

Abstract
Hesperadin, an established human Aurora B inhibitor, was tested against cultures of Trypanosoma brucei, Leishmania major, and Plasmodium falciparum, and was identified to be a potent proliferation inhibitor. A series of analogs was designed and tested to establish the initial structure-activity relationships for each parasite. In this study, we identified multiple non-toxic compounds with high potency against T. brucei and P. falciparum with good selectivity. These compounds may represent an opportunity for continued optimization.
Introduction
Neglected tropical diseases (NTDs) are a collection of debilitating infectious diseases that typically affect the poorest populations in the world, such as human African trypanosomiasis (caused by Trypanosoma brucei), leishmaniasis (caused by Leishmania sp.), and Chagas disease (T. cruzi). When taken together with malaria (caused by Plasmodium sp.), these diseases have a significant and adverse effect on length of quality of life, estimated in 2011 to be over 60 million disability-adjusted life-years.1 Drug discovery for these diseases is cost limited, given that the vast majority of patients cannot pay for treatment. Therefore, pragmatic and cost effective methods for drug discovery are needed.
One such method is target repurposing,2 where essential parasitic targets (such as kinases,3–7 phosphodiesterases,8, 9 and histone deacetylases10) are matched with homologous and druggable targets in humans that have been the subject of previous drug discovery programs. Assessment of chemical matter identified in those programs against the infectious pathogen can often result in identification of new leads for the infectious disease. Furthermore, the medicinal chemistry optimization against the pathogen can be facilitated and accelerated by considering the previous SAR developed for the chemotype.
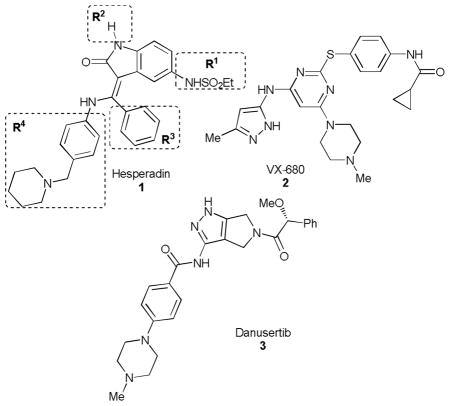
Aurora kinases are central enzymes in cellular division, and have been the focus of drug discovery efforts in oncology, resulting in multiple inhibitors advanced into clinical trials. Both L. major11 (which causes cutaneous leishmaniasis) and P. falciparum12 express Aurora kinases, and established inhibitors of Aurora have been shown to be effective cell proliferation inhibitors for Trypanosoma brucei (including hesperadin (1),13 VX-680 (2)14 and danusertib (3).4
Encouraged by the preliminary results for T. brucei described above, we assessed these three human Aurora kinase inhibitors against other trypanosomatid pathogens L. major (promastigote and intracellular amastigote forms), and the D6 strain of P. falciparum (Table 1). We also tested 1 against the intracellular amastigotes form of T. cruzi, the causative agent of Chagas disease. We counter-screened against the hepatic cancer cell line (HepG2) as a general surrogate for host cell toxicity. We observed a range of potencies, and note that 1 displayed a potent growth inhibitory phenotype against P. falciparum and L. major parasites, though host cell toxicity was apparent. In light of these results, we opted to focus on further exploration of the SAR of this chemotype as a potential antiparasitic agent.
Table 1.
Benchmark screening of human Aurora inhibitors.a
| ||||||
|---|---|---|---|---|---|---|
| EC50 (μM) | TC50 (μM) | |||||
| Compound | T brucei | T. cruzi | L. major Promastig | L. major Amastig | P. falciparum | HepG2 |
| 1 | 0.0613 | 39 | 0.12 | 2.37 | 0.01 | < 0.2 |
| 2 | 1014 | nt | >21 | nt | 0.838 | 3.3 |
| 3 | 0.64 | nt | >21 | nt | 0.857 | 4 |
nt=not tested; all EC50 and TC50 values data are averages of three replicate experiments.
Synthetic strategy
We designed a synthetic strategy to access three regions of the hesperadin molecule, labelled R1-R4 as shown in Table 1, modelled after the synthetic route described in a patent.15 Synthesis initiated with indolone 4, which was nitrated15 and condensed with methyl orthobenzoate to provide 6. Displacement of the vinylogous ester, nitro group reduction, and sulfonylation with a small set of sulfonyl chlorides afforded the R1 analogs 11 following N-deprotection (Table 2). The nitroindolone 5 could be converted to the sulphonamide 12, which was subjected to a similar sequence as above with varied amine nucleophiles (to vary R4) to obtain access the analogs 15 following deprotection. Finally, inclusion of various orthoester reagents in the sequence afforded R3 variations (compound 17).
Table 2.
Potency of analogs of 1 against three protozoan parasites.
| EC50 (μM) | TC50 (μM) | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Cmpd | R1 | R2 | R3 | R4 | T brucei | T. cruzi | L. major Promastig | L. major Amastig | P. falciparum | HepG2 |
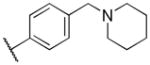
| 1 | NHSO2Et | H | Ph |
|
0.06 | 39 | 0.12 | 2.37 | 0.01 | <0.2 |
| 7a | H | H | Ph | 0.07 | 25 | 0.77 | >4 | 0.230 | > 6.0 | |
| 7b | NO2 | Ac | Ph | 0.13 | 3 | 1.08 | 2.04 | 0.275 | > 6.0 | |
| 8 | NH2 | Ac | Ph | 3.25 | 45 | >4 | >4 | 0.218 | 1.81 | |
| 10 | NH2 | H | Ph | 1.16 | >50 | 0.79 | >4 | 0.199 | > 6.0 | |
| 11a | NHSO2Me | H | Ph | 0.02 | >50 | 0.39 | >4 | 0.052 | 0.2 | |
| 11b | NHSO2Ph | H | Ph | 0.01 | 16 | 0.85 | 1.91 | 0.107 | 3.29 | |
| 11c | NHAc | H | Ph | 0.15 | >50 | 1.52 | >4 | 0.068 | > 6.0 | |
| 17a | NHSO2Et | H | H | 1.32 | >50 | 2.77 | >4 | 0.241 | 5 | |
| 17b | NHSO2Et | H | Me | 3.36 | >50 | >4 | >4 | 1.364 | > 6.0 | |
| 17c | NHSO2Et | H | Et | 1.79 | >50 | 2.8 | >4 | 0.233 | > 6.0 | |
| 17d | NHSO2Et | H | n-Bu | 1.67 | >50 | 2.6 | >4 | 0.167 | > 6.0 | |
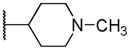
| 15a | NHSO2Et | H | Ph |
|
>50 | >50 | >4 | >4 | >15 | >10.0 |
| 15b | NHSO2Et | H | Ph | Ph | 1.44 | >50 | >4 | >4 | 0.109 | 4.7 |
| 15c | NHSO2Et | H | Ph | p-Tol | 0.37 | >50 | 2.6 | >4 | 1.607 | 3.1 |
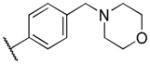
| 15d | NHSO2Et | H | Ph |
|
0.1 | >50 | 0.94 | 1.68 | 0.054 | 1.31 |
| 15e | NHSO2Et | H | Ph |
|
1.53 | >50 | 0.62 | >4 | 0.595 | > 6.0 |
Screening results and discussion
The analogs were tested in parasite cultures, and the results are summarized in Table 2. First, variation of the R1 position (Table 2) revealed a preference for the ethyl sulphonamide moiety present in 1 over the methyl (11a) or phenyl sulphonamide (11b) or replacement with an acetamide (11c). However, 11c afforded reduced potency against HepG2 cells, providing improvement in cellular selectivity over the other analogs. Complete removal of the R1 functionality (7a) led to a significant reduction in antimalarial and anti-leishmanial activity, though 7a was equipotent to 1 against T. brucei and selective over HepG2 cells. The free amine (10) showed marked reduction in activity across pathogens.
In variation of the R4 substituent, substitution of oxygen for carbon of the piperidine ring of 1 provides a slight reduction in antiprotozoan activity (15d), though activity was also reduced in HepG2 cells, maintaining some selectivity. The results in our data set suggest the requirement for a basic nitrogen, noting also that reduction in basicity (e.g. morpholino group) results in loss of potency. Removal of the methylene spacer (15e) caused a strongly adverse effect on potency against all pathogens, as does removal of the piperidinyl moiety altogether (15c). Though the phenyl analog (15b) shows diminished potency against P. falciparum, the selectivity over HepG2 cells is in excess of 50-fold. Finally, removal of the intervening aromatic group (15a) results in complete loss of activity across parasites.
Finally, survey of the R3 phenyl of 1 confirms that replacements with H (17a) or small alkyl chains (17b–d) has a detrimental effect on activities compared to 1. Interestingly, the methyl analog (17b) is far less potent than the others, which are all in a range of ~20 fold less potent than 1, with diminished HepG2 activity. Notably, none of the compounds tested showed appreciable activity against T. cruzi.
Drug resistance is a significant issue for malaria. We therefore opted to test our analogs across three additional strains of P. falciparum besides the drug-sensitive D6 strain: W2 (chloroquine resistant), C235 (chloroquine, mefloquine and pyrimethamine resistant), and C2B (multidrug resistance with atovaquone resistance). The data is tabulated in the Electronic Supplementary Information (ESI), but is presented in Figure 2 as a scatter plot showing D6 versus C235 EC50. Remarkably, the compounds show highly consistent potency values across strains; this is also observed for the W2 and C2B strains (R2=0.95 and 0.95, respectively).
Figure.

Plot of EC50 values of P. falciparum C235 versus D6 strains. R2=0.99
We also note a complete lack of correlation between the compounds’ activities against promastigote and axenic amastigote form of L. major, which is consistent with previous reports.16
Conclusions
In summary, we have identified analogs of 1, an established human Aurora kinase inhibitor, that display modest-to-excellent potency against the protozoan pathogens that cause African sleeping sickness, malaria, and leishmaniasis. Importantly, these compounds are not acting as general cell toxins, as we have observed varying margins of selectivity. Notably, we have also found that these compounds show broad utility as equipotent inhibitors of a range of drug resistant strains of malaria. Though these compounds have not yet been tested against Aurora kinase homologs in the respective parasites, we expect that this will be a useful place to begin to identify molecular mechanisms of action.11–13 This, along with our ongoing optimization of this chemotype against these pathogens will be reported in due course.
Supplementary Material
Scheme.

Synthesis of analogs of hesperadin (1). Reagents and conditions. (a) HNO3, H2SO4 -15 °C; (b) PhC(OCH3)3, Ac2O; (c) 4-(piperidin-1-ylmethyl)aniline, DMF; (d) Zn, NH4Cl, MeOH, H2O; (e) RSO2Cl, Et3N, DMF; (f). NaOH, MeOH; (g) EtSO2Cl, Et3N, DMF; (h) R-NH2, DMF;(i) RC(OCH3)3, Ac2O.
Acknowledgments
We thank Professor Larry Ruben (Southern Methodist University) for helpful discussion and comments on this manuscript. This work was funded in part by NIH R01AI082577 (MPP). We appreciate a free academic license for the OpenEye suite of software.
Footnotes
Electronic Supplementary Information (ESI) available: Synthetic preparations and assay details are described in the ESI. See DOI: 10.1039/b000000x/Compound data is available as a searchable shared data set at http://collaborativedrug.com.
Notes and references
- 1. [Accessed 01/23/2012, 2011.];Metrics: Disability-Adjusted Live Year (DALY) http://www.who.int/healthinfo/global_burden_disease/metrics_daly/en/
- 2.Pollastri MP, Campbell RK. Future Med Chem. 2011;3:1307–1315. doi: 10.4155/fmc.11.92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Diaz-Gonzalez R, Kuhlmann FM, Galan-Rodriguez C, Madeira da Silva L, Saldivia M, Karver CE, Rodriguez A, Beverley SM, Navarro M, Pollastri MP. PLoS Negl Trop Dis. 2011;5:e1297. doi: 10.1371/journal.pntd.0001297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Ochiana SO, Pandarinath V, Wang Z, Kapoor R, Ondrechen MJ, Ruben L, Pollastri MP. Eur J Med Chem. 2013;62:777–784. doi: 10.1016/j.ejmech.2012.07.038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Patel G, Karver CE, Behera R, Guyett PJ, Sullenberger C, Edwards P, Roncal NE, Mensa-Wilmot K, Pollastri MP. J Med Chem. 2013;56:3820–3832. doi: 10.1021/jm400349k. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Woodland A, Grimaldi R, Luksch T, Cleghorn LAT, Ojo KK, Van Voorhis WC, Brenk R, Frearson JA, Gilbert IH, Wyatt PG. ChemMedChem. 2013;8:1127–1137. doi: 10.1002/cmdc.201300072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Ojo KK, Gillespie JR, Riechers AJ, Napuli AJ, Verlinde CLMJ, Buckner FS, Gelb MH, Domostoj MM, Wells SJ, Scheer A, Wells TNC, Van Voorhis WC. Antimicrob Agents Chemother. 2008;52:3710–3717. doi: 10.1128/AAC.00364-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Bland ND, Wang C, Tallman C, Gustafson AE, Wang Z, Ashton TD, Ochiana SO, McAllister G, Cotter K, Fang AP, Gechijian L, Garceau N, Gangurde R, Ortenberg R, Ondrechen MJ, Campbell RK, Pollastri MP. J Med Chem. 2011;54:8188–8194. doi: 10.1021/jm201148s. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.de Koning HP, Gould MK, Sterk GJ, Tenor H, Kunz S, Luginbuehl E, Seebeck T. J Infect Dis. 2012;206:229–237. doi: 10.1093/infdis/jir857. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Kelly JM, Taylor MC, Horn D, Loza E, Kalvinsh I, Björkling F. Bioorg Med Chem Lett. 2012;22:1886–1890. doi: 10.1016/j.bmcl.2012.01.072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Siman-Tov MM, Ivens AC, Jaffe CL. Biochim Biophys Acta Gene Struct. Expression. 2001:1519, 241–245. doi: 10.1016/s0167-4781(01)00240-8. [DOI] [PubMed] [Google Scholar]
- 12.Reininger L, Wilkes JM, Bourgade H, Miranda-Saavedra D, Doerig C. Mol Microbiol. 2011;79:205–221. doi: 10.1111/j.1365-2958.2010.07442.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Jetton N, Rothberg KG, Hubbard JG, Wise J, Li Y, Ball HL, Ruben L. Mol Microbiol. 2009;72:442–458. doi: 10.1111/j.1365-2958.2009.06657.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Li Z, Umeyama T, Wang CC. PLoS Pathog. 2009;5:e1000575. doi: 10.1371/journal.ppat.1000575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Walter R, Heckel A, Roth GJ, Kley J, Schnapp G, Lenter M, Van MJCA, Spevak W, Weyer-Czernilofsky U. 2002036564A1. WO. 2002
- 16.De Muylder G, Ang KKH, Chen S, Arkin MR, Engel JC, McKerrow JH. PLoS Negl Trop Dis. 2011;5:e1253. doi: 10.1371/journal.pntd.0001253. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.