

Abstract
Mips (macrophage infectivity potentiators) are a subset of immunophilins associated with virulence in a range of micro-organisms. These proteins possess peptidylprolyl isomerase activity and are inhibited by drugs including rapamycin and tacrolimus. We determined the structure of the Mip homologue [BpML1 (Burkholderia pseudomallei Mip-like protein 1)] from the human pathogen and biowarfare threat B. pseudomallei by NMR and X-ray crystallography. The crystal structure suggests that key catalytic residues in the BpML1 active site have unexpected conformational flexibility consistent with a role in catalysis. The structure further revealed BpML1 binding to a helical peptide, in a manner resembling the physiological interaction of human TGFβRI (transforming growth factor β receptor I) with the human immunophilin FKBP12 (FK506-binding protein 12). Furthermore, the structure of BpML1 bound to the class inhibitor cycloheximide N-ethylethanoate showed that this inhibitor mimics such a helical peptide, in contrast with the extended prolyl-peptide mimicking shown by inhibitors such as tacrolimus. We suggest that Mips, and potentially other bacterial immunophilins, participate in protein–protein interactions in addition to their peptidylprolyl isomerase activity, and that some roles of Mip proteins in virulence are independent of their peptidylprolyl isomerase activity.
Keywords: Burkholderia pseudomallei, NMR, peptidylprolyl isomerase, small-molecule inhibitor, X-ray crystallography
INTRODUCTION
Melioidosis is a community-acquired infection frequent in SouthEast Asia and Northern Australia [1]. Caused by the Gram-negative bacterium Burkholderia pseudomallei, the disease can range from a localized infection to an acute systemic, chronic or persistent disease. The symptoms of infection are diverse, often resembling tuberculosis or cancer. Consequently, the disease is likely to be often misdiagnosed [2]. The disease is especially pernicious as the bacterium is intrinsically resistant to a wide range of antibiotics [3]: even with optimal treatment, mortality in acute cases of disease ranges from 20 to 40 % [2]. B. pseudomallei is also a potential bioterror agent [4]. Consequently, there has been a recent and concerted effort to develop novel countermeasures against this organism.
One potential target class for novel antimicrobials is the FKBPs (FK506-binding proteins; tacrolimus). These ubiquitous enzymes catalyse the isomerization of preproline peptide bonds between the cis and trans configurations {PPIase (peptidylprolyl isomerase) activity [5]}, and form part of the broader category of PPIase enzymes. Several FKBPs have been found to play a role in virulence in a range of species, including the bacteria Legionella pneumophila [6], Chlamydia trachomatis [7] and Neisseria gonorrhoeae [8], and the protozoan Trypanosoma cruzi [9]. One of the first bacterial FKBPs to be studied in detail is a L. pneumophila protein required for the efficient invasion of macrophages [6]. Consequently, these proteins were labelled as Mip (macrophage infectivity potentiator) proteins. The FKBP domain of the LpMip (L. pneumophila Mip) is required for virulence, and both antibodies binding to the active site and specific inhibitors sufficed to reduce the capacity of Legionella to invade macrophages [10,11]. We recently showed that a Mip-like protein from B. pseudomallei, BPSS1823 [hereafter BpML1 (B. pseudomallei Mip-like protein 1)], has high PPIase activity; and that it is required for efficient invasion of host cells and virulence in a mouse model of infection. Deletion of this gene significantly attenuates B. pseudomallei (I.H. Norville, N.J. Harmer, S. Harding, G. Fischer, K. Keith, K. Brown, M. Sarkar-Tyson and R.W. Titball, unpublished work).
Despite extensive study of Mips in a range of micro-organisms, the true biological targets for these proteins have not been clearly elucidated [8,11,12]. It has been broadly assumed that they act solely as isomerases [11]. Mips have been suggested to be an attractive target class for antimicrobials: there are known high-affinity inhibitors, indicating that the proteins are eminently druggable; and their prokaryotic functions are differentiated from those of higher eukaryotic FKBPs.
We have previously shown that the BpML1 plays a key role in disease progression. In the present study, we report the molecular structure of BpML1, identify common features with other Mips and highlight differences from mammalian FKBPs that could be exploited for drug design. We also identify new approaches to the design of compounds able to block this target. These compounds could be developed as antimicrobials to treat melioidosis, but could also have a much wider utility to treat other diseases where Mips play a key role in infection.
MATERIALS AND METHODS
Preparation of BpML1
Full-length BpML1 from B. pseudomallei strain K96243 was cloned into pET15b using the NdeI and HindIII sites. The vector was transformed into BL21 (DE3) cells. For enzymology, cells were grown in Luria–Bertani medium at 37 °C with agitation until the D600 (attenuance at 600 nm) reached 0.4–0.6. IPTG (isopropyl β-D-thiogalactoside) was added to 1 mM and growth continued at 20 °C for 4 h. Harvested cells were resuspended in 10 mM PBS supplemented with 100 mg/ml DNase I and Complete™ EDTA-free protease inhibitors (Roche), and disrupted by sonication. Clarified lysate was loaded on to a 1 ml Histrap FF column (GE Healthcare) and the recombinant protein was eluted in 10 mM PBS supplemented with 100 mM imidazole. The purified protein was dialysed against 10 mM PBS and samples frozen at −80 °C until use. The protein concentration was determined using a bicinchoninic acid assay (Pierce Biotechnology).
For crystallization, cells were grown in ZYM-5052 medium [13] supplemented with 100 μg/ml ampicillin at 37 °C until the D600 was 0.5, and then at 20 °C for 16 h. Harvested cells were resuspended in 20 mM Tris/HCl, pH 8.0 and 0.5 M NaCl (Buffer A), and lysed using a Soniprep 150 sonicator (MSE). Clarified lysate was purified using a nickel–agarose column (Bioline). Briefly, the loaded protein was washed with Buffer A supplemented with 20 mM imidazole-HCl (pH 8.0), and eluted with Buffer A supplemented with 250 mM imidazole-HCl (pH 8.0). BpML1 was then loaded on to a Superdex 200 16/60 hr column (GE Healthcare), and eluted isocratically with 10 mM Hepes (pH 7.0) and 0.5 M NaCl.
For NMR studies, 15N-labelled and 13C15N-labelled proteins were made by substituting 15NH4Cl and/or [13C]glucose for their unlabelled equivalents in the cell growth medium. 15N-labelled and 13C15N double-labelled proteins were dialysed into 20 mM potassium phosphate (pH 7.0), 100 mM KCl and 2 mM DTT (dithiothreitol), to a final protein concentration of approx. 1 mM. Cycloheximide N-ethylethanoate (>95 %) was purchased from Life Chemicals without further purification. Samples of the complex were prepared by titrating unlabelled cycloheximide N-ethylethanoate into labelled protein samples while monitoring 15N HSQC (heteronuclear single-quantum coherence) chemical shift changes until a previously established ‘saturated’ spectrum was obtained.
Crystallization and structure solution
All crystals were grown using the microbatch method, and were prepared using an Oryx6 crystallization robot (Douglas Instruments). BpML1 at 14 mg/ml was mixed with an equal volume of 2.2 M (NH4)2SO4 and 0.1 M Bis/Tris (pH 5.5), and grown at 20 °C. Prior to flash-freezing in liquid nitrogen, crystals were soaked for 30–60 s in a cryoprotectant solution of 1.1 M (NH4)2SO4, 0.1 M Bis/Tris (pH 5.5) and 30 % (v/v) glycerol. Single-wavelength X-ray diffraction data were collected at a wavelength of 0.861 Å (1 Å = 0.1 nm) at 100 K at beamline I03 of the Diamond synchrotron (Table 1). Data were processed using iMOSFLM version 1.0.3 [14] and SCALA [15]. Initial phases were determined by molecular replacement using the structures of human FKBP12 (PDB code 1FKB) and the N-terminal domain of rabbit FKBP59 (PDB code 1ROT) as a model. These structures were modified to remove non-homologous side chains using CHAINSAW [16]. Molecular replacement was performed using PHASER [17]. One protomer was observed in the asymmetric unit. Model building and refinement of the structures were performed using Coot version 0.6.1 [18] and PHENIX version 1.6.1 [19]. The structure was solved using anisotropic B-factors for all heavy atoms, riding hydrogen atoms, and optimization of stereochemical and ADP restraints. Structures were validated using PHENIX, Coot and MOLPROBITY [20]. The Ramachandran plot for the final model showed 100 % of residues in the favoured region. Structural images were prepared using the PyMOL molecular graphics system, version 1.3 (Schrödinger).
Table 1. Data collection and refinement statistics.
Values in parentheses are for the highest-resolution shell.
| BpML1 (PDB code 2Y78) | |
|---|---|
| Data collection | |
| Space group | P43212 |
| Cell dimensions* | |
| a, b, c (Å) | 54.68, 54.68, 119.2 |
| α, β, γ(°) | 90, 90, 90 |
| Resolution (Å) | 40.3–0.91 (0.96–0.91) |
| Rmerge | 0.080 (0.398) |
| I/σI | 13.8 (2.2) |
| Completeness (%) | 98.3 (88.2) |
| Redundancy | 5.9 (2.7) |
| Refinement | |
| Resolution (Å) | 40.3-0.91 |
| Number of reflections | 125 244 |
| Rwork/Rfree | 0.109/0.116 |
| Number of atoms | |
| Protein | 2032 |
| Ligand/ion | 58 |
| Water | 275 |
| B-factors | |
| Protein | 12.7 |
| Ligand/ion | 27.2 |
| Water | 24.2 |
| Root mean square deviations | |
| Bond lengths (Å) | 0.014 |
| Bond angles (°) | 1.586 |
All data were collected from one crystal. High- and low-resolution passes were merged.
NMR structure solution
All NMR spectra were collected at 298 K. 15N HSQC, triple-resonance spectroscopy, HCCH TOCSY [21], three-dimensional 15N-edited NOESY and three-dimensional 13C-edited NOESY [22] were recorded on a Bruker Avance 500-MHz spectrometer with a cryoprobe. The two-dimensional 1H-1H NOESY and 1H-1H TOCSY spectra were recorded on a Bruker Avance 750-MHz spectrometer. Backbone resonance assignments of H, N, Cα, C′, and side-chain Cβ were performed using triple-resonance HNCA [23], CBCA(CO)NH [24], HNCACB and HN(CO)CA. The remaining side-chain resonance assignments were obtained from the analysis of 15N-edited NOESY, 13C-edited NOESY and HCCH-TOCSY spectra. Aromatic ring protons and protected amides were identified from two-dimensional 1H-1H NOESY, 1H-1H TOCSY and 15N HSQC respectively collected from a sample exchanged into a 100 % 2H2O NMR buffer. The assignments of the ligand were obtained based on the two-dimensional 13C-filtered NOESY and TOCSY spectra of the ‘F1fF2f’ type [25]. The two-dimensional 13C-12C NOESY spectra were collected to facilitate the assignments of protein–ligand intermolecular NOEs (nuclear Overhauser effects) [26].
Spectral data were processed using NMRPipe [27] and analysed using CcpNmr Analysis 1.0 [28]. The structure calculations were conducted automatically by CYANA2.1 [29,30] based on NOEs selected from 15N-edited NOESY, 13C-edited NOESY, 1H-1H NOESY and 13C-12C NOESY, as well as additional dihedral-angle and hydrogen-bond constraints generated from the NMR data.
Inhibitor assay
PPIase activity of BpML1 protein was determined by a protease-coupled assay as described previously [31]. Briefly, 10 nM BpML1 protein was incubated for 6 min at 6 °C in 1.2 ml of 35 mM Hepes buffer, pH 7.8 with succinyl-Ala-Phe-Pro-Phe-p-nitroanilide (10 mg/ml; Bachem). α-chymotrypsin (Sigma) was added to the cuvette to a final concentration of 1.25 mg/ml and mixed. Hydrolysis of the substrate was measured at 390 nm using a Shimadzu 1800 UV–visible spectrophotometer at 1 s intervals until there was no further change in absorbance. For inhibition measurements, BpML1 protein was pre-incubated with various concentrations of cycloheximide-N-ethylethanoate from 1 to 500 μM for 6 min prior to addition of chymotrypsin. At least three independent readings were taken at each data point. All data fitting and statistical analyses were performed using SPSS v16.0 (IBM). The pseudo first-order rate constant was calculated using eqn (1) [32]; data from 10 to 50 s (following the lag phase, and before substrate became limiting) were taken, and kobs was calculated by linear regression.
| (1) |
The enzymatic rate was determined by comparing the observed rate with the uncatalysed rate (eqn 2):
| (2) |
Data for inhibitor assays were fitted to eqn (3) [33] using least squares non-linear fitting. [E] was treated as a constant (10 nM); v0 and KIapp were fitted, using initial estimates based on the raw data.
| (3) |
RESULTS AND DISCUSSION
The BpML1 fold and active-site residue configuration are highly similar to LpMip
We solved the X-ray structure of BpML1 to 0.91 Å (Table 1). The structure confirmed that BpML1 adopts a classical FKBP fold (Figure 1A). Closer inspection of the active site in the crystal revealed that key side chains adopt a conformation that closely resembles that of the ligand-free L. pneumophila Mip FKBP domain (Figure 1B). NMR structures of BpML1 that were determined independently show an excellent similarity in overall fold with the crystal structure. They also demonstrate that there is considerable flexibility in both peripheral loops and side chains (Figure 1C) in the well-defined active site (see Supplementary Figure S1 available at http://www.BiochemJ.org/bj/437/bj4370413add.htm [34,35]). In particular, the β4–β5 loop, which contributes the key Tyr89 side chain to the active site, shows two distinct conformations in solution; and several of the key conserved active-site side chains have considerable flexibility. These observations are in contrast with Mip from L. pneumophila and T. cruzi, which show a remarkable commonality in the conformations adopted by these regions (see Supplementary Figure S2 available at http://www.BiochemJ.org/bj/437/bj4370413add.htm [34,36,37]). These observations suggest that previous structures (especially crystal structures) might be trapping a preferred low-energy conformation that may be catalytically relevant, but which does not capture the full range of dynamics available to the enzyme in solution.
Figure 1. Structures of BpML1.

(A) Cartoon of the 0.91 Å structure of BpML1 (left, green; PDB code 2Y78) compared with hFKBP12 (right, cyan; PDB code 2DG3 [48]). The overall fold is identical, with BpML1 containing all of the secondary structural elements, and an additional N-terminal β-strand (arrowed). (B) BpML1 shows an active-site conformation nearly identical with that of LpMip (purple; PDB code 1FD9). All of the most conserved side chains are identical in conformation, except for Phe46 (arrow), which is replaced functionally in LpMip with a neighbouring phenylalanine residue. (C) The NMR structure of BpML1 confirms the crystallographic data and identifies flexibility within the active site. Left: cartoon of the NMR structure of BpML1 bound to ligand (yellow; PDB code 2KO7) shows a very similar architecture to the crystal structure (A). Right: the comparison of bound (yellow) and apo protein (pink: PDB code 2KE0) shows significant conformational flexibility in the ligand-binding β3a strand (arrow) and the β4–β5 loop (arrowhead). Both structures shown are representative for an ensemble of 20 structures consistent with the data.
BpML1 forms protein–protein interactions reminiscent of other FKBPs
The protein sample used for crystallization of BpML1 had been cloned with a His6 tag, linked to the protein by a thrombin cleavage site (sequence MSSHHHHHHSSGLVPRGSHM …, thrombin site underlined). The thrombin cleavage peptide was clearly resolved in the electron density, and forms an interaction with a neighbouring molecule in the crystal. This interaction placed the peptide directly in the active site of the neighbouring molecule, forming an apparently tight interaction, burying 890 Å2 of surface (Figure 2A). Intriguingly, although BpML1 is a very active PPIase (I.H. Norville, N.J. Harmer, S. Harding, G. Fischer, K. Keith, K. Brown, M. Sarkar-Tyson and R.W. Titball, unpublished work), the conformation of the peptide is inconsistent with the expected binding for a proline-containing peptide. The proline side chain does not sit in the deep pocket of the enzyme: complexes with the inhibitor FK506 (see Supplementary Figure S1B [38]) and with a cis-peptide [35] have defined this as the likely site for cis–trans isomerization. In contrast, this site is occupied by a valine side chain (Figure 2B). Furthermore, the overall conformation of the peptide is highly diverged from the expected peptide orientation, suggested by structures of rapamycin or peptide-bound FKBPs (Figure 2C [34,35,38]). We conclude that, although BpML1 is a bona fide PPIase, this peptide binding is not representative of the binding of substrates for pre-prolyl-peptide cis–trans isomerization.
Figure 2. BpML1 binding to thrombin recognition peptide suggests that the protein is involved in protein–protein interactions.

(a) Analysis of crystal packing in the X-ray structure of BpML1 (PDB code 2Y78) shows a helical peptide binding to the active site. Cartoon is shown of BpML1 (green) and the peptide derived from cloning (orange). (b) The peptide binds with a valine side chain (black arrow) in the heart of the enzyme active site. The peptide is represented by orange sticks, with BpML1 as a green cartoon. The black open-headed arrow indicates the direction of the peptide, N- to C-terminus. (c) The peptide binding is unlikely to represent a substrate for the PPIase activity. Comparison with the structure of a cis-prolyl-peptide bound to SlyD from T. thermophilus (left, yellow; PDB code 3LUO) and FK-506 bound to hFKBP12 (right, cyan; FK-506 pseudopeptide shown in yellow; PDB code 1FKJ) shows that a bona fide cis-proline peptide lying in the opposite orientation (black open-headed arrows); and that a prolyl or pseudoprolyl side chain occupies the active site (black arrowheads). (d) BpML1 peptide binding mimics hFKBP12 binding to TGFβRI. Left: BpML1 (green) binding to peptide (white sticks). Right: hFKBP12 (cyan) binding to a helix from TGFβRI (pink sticks). The side chains binding to the heart of the active sites are coloured yellow. Note the similarity of the positions of the carbonyls in the red boxes. Six peptide main chain atoms making direct or through single water-mediated hydrogen bonds with BpML1 are shown as spheres on top of the sticks, and are further indicated by black arrowheads.
However, the peptide binding is strikingly similar to the structure of hFKBP12 (human FKBP12) in complex with the TGFβRI (transforming growth factor β receptor I), bone morphogenetic protein receptor 1B and the activin type I receptor (PDB code 3H9R) [39] (Figure 2D; see Supplementary Figure S3 available at http://www.BiochemJ.org/bj/437/bj4370413add.htm). Eukaryotic FKBPs form physiologically important complexes with other proteins, binding partner proteins to modulate their activity [40,41], in addition to their roles in protein folding. In particular, the BpML1 peptide forms a short helix, in a similar conformation to the receptor helices that binds to hFKBP12; and each of these receptors places a leucine side chain into the deep pocket of hFKBP12, reminiscent of the valine residue observed in BpML1. These observations suggest that, like hFKBP12, BpML1 is likely to form complexes with other proteins in the cell in addition to its role as a PPIase. hFKBP12 masks a multiple phosphorylation peptide (the Gly-Ser region) in TGFβRI, and locks the receptor into an inactive conformation to ensure that there is no activation in the absence of an extracellular signal [41]. As bacteria require robust signalling networks to respond appropriately to their surroundings, similar roles in dampening signalling noise are conceivable for BpML1.
X-ray and NMR structures of BPSS1823 reveal intrinsic flexibility in key active-site residues
The X-ray structure presented here diffracted to 0.91 Å. This resolution is high enough to reveal details of structural flexibility that cannot be observed at lower resolution. At this resolution, we can observe that 18 % of the amino acid side chains (not including alanine and glycine residues) display multiple conformations. The majority of these residues are distant from the active site, in largely solvent exposed parts of the molecule. In addition to these amino acids, four alternative conformations are observed in the active site. First, the pair of Leu−6 from the binding peptide and Ile93 displays correlated alternative conformers (see Supplementary Figure S4 available at http://www.BiochemJ.org/bj/437/bj4370413add.htm). More importantly, both Asp44 and Tyr89 show significant flexibility, with hydrogen-bond donor and acceptor atoms moving by up to 1.5 Å between the observed extremes (Figure 3). The refined positions probably represent maxima within the electron density: the two residues therefore probably sample a larger range of positions. Similar flexibility is not observed in the 0.92 Å structure of unliganded hFKBP12 (see Supplementary Figure S5 available at http://www.BiochemJ.org/bj/437/bj4370413add.htm [42]), indicating that this is not an artefact of the software used. These two amino acids provide the only sources of hydrogen-bond donors and acceptors in the BpML1 active site; and mutations of these residues in LpMip significantly reduce the activity of the enzyme [43]. As many proposed mechanisms of action of the enzyme involve these amino acids, this unexpected flexibility is highly suggestive of a role in catalysis. Accordingly, we examined the NMR structures of free and inhibitor-bound BpML1 to examine whether these also are consistent with a pronounced flexibility for Asp44 and Tyr89. Strikingly, the side chain of Asp44 shows a wide range of conformations (see Supplementary Figure S6 available at http://www.BiochemJ.org/bj/437/bj4370413add.htm). This is quite unusual for a residue that is apparently making a hydrogen-bond interaction with the bound ligand. In contrast, the side chain of Tyr89 adopts two preferred rotamers, showing a classical alternative conformation. This contrasts with other well-conserved ligand-interacting residues (e.g. Trp66), which show only a single conformation across 20 models, consistent with the data.
Figure 3. The ultra-high-resolution X-ray structure of BpML1 (PDB code 2Y78) shows multiple conformers of key ligand-binding residues.

(a) Asp44, which accepts a key hydrogen bond from the bound peptide backbone, shows two conformers for the side chain. The distance between the two conformations for Oδ2 is 0.9 Å. The higher-occupancy conformation makes an additional hydrogen bond to the bound peptide backbone, while the other makes two additional hydrogen bonds to other residues of BpML1. (b) Tyr89, the only hydrogen-bond donor in the active-site pocket, also shows a range of conformations. The distance between the Oη atoms is 1.5 Å. One conformation is consistent with a direct (3.0 Å) hydrogen bond to the peptide, while the other makes a water-mediated hydrogen bond to the peptide. Lower-occupancy forms are indicated by a black arrow. (c) Representative electron density for a multiple conformer residue (Tyr89, left) and the peptide binding to the active site (right). Colours: BpML1, yellow; binding peptide, orange; multiple conformation at lower occupancy, cyan; mFo − DFc simulated annealing omit map (side chains truncated to Cβ) contoured at 3σ, green. 2mFo − DFc map for the final structure contoured at 1σ, blue.
This unusual flexibility of these highly conserved side chains in the active site strongly suggests a role for this flexibility in function. The side chains make hydrogen-bonding interactions in at least one of their conformers, indicating that they do not lack a stable conformation. The conformations observed in the structures offer a tantalizing possible role in catalysis: superimposition of the extended prolyl-peptide from the recently solved FKBP-peptide structure [35] shows that one of the Tyr89 conformers would be perfectly placed to stabilize the peptide nitrogen in an intermediate state between the cis and trans peptide. One alternative conformation of Asp44 could interact with and stabilize the peptide backbone of the substrate. These observations suggest that Tyr89 and Asp44 are natively flexible in BpML1, and that this flexibility is likely to be relevant to catalysis.
The structure of BpML1 bound to cycloheximide N-ethylethanoate reveals a novel mode of inhibitor binding
We previously showed that BpML1 binds to rapamycin, which inhibits all known FKBPs (I.H. Norville, N.J. Harmer, S. Harding, G. Fischer, K. Keith, K. Brown, M. Sarkar-Tyson and R.W. Titball, unpublished work). Cycloheximide, an inhibitor of eukaryotic protein synthesis, is an unrelated compound that has previously been shown to inhibit the PPIase activity of both hFKBP12 and LpMip (Figure 4A) [44]. Cycloheximide N-ethylethanoate, which is also active against hFKBP12, inhibited the PPIase activity of BpML1 (Figure 4B): the observed KI of 6.5 ± 1.0 μM is similar to the KI reported for this compound towards FKBP12 (4.4 μM). As there is no extant structure of cycloheximide N-ethylethanoate with an FKBP, we determined the structure of BpML1 bound to cycloheximide N-ethylethanoate by NMR. This structure reveals that the compound binds to the active site (Figure 4C). However, although the compound binds to most of the highly conserved side chains, the major interactions occur with the β3b–α1 and β4–β5 loops (Figure 4D). In fact, hydrophobic contacts are formed with residues Ala94 and Ile98, outside the core active site. No significant interactions take place with the other sides of the catalytic bowl of the FKBP, with the highly conserved active-site residues Tyr33 and Phe57 in particular having no contact with the inhibitor. In comparison, previously observed inhibitors such as FK506, rapamycin and other synthetic inhibitors [38,45,46] tend to bind more broadly across the catalytic bowl, and contact conserved active-site residues across it (Figure 5). Many of these inhibitors also make extensive contacts with the same two loops as cycloheximide N-ethylethanoate.
Figure 4. Cycloheximide N-ethylethanoate identifies a novel binding mode for FKBP inhibitors.

(a) Structures of cycloheximide (left) and its derivative cycloheximide N-ethylethanoate (right). (b) Inhibition of BpML1 by cycloheximide N-ethylethanoate: cycloheximide N-ethylethanoate inhibited the PPIase activity of BpML1 in a dose-dependent manner, showing a KI of 6.5 ± 1.0 μM. All experiments were performed in triplicate, and S.E.M. are shown. The continuous line shows the inhibition curve for fitted values of the inhibition parameters. (c) Overview of the NMR structure of cycloheximide N-ethylethanoate (orange spheres) bound to BpML1 (PDB code 2KO7). Left: BpML1 shown as a yellow cartoon. Right: surface of BpML1 shown, coloured by charge density (red: negative; blue: positive). Charge density was calculated using APBS [49] and PDB2PQR [50]. (d) Close-up view of active-site residues binding to cycloheximide N-ethylethanoate. Residues within 3.5 Å of cycloheximide N-ethylethanoate are shown as sticks. Left: BpML1 shown as a cartoon, cycloheximide N-ethylethanoate shown as sticks. The hydrogen bond between the Cycloheximide N-ethylethanoate hydroxyl and Asp44 is especially noteworthy; and that some functional groups of cycloheximide N-ethylethanoate (arrowed) do not make any interactions with the protein, and could be altered. Right: BpML1 is shown as semi-transparent surface, cycloheximide N-ethylethanoate as spheres. Cycloheximide N-ethylethanoate shows good shape complementarity to BpML1, consistent with its micromolar inhibitory activity. (e) Cycloheximide N-ethylethanoate mimics the binding of peptides to FKBPs. Left: cycloheximide N-ethylethanoate (light-orange sticks; atoms mimicking the peptide coloured bolder and shown thicker) bound to BpMip (yellow cartoon; PDB code 2KO7). Centre: peptide (white sticks; atoms equivalent to those highlighted left shown light grey and thicker) bound to BpMip (green cartoon; PDB code 2Y78). Right: helix from TGFβRI (pink sticks) binding to hFKBP12 (cyan cartoon; PDB code 1B6C). The relevant portions of each peptide are highlighted with black arrowheads, while hydrophobic side chains/cycloheximide N-ethylethanoate atoms binding to the protein are highlighted with white arrowheads. The black arrow indicates a section of cycloheximide N-ethylethanoate that binds to a BpML1 pocket that is not accessed by the peptides. Note how the cycloheximide N-ethylethanoate molecule binds closer to the β4–β5 loop than the peptides.
Figure 5. Comparison of the binding of FKBP inhibitors to FKBPs indicates that cycloheximide N-ethylethanoate represents a novel class of inhibitor.
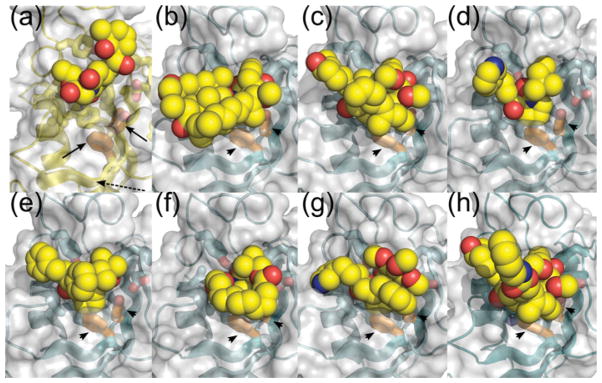
Inhibitors are shown as all heavy atom spherical models, with carbon coloured yellow, oxygen coloured red and nitrogen coloured blue; proteins are shown as white semi-transparent surfaces, with cartoon beneath. Cartoon colours: BpMip, yellow; FKBP12, cyan. The nine most conserved active-site residues of FKBPs are shown as sticks, with carbon coloured as for the main chain, and other atoms as above. Side chains Tyr33 and Phe57 of BpMip do not contact the inhibitor, and are highlighted with carbon coloured orange and with black arrows; equivalent FKBP12 residues are highlighted with carbon coloured orange and with black arrowheads. (a) BpMip binding to cycloheximide N-ethylethanoate (the present study; PDB code 2KO7). (b) FKBP12 binding to rapamycin (PDB code 2DG3). (c) FKBP12 binding to FK-506 (PDB code 1FKJ). (d) FKBP12 binding to the neurotrophic ligand GPI-1046 (PDB code 1F40). (e–g) FKBP12 binding to synthetic high-affinity ligands (PDB codes: 1FKG, 1FKI, 1J4R). (h) FKBP12 binding to a synthetic dimeric ligand (PDB code 1A7X). Note that cycloheximide N-ethylethanoate occupies a distinct area of the protein, and in particular does not interact with the β3b strand (black broken arrow in a).
Remarkably, the conformation of cycloheximide N-ethylethanoate mimics that of the peptide found in the BpML1 crystal structure, and the structure of hFKBP12 bound to TGFβRI (Figure 4E). Thus cycloheximide N-ethylethanoate represents a new class of FKBP inhibitors, mimicking the binding of FKBPs to proteins to modulate activity, rather than proline-peptides as rapamycin and FK506 do.
Conclusions
In the present paper, we report the structure of BpML1, a Mip homologue of B. pseudomallei, which was determined by X-ray crystallography and NMR in parallel. We show that this enzyme is competent in binding to peptides or the class inhibitor cycloheximide N-ethylethanoate. Intriguingly, we show that the peptide bound in the structure is not a cis-proline, but a small helical peptide that mimics TGFβRs bound to hFKBP12; and that cycloheximide N-ethylethanoate binds in the active site in a way that mimics this peptide, rather than the catalytic peptides that are mimicked by other class inhibitors. These results suggest that Mips (and potentially other bacterial FKBPs), like their eukaryotic cousins, are competent to bind peptides in a helical conformation. This suggests that BpML1 and other Mips have roles in protein–protein interactions, perhaps as modulators of signalling, in addition to their chaperone functions. Consistent with this observation, deletion of BpML1 abolished B. pseudomallei showing a pleiotrophic phenotype, including loss of motility which might suggest a role in the control of the flagellar motors (I. Norville, N. Harmer, S. Harding, G. Fischer, K. Keith, K. Brown, M. Sarkar-Tyson and R. Titball, unpublished work).
The ultra-high-resolution crystal structure of BpML1 revealed that two highly conserved active-site side chains, Asp44 and Tyr89, show unusual flexibility. These observations were confirmed by NMR, and suggest that these side chains require flexibility, even in a ligand-bound form, for performing their roles in catalysis. However, it is not clear as to whether this represents a general mechanism for FKBPs or a specific case of BpML1: this enzyme is up to 6-fold more active than previously described FKBPs, and so the flexibility that we observed may be an adaptation of this enzyme to provide it with a greater rate enhancement.
The structure of the cycloheximide N-ethylethanoate complex with BpML1 suggests that this compound might provide an excellent starting point for novel drug development. The binding site for cycloheximide N-ethylethanoate, uniquely for FKBP inhibitors of known structure, is focused on the β3b–α1 and β4–β5 loops, and contacts residues outside the active site in the β4–β5 loop. This new interaction offers a greater possibility of obtaining a selective compound that would not inhibit human proteins: the β4–β5 loop, in particular, shows much greater diversity between species than the active site. Furthermore, we show in the present study that both of these loops show considerable flexibility, increasing the likelihood of a species-specific-induced fit.
We conclude that the bacterial FKBPs are likely to be a more diverse and functionally rich protein family than has previously been appreciated. In consequence of this, their validated role in the pathogenesis of several micro-organisms, and the availability of multiple inhibitors for this protein class, they show great promise for antimicrobial drug discovery.
Supplementary Material
Table 2.
NMR and refinement statistics
| Parameter | Apo-BpML1 (PDB code 2KE0) | BpML1–CNE complex (PDB code 2KO7) |
|---|---|---|
| Structure constraints | ||
| Distance constraints* | ||
| Total meaningful NOE | 1792 | 1757 |
| Restraints | ||
| Short range (|i–j|≤1) | 777 | 808 |
| Medium range (1<|i–j|<5) | 216 | 205 |
| Long range (|i–j|≥5) | 799 | 744 |
| Hydrogen-bond restraints | 78 | 92 |
| Dihedral angle restraints (ϕ, ψ)† | 134 | 136 |
| Total number of constraints | 2004 | 1985 |
| Constraints per residue | 17.1 | 16.8 |
| Intermolecular NOE restraints | 39 | |
| Residual constraint violations‡ | ||
| CYANA target function | 0.84 | 1.54 |
| Distance violations (>0.2 Å) | 3 | 4 |
| Torsion angles (>5.0°) | 0 | 0 |
| Ramachandran statistics (%)§ | ||
| Favoured | 80.8 | 80.7 |
| Additionally allowed | 19.0 | 19.0 |
| Generously allowed | 0.1 | 0.3 |
| Disallowed | 0.0 | 0.0 |
| Average root mean square deviation to the mean (Å)|| | ||
| Backbone | 0.39 ± 0.05 | 0.52 ± 0.15 |
| Heavy atom | 0.89 ± 0.08 | 0.98 ± 0.15 |
| Ligand heavy atom | 1.11 ± 0.14 | |
Final 20/100 structures selected based on the lowest CYANA target function.
Dihedral angle restraints derived from TALOS [47].
Maximum violation shown in parentheses.
Ramachandran analysis was performed with PROCHECKNMR.
The average root mean square deviation to the mean of the protein was calculated over regions 13–117.
Acknowledgments
N.H. acknowledges the Diamond Synchrotron (Oxford, U.K.) for X-ray data collection. G.V. acknowledges the protein expression team at the SSGCID (Seattle Structural Genomics Center for Infectious Disease), and access to facilities at Pacific Northwest National Laboratory (Richland, WA, U.S.A.) for NMR data collection.
FUNDING
This work was supported by the University of Exeter (to K.O’S. and N.H.); the UK Ministry of Defence (to I.N. and M.S.-T.); and the National Institutes of Health Institute of Allergy and Infectious Disease [Federal Contract HHSN272200700057C (to S.Z. and G.V.)]
Abbreviations used
- BpML1
Burkholderia pseudomallei Mip-like protein 1
- FKBP
FK506-binding protein
- HSQC
heteronuclear single-quantum coherence
- LpMip
Legionella pneumophila Mip
- Mip
macrophage infectivity potentiator
- NOE
nuclear Overhauser effect
- PPIase
peptidylprolyl isomerase
- TGFβRI
transforming growth factor β receptor I
Footnotes
AUTHOR CONTRIBUTION
Isobel Norville, Katherine O’Shea and Suxin Zheng designed and performed experiments; Mitali Sarkar-Tyson and Richard Titball, together with Gabriele Varani and Nicholas Harmer conceived and designed the study, and edited the paper before submission. Gabriele Varani and Nicholas Harmer supervised the project and wrote the paper.
The structural co-ordinates reported for Burkholderia pseudomallei peptidylprolyl cis–trans isomerase, B. pseudomallai peptidylprolyl cis–trans isomerase in complex with cycloheximide N-ethylethanoate and BPSS1823, a B. pseudomallei Mip-like chaperone, have been deposited in the PDB under codes 2KE0, 2KO7 and 2Y78 respectively.
References
- 1.Wiersinga WJ, van der Poll T, White NJ, Day NP, Peacock SJ. Melioidosis: insights into the pathogenicity of Burkholderia pseudomallei. Nat Rev Microbiol. 2006;4:272–282. doi: 10.1038/nrmicro1385. [DOI] [PubMed] [Google Scholar]
- 2.White N. Melioidosis. Lancet. 2003;361:1715–1722. doi: 10.1016/s0140-6736(03)13374-0. [DOI] [PubMed] [Google Scholar]
- 3.Cheng AC, Currie BJ. Melioidosis: epidemiology, pathophysiology, and management. Clin Microbiol Rev. 2005;18:383–416. doi: 10.1128/CMR.18.2.383-416.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Rotz LD, Khan AS, Lillibridge SR, Ostroff SM, Hughes JM. Public health assessment of potential biological terror agents. Emerg Infect Dis. 2002;8:225–230. doi: 10.3201/eid0802.010164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Kang CB, Hong Y, Dhe-Paganon S, Yoon HS. FKBP family proteins: immunophilins with versatile biological functions. Neurosignals. 2008;16:318–325. doi: 10.1159/000123041. [DOI] [PubMed] [Google Scholar]
- 6.Cianciotto NP, Eisenstein BI, Mody CH, Toews GB, Engleberg NC. A Legionella pneumophila gene encoding a species-specific surface protein potentiates initiation of intracellular infection. Infect Immun. 1989;57:1255–1262. doi: 10.1128/iai.57.4.1255-1262.1989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Lundemose AG, Kay JE, Pearce JH. Chlamydia trachomatis Mip-like protein has peptidyl-prolyl cis/trans isomerase activity that is inhibited by FK506 and rapamycin and is implicated in initiation of chlamydial infection. Mol Microbiol. 1993;7:777–783. doi: 10.1111/j.1365-2958.1993.tb01168.x. [DOI] [PubMed] [Google Scholar]
- 8.Leuzzi R, Serino L, Scarselli M, Savino S, Fontana MR, Monaci E, Taddei A, Fischer G, Rappuoli R, Pizza M. Ng-MIP, a surface-exposed lipoprotein of Neisseria gonorrhoeae, has a peptidyl-prolyl cis/trans isomerase (PPIase) activity and is involved in persistence in macrophages. Mol Microbiol. 2005;58:669–681. doi: 10.1111/j.1365-2958.2005.04859.x. [DOI] [PubMed] [Google Scholar]
- 9.Moro A, Ruiz-Cabello F, Fernandez-Cano A, Stock RP, Gonzalez A. Secretion by Trypanosoma cruzi of a peptidyl-prolyl cis–trans isomerase involved in cell infection. EMBO J. 1995;14:2483–2490. doi: 10.1002/j.1460-2075.1995.tb07245.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Helbig JH, Konig B, Knospe H, Bubert B, Yu C, Luck CP, Riboldi-Tunnicliffe A, Hilgenfeld R, Jacobs E, Hacker J, Fischer G. The PPIase active site of Legionella pneumophila Mip protein is involved in the infection of eukaryotic host cells. Biol Chem. 2003;384:125–137. doi: 10.1515/BC.2003.013. [DOI] [PubMed] [Google Scholar]
- 11.Wagner C, Khan AS, Kamphausen T, Schmausser B, Unal C, Lorenz U, Fischer G, Hacker J, Steinert M. Collagen binding protein Mip enables Legionella pneumophila to transmigrate through a barrier of NCI-H292 lung epithelial cells and extracellular matrix. Cell Microbiol. 2007;9:450–462. doi: 10.1111/j.1462-5822.2006.00802.x. [DOI] [PubMed] [Google Scholar]
- 12.Zang N, Tang DJ, Wei ML, He YQ, Chen B, Feng JX, Xu J, Gan YQ, Jiang BL, Tang JL. Requirement of a mip-like gene for virulence in the phytopathogenic bacterium Xanthomonas campestris pv. campestris . Mol Plant Microbe Interact. 2007;20:21–30. doi: 10.1094/MPMI-20-0021. [DOI] [PubMed] [Google Scholar]
- 13.Studier FW. Protein production by auto-induction in high-density shaking cultures. Protein Expression Purif. 2005;41:207–234. doi: 10.1016/j.pep.2005.01.016. [DOI] [PubMed] [Google Scholar]
- 14.Leslie AGW. Recent changes to the MOSFLM package for processing film and image plate data. Joint CCP4 and ESF-EAMCB Newsletter on Protein Crystallography. 1992;(26) [Google Scholar]
- 15.Evans PR. Scaling and assessment of data quality. Acta Crystallogr Sect D Biol Crystallogr. 2005;62:72–82. doi: 10.1107/S0907444905036693. [DOI] [PubMed] [Google Scholar]
- 16.Stein N. CHAINSAW: a program for mutating pdb files used as templates in molecular replacement. J Appl Crystallogr. 2008;41:641–643. [Google Scholar]
- 17.McCoy AJ, Grosse-Kunstleve RW, Adams PD, Winn MD, Storoni LC, Read RJ. Phaser crystallographic software. J Appl Crystallogr. 2007;40:658–674. doi: 10.1107/S0021889807021206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Emsley P, Lohkamp B, Scott WG, Cowtan K. Features and development of Coot. Acta Crystallogr Sect D Biol Crystallogr. 2010;66:486–501. doi: 10.1107/S0907444910007493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Adams PD, Grosse-Kunstleve RW, Hung L-W, Ioerger TR, McCoy AJ, Moriarty NW, Read RJ, Sacchettini JC, Sauter NK, Terwilliger TC. PHENIX: building new software for automated crystallographic structure determination. Acta Crystallogr Sect D Biol Crystallogr. 2002;58:1948–1954. doi: 10.1107/s0907444902016657. [DOI] [PubMed] [Google Scholar]
- 20.Davis IW, Leaver-Fay A, Chen VB, Block JN, Kapral GJ, Wang X, Murray LW, Arendall B, III, Snoeyink J, Richardson JS, Richardson DC. MOLPROBITY: all-atom contacts and structure validation for proteins and nucleic acids. Nucleic Acids Res. 2007;35:W375–W383. doi: 10.1093/nar/gkm216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Kay LE, Xu GY, Singer AU, Muhandiram DR, Formankay JD. A gradient-enhanced HCCH TOCSY experiment for recording side-chain H-1 and C-13 correlations in H2 O samples of proteins. J Magn Reson B. 1993;101:333–337. [Google Scholar]
- 22.Muhandiram DR, Xu GY, Kay LE. An enhanced-sensitivity pure absorption gradient 4d N-15, C-13-edited NOESY experiment. J Biomol NMR. 1993;3:463–470. [Google Scholar]
- 23.Yamazaki T, Lee W, Revington M, Mattiello DL, Dahlquist FW, Arrowsmith CH, Kay LE. An HNCA pulse scheme for the backbone assignment of N-15,C-13,H-2-labeled proteins-application to a 37-kDa Trp repressor DNA complex. J Am Chem Soc. 1994;116:6464–6465. [Google Scholar]
- 24.Muhandiram DR, Kay LE. Gradient-enhanced triple-resonance 3-dimensional NMR experiments with improved sensitivity. J Magn Reson B. 1994;103:203–216. [Google Scholar]
- 25.Peterson RD, Theimer CA, Wu HH, Feigon J. New applications of 2D filtered/edited NOESY for assignment and structure elucidation of RNA and RNA-protein complexes. J Biomol NMR. 2004;28:59–67. doi: 10.1023/B:JNMR.0000012861.95939.05. [DOI] [PubMed] [Google Scholar]
- 26.Zwahlen C, Legault P, Vincent SJF, Greenblatt J, Konrat R, Kay LE. Methods for measurement of intermolecular NOEs by multinuclear NMR spectroscopy: application to a bacteriophage lambda N-peptide/boxB RNA complex. J Am Chem Soc. 1997;119:6711–6721. [Google Scholar]
- 27.Delaglio F, Grzesiek S, Vuister GW, Zhu G, Pfeifer J, Bax A. NMRPipe: a multidimensional spectral processing system based on Unix pipes. J Biomol NMR. 1995;6:277–293. doi: 10.1007/BF00197809. [DOI] [PubMed] [Google Scholar]
- 28.Vranken WF, Boucher W, Stevens TJ, Fogh RH, Pajon A, Llinas P, Ulrich EL, Markley JL, Ionides J, Laue ED. The CCPN data model for NMR spectroscopy: development of a software pipeline. Proteins. 2005;59:687–696. doi: 10.1002/prot.20449. [DOI] [PubMed] [Google Scholar]
- 29.Güntert P. Automated NMR protein structure calculation. Prog Nucl Magn Reson Spectrosc. 2003;43:105–125. [Google Scholar]
- 30.Herrmann T, Güntert P, Wuthrich K. Protein NMR structure determination with automated NOE assignment using the new software CANDID and the torsion angle dynamics algorithm DYANA. J Mol Biol. 2002;319:209–227. doi: 10.1016/s0022-2836(02)00241-3. [DOI] [PubMed] [Google Scholar]
- 31.Fischer G, Bang H, Mech C. Detection of enzyme catalysis for cis–trans-isomerization of peptide-bonds using proline-containing peptides as substrates. Biomed Biochim Acta. 1984;43:1101–1111. [PubMed] [Google Scholar]
- 32.Kullertz G, Luthe S, Fischer G. Semiautomated microtiter plate assay for monitoring peptidyl-prolyl cis/trans isomerase activity in normal and pathological human sera. Clin Chem. 1998;44:502–508. [PubMed] [Google Scholar]
- 33.Williams JW, Morrison JF. The kinetics of reversible tight-binding inhibition. Methods Enzymol. 1979;63:437–467. doi: 10.1016/0076-6879(79)63019-7. [DOI] [PubMed] [Google Scholar]
- 34.Ceymann A, Horstmann M, Ehses P, Schweimer K, Paschke AK, Steinert M, Faber C. Solution structure of the Legionella pneumophila Mip-rapamycin complex. BMC Struct Biol. 2008;8:17. doi: 10.1186/1472-6807-8-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Löw C, Neumann P, Tidow H, Weininger U, Haupt C, Friedrich-Epler B, Scholz C, Stubbs MT, Balbach J. Crystal structure determination and functional characterization of the metallochaperone SlyD from Thermus thermophilus. J Mol Biol. 2010;398:375–390. doi: 10.1016/j.jmb.2010.03.014. [DOI] [PubMed] [Google Scholar]
- 36.Pereira PJ, Vega MC, Gonzalez-Rey E, Fernandez-Carazo R, Macedo-Ribeiro S, Gomis-Ruth FX, Gonzalez A, Coll M. Trypanosoma cruzi macrophage infectivity potentiator has a rotamase core and a highly exposed α-helix. EMBO Rep. 2002;3:88–94. doi: 10.1093/embo-reports/kvf009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Riboldi-Tunnicliffe A, Konig B, Jessen S, Weiss MS, Rahfeld J, Hacker J, Fischer G, Hilgenfeld R. Crystal structure of Mip, a prolylisomerase from Legionella pneumophila. Nat Struct Biol. 2001;8:779–783. doi: 10.1038/nsb0901-779. [DOI] [PubMed] [Google Scholar]
- 38.Wilson KP, Yamashita MM, Sintchak MD, Rotstein SH, Murcko MA, Boger J, Thomson JA, Fitzgibbon MJ, Black JR, Navia MA. Comparative X-ray structures of the major binding protein for the immunosuppressant FK506 (tacrolimus) in unliganded form and in complex with FK506 and rapamycin. Acta Crystallogr Sect D Biol Crystallogr. 1995;51:511–521. doi: 10.1107/S0907444994014514. [DOI] [PubMed] [Google Scholar]
- 39.Huse M, Chen YG, Massague J, Kuriyan J. Crystal structure of the cytoplasmic domain of the type I TGFβ receptor in complex with FKBP12. Cell. 1999;96:425–436. doi: 10.1016/s0092-8674(00)80555-3. [DOI] [PubMed] [Google Scholar]
- 40.Chelu MG, Danila CI, Gilman CP, Hamilton SL. Regulation of ryanodine receptors by FK506 binding proteins. Trends Cardiovasc Med. 2004;14:227–234. doi: 10.1016/j.tcm.2004.06.003. [DOI] [PubMed] [Google Scholar]
- 41.Huse M, Muir TW, Xu L, Chen YG, Kuriyan J, Massague J. The TGFβ receptor activation process: an inhibitor- to substrate-binding switch. Mol Cell. 2001;8:671–682. doi: 10.1016/s1097-2765(01)00332-x. [DOI] [PubMed] [Google Scholar]
- 42.Szep S, Park S, Boder ET, Van Duyne GD, Saven JG. Structural coupling between FKBP12 and buried water. Proteins. 2009;74:603–611. doi: 10.1002/prot.22176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Wintermeyer E, Ludwig B, Steinert M, Schmidt B, Fischer G, Hacker J. Influence of site specifically altered Mip proteins on intracellular survival of Legionella pneumophila in eukaryotic cells. Infect Immun. 1995;63:4576–4583. doi: 10.1128/iai.63.12.4576-4583.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Christner C, Wyrwa R, Marsch S, Kullertz G, Thiericke R, Grabley S, Schumann D, Fischer G. Synthesis and cytotoxic evaluation of cycloheximide derivatives as potential inhibitors of FKBP12 with neuroregenerative properties. J Med Chem. 1999;42:3615–3622. doi: 10.1021/jm991038t. [DOI] [PubMed] [Google Scholar]
- 45.Sich C, Improta S, Cowley DJ, Guenet C, Merly JP, Teufel M, Saudek V. Solution structure of a neurotrophic ligand bound to FKBP12 and its effects on protein dynamics. Eur J Biochem. 2000;267:5342–5355. doi: 10.1046/j.1432-1327.2000.01551.x. [DOI] [PubMed] [Google Scholar]
- 46.Sun F, Li P, Ding Y, Wang L, Bartlam M, Shu C, Shen B, Jiang H, Li S, Rao Z. Design and structure-based study of new potential FKBP12 inhibitors. Biophys J. 2003;85:3194–3201. doi: 10.1016/S0006-3495(03)74737-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Cornilescu G, Delaglio F, Bax A. Protein backbone angle restraints from searching a database for chemical shift and sequence homology. J Biomol NMR. 1999;13:289–302. doi: 10.1023/a:1008392405740. [DOI] [PubMed] [Google Scholar]
- 48.Fulton KF, Jackson SE, Buckle AM. Energetic and structural analysis of the role of tryptophan 59 in FKBP12. Biochemistry. 2003;42:2364–2372. doi: 10.1021/bi020564a. [DOI] [PubMed] [Google Scholar]
- 49.Baker N, Sept D, Joseph S, Holst M, McCammon J. Electrostatics of nanosystems: application to microtubules and the ribosome. Proc Natl Acad Sci USA. 2001;98:10037–10041. doi: 10.1073/pnas.181342398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Dolinsky TJ, Czodrowski P, Li H, Nielsen JE, Jensen JH, Klebe G, Baker NA. PDB2PQR: expanding and upgrading automated preparation of biomolecular structures for molecular simulations. Nucleic Acids Res. 2007;35:W522–W525. doi: 10.1093/nar/gkm276. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.