patterson C. Muscle ring finger protein-1 inhibits PKC{epsilon} activation and prevents cardiomyocyte hypertrophy. J Cell Biol. 2004;167:1147–1159. doi: 10.1083/jcb.200402033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Li HH, Kedar V, Zhang C, McDonough H, Arya R, Wang DZ, Patterson C. Atrogin-1/muscle atrophy F-box inhibits calcineurin-dependent cardiac hypertrophy by participating in an SCF ubiquitin ligase complex. J Clin Invest. 2004;114:1058–1071. doi: 10.1172/JCI22220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Niemeyer CM, Kang MW, Shin DH, Furlan I, Erlacher M, Bunin NJ, Bunda S, Finklestein JZ, Sakamoto KM, Gorr TA, Mehta P, Schmid I, Kropshofer G, Corbacioglu S, Lang PJ, Klein C, Schlegel PG, Heinzmann A, Schneider M, Stary J, van den Heuvel-Eibrink MM, Hasle H, Locatelli F, Sakai D, Archambeault S, Chen L, Russell RC, Sybingco SS, Ohh M, Braun BS, Flotho C, Loh ML. Germline CBL mutations cause developmental abnormalities and predispose to juvenile myelomonocytic leukemia. Nat Genet. 2010;42:794–800. doi: 10.1038/ng.641. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.