

Abstract
We performed unbiased, comprehensive immunophenotyping of cerebrospinal fluid (CSF) and blood leukocytes in 221 subjects referred for the diagnostic work-up of neuroimmunological disorders in order to obtain insight about disease-specific phenotypes of intrathecal immune responses. Quantification of 14 different immune cell subsets, coupled with the assessment of their activation status, revealed physiological differences between intrathecal and systemic immunity, irrespective of final diagnosis. Our data are consistent with a model, where the central nervous system shapes intrathecal immune responses to provide effective protection against persistent, especially by memory T cells, plasmacytoid dendritic cells and CD56bright NK cells. Our data also argue that CSF immune cells do not simply reflect cells recruited from the periphery. Instead, they represent a mixture of cells that are recruited from the blood, have been activated intrathecally and leave the CNS after performing effector functions.
Diagnosis-specific differences provide mechanistic insight into the disease process in the defined subtypes of multiple sclerosis (MS), neonatal onset multisystem inflammatory disease and Aicardi-Goutieres syndrome. This analysis also determined that secondary-progressive MS patients are immunologically closer to relapsing-remitting patients as compared to patients with primary-progressive MS. Because CSF immunophenotyping captures the biology of the intrathecal inflammatory processes, it has the potential to guide optimal selection of immunomodulatory therapies in individual patients and monitor their efficacy. Our study adds to the increasing number of publications that demonstrate poor correlation between systemic and intrathecal inflammatory biomarkers in patients with neuroimmunological diseases and stresses the importance of studying immune responses directly in the intrathecal compartment.
Keywords: multiple sclerosis, cerebrospinal fluid, flow cytometry, neurological disorders, neuroinflammation
INTRODUCTION
Neuroimmunological disorders of the central nervous system (CNS) are an expanding group of diseases affecting all age groups. The most extensively studied disorder is multiple sclerosis (MS). Although the pathogenic role of inflammation is no longer disputed in MS (at least in the most frequent form called relapsing-remitting MS [RR-MS]), neither the antigenic target, nor the pathogenic cell population(s) have been defined. Heterogeneity in pathological MS specimens suggests that diverse mechanisms may be driving the development of CNS lesions in different patients (1). Furthermore, decreased efficacy of immunomodulatory disease-modifying therapies (DMTs) in the later stages of MS raises the possibility that neurodegenerative mechanisms drive disability in primary-progressive (PP-MS) and secondary-progressive MS (SP-MS) (2, 3). This hypothesis is supported by a paucity of contrast-enhancing lesions (CEL) in neuroimaging studies (4) and presence of diffuse CNS tissue injury (5). However, pathological studies also demonstrate continuous presence of inflammatory cells in progressive MS, especially in the meninges (6–8). It is likely that patients with progressive disease who retain a prominent inflammation experience partial benefit from DMTs (9).
Thus, understanding the heterogeneity of intrathecal immune responses and their relationship to disease phenotype is a necessary prerequisite for rational selection of optimal therapy in patients with MS, as well as other neuroimmunological diseases for which pathogenic mechanisms are even less understood. Moreover, when targeted therapies are applied, it often remains unclear whether residual inflammation persists. This is exemplified by use of interleukin-1 (IL-1) receptor antagonist anakinra for the treatment of neonatal onset multisystem inflammatory disease (NOMID), characterized by excessive activation of innate immunity secondary to genetic defect in NLRP3 inflammasome pathway (10).
Immunophenotyping of cerebrospinal fluid (CSF) cells by flow cytometry is a powerful tool that provided important insights into the MS. It revealed that RR-MS patients have intrathecal expansion of B cells and plasma blasts, in comparison to non-inflammatory neurological diseases (NIND) controls (11). Others demonstrated a link between CSF B cells and intrathecal production of CXCL13 (12, 13) and between CSF B cells and CEL (14, 15). Additional cellular abnormalities involved decreased proportion of monocytes (11, 13), an elevated number of dendritic cells (DCs) (16), especially plasmacytoid DCs (17) and an increase in activated T cells (18–20). The latter abnormalities have not been confirmed in independent cohorts. As CSF evaluation of patients with progressive MS is rarely done in contemporary clinical practice, it is unclear if afore-mentioned findings apply also to these patients. The abnormalities summarized above are also found in other inflammatory neurological disease (OIND) patients and therefore raise the question whether immunophenotyping can reveal differences in the phenotype of inflammatory responses and thus provide insight into disease pathogenesis.
We performed comprehensive immunophenotyping in a sizable cohort of prospectively acquired pediatric and adult patients (N=221) who presented to NIH for diagnostic work-up of a neuroimmunological diseases with the following goals: 1. Develop a standardized 12 color, single staining flow cytometry immunophenotyping panel that can be applied to CSF specimens containing of as few as 5,000 CSF cells; 2. Define differences in the number and activation status of major immune subpopulations between blood and CSF; 3. Define immunophenotyping differences between different inflammatory disorders, including MS subtypes and 4. Assess the value of this method in defining disease pathogenesis and therapeutic management.
METHODS
Subjects
The study was approved by the NIH Institutional Review Board and all patients provided written consent (or assent). Patients were prospectively recruited between February 2011 and August 2013 from multiple NIH groups that provide care for patients with neuroimmunological or neuro-infectious disorders. Adult subjects had not received immunomodulatory therapies for a minimum of 3 months before immunophenotyping. As untreated pediatric patients could not be readily recruited, all pediatric patients were included irrespective of treatment.
All patients underwent a thorough diagnostic work-up evaluating infectious and auto-immune causes, magnetic resonance imaging (MRI) and CSF studies. When indicated by history or serological studies, CSF work-up included serological and qPCR search for infectious etiologies. Diagnosis of MS was based on 2010 revisions to the McDonald diagnostic criteria (21). Patients who presented with clinically isolated syndrome (CIS) were followed for a minimum of 1 year and reclassified as MS if they fulfilled diagnostic criteria. Patients who did not convert to MS were grouped with OIND subjects, because based on phenotypical and MRI characteristics, these patients were deemed to have low probability of converting to definite MS in the future.
Non-MS patients were grouped into diagnostic categories of adult or pediatric inflammatory (OIND) or non-inflammatory neurological diseases (NIND). Patients with a defined diagnosis were grouped based on whether the CNS disease was thought to be immune-mediated or not. Patients whose diagnosis remained unclear after diagnostic work-up were classified into OIND subgroup based on at least one of the following accepted laboratory and imaging markers of intrathecal inflammation: CSF pleiocytosis, increased IgG index, CSF oligoclonal bands (OCB) or presence of CEL on brain or spinal cord MRI. Patients who did not fulfill these criteria were classified as NIND.
Because NIH is a highly specialized international referral center, difficult-to diagnose patients are overrepresented in our cohort in comparison to regular clinical practice. The demographic data and diagnoses are summarized in Table 1.
Table 1. Patient Demographics and diagnosis.
221 patients were prospectively recruited between February 2011 and August 2013 from the Neuroimmunological Diseases Unit (NDU) natural history protocol 09-N-0032 that provides diagnostic work-up for untreated patients with putative neuroimmunological disorder. Statistically-significant differences in demographic and clinical parameters are outlined in the table.
| Demographics | PP-MS n=61 | RR-MS n=51 | SP-MS n=30 | NIND n=21 | OIND n=29 | p-NIND n=12 | p-OIND n=17 | Health donor |
|---|---|---|---|---|---|---|---|---|
| Age (year) | 55.29 a [31.00–70.43] | 38.92 b [18.00–65.80] | 54.89 a [43.06–65.99] | 49.94 a [19.94–70.56] | 51.20 a [19.80–74.86] | 6.10 c [1.48–17.21] | 8.85 c [0.80–21.58] | 38.50 [34.15–44.14] |
| Disease duration (year) | 2.40 a [0.10–36.60] | 9.8 a [0.52–38.30] | 25.00 b [10.90–42.11] | 2.90 a [0.56–31.33] | 7.60ab [0.20–25.69] | 2.71 a [1.02–5.72] | 5.36 ab [0.80–9.32] | 0 |
| EDSS | 5.60 a [1.50–7.50] | 1.90 a [1.00–6.50] | 6.40 a [6.00–7.00] | 1.20 a [0.00–4.50] | 3.30 a [1.00–6.50] | Not available | Not available | Not available |
| SNRS | 66.40 a [45.00–90.00] | 91.70 c [72.00–100.00] | 60.3 a [47.00–82.00] | 92.00 b [57.00–100.00] | 84.70 ab [68.00–98.00] | Not available | Not available | Not available |
| CSF WBC (per mL) | 3.10 c [1.00–15.00] | 5.30 c [1.00–21.00] | 2.7 c [1.00–10.00] | 1.59 c [0.00–2.00] | 14.20 b [1.00–92.00] | 0.90 c [0.00–4.00] | 48.30 a [0.00–905.00] | 1.73 [1.00–2.00 |
| CSF Lymphocyte (%) | 92.50 a [88.70–97.00] | 91.00 a [68.00–97.00] | 92.50 a [88.0–97.00] | Not available | 74.00 ab [39.00–92.00] | Not available | 61.70 b [29.00–81.00] | Not available |
| Oligoclonal band pattern % (l/ ll/ lll/ IV)* | 1.80 / 53.50 / 44.70 / 0 | 4.30 / 82.79 / 10.80 / 2.20 | 0 / 50.00 / 50.00 / 0 | 46.20 / 0 / 23.01 / 15.42 | 20.00 / 53.30 / 13.30 /13.03 | 100.00 / 0 / 0 / 0 | 100.00 / 0 / 0 / 0 | 66.67 / 0 / 0 / 33.33 |
| IgG index | 2.30 a [1.00–3.00] | 2.00 a [0.60–3.00] | 2.60 a [2.00–4.00] | 1.80 a [1.00–4.00] | 2.00 a [1.00–4.00] | 0.5 a [0.48–0.57] | 0.6 a [0.37–1.24] | 0.50 [0.37–0.53] |
| Treatment | untreated | untreated | untreated | untreated | untreated | untreated | untreated (n=8) Anakinra (n=9) |
untreated |
median [range]
EDSS: Expanded Disability Status Scale
SNRS: Scripps Neurologic Rating Scale
I- No bands in CSF and Serum; normal profile / II-Oligoclonal bands in CSF only; consistent with intrathecal IgG synthesis / III- Partially identical Oligoclonal bands in CSF and serum; consistent with systemic disease / IV- Identical Oligoclonal bands in CSF and serum; consistent with systemic inflammation
means with different letters are significantly different with p<0.001.
Sample preparation and Flow cytometry
All samples were labeled with a prospectively assigned alpha-numeric code and personnel preforming the studies were blinded to the diagnosis of the subject. Specimen collection, handling and processing was performed according to a written standard operating procedures (SOPs).
Immunophenotyping of peripheral blood cells was performed on anticoagulated blood within 60 minutes of ex vivo collection after osmotic lysis of erythrocytes. CSF samples were placed on ice immediately after collection. Within 15 minutes the CSF (usually 20ml) was spun and cell pellets were resuspended in 400 μL ice-cold X-Vivo media (Lonza). Concentrated CSF cells were counted by hemocytometer (Neubauer; Hausser Scientific) at high magnification to allow differentiation of erythrocytes from nucleated cells. Concentration of CSF leukocytes per 1ml of CSF was calculated by dividing the total number of CSF leukocytes by volume of collected CSF.
The 12 color immunophenotyping panel is described in Table 2. A minimum of 106 blood cells and 5000 CSF cells were stained according to a previously established protocol (22), which included blocking of Fc receptors by 2% intravenous immunoglobulin (IVIg). Cells were immediately acquired on a BD LSR II with High Throughput Sampler (HTS) delivery system and analyzed with FACSDiva 6.1 software (all BD Biosciences). Gating was based on isotype controls. Sample acquisition, gating and sample exclusion (based on the review of quality of the staining and of absolute numbers of acquired events to assess reliability of data) was done on coded samples.
Table 2. Optimized combination of twelve commercially-available flourochrome-conjugated antibodies to reliably quantify 14 subpopulations of immune cells.
| Conjugation | Name | Company (Clone) |
|---|---|---|
| FITC | Anti-human CD56 antibody | BD (Clone: MEM188) |
| PE | Anti-human CD80 antibody | BD (Clone: M-A712) |
| PE | Mouse IgG1 isotype control | BD (Clone: G18–145) |
| PerCP-Cy5.5 | Anti-human CD123 antibody | eBioscience (Clone: 7G3) |
| PE-Cy7 | Anti-human CD11c antibody | eBioscience (Clone: 3.9) |
| V450 | Anti-human CD45 antibody | BD (Clone: HI30) |
| AmCyan | Anti-human CD8 antibody | BD (Clone: SK1) |
| eFluor 605 Nanocrystal | Anti-human CD19 antibody | eBioscience (Clone: HIB19) |
| eFluor 655 Nanocrystal | Anti-human CD3 antibody | eBioscience (Clone: OKT3) |
| Qdot 705 | Anti-human CD4 antibody | Invitrogen (Clone: S3.5) |
| APC | Anti-human CD25 antibody | BD (Clone: M-A251) |
| APC | Mouse IgG1 isotype control | BD (Clone: MOPC-21) |
| Alexa Fluor 700 | Anti-human CD14 antibody | BioLegend (Clone: HCD14) |
| APC-Cy7 | Anti-human HLA-DR antibody | eBioscience (Clone: LN3) |
Statistical analysis
Appropriate transformations were applied to the 66 markers based on the results of the Box-Cox method. To evaluate the association of the markers with the factor of diagnosis, analysis of covariance (ANCOVA) with unequal variance model was performed with gender as covariate. Since age was related to the factor of diagnosis, it could not be used as a covariate. To assess the effect of age, ANCOVA with both age and gender as covariates was applied to a subset of four patient groups (PP-MS, SP-MS, OIND and NIND) in which there was no significant difference in age. To distinguish age-related from the disease-related effects in pediatric group, we searched PubMed for articles describing age-related effects on the immune system in healthy donors (HDs) and compiled data from these articles into Supplementary Table 1. We also performed analysis of correlations between measured markers and age within three age-homogeneous cohorts in our study (i.e. “Pediatric”, “Young adult” and “Older adult” cohorts [Supplementary Table 1]). When congruency in correlation between the marker and the age was observed within several cohorts (including published data) we attributed the observed difference to age-related change and highlighted associated statistical annotations by grey shading in the relevant figures.
To evaluate the relationship between markers, pair-wise Spearman correlation coefficients were calculated for each cohort. To visualize the combined marker effect on diagnosis, a heat map was created from cluster analysis (Ward method) based on the markers with p-value of F-test (df=6) less than 0.015 in ANCOVA.
To examine the difference between CSF and blood, repeated measures ANOVA was performed with two factors, diagnosis (between-subject factor), type (within-subject factor) and their interaction in the model. Statistical analyses were performed using SAS version 9.2.
Because we were able to recruit only 5 HDs, this group was too small to use for statistical analysis. Instead, we plotted mean ± 2SD for HD group as approximate reference range, with the understanding that SD may be artificially inflated due to small cohort size.
RESULTS
Development of a 12 color flow cytometry immunophenotyping panel
In pilot experiments we have optimized the combination of commercially-available flourochrome-conjugated antibodies (Table 2) that allowed us to reliably quantify 14 subpopulations of immune cells (see gating strategy in Supplementary Fig. 1) and assess their in vivo activation. We used several activation markers (HLA-DR: activated “effector” T cells, CD25: activated T cells, B cells, monocytes and DCs, and CD80: activated monocytes, DCs and B cells) combined with cell-specific measurements of size and granularity.
We observed significant differences between blood and CSF samples in proportion and activation status of virtually all immune cells analyzed. These changes were seen across all diagnostic categories. In Figure 1 we highlight markers for which there was interaction between sample type and diagnosis. In Supplementary Figure 2 we provide plots of differences between diagnostic categories for the highlighted markers. Diagnosis-specific changes will be discussed later; here we focus on global changes within the entire cohort and highlight only statistically significant changes.
Figure 1. The differences between the blood and CSF samples in the proportion and activation status of immune cells for all patients.

(a) Differences in the cells of the innate immune system: monocytes, granulocytes, plasmacytoid DCs (PlDC), myeloid DCs (MyDC), CD56dim and CD56bright NK cells and basophils. (b) Differences in the cells belonging to adaptive immune system: CD4+ and CD8+ T cells and their subsets (HLA-DR+ effector cells and CD56+ cytotoxic cells) and CD19+ B cells. Left panels in each row demonstrate differences in the proportions of specified cell population among all CD45+ leukocytes between blood (red) and CSF (blue). Next two panels in each row show representative raw FACS images of the size (forward scatter; FSC on x axis) and granularity (side scatter; SSC on y axis) for specified cell population from MS patient. The right 2 panels in each row represent group comparisons between blood and CSF of the size and granularity for specified subpopulation of immune cells. Statistically significant differences are depicted as follows: *: P<0.05, **: 0.001<P<0.05, ***: P<0.001. Mean values are shown ± SD. Red edge highlights those markers for which statistical interaction was identified between sample type and diagnosis. For these markers the diagnosis – specific plots can be found in Supplementary Figures 2a and 2b.
Compared to blood (Fig 1A), the proportion innate immune cells is lower in the CSF; monocytes were decreased by 45.18±7.51%, granulocytes by 71.73±1.69% and basophils by 53.89±36.83%. Subsets of DC and NK cells were differentially represented: myeloid DCs (MyDC) and CD56dim NK cells were under-represented in the CSF compared to blood (MyDC by 3.89±1.22% and CD56dim NK cells by 47.85±2.99%), while plasmacytoid DC (PlDC) and CD56bright NK cells were over-represented in the CSF (PlDC by 143.57±0.18% and CD56bright NK cells by 109.93±40.09%).
Several innate immune subsets in the CSF had phenotypes consistent with recent activation: monocytes were significantly smaller and dramatically degranulated (SSC decreased by 17.32±9.15%). Granulocytes were also smaller in the CSF but had comparable granularity to blood cells (SSC smaller by 0.94±1.58% in the CSF compared to blood) and basophils did not have altered size or granularity. In contrast, DC and NK cells were significantly larger in the CSF, indicating their activated status, but only those DC and NK cells that were over-represented in the CSF were also significantly degranulated in comparison to blood: i.e. PlDC were degranulated by 18.64±6.49% and CD56bright NK cells were degranulated by 12.50±2.19%.
Among adaptive immune cells (Fig 1B), proportions of all T cell subsets were significantly increased in the CSF, while B cells were dramatically reduced (by 75.79±37.18%). CD4+ T cells were proportionally more expanded in the CSF than CD8+ T cells (by 38.30±8.14% for CD4+ and by 13.03±4.72% for CD8+ T cells), which resulted in increased CD4/CD8 T cell ratio in the CSF, consistent with a previous reports (23). Compared to blood, T cell populations had activated phenotypes in the CSF: they were significantly larger (CD4+ T cells by 15.09±3.31% and CD8+ T cells by 13.68±7.31%) and degranulated (CD4+ T cells by 4.92±3.31% and CD8+ T cells by 3.56±1.39%). Interestingly, while HLA-DR+ T cells are larger and more granular than HLA-DR-T cells (consistent with recent-activation) they were even larger and more strongly degranulated in the CSF compared to their counterparts in the blood (HLA-DR/CD4+: +4.45±1.02% size and −11.04±3.98% granularity; and HLA-DR/CD8+ T cells: +3.07±1.47% size and −8.02±2.08% granularity). These effectors were dramatically expanded in the CSF (Fig 1B). We also noted over-representation of “cytotoxic” T cells, CD4+ and CD8+ T cells expressing NK marker CD56 in the CSF, which were also larger than their blood counterparts. However, only CD8+/CD56+ T cells were degranulated in the CSF.
B cells were significantly larger in the CSF (28.90±3.09%) and were the only cellular subpopulation with higher granularity in the CSF (4.3±0.91%). This was likely due to the fact that our immunophenotyping panel could not differentiate B cells from plasma blasts, which are larger and more granular than B cells, but still express CD19 (14).
Differences in the immune cells among diagnostic categories
Overall, we observed prominent overlap between diagnostic categories for both blood and CSF markers (Fig 2–5). In general, the two pediatric cohorts were more dissimilar compared to the adult subgroups. Although we show all significant differences in the Figures 2–5, we only highlight those that we consider to be biologically meaningful based on consistency and high levels of significance. Differences that could be age-related (and thus were not considered to be disease specific) are highlighted by grey shading of the statistical annotations within Fig 2–5.
Figure 2. Differences in the proportions and absolute numbers of immune cells among diagnostic categories: CD3 T cells, CD4+ T cells and their subtypes.

Two left panels in each row represent proportions of specific cell populations in blood and CSF, while two right panels represent absolute numbers of the same cell population in the blood and CSF. Each diagnostic category is represented by one vertical box blot, while data from healthy donors (HD) are depicted as grey shading, with horizontal line representing mean, dark shade of grey representing +/− 1SD and lighter shade of grey representing +/− 2SD of HD cohort. Each box plot shows median, 25–75%-tile and whisker blots represent minimum-maximum-tile for each diagnostic category. *: P<0.05, **: 0.001<P<0.05, ***: P<0.001.
Figure 5. Differences in the proportions and absolute numbers of immune cells among diagnostic categories: Myeloid (MyDC) and plasmacytoid dendritic cell (PlDC), granulocytes and basophils.

Two left panels in each row represent proportions of specific cell populations in blood and CSF, while two right panels represent absolute numbers of the same cell population in the blood and CSF. Each diagnostic category is represented by one vertical box blot, while data from healthy donors (HD) are depicted as grey shading, with horizontal line representing mean, dark shade of grey representing +/− 1SD and lighter shade of grey representing +/− 2SD of HD cohort. Each box plot shows median, 25–75%-tile and whisker blots represent minimum-maximum-tile for each diagnostic category. *: P<0.05, **: 0.001<P<0.05, ***: P<0.001.
Differences in T cells
While no differences among adult subgroups were observed for blood T cells, in the CSF the OIND patients had significantly higher absolute numbers of all T cells (CD3+, CD4+, CD8+), including HLA-DR+ effector CD4+ T cells (Fig 2) in comparison to NIND patients and the two progressive MS subgroups. Additionally, RR-MS patients had significantly higher absolute numbers of the majority of CSF T cell subpopulations (CD3+, CD4+, CD8+ and CD4−/CD8− “double negative” T cells (Tdn; Fig 3)) in comparison to PP-MS, but not SP-MS patients.
Figure 3. Differences in the proportions and absolute numbers of immune cells among diagnostic categories: double negative T cells, CD8+ T cells and their subtypes.

Two left panels in each row represent proportions of specific cell populations in blood and CSF, while two right panels represent absolute numbers of the same cell population in the blood and CSF. Each diagnostic category is represented by one vertical box blot, while data from healthy donors (HD) are depicted as grey shading, with horizontal line representing mean, dark shade of grey representing +/− 1SD and lighter shade of grey representing +/− 2SD of HD cohort. Each box plot shows median, 25–75%-tile and whisker blots represent minimum-maximum-tile for each diagnostic category. *: P<0.05, **: 0.001<P<0.05, ***: P<0.001.
Multiple differences were observed between pediatric and adults subgroups. In the blood, pediatric patients had elevations in absolute numbers of T cells (Fig 2), especially CD8+ and Tdn (Fig 3), as compared to adult patients. Based on literature review and our age-homogenous subgroup analysis (Supplementary Table 1), these results reflect physiological elevations of lymphocytes in pediatric subjects.
The increases in Tdn in both pediatric cohorts compared to adults was also seen in the CSF. The uniformity of this change and its congruency with our age-homogenous subgroup analysis suggests that this difference is likely age-related. Both pediatric cohorts also had elevated absolute numbers of CD3+ and CD8+ in the CSF in comparison to NIND, PP-MS and SP-MS subjects. For CD4+ T cells, the difference was only significant for absolute numbers between pediatric OIND (pOIND) cohort and NIND, PP-MS and SP-MS patients, which was likely driven by the overall CSF pleiocytosis observed in the pOIND cohort. The pOIND patients had actually significantly lower %-age of CD4+ T cells in the CSF. Because this change is inconsistent with the physiological enrichment of T cell in pediatric patients, it is likely disease-related. The proportional decline in CSF T cells in the pOIND patients can be explained by substantial enrichment of monocytes and granulocytes in the CSF of this cohort (see below), consisting mostly of NOMID patients. Our interpretation that adaptive immunity does not play dominant role in this disorder is further supported by additional observations: pOIND cohort had decreased proportions of HLA-DR+ T cells (Figs 2&3) and it was the cohort that was driving the statistical interactions with the diagnosis identified in Fig 1. Specifically, in comparison to all other cohorts, pOIND patients had the smallest enrichment of T cells in the CSF in comparison to blood (Supplementary Fig 2) and in fact, for CD8+ T cells, the CSF/blood ratio was inverted (Supplementary Fig 2B). Only absolute numbers of cytotoxic CD56+/CD8+ T cells were significantly enriched in the CSF of the pOIND cohort, but these cells cannot be distinguished by our immunophenotyping panel from NKT cells, which are part of the innate immune system.
We did not observe significant differences in the remaining activation markers, such as size, granularity and CD25 expression among other patient subgroups (data not shown).
Differences in B cells
In adults, we did not observe any B cell related differences in the blood. In contrast, B cells were over-represented in the CSF of RR-MS and SP-MS (but not PP-MS) patients in comparison to NIND controls (Fig 4, top panels). When considering absolute numbers of CSF B cells, RR-MS patients had significantly higher numbers in comparison to both NIND and PP-MS (but not SP-MS) patients. Similarly, OIND patients had elevated CSF B cell numbers in comparison to NIND and PP-MS cohorts.
Figure 4. Differences in the proportions and absolute numbers of immune cells among diagnostic categories: B cells, Monocytes and NK cell.

Two left panels in each row represent proportions of specific cell populations in blood and CSF, while two right panels represent absolute numbers of the same cell population in the blood and CSF. Each diagnostic category is represented by one vertical box blot, while data from healthy donors (HD) are depicted as grey shading, with horizontal line representing mean, dark shade of grey representing +/− 1SD and lighter shade of grey representing +/− 2SD of HD cohort. Each box plot shows median, 25–75%-tile and whisker blots represent minimum-maximum-tile for each diagnostic category. *: P<0.05, **: 0.001<P<0.05, ***: P<0.001.
We also observed a significant increase in CD80 expression on CSF B cells in RR-MS patients as compared to NIND subjects (p=0.02257, data not shown).
B cells were dramatically enriched in the blood of pediatric NIND (pNIND) patients in comparison to all adult subgroups, both as a proportion and absolute numbers. This change is consistent with physiological enrichment of lymphocytes in the pediatric subjects (Supplementary Table 1). However, we cannot rule out disease-related contributions, because the pNIND group exhibited dramatic enrichment of blood B cells in comparison to CSF B cells among all diagnostic categories, including HDs (Fig 4 upper panels and Supplementary Fig 2B). In contrast, no statistically-significant differences from adult cohorts were observed in pOIND patients. Pediatric patients had increased absolute numbers of CSF B cells in comparison to NIND and PP-MS adult patients, which is likely age-related.
Differences in monocytes
RR-MS patients had a significantly lower CSF proportion of monocytes in comparison to NIND and PP-MS, but not SP-MS patients (Fig 4, second raw). Due to relative CSF pleiocytosis in RR-MS cohort, this change was no longer significant when considering absolute numbers of monocytes.
pNIND patients had robust decrease in the proportion of monocytes in the blood in comparison to all adult patients. Because this difference disappeared when absolute numbers were considered, we believe that this proportional decrease of monocytes in the blood is linked to robust proportional enrichment of lymphocytes (especially B cells) in the same patients. In the CSF, pOIND patients exhibited strong increase in the absolute numbers of monocytes compared to all subgroups, including pNIND.
Differences in NK cells
Among adult patients, the OIND group had significantly higher absolute numbers of NK cells in the CSF in comparison to NIND and PP-MS patients (Fig 4, last 2 rows). No significant differences were noted among adult subjects in the blood.
In the blood, pediatric patients had overall higher numbers of NK cells in comparison to adult patients; this difference was more pronounced for CD56bright NK cells and especially in the pNIND cohort. While this is consistent with physiological enrichment of lymphocytes in the blood of pediatric patients, the difference between pNIND and pOIND subjects in numbers of CD56bright NK cells suggests the possibility of disease-related change.
Absolute numbers of CD56dim NK cells were increased in the CSF of pediatric patients in comparison to all adult subgroups, except OIND. Absolute numbers of CD56bright NK cells were increased only in pOIND subgroup and only in comparison to NIND, PP-MS and SP-MS cohorts. Because of lack of normative pediatric data, it is unclear if this represents physiological or disease-related difference.
Differences in DCs
The more prevalent MyDC were proportionally under-represented in the blood of pediatric cohorts (Fig 5, upper row); this difference was significant only in pNIND in comparison to adult subgroups. The difference was less robust for pOIND group and disappeared when absolute numbers of MyDC were considered. However, both pediatric cohorts had significantly higher absolute numbers of MyDC in the CSF in comparison to NIND, PP-MS and SP-MS subgroups. In contrast, no differences in the proportion, activation status or absolute numbers of MyDC were observed among adult patients.
PlDC were conspicuously elevated in the CSF of the pediatric cohort, both in terms of proportions and absolute numbers (Fig 5, second raw). The prominent increase in the proportion and absolute numbers of CSF PlDC in a subgroup of pNIND patients (in AGS patients; Supplementary Fig 2A), strongly suggests disease-related process.
In contrast, among adult subjects, only OIND and RR-MS patients had higher absolute numbers of PlDC in the CSF in comparison to NIND group.
Differences in granulocytes and basophils
Granulocytes were proportionally decreased in the blood of pNIND cohort in comparison to all adult patients (Fig 5), consistent with age-related normative data (Supplementary Table 1). In contrast, pOIND cohort did not have physiological proportional decrease in blood granulocytes. Furthermore, pOIND patients had prominent expansion of absolute numbers of granulocytes in the CSF, in comparison to adult subgroups and pNIND cohort. There was similarly robust expansion of absolute numbers of basophils in the CSF of pOIND cohort as compared to all adult subgroups, except OIND. Overall these data indicate that the primary immune alteration in the pOIND cohort resides in innate immune cells, such as monocytes, granulocytes and basophils. This interpretation is supported by the observation that the majority of statistical interactions with the diagnosis identified in Fig 1A were driven by pOIND NOMID patients. Specifically, these patients had inverted blood/CSF ratio of monocytes (i.e. had higher proportion of monocytes in the CSF than in the blood) and had also inverted ratios of monocyte size and granulocyte granularity (Supplementary Fig 2A). In other words, CSF monocytes of NOMID patients were larger and CSF granulocytes were significant more degranulated in comparison to their counterparts in the blood.
No significant differences in the proportion, numbers or activation status of granulocytes and basophils were identified among adult patients, with the exception of increased proportion of granulocytes in the CSF of PP-MS cohort in comparison to RR-MS.
Correlations between immune subpopulations in the blood and CSF
Virtually all non-physiological differences between diagnostic categories were related to CSF and not blood biomarkers, suggesting that immune cells in the CSF are not simply recruited from the periphery. To support this interpretation, we analyzed correlations between blood and CSF biomarkers using Spearman correlation coefficient higher than 0.5 as an indication of biologically meaningful correlation (i.e. explaining >25% of variance).
We found significant correlations between immune cell subsets in the blood and CSF only in two non-inflammatory cohorts: the NIND and pNIND (Table 3). In NIND, the strongest correlations were observed for DnT (r = 0.72 p<0.0001), CD8+ T cells r = 0.55; p = 0.0035) and their “effectors” (HLA-DR+ CD8+ T cells; r = 0.68; p = 0.0001) and for B cells (r = 0.67; p = 0.0002). Within the same cohort, the activation status of B cells, as measured by levels of CD80, also correlated strongly between blood and CSF compartment (r = 0.84; p = 0.0001). In pNIND cohort we observed significant correlations between blood and CSF B cells (r = 0.61; p = 0.0358) and between subsets of CD4+ T cells (CD56+/CD4+; r = 0.73; p = 0.0065 and HLA-DR+/CD4+; r = 0.66; p = 0.0202) and MyDC (r = 0.59; p = 0.0446). Additionally, although monocyte numbers in the CSF did not correlate with monocyte numbers in the blood for any cohort, we observed statistically significant correlation between CD80 expression on monocytes in the blood and CSF for both OIND (r = 0.62; p = 0.0036) and pOIND (r = 0.53; p = 0.0432) patients.
Table 3. Correlations between immune subpopulations in the blood and CSF.
The immune cell subsets in the blood and CSF showed significant correlations only in two non-inflammatory cohorts: the adult NIND and pediatric pNIND.
| Abs | PP-MS | RR-MS | SP-MS | NIND | OIND | p-NIND | p-OIND |
|---|---|---|---|---|---|---|---|
| CD3 | 0.1112 | −0.0294 | −0.0753 | 0.4550* | −0.0051 | 0.0559 | −0.2478 |
| CD4T | 0.1031 | 0.0628 | −0.0974 | 0.2950 | −0.0191 | 0.0140 | −0.2748 |
| HLA-DR+CD4T | 0.1387 | −0.2536 | 0.0013 | 0.4715* | −0.0271 | 0.6573* | 0.2061 |
| CD56+CD4T | 0.1116 | −0.1052 | 0.2519 | 0.2568 | 0.1221 | 0.7343** | −0.0113 |
| CD8T | 0.2419 | 0.0882 | −0.0299 | 0.5508** | 0.1147 | 0.1608 | 0.1183 |
| HLA-DR+CD8T | 0.3393* | −0.0118 | 0.2104 | 0.6807*** | 0.1888 | 0.4965 | 0.1148 |
| CD56+CD8T | 0.1468 | −0.0707 | 0.1519 | 0.1166 | −0.0510 | −0.3287 | 0.1730 |
| DN T | 0.3066* | −0.0002 | 0.2208 | 0.7176*** | 0.3717* | 0.3986 | 0.1104 |
| Cd56dimNK | 0.1232 | 0.2531 | 0.1091 | 0.3422 | 0.1902 | 0.3147 | 0.3304 |
| CD56briNK | 0.2002 | 0.1050 | 0.3532 | 0.4489* | −0.0136 | −0.3986 | −0.0461 |
| Monocytes | 0.2999* | 0.3277* | 0.3078 | 0.3128 | 0.0477 | 0.1119 | 0.0478 |
| B cells | 0.1608 | 0.1748 | −0.2312 | 0.6677*** | 0.2394 | 0.6084* | 0.0557 |
| PlDC | 0.2013 | 0.3083* | −0.0819 | 0.4346* | −0.0422 | 0.1399 | 0.0074 |
| MyDC | 0.2408 | 0.0312 | 0.3468 | 0.0947 | 0.1503 | 0.5874* | 0.0504 |
| Basophils | 0.2109 | 0.4087** | 0.1612 | 0.4822* | 0.4179* | 0.4336 | 0.3414 |
| Granulocytes | 0.0771 | 0.0704 | −0.1987 | 0.2807 | 0.2786 | 0.2727 | 0.2052 |
| Monocytes CD80 MFI | 0.3919* | 0.2665 | 0.6000 | 0.3664 | 0.6198*** | 0.4692 | 0.5277* |
| B cell CD80 MFI | 0.0227 | 0.1084 | 0.2909 | 0.8365*** | −0.1504 | 0.1321 | 0.2198 |
: P<0.05,
: 0.001<P<0.05,
P<0.001,
light green: 0.5<R<0.6, green: 0.6<R<0.7, dark green: R>0.7.
Unsupervised clustering based on CSF immunophenotyping data
Because the biggest challenge for clinicians is to determine extent of intrathecal inflammation in diagnostically uncertain cases, we wanted to assess relationship between immunophenotyping data and the diagnostic categories by unsupervised clustering (Fig. 6).
Figure 6. Unsupervised clustering of diagnostic codes based on CSF immunophenotyping data.
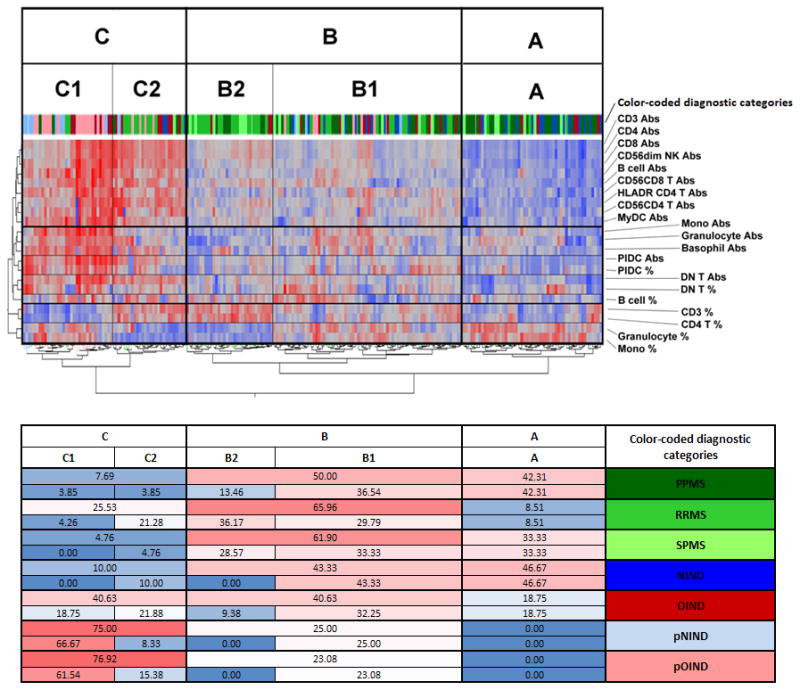
A two-way, unsupervised hierarchical clustering dendrogram of subjects and biomarkers based on selection of CSF immunophenotyping markers that were discriminatory in subgroup analyses. Red: relatively high expression; blue: relatively low expression. The proportions of patients from different diagnostic categories that are classified to distinct clustering subgroups are outlined below the dendrogram. Color-coded diagnostic categories are identical for both panels.
On the cell level, the algorithm clustered absolute numbers of adaptive immune cells, such as T and B cells subsets. Interestingly, CD56dim NK cells and myeloid DCs also clustered with this large group. Based on this largest discriminatory group, patients could be separated into 3 clusters: A: low, B: medium and C: high absolute numbers of immune cells. A smaller cluster contained absolute numbers of monocytes, granulocytes and basophils. The next cluster consisted of proportions and numbers of PlDCs and DnT, which contain high proportion of γ/δ T cells (unpublished observations). Proportions of B cells represented a unique cluster, but with close proximity to the DnT cluster. The final two clusters consisted of proportions of CD3+ and CD4+ T cells and proportions of granulocytes and monocytes and these subdivided patient categories with moderate to high CSF pleiocytosis (i.e. groups B&C) into 2 distinct subgroups: B1 and C1 had proportional predominance of monocytes and granulocytes (which also dominated the entire group A), whereas B2 and C2 had proportional predominance of T cells, especially CD4+. Thus, the unsupervised clustering actually reflected the biology of the immune responses, by clustering in proximity those elements that are usually activated together.
On the patient level, group A, characterized by low numbers of immune cells and relative dominance of innate immunity, contained most patients with non-inflammatory etiology. The majority of HDs (80%) and NIND (46.7%) patients fell into this group. More than a third of progressive MS patients also fell into this group (42.31% of PP-MS and 33.33% of SP-MS).
Group B contained intermediate numbers of immune cells. RR-MS patients clearly dominated this group (65.96%), followed by a large proportion of SP-MS (61.90%) and PP-MS (50.00%) patients. Although 43.3% of NIND patients and 20% of HD also fell into this group, all of them were classified into group B1: with relative predominance of innate immune cells. Similarly, out of 40.6% of OIND patients who clustered here, ¾ clustered to the B1 sub-group. Likewise, pNIND and pOIND patients clustered exclusively to B1 subgroup. In contrast, MS patients, especially RR-MS and SP-MS clustered preferentially to B2 subgroup.
Finally, group C was characterized by high numbers of immune cells. Not surprisingly, the largest proportions of patients with intrathecal inflammation (40.63% of OIND, 76.92% of pOIND, but also 25.53% of RR-MS and 75.00% pNIND) clustered here, whereas NIND patients, as well as patients with progressive MS were almost completely excluded. As would be expected for NOMID disease process, the majority of pOIND patients (61.54%) clustered to C1. Interestingly, a large portion of pNIND patients (66.7%) also clustered to C1. In addition to AGS patients, who had the highest proportions and absolute numbers of PlDC with relative lack of monocytes and granulocytes (and thus clustered at the R-edge of C1 category, Fig 6), pNIND category contained mostly children with autoinflammatory syndromes other than NOMID.
While OIND patients were equally distributed between C1 (21.88%) and C2 (18.75%), almost all RR-MS patients clustered to group C2, consistent with predominance of adaptive immunity in this disease.
DISCUSSION
Results of the current study can be conceptually divided into two categories:
The first relates to the physiological relationship between systemic and intrathecal immune responses: CSF leukocytes differ from those from blood in a surprisingly uniform manner, irrespective of patient diagnosis. Most of innate immune cells (i.e. granulocytes, monocytes, MyDCs, basophils and CD56dim NK cells) and B cells are proportionally under-represented in the CSF, while T cells (especially CD4+), immunoregulatory CD56bright NK cells and PlDCs are over-represented. We confirmed that T and B cells in the CSF have more activated phenotype than analogous cells in the blood, an observation that was previously attributed to selective ability of activated lymphocytes to cross the blood brain barrier (BBB) (24). We expand these findings by demonstrating that CSF T cells not only express more activation markers such as HLA-DR, but they are also significantly and uniformly degranulated in comparison to their blood counterparts. Our data argue that CSF immune cells do not simply reflect cells recruited from the periphery. Instead, they represent a mixture of cells that are recruited from the blood, are activated intrathecally and leave the CNS after performing effector functions. This explanation is consistent with our observation that the only biologically-meaningful correlations between numbers of immune cells in the blood and CSF were observed in patients with non-inflammatory CNS processes, where selective expansion and/or retention of immune cells in the intrathecal compartment was minimized. “Finally, this explanation is also consistent with observations from animal models, which demonstrated that activated T cells enter the CNS compartment irrespective of their antigen specificity, but only those T cells that recognize antigen(s) expressed in the CNS are retained and expanded (25, 26). While the majority of T cells that expand in CNS tissue undergo apoptotic death leading to termination of the intrathecal immune response(s), some of them leave the CNS compartment to become memory T cells, as evidenced by increased precursor frequency of autoreactive T cells in the blood several weeks/months after experimentally-induced stroke (27).
The uniformity of the phenotypical changes affecting immune cell subsets in the CSF supports the notion that the CNS microenvironment shapes intrathecal immune responses: e.g. we observed simultaneous activation and degranulation of the PlDCs and CD56bright NK cells in the CNS, the two cell populations that are also selectively enriched in the CSF in comparison to blood. In contrast, MyDCs, CD56dim NK cells, but also granulocytes and basophils (all cell types that are under-represented in the CSF) had comparable granularity in both compartments. It is likely that recruitment to the CNS and local activation of these cells is physiologically restricted, because they can be highly destructive for CNS tissue (28). This explanation is supported by our observations that in NOMID patients, with genetically-determined aberrant activation of inflammasome pathway, granulocytes (and monocytes) in the CSF are both abundant and degranulated.
Out of the common innate effectors, monocytes behave uniquely in their decreased granularity in the CSF indicating intrathecal engagement of effector functions. Interestingly, while some studies have highlighted a pathogenic role of monocytes in CNS tissue destruction (28) others have shown that under physiological conditions monocytes promote repair of CNS tissue, including remyelination (29–31).
In contrast to constrained innate immune responses, CSF is highly enriched for memory/effector T cells, especially CD4+. It is known that T cells are important for immunosurveilance against persisting neurotrophic viruses (32, 33). Physiological intrathecal immunity controls inadvertent activation of such opportunistic pathogens and memory T cells in conjunction with PlDCs (34) and NK cells (35, 36) are uniquely suited to play this role. However the humoral part of the adaptive immune responses is profoundly under-represented in the CSF. One wonders whether an abundance of B cells poses a special threat for CNS tissue. It is informative to recall that transgenic animals, in which the majority of T cells (or B cells) recognize CNS auto-antigen, rarely develop spontaneous CNS autoimmunity. However, when they are crossed to animals, in which transgenic T and B cells recognize the same auto-antigen, spontaneous CNS inflammation is frequent (37, 38). Thus, autoreactive T cells need help from autoreactive B cells in order to mediate CNS tissue injury, explaining the high efficacy of B cell-depleting therapies in MS (39).
However, one cannot forget that CSF immunophenotyping reflects only the “mobile” pool of immune cells that reside in the CNS. For example, it has been demonstrated previously that despite predominance of CD4+ T cells in the CSF, CD8+ T cells actually represent the majority of T cells infiltrating the CNS tissue (40). Nevertheless, pathology studies that utilized NIND controls generally found very few immune cells (other than microglia) infiltrating CNS parenchyma or meninges and therefore in HD and NIND subjects, the CSF immunophenotyping likely reflects physiological status of intrathecal immunity (or healthy immunosurveilance function of the CNS) and this is what we are focusing on in this first part of the discussion. To summarize: our data point to the existence of physiological regulation of intrathecal immune responses, which likely aim to provide immunity against persistent pathogens and to enhance immune-mediated repair, while limiting the potential for immune-mediated destruction of CNS tissue (by enhanced presence of regulatory immune cells such as FoxP3+ T-regs (41, 42) and CD56bright NK cells (43) and by limited recruitment of granulocytes, CD56dim NK cells, myeloid DCs and B cells).
Nevertheless, our data also indicate that despite physiological regulation, in-situ inflammation-driven changes shape cellular composition of CSF in a disease-specific manner. Referring back to previously mentioned “mobile” versus “static” pools of intrathecal inflammatory cells, we recognize that CSF immunophenotyping may be significantly under-representing disease-specific differences, especially as they relate to immune cells infiltrating CNS tissue.
With this drawback in mind, we reproduced previously-validated enrichment of CSF B cells and relative decrease of CSF monocytes in RR-MS patients in comparison to NIND controls, leading to profound decrease in monocyte/B cell ratio in the RR-MS cohort (Supplementary Fig 3a). We have put un-validated reports of other CSF abnormalities observed in MS patients into the perspective of properly-controlled large datasets, analyzed in a blinded fashion using identical SOPs. Thus, we conclude that the most conspicuous and reproducible intrathecal immune abnormality in RR-MS resides in abnormal adaptive immune responses, including humoral immunity, in accordance to previously-described presence of OCB and high IgG index (44), high intrathecal concentrations of CXCL13 (12, 45, 46) and high CSF numbers of B cells and plasma cells. MS has been traditionally viewed as T cell-mediated disease and our data do not disprove this notion. Instead, they indicate that both parts of the adaptive immune responses, i.e. T cell and B cells, play important role in MS disease process, likely through potentiation of each other’s functions. In contrast, we reason that the scarcity of CSF monocytes in RR-MS is likely secondary to their preferential recruitment to actively demyelinating lesions, where they clear myelin debris and potentially promote remyelination (30). This explanation is consistent with observations that RR-MS patients have higher (not lower) CSF levels of IL-12p40 (the cytokine preferentially released by activated monocytes/macrophages) and that levels of IL-12p40 peak after development of MRI CEL (47). The rapid filling of MS plaques with intravenous contrast likely signifies that the tissue integrity inside the CEL has already been damaged; otherwise the fluid would slowly propagate along the white matter tracks, as it does in vasogenic edema associated with brain tumors. It is into this damaged tissue that monocytes are recruited to phagocytose myelin and activated to produce IL-12p40.
We also provide a comprehensive comparison between patients with different MS subtypes: our data show decidedly that on a group level, SP-MS patients are immunologically closer to RR-MS than PP-MS patients are. RR-MS patients have significantly higher numbers of CD4+ and CD8+ T cells and B cells, in comparison to PP-MS only. Both RR-MS and SP-MS (but not PP-MS) patients have elevated proportions of CSF B cells in comparison to NIND. Finally, PP-MS patients have higher proportions of monocytes and granulocytes as compared to RR-MS, but not SP-MS subjects. Unsupervised clustering also grouped SP-MS closer to RR-MS in comparison to PP-MS. Having said that, we also observed substantial overlap between the three MS groups, supporting the notion of heterogeneity of disease mechanisms across clinical diagnostic categories. We will get back to this point later in the discussion.
Genetically confirmed NOMID and AGS patients represent clear examples of the potential of CSF immunophenotyping to provide insight into disease processes. Despite the fact that the vast majority of studied NOMID patients were treated with anakinra, we found significant elevations in their intrathecal levels of monocytes and granulocytes, even in comparison to the pNIND cohort. Furthermore, intergroup comparison in the size and granularity of monocytes and granulocytes provided strong evidence for their intrathecal activation in NOMID patients. Thus, we conclude that therapy with anakinra does not completely normalize intrathecal immune abnormalities in this cohort, perhaps due to persistent activation of IL-18 arm of the inflammasome pathway (48) or inability of anakinra to access intrathecal compartment after therapeutic closure of the blood brain barrier (BBB).
Similarly, we observed homogeneous abnormalities in AGS patients, which were grouped into pNIND cohort based on the lack of CSF abnormalities on clinical laboratory tests. However, research laboratory CSF counts were consistently elevated in this group, in accordance with published reports (49). We observed prominent expansion of PlDCs in their CSF (Supplementary Fig 2A), consistent with the proposed disease mechanism, where genetic defect in the metabolism of nucleic acids leads to activation of innate immune system and intrathecal production of α-interferon (IFN-α) (50). PlDCs are the best known cellular producers of IFN-α (51). AGS might be more appropriately classified into pOIND group despite lack of clinical laboratory biomarkers of CNS inflammation. AGS may be considered another auto-inflammatory disorder, especially because AGS-associated genetic defects can also aberrantly activate the inflammasome pathway (50). This brings us to the final topic of the discussion.
We acknowledge that our study has important limitations: we consider the lack of normative data on HDs the most imperative. We were able to collect only 5 HDs. The concentration of CSF cells (median 2472.73, range: 866.67–4740.64) obtained from this small cohort is slightly higher than the only other published cohort of HDs we are aware of (median 968 cells/ml of CSF, range 413–2616; (52)). Our strict SOPs designed to limit CSF cells loss are the likely explanation for the higher numbers of CSF leukocytes in our cohort. Accordingly, we observed a higher proportion of granulocytes and monocytes, which are most susceptible to lysis or adherence to plastic in CSF samples that remain unprocessed for extended time. If this small cohort is truly representative, then all patient cohorts differ from HDs in many aspects (Figs 2–5).
In this regard, inclusion of a broad range of patients with putative neuroimmunological disorder represents both a strength and noteworthy challenge of the current study. In contrast to previous studies, where control groups were clearly different from MS (e.g. patients with normal pressure hydrocephalus), NIND and OIND patients included in this study were all referred for evaluation of possible neuroimmunological disorder. Patients classified as OIND have infectious, autoimmune and autoinflammatory CNS diseases with diverse phenotypes of the intrathecal immune responses. This diversity contributes to the broad spread of immunophenotyping values and diminishes statistical significance of the inter-group comparisons. Furthermore, because of our strict adherence to currently approved CSF laboratory tests in diagnostic classification, it is likely that the OIND cohort includes patients with history of past intrathecal inflammatory process, but without active CNS inflammation. This is due to the fact that IgG index or OCB may remain elevated for years after the intrathecal inflammatory process has subsided, because of the longevity of plasma cells (53) and that contrast-enhancement on brain MRI represents opening of the BBB, which may, or may not be due to inflammatory process.
A similar degree of heterogeneity and diagnostic uncertainty applies to NIND patients. Classification of NIND category was based on negative systemic work-up for neuroimmunological disorder, the absence of CEL on MRI and benign CSF profile. Unfortunately, these standard markers, developed decades ago, are not of sufficient sensitivity to unambiguously exclude intrathecal inflammation. For example, counting in a Neubauer hemocytometer is (according to manufacturer insert) unreliable under cell concentrations below 250,000 cells/ml. This represents the vast majority of unspun CSF samples. In contrast, our research laboratory effectively concentrated CSF cells 50 fold and thus increased the reliable range of hemocytometer counts to specimens with more than 5000 cells/ml of CSF. This represents the majority of CSF samples processed in this study. Not surprisingly therefore, we observed poor, albeit statistically significant (r=0.4636; p<0.001; Supplementary Fig 3b) correlations between CSF cell counts generated in the NIH clinical laboratory, versus our research laboratory. Most importantly, this enhanced counting of CSF cells demonstrated that many patients from the NIND category had absolute counts above 2SD of HD range. While we do not dare to re-classify patients from NIND to OIND category solely based on the CSF counts obtained in our research laboratory, we remain open to the possibility that current diagnostic processes are insensitive to intermediate levels of intrathecal inflammation, which may nevertheless be pathophysiologically important.
That is the openness with which we also interpret results of unsupervised clustering. By itself, CSF immunophenotyping cannot represent a diagnostic test. However, our data on NOMID, AGS patients and MS subtypes indicate that CSF immunophenotyping captures the biology of the immune process extremely well, probably better than clinical diagnostic classification of polygenic diseases. Therefore, the question we should be asking is “what kind of disease characteristics do patients who cluster together have in common”? Can clustering based on immunophenotyping data identify those patients with progressive MS who have remaining intrathecal inflammatory process amenable to therapy with current DMTs? Will NIND patients, who cluster with OIND patients show abnormal levels of other neuroinflammatory biomarkers? Undoubtedly, these questions are beyond the scope of the current paper. Nevertheless, each clinical collaborator plans to address in future studies whether CSF immunophenotyping relates to the phenotype, genotype or severity of the disease process. Our anecdotal observations suggest that this may be the case; e.g. when immunophenotyping profile of PP-MS patient cluster with RR-MS patients, such patient may have phenotypical aspects of disease that are more typical for RR-MS, such as CEL or a large MS lesion load in the brain, as opposed to predominant involvement of the brainstem or spinal cord, which is more typical for PP-MS. But we obviously need to perform this analysis in an unbiased way, where rating of the imaging and clinical disease characteristics is done by an evaluator blinded to the immunological data and by using pre-defined, reproducible outcomes (54).
Lastly, our study adds to the increasing list of publications that demonstrate poor correlation between systemic and intrathecal immune responses in patients with neuroinflammatory diseases. For example, soluble inflammatory markers do not correlate between blood and CSF and sometimes may have even opposing trends (46, 55, 56). Similarly, a study which evaluated B cell exchange between peripheral blood and CSF by deep sequencing of IgG heavy chain variable region genes (57) identified on average less than 5% sharing of B cell clonotypes between these two compartments. Together with current data, these studies indicate that the CSF represents an unique window into CNS pathology (58) and that assessment of the phenotype or severity of neuroinflammatory process from blood biomarkers may lead to unreliable conclusions, especially when such studies use methodology susceptible to biases (59). At best, the signature of the intrathecal process is extremely “diluted” in the systemic circulation and until we fully understand what we are looking for, we should focus our search for mechanistic insight into CNS diseases by studying CSF, or non-invasively, CNS tissue.
Supplementary Material
Acknowledgments
We thank Dr. Dennis Landis for providing neuro exams for those patients from NIH Undiagnosed Diseases Program (UDP) who were not also seen by Neuroimmunological Diseases Unit (NDU) clinicians. We thank Jenifer Dwyer, Rosalind Hayden and Kaylan Fenton for expert clinical/nursing assistance and Anne Mayfield and Freddy Reyes for patient scheduling.
The study was supported by the intramural research program of the US National Institute of Neurological Disorders and Stroke (NINDS)/US National Institutes of Health (NIH).
References
- 1.Lucchinetti CF, Bruck W, Rodriguez M, Lassmann H. Distinct patterns of multiple sclerosis pathology indicates heterogeneity on pathogenesis. Brain Pathol. 1996;6:259–274. doi: 10.1111/j.1750-3639.1996.tb00854.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Miller DH, Leary SM. Primary-progressive multiple sclerosis. Lancet neurology. 2007;6:903–912. doi: 10.1016/S1474-4422(07)70243-0. [DOI] [PubMed] [Google Scholar]
- 3.Mahad D, Lassmann H, Turnbull D. Review: Mitochondria and disease progression in multiple sclerosis. Neuropathol Appl Neurobiol. 2008;34:577–589. doi: 10.1111/j.1365-2990.2008.00987.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Sastre-Garriga J, Ingle GT, Chard DT, Cercignani M, Ramio-Torrenta L, Miller DH, Thompson AJ. Grey and white matter volume changes in early primary progressive multiple sclerosis: a longitudinal study. Brain. 2005;128:1454–1460. doi: 10.1093/brain/awh498. [DOI] [PubMed] [Google Scholar]
- 5.Lucchinetti C, Bruck W. The pathology of primary progressive multiple sclerosis. Mult Scler. 2004;10(Suppl 1):S23–30. doi: 10.1191/1352458504ms1027oa. [DOI] [PubMed] [Google Scholar]
- 6.Choi SR, Howell OW, Carassiti D, Magliozzi R, Gveric D, Muraro PA, Nicholas R, Roncaroli F, Reynolds R. Meningeal inflammation plays a role in the pathology of primary progressive multiple sclerosis. Brain. 2012;135:2925–2937. doi: 10.1093/brain/aws189. [DOI] [PubMed] [Google Scholar]
- 7.Androdias G, Reynolds R, Chanal M, Ritleng C, Confavreux C, Nataf S. Meningeal T cells associate with diffuse axonal loss in multiple sclerosis spinal cords. Ann Neurol. 2010;68:465–476. doi: 10.1002/ana.22054. [DOI] [PubMed] [Google Scholar]
- 8.Magliozzi R, Howell OW, Reeves C, Roncaroli F, Nicholas R, Serafini B, Aloisi F, Reynolds R. A Gradient of neuronal loss and meningeal inflammation in multiple sclerosis. Ann Neurol. 2010;68:477–493. doi: 10.1002/ana.22230. [DOI] [PubMed] [Google Scholar]
- 9.Hawker K, O’Connor P, Freedman MS, Calabresi PA, Antel J, Simon J, Hauser S, Waubant E, Vollmer T, Panitch H, Zhang J, Chin P, Smith CH. Rituximab in patients with primary progressive multiple sclerosis: Results of a randomized double-blind placebo-controlled multicenter trial. Ann Neurol. 2009;66:460–471. doi: 10.1002/ana.21867. [DOI] [PubMed] [Google Scholar]
- 10.Almeida de Jesus A, Goldbach-Mansky R. Monogenic autoinflammatory diseases: Concept and clinical manifestations. Clinical immunology. 2013;147:155–174. doi: 10.1016/j.clim.2013.03.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Cepok S, Jacobsen M, Schock S, Omer B, Jaekel S, Boddeker I, Oertel WH, Sommer N, Hemmer B. Patterns of cerebrospinal fluid pathology correlate with disease progression in multiple sclerosis. Brain. 2001;124:2169–2176. doi: 10.1093/brain/124.11.2169. [DOI] [PubMed] [Google Scholar]
- 12.Krumbholz M, Theil D, Cepok S, Hemmer B, Kivisakk P, Ransohoff RM, Hofbauer M, Farina C, Derfuss T, Hartle C, Newcombe J, Hohlfeld R, Meinl E. Chemokines in multiple sclerosis: CXCL12 and CXCL13 up-regulation is differentially linked to CNS immune cell recruitment. Brain. 2006;129:200–211. doi: 10.1093/brain/awh680. [DOI] [PubMed] [Google Scholar]
- 13.Kowarik MC, Cepok S, Sellner J, Grummel V, Weber MS, Korn T, Berthele A, Hemmer B. CXCL13 is the major determinant for B cell recruitment to the CSF during neuroinflammation. Journal of neuroinflammation. 2012;9:93. doi: 10.1186/1742-2094-9-93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Cepok S, Rosche B, Grummel V, Vogel F, Zhou D, Sayn J, Sommer N, Hartung HP, Hemmer B. Short-lived plasma blasts are the main B cell effector subset during the course of multiple sclerosis. Brain. 2005;128:1667–1676. doi: 10.1093/brain/awh486. [DOI] [PubMed] [Google Scholar]
- 15.Kuenz B, Lutterotti A, Ehling R, Gneiss C, Haemmerle M, Rainer C, Deisenhammer F, Schocke M, Berger T, Reindl M. Cerebrospinal fluid B cells correlate with early brain inflammation in multiple sclerosis. PLoS ONE. 2008;3:e2559. doi: 10.1371/journal.pone.0002559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Pashenkov M, Huang YM, Kostulas V, Haglund M, Soderstrom M, Link H. Two subsets of dendritic cells are present in human cerebrospinal fluid. Brain. 2001;124:480–492. doi: 10.1093/brain/124.3.480. [DOI] [PubMed] [Google Scholar]
- 17.Longhini AL, von Glehn F, Brandao CO, de Paula RF, Pradella F, Moraes AS, Farias AS, Oliveira EC, Quispe-Cabanillas JG, Abreu CH, Damasceno A, Damasceno BP, Balashov KE, Santos LM. Plasmacytoid dendritic cells are increased in cerebrospinal fluid of untreated patients during multiple sclerosis relapse. Journal of neuroinflammation. 2011;8:2. doi: 10.1186/1742-2094-8-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Heinrich A, Ahrens N, Schmidt S, Khaw AV. Immunophenotypic patterns of T-cell activation in neuroinflammatory diseases. Acta neurologica Scandinavica. 2006;113:248–255. doi: 10.1111/j.1600-0404.2005.00562.x. [DOI] [PubMed] [Google Scholar]
- 19.Scolozzi R, Boccafogli A, Tola MR, Vicentini L, Camerani A, Degani D, Granieri E, Caniatti L, Paolino E. T-cell phenotypic profiles in the cerebrospinal fluid and peripheral blood of multiple sclerosis patients. Journal of the neurological sciences. 1992;108:93–98. doi: 10.1016/0022-510x(92)90193-o. [DOI] [PubMed] [Google Scholar]
- 20.de Graaf MT, Smitt PA, Luitwieler RL, van Velzen C, van den Broek PD, Kraan J, Gratama JW. Central memory CD4+ T cells dominate the normal cerebrospinal fluid. Cytometry Part B, Clinical cytometry. 2011;80:43–50. doi: 10.1002/cyto.b.20542. [DOI] [PubMed] [Google Scholar]
- 21.Polman CH, Reingold SC, Banwell B, Clanet M, Cohen JA, Filippi M, Fujihara K, Havrdova E, Hutchinson M, Kappos L, Lublin FD, Montalban X, O’Connor P, Sandberg-Wollheim M, Thompson AJ, Waubant E, Weinshenker B, Wolinsky JS. Diagnostic criteria for multiple sclerosis: 2010 revisions to the McDonald criteria. Ann Neurol. 2011;69:292–302. doi: 10.1002/ana.22366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Bielekova B, Catalfamo M, Reichert-Scrivner S, Packer A, Cerna M, Waldmann TA, McFarland H, Henkart PA, Martin R. Regulatory CD56(bright) natural killer cells mediate immunomodulatory effects of IL-2Ralpha-targeted therapy (daclizumab) in multiple sclerosis. Proceedings of the National Academy of Sciences of the United States of America. 2006;103:5941–5946. doi: 10.1073/pnas.0601335103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Stuve O, Marra CM, Bar-Or A, Niino M, Cravens PD, Cepok S, Frohman EM, Phillips JT, Arendt G, Jerome KR, Cook L, Grand’Maison F, Hemmer B, Monson NL, Racke MK. Altered CD4+/CD8+ T-cell ratios in cerebrospinal fluid of natalizumab-treated patients with multiple sclerosis. Arch Neurol. 2006;63:1383–1387. doi: 10.1001/archneur.63.10.1383. [DOI] [PubMed] [Google Scholar]
- 24.Chofflon M, Gonzalez V, Weiner HL, Hafler DA. Inflammatory cerebrospinal fluid T cells have activation requirements characteristic of CD4+CD45RA− T cells. European journal of immunology. 1989;19:1791–1795. doi: 10.1002/eji.1830191005. [DOI] [PubMed] [Google Scholar]
- 25.Kawakami N, Nagerl UV, Odoardi F, Bonhoeffer T, Wekerle H, Flugel A. Live imaging of effector cell trafficking and autoantigen recognition within the unfolding autoimmune encephalomyelitis lesion. The Journal of experimental medicine. 2005;201:1805–1814. doi: 10.1084/jem.20050011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Grundtner R, Dornmair K, Dahm R, Flugel A, Kawakami N, Zeitelhofer M, Schoderboeck L, Nosov M, Selzer E, Willheim M, Kiebler M, Wekerle H, Lassmann H, Bradl M. Transition from enhanced T cell infiltration to inflammation in the myelin-degenerative central nervous system. Neurobiol Dis. 2007;28:261–275. doi: 10.1016/j.nbd.2007.05.006. [DOI] [PubMed] [Google Scholar]
- 27.Becker KJ. Activation of immune responses to brain antigens after stroke. Journal of neurochemistry. 2012;123(Suppl 2):148–155. doi: 10.1111/j.1471-4159.2012.07953.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Kim JV, Kang SS, Dustin ML, McGavern DB. Myelomonocytic cell recruitment causes fatal CNS vascular injury during acute viral meningitis. Nature. 2009;457:191–195. doi: 10.1038/nature07591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Kotter MR, Zhao C, van Rooijen N, Franklin RJ. Macrophage-depletion induced impairment of experimental CNS remyelination is associated with a reduced oligodendrocyte progenitor cell response and altered growth factor expression. Neurobiol Dis. 2005;18:166–175. doi: 10.1016/j.nbd.2004.09.019. [DOI] [PubMed] [Google Scholar]
- 30.Ruckh JM, Zhao JW, Shadrach JL, van Wijngaarden P, Rao TN, Wagers AJ, Franklin RJ. Rejuvenation of regeneration in the aging central nervous system. Cell stem cell. 2012;10:96–103. doi: 10.1016/j.stem.2011.11.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Miron VE, Boyd A, Zhao JW, Yuen TJ, Ruckh JM, Shadrach JL, van Wijngaarden P, Wagers AJ, Williams A, Franklin RJ, Ffrench-Constant C. M2 microglia and macrophages drive oligodendrocyte differentiation during CNS remyelination. Nature neuroscience. 2013 doi: 10.1038/nn.3469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Hislop AD, Taylor GS, Sauce D, Rickinson AB. Cellular responses to viral infection in humans: lessons from Epstein-Barr virus. Annual review of immunology. 2007;25:587–617. doi: 10.1146/annurev.immunol.25.022106.141553. [DOI] [PubMed] [Google Scholar]
- 33.Stohlman SA, Hinton DR, Parra B, Atkinson R, Bergmann CC. CD4 T cells contribute to virus control and pathology following central nervous system infection with neurotropic mouse hepatitis virus. J Virol. 2008;82:2130–2139. doi: 10.1128/JVI.01762-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Donaghy H, Bosnjak L, Harman AN, Marsden V, Tyring SK, Meng TC, Cunningham AL. Role for plasmacytoid dendritic cells in the immune control of recurrent human herpes simplex virus infection. J Virol. 2009;83:1952–1961. doi: 10.1128/JVI.01578-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Kim M, Osborne NR, Zeng W, Donaghy H, McKinnon K, Jackson DC, Cunningham AL. Herpes simplex virus antigens directly activate NK cells via TLR2, thus facilitating their presentation to CD4 T lymphocytes. Journal of immunology. 2012;188:4158–4170. doi: 10.4049/jimmunol.1103450. [DOI] [PubMed] [Google Scholar]
- 36.Chew T, Taylor KE, Mossman KL. Innate and adaptive immune responses to herpes simplex virus. Viruses. 2009;1:979–1002. doi: 10.3390/v1030979. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Bettelli E, Baeten D, Jager A, Sobel RA, Kuchroo VK. Myelin oligodendrocyte glycoprotein-specific T and B cells cooperate to induce a Devic-like disease in mice. The Journal of clinical investigation. 2006;116:2393–2402. doi: 10.1172/JCI28334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Krishnamoorthy G, Lassmann H, Wekerle H, Holz A. Spontaneous opticospinal encephalomyelitis in a double-transgenic mouse model of autoimmune T cell/B cell cooperation. The Journal of clinical investigation. 2006;116:2385–2392. doi: 10.1172/JCI28330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Hauser SL, Waubant E, Arnold DL, Vollmer T, Antel J, Fox RJ, Bar-Or A, Panzara M, Sarkar N, Agarwal S, Langer-Gould A, Smith CH. B-cell depletion with rituximab in relapsing-remitting multiple sclerosis. N Engl J Med. 2008;358:676–688. doi: 10.1056/NEJMoa0706383. [DOI] [PubMed] [Google Scholar]
- 40.Babbe H, Roers A, Waisman A, Lassmann H, Goebels N, Hohlfeld R, Friese M, Schroder R, Deckert M, Schmidt S, Ravid R, Rajewsky K. Clonal expansions of CD8(+) T cells dominate the T cell infiltrate in active multiple sclerosis lesions as shown by micromanipulation and single cell polymerase chain reaction. The Journal of experimental medicine. 2000;192:393–404. doi: 10.1084/jem.192.3.393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Tsakiri A, Kjaersgaard E, Grigoriadis N, Svane IM, Frederiksen JL. Effector and regulatory T cells in patients with acute optic neuritis. Neuroimmunomodulation. 2012;19:111–120. doi: 10.1159/000330242. [DOI] [PubMed] [Google Scholar]
- 42.Fritzsching B, Haas J, Konig F, Kunz P, Fritzsching E, Poschl J, Krammer PH, Bruck W, Suri-Payer E, Wildemann B. Intracerebral human regulatory T cells: analysis of CD4+ CD25+ FOXP3+ T cells in brain lesions and cerebrospinal fluid of multiple sclerosis patients. PLoS One. 2011;6:e17988. doi: 10.1371/journal.pone.0017988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Bielekova B, Catalfamo M, Reichert-Scrivner S, Packer A, Cerna M, Waldmann TA, McFarland H, Henkart PA, Martin R. Regulatory CD56bright natural killer cells mediate immunomodulatory effects of IL-2R-alpha-targeted therapy (daclizumab) in multiple sclerosis. PNAS. 2006;103:5941–5946. doi: 10.1073/pnas.0601335103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Cross AH, Trotter JL, Lyons J. B cells and antibodies in CNS demyelinating disease. J Neuroimmunol. 2001;112:1–14. doi: 10.1016/s0165-5728(00)00409-4. [DOI] [PubMed] [Google Scholar]
- 45.Krumbholz M, Theil D, Derfuss T, Rosenwald A, Schrader F, Monoranu CM, Kalled SL, Hess DM, Serafini B, Aloisi F, Wekerle H, Hohlfeld R, Meinl E. BAFF is produced by astrocytes and up-regulated in multiple sclerosis lesions and primary central nervous system lymphoma. The Journal of experimental medicine. 2005;201:195–200. doi: 10.1084/jem.20041674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Sellebjerg F, Bornsen L, Khademi M, Krakauer M, Olsson T, Frederiksen JL, Sorensen PS. Increased cerebrospinal fluid concentrations of the chemokine CXCL13 in active MS. Neurology. 2009;73:2003–2010. doi: 10.1212/WNL.0b013e3181c5b457. [DOI] [PubMed] [Google Scholar]
- 47.Bielekova B, Komori M, Xu Q, Reich DS, Wu T. Cerebrospinal Fluid IL-12p40, CXCL13 and IL-8 as a Combinatorial Biomarker of Active Intrathecal Inflammation. PLoS One. 2012;7:e48370. doi: 10.1371/journal.pone.0048370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Sedimbi SK, Hagglof T, Karlsson MC. IL-18 in inflammatory and autoimmune disease. Cellular and molecular life sciences : CMLS. 2013 doi: 10.1007/s00018-013-1425-y. [DOI] [PMC free article] [PubMed]
- 49.Crow YJ. Aicardi-Goutieres syndrome. Handbook of clinical neurology. 2013;113:1629–1635. doi: 10.1016/B978-0-444-59565-2.00031-9. [DOI] [PubMed] [Google Scholar]
- 50.Crow YJ, Rehwinkel J. Aicardi-Goutieres syndrome and related phenotypes: linking nucleic acid metabolism with autoimmunity. Human molecular genetics. 2009;18:R130–136. doi: 10.1093/hmg/ddp293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Reizis B, Bunin A, Ghosh HS, Lewis KL, Sisirak V. Plasmacytoid dendritic cells: recent progress and open questions. Annual review of immunology. 2011;29:163–183. doi: 10.1146/annurev-immunol-031210-101345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Ho EL, Ronquillo R, Altmeppen H, Spudich SS, Price RW, Sinclair E. Cellular Composition of Cerebrospinal Fluid in HIV-1 Infected and Uninfected Subjects. PLoS One. 2013;8:e66188. doi: 10.1371/journal.pone.0066188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Amanna IJ, Carlson NE, Slifka MK. Duration of humoral immunity to common viral and vaccine antigens. N Engl J Med. 2007;357:1903–1915. doi: 10.1056/NEJMoa066092. [DOI] [PubMed] [Google Scholar]
- 54.Landis SC, Amara SG, Asadullah K, Austin CP, Blumenstein R, Bradley EW, Crystal RG, Darnell RB, Ferrante RJ, Fillit H, Finkelstein R, Fisher M, Gendelman HE, Golub RM, Goudreau JL, Gross RA, Gubitz AK, Hesterlee SE, Howells DW, Huguenard J, Kelner K, Koroshetz W, Krainc D, Lazic SE, Levine MS, Macleod MR, McCall JM, Moxley RT, 3rd, Narasimhan K, Noble LJ, Perrin S, Porter JD, Steward O, Unger E, Utz U, Silberberg SD. A call for transparent reporting to optimize the predictive value of preclinical research. Nature. 2012;490:187–191. doi: 10.1038/nature11556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Kuehne LK, Reiber H, Bechter K, Hagberg L, Fuchs D. Cerebrospinal fluid neopterin is brain-derived and not associated with blood-CSF barrier dysfunction in non-inflammatory affective and schizophrenic spectrum disorders. Journal of psychiatric research. 2013 doi: 10.1016/j.jpsychires.2013.05.027. [DOI] [PubMed] [Google Scholar]
- 56.Pranzatelli MR, Tate ED, McGee NR, Travelstead AL, Colliver JA, Ness JM, Ransohoff RM. BAFF/APRIL system in pediatric OMS: relation to severity, neuroinflammation, and immunotherapy. Journal of neuroinflammation. 2013;10:10. doi: 10.1186/1742-2094-10-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.von Budingen HC, Kuo TC, Sirota M, van Belle CJ, Apeltsin L, Glanville J, Cree BA, Gourraud PA, Schwartzburg A, Huerta G, Telman D, Sundar PD, Casey T, Cox DR, Hauser SL. B cell exchange across the blood-brain barrier in multiple sclerosis. The Journal of clinical investigation. 2012;122:4533–4543. doi: 10.1172/JCI63842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Lovato L, Willis SN, Rodig SJ, Caron T, Almendinger SE, Howell OW, Reynolds R, O’Connor KC, Hafler DA. Related B cell clones populate the meninges and parenchyma of patients with multiple sclerosis. Brain. 2011 doi: 10.1093/brain/awq350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Ioannidis JP. Why most published research findings are false. PLoS medicine. 2005;2:e124. doi: 10.1371/journal.pmed.0020124. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.