

Abstract
Protein-DNA interactions are essential for many biological processes, X-ray crystallography can provide high-resolution structures, but protein-DNA complexes are difficult to crystallize and typically contain only small DNA fragments. Thus, there is a need for computational methods that can provide useful predictions to give insights into mechanisms and guide the design of new experiments. We used the program DOT, which performs an exhaustive, rigid-body search between two macromolecules, to investigate four diverse protein-DNA interactions. Here, we compare our computational results with subsequent experimental data on related systems. In all cases, the experimental data strongly supported our structural hypotheses from the docking calculations: a mechanism for weak, non-sequence-specific DNA binding by a transcription factor, a large DNA-binding footprint on the surface of the DNA-repair enzyme uracil-DNA-glycosylase, viral and host DNA-binding sites on the catalytic domain of HIV integrase, and a three-DNA-contact model of the linker histone bound to the nucleosome. In the case of uracil-DNA-glycosylase, the experimental design was based on the DNA-binding surface found by docking, rather than the much smaller surface observed in the crystallographic structure. These comparisons demonstrate that the DOT electrostatic energy gives a good representation of the distinctive electrostatic properties of DNA and DNA-binding proteins. The large, favorably-ranked clusters resulting from the dockings identify active sites, map out large DNA-binding sites, and reveal multiple DNA contacts with a protein. Thus, computational docking can not only help to identify protein-DNA interactions in the absence of a crystal structure, but also expand structural understanding beyond known crystallographic structures.
Keywords: protein-DNA structure, HIV integrase, uracil DNA-glycosylase, linker histone, transcription factor, Poisson-Boltzmann electrostatics, hydrogen/deuterium exchange
Introduction
Protein-DNA interactions are involved in many essential biological processes, such as gene regulation, DNA repair, and chromatin structure. The determination of the structures of protein-DNA complexes can be instrumental for understanding function, for designing experiments to probe biological mechanisms, and for developing new drugs. X-ray crystallography provides high-resolution structures of protein-DNA complexes, but these complexes are difficult to crystallize. When high quality crystals of protein-DNA complexes are obtained, they typically contain only small DNA fragments due to constraints imposed by crystal packing.1 NMR studies can be done on DNA-binding proteins, but they are limited to small proteins that are soluble at high concentrations. Other methods for probing specific interactions between proteins and DNA, such as mutagenesis or cross-linking, require chemical modifications that may influence the interaction. These experimental difficulties present an opportunity for computational methods as a predictive tool for developing structural hypotheses that can provide insights into mechanisms and guide the design of new experiments.
Despite significant progress in applying macromolecular docking methods to protein-protein complexes,2–4 the prediction of protein-DNA interactions remains a largely unaddressed challenge.5 DNA as one of the interacting partners poses a particular problem because global conformational changes, such as bending or kinking, are often induced upon protein binding. Methods have been developed to predict these global changes. The program HADDOCK performs rigid-body dockings on an ensemble of protein and DNA structures, followed by semi-flexible refinement of the best rigid-body solutions.6, 7 The Monte Carlo simulation program MONTY permits flexibility of protein side chains and DNA during docking.8, 9 Linear DNA fragments placed out a distance from a protein were driven towards the protein surface in a series of molecular mechanics and dynamics steps.10 Another method uses a library of pre-bent DNA models11 but is limited to proteins with a 2-fold symmetry. All of these methods allow for conformational change in the dsDNA structures, but assume that base pairing is maintained and require experimental knowledge of the DNA/protein interaction, either for initial positioning of the DNA or to derive restraints.
When the DNA-binding site is unknown and little or no experimental data is available, full search methods are needed. To make the full search manageable, the problem can be simplified by treating the individual macromolecules as rigid bodies and searching over the three translational and three rotational degrees of freedom. These searches can be performed efficiently using convolution techniques, in which molecular properties are mapped onto grids. Convolutions calculated using Fast Fourier Transforms rapidly evaluate energies for all relative positions of the two molecules. 12–26 An alternative formulation based on computer vision methods has also been developed.27, 28 There have now been several applications of these techniques to the prediction of protein-DNA complexes.29–38 Our global, systematic search program DOT15–17 uses convolution methods to calculate interaction energies as the sum of electrostatic and van der Waals components. Because of our interest in biological systems in which electrostatic forces play an important role, we implemented a detailed electrostatic energy model. DOT calculates the electrostatic energy as the set of partial atomic charges of one molecule moving in the electrostatic potential field of a stationary molecule. The electrostatic potential is calculated by Poisson-Boltzmann methods, which takes into account dielectric, solvation, and ionic strength effects. We found that the DOT energy term is a good approximation of the Poisson-Boltzmann calculation on the full protein-DNA complex.33
Here, we compare predictions from four computational docking studies on diverse protein-DNA systems with subsequent experimental evidence. In all studies we used the exhaustive, rigid-body search method implemented in DOT. The predicted interactions were based on configurations with the most favorable DOT energies; no experimental knowledge of the systems was used in the calculations. For three of these systems — the transcription factor FadR, the linker histone/nucleosome complex, and the core catalytic domain (CCD) of integrase - key experimental findings on related systems were published months to years later. In the fourth system — the DNA-repair enzyme uracil-DNA-glycosylase (UNG) - we used the computational results to design an experimental approach that provided new structural information on this well-studied system.
Materials and Methods
Coordinate preparation
An essential step in computational docking is constructing biologically relevant starting models. In all studies, we evaluated coordinates from the Protein Data Bank (PDB) (http://www.rcsb.org/pdb), including aspects such as crystal packing effects. Key changes to the original PDB coordinates included building full side chains in protein residues with missing atoms and applying symmetry operations to construct the biological oligomerization state. In addition, non-protein components, such as metal ions, were included in the coordinates if they were essential structural elements. The catalytic core domain (CCD) of integrase presented a special problem because all existing crystallographic structures lacked the loop region adjacent to the active site and one or both active-site metal ions. Therefore, we used coordinates from a molecular dynamics model39, 40 into which both of these features had been built. The nucleosome also presented a special problem because of its large size and complexity. We selected coordinates from the structure of the full nucleosome41 to obtain a model containing about half of the structure. This model was small enough to be computationally feasible, yet still retained all the proposed linker-histone-binding sites.35 Analysis of the full crystal environment of the nucleosome identified histone tails that were involved in crystal contacts. These regions are likely to be disordered in solution, and therefore were removed from the model. The nucleosomal DNA was extended to create linker DNA entering and exiting the nucleosome, which is known to interact with the linker histone. The crystallographic structure of the globular domain of linker histone H5, GH5,42 contains two molecules in the asymmetric unit with significantly different structures. Analysis35 suggested that molecule A was perturbed by crystal packing interactions, so molecule B was selected as the better model of the biological structure.
Linear B-form DNA (B-DNA) models were built with the Nucleic Acid Builder (NAB) program.43 In most studies, these models were used without further modification. In the UNG-DNA study,44 we used DNA with a G:U mismatch pair as well as two undamaged DNA fragments with G:U replaced by A:T and G:C. To obtain the correct wobble-pair geometry for the G:U mismatch, the B-DNA was minimized with AMBER 8 using the generalized Born model.45 The undamaged DNA fragments were also minimized. Minimization caused small changes of the phosphate backbone geometry in all three DNA fragments. The minimized and starting NAB B-DNA fragments all gave very similar results when docked to UNG. B-DNA built with NAB and B-DNA built with the program 3DNA (rutchem.rutgers.edu/xiangjun/3DNA/) vary in the sugar conformation, but gave similar results when docked to a transcription factor.33 Thus, small structural differences in B-DNA structure have little effect on the docking outcome.
Docking calculations with the DOT program
In the DOT calculation, one molecule (the moving molecule) is systematically translated and rotated about a second molecule (the stationary molecule) in a exhaustive search 16, 17. Interaction energies for all configurations are evaluated as convolution functions, which are efficiently computed with Fast Fourier Transforms. Electrostatic and shape components, described below, were mapped onto cubic grids 128 Å on a side with 1 Å grid spacing. The size of the grid was sufficiently large to ensure that the moving molecule fit within the grid when it was close to the stationary molecule. In addition, the stationary potentials were close to zero at the grid boundaries so that artifacts from the periodic Fourier calculation were negligible. In the efficient translational search, the moving molecule is centered at all grid points and the electrostatic and van der Waals energy terms are calculated. The moving molecule is then rotated and the translational search repeated. Rotational sets of 28,800 (7.5°) and 54,000 (6°) were used in these studies. Based on input coordinates for two molecules, the DOT2 program suite17 (http://www.sdsc.edu/CCMS/DOT) provides an automated script that selects the optimal grid size, constructs the potentials described below, and creates the DOT input files.
Van der Waals energy term for the DOT calculation
The van der Waals energy for each configuration is proportional to the number of moving molecule atoms that lie within a favorable interaction layer surrounding the stationary molecule.15 During the time that these reported protein-DNA studies were done, the descriptions of the molecular shape properties have been refined. The stationary molecule potential is described as an excluded volume surrounded by a 3.0 Å favorable layer. Initially the excluded volume was defined as the volume inside van der Waals spheres around each heavy atom of the stationary molecule, but this created tunnels of favorable values that extended deep into the molecule. Because the moving molecule shape is represented only by its atomic positions, long side chains could deeply penetrate these regions in the stationary molecule, leading to incorrect, favorably ranked configurations. The excluded volume is now bounded by the molecular surface calculated with the program MSMS, which is the contact surface of a 1.4 Å probe sphere rolled over the van der Waals surface. As a further refinement, we now only count the heavy atoms of the moving molecule in the energy term. Previously, polar hydrogen atoms were included in the count, but this overemphasized polar interfaces over hydrophobic ones. These two refinements significantly improved protein-protein docking, but only slightly improved protein-DNA docking, probably because DNA has no long side chains and does not bind to hydrophobic protein surfaces.
Electrostatic energy term for the DOT calculation
The electrostatic energy term is calculated as the set of atomic point charges of the moving molecule placed in the electrostatic potential of the stationary molecule. For these protein-DNA docking studies, the electrostatic potential was calculated with UHBD,46 which solves the linearized Poisson-Boltzmann equation by finite difference methods, providing a continuum solvent treatment that takes dielectric and salt effects into account. We now use APBS47 for this calculation in DOT2.17 Both programs give similar results. To make the continuous electrostatic potential compatible with the lenient shape potential, the electrostatic potential values close to the stationary molecule surface are modified to be no greater than the largest values a moving molecule can realistically see.16, 17, 48 Electrostatic potential clamping is essential for protein-DNA docking because DNA-binding surfaces often include clusters of positively charged residues that can create large values (up to about 15 kcal/mol/e) very close to the molecular surface.
Analysis of docking results
Analysis of protein-DNA dockings presents problems not found in protein-protein docking. Where there is no sequence specific recognition, B-DNA placements often are shifted by one or more base pairs along the DNA axis. Clustering based on RMSD values of corresponding atoms would not recognize these closely related placements.29, 33 To evaluate B-DNA docking, we used computer graphics to analyze the 30 top-ranked B-DNA placements, examining the variation of the orientation of the DNA axis and the contacts of the DNA major and minor grooves. We also examined the distribution of the centers of the 2000 top-ranked DNA placements over the protein surface.
Results
FadR: Mechanism for non-sequence-specific DNA binding
We examined the prokaryotic transcription factor FadR, which controls the expression of bacterial fatty acid metabolic genes,49 to explore how conformational change of the protein influences its interactions with DNA. FadR is a homodimer of a two-domain protein: the two N-terminal domains bind DNA and the two C-terminal domains bind long-chain acyl-CoA, an effector molecule that causes loss of specific DNA binding.50 Crystallographic structures for the FadR homodimer have been determined in three states: free,51, 52 DNA-bound,52, 53 and bound to the effector molecule myristoyl-CoA.53 In the free FadR structure, the two recognition helices, one from each N-terminal domain, are adjacent to each other. A model of the DNA-bound complex was attempted based on structural similarity of the free FadR monomer to other winged-helix transcription factors, but this gave a model inconsistent with the relative orientation of the two N-terminal domains. It was concluded that the free protein was in a different conformation than the DNA-bound structure. Subsequent structure determination of the DNA-bound protein, however, revealed that the two N-terminal domains are oriented as in the free structure and that the predicted DNA-binding mode was incorrect.52, 53 The palindromic cognate DNA is bent about 20° over the protein surface, aligning major groove contacts with side chains in each monomer that confer specific binding. The structure of myristoyl-CoA-bound FadR revealed a large allosteric shift of the N-terminal domains, increasing the separation of the recognition helices by over 7 Å.53
Our study of FadR addressed two questions. First, could computational docking identify the correct binding mode of cognate DNA based on the structure of the free protein, where homology modeling failed? The DNA-bound and free FadR structures have the same relative geometry of the two N-terminal domains, but they differ in the conformations of key Arg side chains involved in DNA sequence recognition. This could present problems for rigid-body docking. Second, could docking DNA to the effector-bound structure identify the changes in the interaction that lead to weaker DNA binding and loss of specificity? Nonspecific binding may play an important physiological role in efficient sequence-specific recognition and DNA translocation, but, at the time of our study, there was no structural data on nonspecific DNA binding to transcription factors.
We found that rigid-body docking of linear B-DNA to the free FadR structure successfully identified the DNA-binding surface on the N-terminal dimer, revealing the key side chains likely to be important for specific recognition.33 The centers of the top 500 docked B-DNA fragments followed the alignment of the DNA axis over the protein surface and indicated the need for DNA to be bent over the surface (Fig. 1A,B). The axes of all 30 top-ranked solutions were aligned with that of the crystallographic DNA, but they showed a mixture of major and minor groove binding over the FadR recognition helices, with the correct binding of the DNA major groove predominating (19/30). These 19 solutions were translated along the DNA-binding site, with a cluster of four showing the sequence-specific interactions of the Arg 35 and 45 side chains with the GG sequence in the DNA.
Figure 1.
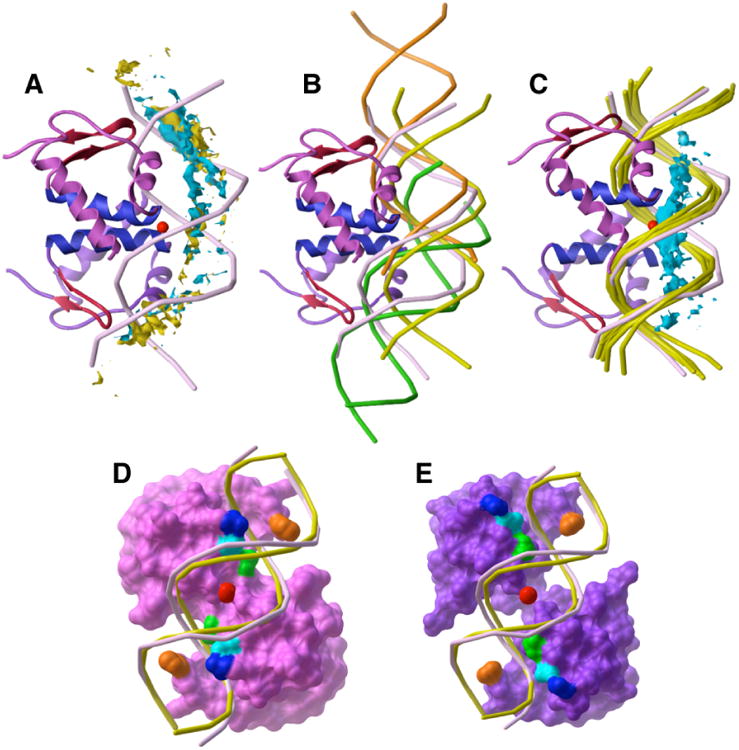
Linear B-DNA docked to FadR (monomer A, magenta ribbons; monomer B, purple ribbons; blue recognition helices and red wing structures). (A) The 500 best-ranked B-DNA fragments docked to free FadR (centers shown in turquoise) show tighter clustering than the 500 best-ranked B-DNA fragments docked to the DNA-bound FadR (centers shown in yellow). Both clusters follow the 20° bend of the DNA (light purple) from the FadR-DNA complex. The geometric center of the crystallographic DNA (red sphere) demonstrates the substantial bend induced upon DNA when bound to FadR. (B) Top-ranked DNA solutions docked to the free FadR structure indicate the curvature of the bound DNA (light purple). Three clusters are represented by rank #2 (yellow), rank #1 (orange), and rank #30 (green). All three position the DNA major groove against the N-termini of the two recognition helices and contact both FadR monomers. The yellow structure is centered over the recognition helices, but is farther from the wing structures (red) than the crystallographic DNA. Both the orange and green structures have a good fit with the wings and the N-termini of the two recognition helices. Together, the ensemble of these three clusters identifies the three main protein contacts and the need for the DNA to bend to accommodate all three simultaneously. (C) Docking of B-DNA to the acyl-CoA-bound FadR structure indicates that the bound DNA is not bent. The centers of the best-ranked 500 docked B-DNA solutions (turquoise) lie along a straight line, rather than along the axis of the FadR-bound DNA (light purple). The top 5 linear B-DNA solutions (yellow) fit better into the large channel between the FadR N-terminal monomers than the FadR-bound DNA. For this analysis, the DNA-bound and acyl-CoA-bound FadR structures were superposed by the Cα atoms of their N-terminal domains. (D) Requirement for DNA bending over the convex surface of the DNA-binding site in DNA-bound FadR. The DNA-bound FadR surface (magenta) is colored to show the guanidinium groups of Arg 35 (blue), Arg 45 (cyan), and Arg 49 (green) bound in the DNA major groove and the His 65 side chains (orange) in the DNA minor groove. The geometric center (red sphere) of the bent FadR-bound DNA (light purple) slightly penetrates the FadR molecular surface (magenta). The top-ranked B-DNA placement (yellow) docked to acyl-CoA-bound FadR penetrates into the DNA-bound FadR molecular surface in the center of the binding site. (E) Opening of a large channel between the N-terminal monomers in acyl-CoA-bound FadR (purple molecular surface). The guanidinium groups of Arg 35 (blue) and Arg 45 (light blue) move so that they now contact the B-DNA phosphate backbone (yellow), removing the major groove contacts needed for sequence-specific DNA recognition. The B-DNA phosphate backbone also fits into the slots created by His 65 (orange) and Arg 49 (green) in both monomers.
In contrast, the ensemble of B-DNA fragments docked to myristoyl-CoA-bound FadR indicated an unbent structure for bound DNA (Fig. 1C). The allosteric shift of the N-terminal domains53 creates a deeper channel between them than seen in the free FadR structure (Fig. 1D,E), allowing optimal fit of linear B-DNA. Arg side chains involved in sequence recognition no longer interact with bases in the major groove, but instead interact with the DNA phosphate backbone. The docking was repeated with DNA in which all A:T pairs were replaced with G:C and vice versa. Docking with this DNA sequence showed the same linear binding mode and interactions with Arg side chains, but favorable B-DNA placements had a greater degree of translation over the protein surface.33 We concluded that the allosteric N-terminal domain movement upon acyl-CoA binding had two effects: loss of the ability to bend DNA and replacement of sequence-specific interactions with non-specific electrostatic interactions.
A few months after this study was published, the first definitive structural data was published on the interaction of nonspecific DNA with the DNA-binding domain of lac repressor.54 Significant conformational changes were found in both protein and DNA depending on the DNA sequence. NMR studies showed that the lac repressor dimer induces significant conformational change in bound cognate DNA, with an overall bending of about 36°. In contrast, little conformational change was found for non-cognate DNA, which binds as linear, canonical B-DNA. Further, several side chains that interact with DNA bases in the specific complex now interact with the DNA phosphate backbone in the nonspecific complex, strongly supporting our hypothesis from computational docking.
UNG: Characterization of a full DNA-binding footprint
The DNA-repair enzyme uracil-DNA-glycosylase (UNG) cleaves uracil from ssDNA and dsDNA by hydrolysis of the N-glycosylic bond between uracil and the deoxyribose. The UNG mechanism55, 56 and structure57–60 have been extensively studied, making UNG an excellent system for testing computational docking. In the crystallographic structure of the catalytic domain of human UNG bound to a 10-base-pair DNA with a one-nucleotide overhang, the uracil base has been cleaved. The UNG Leu 272 side chain inserts through the DNA minor groove into the base stack to replace the flipped-out uracil. Docking44 using coordinates from the crystallographic UNG-DNA complex58 reproduced the complex.
It was unclear if B-form, fully base-paired DNA could fit into the active site with the Leu 272 side chain protruding into the DNA-binding groove. We found that docking B-DNA to the DNA-bound UNG structure indicated a much longer DNA-binding surface than observed in the crystallographic structure of the UNG-DNA complex (Fig. 2A).44 The active site was identified as the largest cluster in the 30 top-ranked B-DNA placements, which all showed correct positioning of the DNA minor groove over Leu 272 (Fig. 2B). Unexpectedly, a second, well-organized cluster of favorable B-DNA placements (Fig. 2A, green) was found at a site about 30 Å from the active site. The distribution of the centers of the 2000 most favorable B-DNA placements showed a continuous DNA-binding surface extending from the active site to this secondary site. Docking B-DNA to the free UNG structure showed a similar distribution for the 2,000 top-ranked B-DNA placements (Fig. 2C). The 30 top-ranked B-DNA placements occured at the active site, the secondary site, or between the two sites (Fig. 2C). The axes of the favorable B-DNA fragments at the active site were generally aligned with that of the crystallographic DNA, but most positioned the DNA major groove, rather than the minor groove, over Leu 272. The free UNG structure has a more open active-site groove than the DNA-bound UNG structure and there are small conformation differences in the Leu 272 loop. Thus, both large-scale and small-scale differences in the protein structure may contribute to incorrect DNA groove placements in the docking to free UNG.
Figure 2.
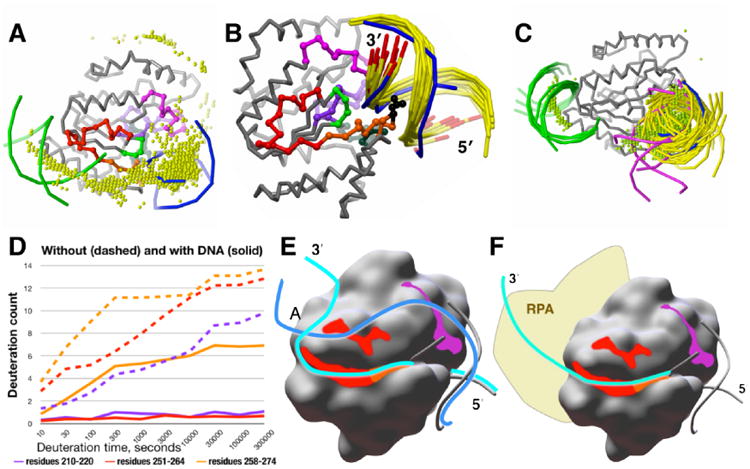
A model for the full DNA-binding site on UNG from computational docking and DXMS. (A) B-DNA docked to the DNA-bound structure of UNG (gray Cα backbone). The 2000 top-ranked B-DNA placements, represented by their geometric centers (yellow spheres), are concentrated over the active site (indicated by the crystallographic DNA, blue, right), at the secondary site (indicated by docked B-DNA, green, left), and between the two sites. Residues 210-220 (magenta), 251-264 (red), and 265-274 (orange) are highlighted on the UNG backbone. (B) The active-site cluster of B-DNA (yellow, with 3′-ends red) docked to the DNA-bound structure of UNG (gray Cα backbone) replicates the crystallographic DNA position (blue, 3′-ends red), including the correct 5′ to 3′ direction and insertion of Leu 272 (black) into the DNA minor groove. These dockings also show direct contact of the complementary strand with residues 210-220 (magenta). (C) B-DNA docked to the unbound structure of UNG (gray). The 30 top-ranked B-DNA placements compared with the crystallographic DNA (blue phosphate backbone): 23 (yellow) at the DNA-binding site; 5 (green) at the secondary site; and 2 (magenta) between the two sites. The 2000 top-ranked B-DNA placements, represented by their geometric centers (green spheres), show the same distribution. (D) DNA binding strongly protects residues 210-220 (magenta) and 251-264 (red) from solvent. A decrease in the amount of deuteration indicates increased protection of protein residues from the deuterated solvent. Over the entire UNG sequence, peptides 210-220 (magenta) and 251-264 (red) showed the greatest solvent protection in the presence of a 30-bp DNA substrate, acquiring no more than one deuteron even at the longest deuteration time of 300,000 s (more than 3 days). In the absence of DNA, both peptides become almost fully deuterated in 300,000 s. Neither region has direct contacts with the 10-bp DNA in the crystallographic structure of the UNG-DNA complex. Active site peptide 258-274 (orange), which includes Leu 272, shows significant protection, but part of this is due to its overlap with peptide 251-264 (red). (E) Model of the 30-bp product dsDNA bound to UNG. Both DNA strands in the model align with the crystallographic DNA (gray phosphate backbone) on the 5′ side of the active site. On the 3′ side of the active site, the active-site strand (light blue) contacts the groove created by residues 251-274, including the continuous surface formed by main-chain atoms of residues 251-264 (red) and 265-274 (orange). The complementary strand (blue) contacts the groove created by residues 210-220, including the surface created by the main-chain atoms (magenta), and may also contact the surface created by main-chain atoms of residues 251-258 (red) protected by bound DNA. (F) Model of single-strand DNA binding. The 3′-end of the active-site DNA strand extends from the active site over the surface groove created by residues 251-264, and continues into the predicted secondary binding site. This places the strand near the N-terminus of the catalytic domain, where it would be near RPA, which binds to a UNG recognition motif just N-terminal of the catalytic domain in nuclear UNG.63
To test the possibility of a longer DNA-binding site, we examined the UNG-DNA interaction in solution with hydrogen/deuterium exchange mass spectrometry (DXMS).44 In DXMS, hydrogen/deuterium exchange is followed by protein proteolysis and characterization of the resulting peptides by mass spectrometry, revealing the degree of solvent exposure for backbone amide hydrogen atoms throughout the protein chain. By measuring the change in solvent exposure on going from unbound to DNA-bound protein, DXMS has the potential to reveal the DNA footprint on the protein surface.61, 62 We designed a 30-bp DNA that included the DNA sequence of the crystallographic DNA and had the potential to reach from the active site to the secondary DNA-binding site. Peptides making up the active site showed greater protection in the presence of DNA. We saw the strongest increase in protection for two peptides (Fig. 2D): peptide 210-220 (magenta), which is adjacent to the active site, and peptide 251-264 (red), which is farther from the active site and forms part of the secondary DNA-binding site predicted by docking. Both peptide regions lie on the same side of the active site, but are not adjacent to each other. The almost complete protection observed by DXMS means that both must be simultaneously occupied by the bound DNA, but neither contact DNA in the crystallographic structure.
To develop a hypothesis for how bound DNA could contact both regions, we examined the B-DNA placements docked to the active site of DNA-bound UNG (Fig. 2B) in more detail. The 3′-end of the DNA strand that occupies the active-site groove also was oriented appropriately to extend over residues 251-264. The complementary DNA strand had direct contacts with residues 210-220. Thus, the two strands of the 30-bp bound dsDNA product can contact both positions simultaneously, but this requires separation of the two DNA strands on the protein surface. In our proposed model (Figure 2E), the active-site strand (light blue) extends from the active site toward the predicted secondary site, contacting the shallow surface groove created by residues 251-274. The complementary strand (blue) contacts the shallow groove created by residues 210-220 and then extends over residues 251-258 to meet the active-site strand.
For ssDNA, the DNA strand follows the path of the active-site strand of dsDNA through the shallow surface groove created by residues 251-264, possibly continuing into the predicted secondary DNA-binding site (Figure 2F). This positions the strand to extend toward replication protein A (RPA), which is bound to nuclear UNG a few residues before the N-terminus of the catalytic domain.63–65 RPA is the nuclear ssDNA-binding protein in eukaryotes that is essential to DNA replication, recombination, and repair, so RPA binding by UNG links its base-excision repair activity with DNA replication. Indirect evidence for this mode of ssDNA binding comes from comparison with family 5 UNG, which processes only dsDNA. The crystallographic structure with bound dsDNA 66 shows that the region of family 5 UNG corresponding to residues 251–264 shares the helix-loop-strand structure, but the loop is much longer. The added residues fold over the groove created by the helix and β-strand, eliminating the groove and explaining the inability of family 5 UNG to process ssDNA. In addition, the bound DNA contacts the surface corresponding to residues 210–220, supporting our prediction that this region is important for DNA binding.
Strand separation of dsDNA on the UNG surface has been previously proposed,67, 68 partly because UNG could then use the same search mechanism for both dsDNA and ssDNA substrates,68 explaining their similar rates of uracil cleavage.67 This idea was discarded when evidence from crystallography57, 58, 60 and NMR,69 all based on 10-bp DNA fragments, found no evidence for strand separation. To probe this question, we performed DXMS studies on the 11-bp DNA fragment containing the sequence of the crystallographic DNA. With the short DNA fragment, most of UNG showed the same degree of protection as free UNG, including peptides 210-220, 251-264, and most active-site peptides. Only peptides that contain Leu 272 showed increased protection, suggesting that, although Leu 272 is inserted into the base stack of the product DNA, this short DNA is not held tightly in the active-site groove. In the UNG-DNA crystallographic complex, extensive crystal contacts 44 trap a single configuration from the large ensemble available in solution. In solution, the 30-bp DNA is sufficiently long to form a stable complex, allowing the identification of the DNA footprint on the UNG surface. Thus, the 30-bp DNA better represents the very long natural substrate.
HIV Integrase: Contacts with two DNA duplexes
HIV integrase, one of just three enzymes encoded in the retroviral genome, has promise as a drug target for the treatment of AIDS. This 3-domain enzyme first removes two nucleotides from each end of the viral DNA, then inserts the two processed ends into host DNA (integration).70 Bound viral DNA is necessary to organize the integrase multimer needed for the integration step, but as of 2003 no crystallographic structures had been obtained for any integrase or integrase fragment with bound DNA. We examined the interaction of DNA with the HIV IN catalytic core domain (CCD) using computational docking.32 The resulting models were unconfirmed until 2010 when the first IN crystal structures with bound DNA were reported: those of full-length prototype foamy virus (PFV) IN with bound viral DNA71 and with both viral and host DNA.72
In 2003, several crystallographic structures of the HIV CCD were available,70 but all had a disordered active-site loop and were missing one or both active-site metal ions. We used CCD coordinates from a molecular dynamics model39, 40 that included the disordered active-site loop and both metal ions as our starting point in the docking calculation. We built a linear B-DNA fragment from the sequence of the viral DNA that included removal of 2 nucleotides from the 3′-end of one strand to mimic the 3′-processed viral DNA. Docking this DNA fragment to the CCD model identified the active-site region as the most favored DNA-binding region. Two distinct orientations occurred at the active site. The most heavily populated orientation lay over a helix containing Lys residues 156 and 159, which have been implicated in DNA binding.73 A 5′-end of one strand lay over the active site, suggesting that this orientation represented the host DNA. The less populated orientation positioned the cleaved 3′-end adjacent to an active-site metal ion, possibly representing the viral DNA.
Comparison with the PFV IN structures revealed that the most populated orientation actually corresponds to the bound viral DNA in the PFV IN structure (Fig. 3A). The axis of the docked DNA aligns well with that of the viral DNA and the centers of the most favorable top-ranked docked placements cluster along the viral DNA axis (Fig. 3B). This good alignment is remarkable given that the disordered active-site residues 139-153 (magenta, Fig. 3A) in the HIV CCD model have a very different conformation than the corresponding residues (black) in the PFV IN structure. The modeled HIV IN residues fill the groove that binds the unprocessed 5′ end of the viral DNA in the PFV crystal structure, preventing correct docking at the active site. Instead, the 5′ end of the docked structure overlaps the region that is occupied by the residues corresponding to 139-153 in the PFV IN structure. This causes the two DNA strands to be switched, so that the DNA major groove rather than the minor groove is positioned over the helix containing Lys 156 and the 5′-end rather than the 3′-end contacts the active site.
Figure 3.
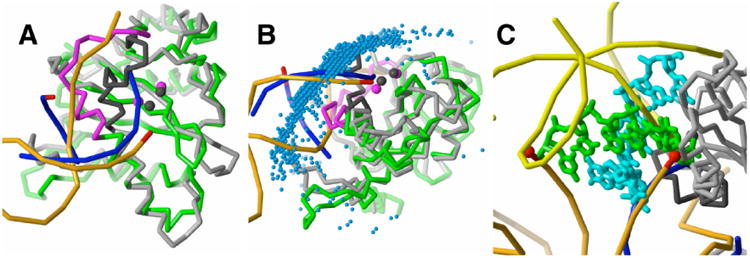
Comparison of the HIV-1 CCD complex with docked DNA to the PFV CCD structure with bound vDNA. The HIV-1 (green) and PFV CCD (gray) structures were superposed by the Cα atoms of conserved β-strand and α-helix secondary structure. (A) Computational docking locates the CCD binding site for viral DNA. The best-energy DNA placement (blue phosphate backbone) is representative of the largest favorable-energy cluster. One strand of the docked DNA shows good alignment with the active-site strand of the vDNA (orange with red 3′ end) bound to PFV IN, but it also penetrates into the active site loop (black) of the PFV CCD. In the HIV CCD coordinates, this loop corresponds to residues 139-153 (magenta). The incorrect conformation of this modeled active site loop fills the groove occupied by unprocessed 5′-end of the viral DNA, preventing correct positioning of the docked DNA at the active site. The two metal ions (magenta spheres) of the HIV-1 CCD model show good agreement with those of the PFV CCD (black spheres). (B) Docked DNA fragments follow the axis of vDNA. The most favorable 2000 DNA placements, represented by their geometric centers (blue spheres), form a large cluster over the CCD active site and extend over the full surface contacted by vDNA. (C) Second active-site orientation of docked DNA corresponds to the host DNA binding site. The docked DNA (green and light blue) overlaps the host DNA (yellow, bound to PFV IN) and is within 4Å of the two vDNA 3′ processed ends (red) in the PFV IN tetramer.
The less populated DNA orientation partially overlaps the host DNA bound to PFV IN and is near the two 3′-ends of viral DNA bound in the PFV IN tetramer71 (Fig. 3C). This overlap is remarkable given that the single CCD domain used in the docking calculation contains only a partial DNA-binding site for the host DNA. In the PFV IN structure, the host DNA contacts two CCD and two C-terminal domains. Thus, even with an incorrectly modeled active site and a partial host DNA-binding surface, computational docking gave a surprisingly good fit to both viral and host DNA positions.
It is unclear if the PFV IN structure fully represents the arrangement of the active complex of HIV IN. The two linker regions connecting the CCD with the N-terminal and C-terminal domains are much longer in PFV IN. The linker regions and the N- and C-terminal domains of PFV IN have little sequence similarity with HIV IN.70, 71 To examine the structure of HIV IN in solution, we are currently applying the combined computational/DXMS approach used for UNG. With such a complex system, computational and structural analysis will be essential for interpreting DXMS results.
Linker histone H5: Contacts with three DNA duplexes
Linker histones are essential for chromatin filament formation, and they play key roles in the regulation of gene expression. The location of the linker histone on the nucleosome is still a matter of debate.74, 75 Of the two linker histones, H1 and H5, H5 induces greater compaction of chromatin and is more inhibitory toward transcription.76 H5 consists of a central globular domain (GH5) that is required for nucleosome binding and basic N- and C-terminal tails. Binding of H5 or GH5 to nucleosomes protects an additional 20 bp of linker DNA (DNA entering or exiting the nucleosome) from micrococcal nuclease digestion.77 At the time of our study, several models for GH5 binding to the nucleosome had been suggested by experimental studies. Based on early studies,77, 78 a symmetrical model was proposed in which GH5 contacted the central region of the bound nucleosomal DNA (the nucleosome dyad) and both linker DNA regions entering and exiting the nucleosome. Zhou et al.79 proposed a two-contact model in which the linker histone forms a bridge between one arm of linker DNA and the dyad. Hayes et al.80 proposed a very different model in which GH5 is positioned ≈65 bp away from the dyad and is bound inside the DNA superhelix. The bridging and off-axis models could allow simultaneous binding at two equivalent sites per nucleosome, but a 1:1 ratio of linker histone to nucleosome is observed. GH5 binding models were also proposed based on comparisons with the DNA-binding motifs of structurally similar transcription factors, but radiolabeling,81 mutagenesis,75, 82 and crosslinking experiments79, 83 found that DNA binding involves many side chains widely dispersed over the GH5 surface rather than a single, localized site.
To investigate the interactions of GH5, we first docked DNA fragments to GH5 and then docked GH5 to models of the nucleosome structure.35 DNA-fragment docking found three distinct DNA-binding sites on GH5. Together these sites encompassed all of the GH5 side chains implicated in nucleosomal DNA binding. To investigate the interaction of GH5 with the nucleosome, several modifications of the nucleosome coordinates from the PDB41 were needed (see Methods). An essential modification was extension of the nucleosomal DNA because GH5 is known to protect about 20 bp of linker DNA beyond the nucleosomal DNA. The positioning of the two linker DNA arms built into our final model was close to that found in a low resolution structure of the tetranucleosome.84 Docking GH5 to the nucleosome model resulted in a large cluster within the 1000 most favorable configurations that was centered over the nucleosome dyad and contacted both linker DNA arms (Fig. 4A). GH5 used the same DNA-binding sites identified in the DNA fragment dockings, but the orientation of the DNA at each site was different, reflecting the more complex nucleosomal environment. The 30 most favorable configurations revealed two symmetry-related binding modes. In both, GH5 residues implicated in nucleosome binding contacted DNA, including essential Lys 8581, 82 that inserts into the DNA at the nucleosomal dyad (Fig. 4B). Together, these contacts account for the observed protection of 20 bp of linker DNA against nuclease digestion. Because of the sequence conservation of the residues involved in nucleosomal DNA binding within the H1/H5 linker histone family,85 our results are likely to apply to linker histone-nucleosome interactions in general.
Figure 4.
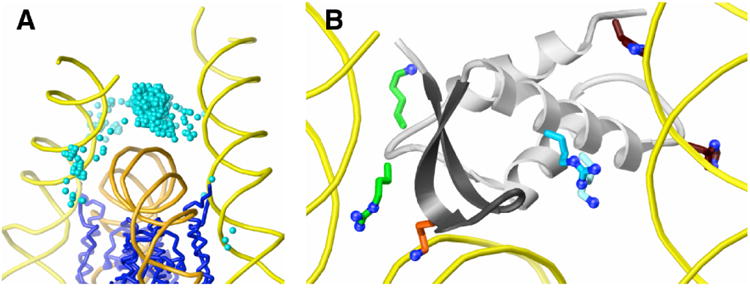
GH5 docked to the nucleosome. (A) The 1000 top-ranked GH5 solutions (centers shown as aqua spheres) are concentrated between the DNA (orange) at the dyad axis and the linker DNA (yellow) entering and exiting the nucleosome. The Cα backbone of the histone core is shown in blue. (B) Interactions of GH5 side chains with the nucleosome. In the top-ranked GH5 solution (gray ribbons), Lys 69 (light blue), Arg 74 (blue), and His residues 25 and 62 (brown) contact one arm of the linker DNA (right), Arg 40 and Lys 72 (green) contact the other linker DNA arm (left), and essential residue Lys 85 (orange) interacts with dyad DNA.
Recent hydroxyl-radical footprinting techniques86, 87 show the pattern of nucleosomal DNA protection at single-base resolution. In the absence of H1, linker DNA is unprotected from radical attack and the nucleosomal DNA shows a periodic 10-bp protection due to contacts with the histone core of the nucleosome. In the presence of H1 or its central globular domain (GH1), the nucleosome dyad and the first helical term of the linker DNA are also protected. This pattern of DNA protection was compared with structure-derived patterns calculated for three GH1-nucleosome models. One model was based on our 3-DNA-contact model, but GH5 was replaced by GH1. This model retained our predicted interactions with the nucleosomal dyad and both linker DNA arms. A second model was based on the two-contact model of Zhou et al.79 in which GH1 is displaced about 2 bp off the dyad and contacts one linker DNA arm. A third model was based on another two-contact proposal in which GH1 is displaced about 5 bp from the dyad.80 Both two-contact models failed to reproduce the strong protection found at the dyad. The three-contact model reproduced both the distinctive double-peak protection at the dyad and the protection observed for the first helical turn of the linker DNA. Therefore this model was selected to be the root of a detailed model of the full nucleosomal stem that explains the pattern of added protection conferred by bound H1 on linker DNA at least 40-bp away from the nucleosome.87
This new, detailed protection data strongly supports our model of the GH5-nucleosome interaction. More importantly, our model was directly useful for the modeling of larger assemblies within chromatin. Functional differences among the linker histones influence chromatin packing. Unlike H1, H5 and GH5 can form dimers in solution. The crystallographic structure of GH5 also contains a dimer. We found that forming this dimer with two of our predicted GH5-nucleosome complexes gave a dinucleosome complex with no steric clashes.35 Our proposal that formation of H5-nucleosome dimers contributes to the greater chromatin compaction ability of H5 has yet to be tested.
Discussion
The interactions between DNA-binding proteins and DNA involve molecular properties not typically found in stable protein-protein interactions. These molecular properties temper approaches for the prediction of protein-DNA complexes and the analysis and interpretation of computational results. DNA-binding proteins must have distinctive electrostatic properties that allow them to pull the highly charged DNA substrate out of solution.88–90 DNA-binding sites are not necessarily obvious from the electrostatic potential at the molecular surface, which is largely determined by the very local environment. Instead, the electrostatic potential field that extends out from the protein is responsible for attracting and binding the DNA.
The highly charged, relatively rigid, repeating structure of dsDNA results in distinctive properties. One consequence of the repeating structure is its involvement in biologically important, non-sequence-specific interactions. All of the systems in this study include these interactions. Although FadR has extensive sequence-specific DNA contacts when it acts as a transcriptional regulator, they become nonspecific in the acyl-CoA-bound state. In UNG and other DNA repair enzymes, non-sequence-specific interactions allow the search of genomic DNA for damage. Nonspecific interactions also contribute to binding of substrate and product once the damage is found. In integrase, only the region local to the CCD active site interacts with a conserved DNA sequence; the rest of the CCD binding site has non-sequence-specific interactions with viral DNA. These nonspecific interactions must be responsible for the good alignment of docked DNA fragments to the crystallographic DNA, since the active site surface was incorrectly modeled. The linker histone is not involved in any sequence-specific interactions. Instead, the bound position of its globular domain is determined by the arrangement of three DNA segments within the nucleosome-linker DNA complex.
DOT appears to be particularly effective for investigating protein interactions with dsDNA. Key for revealing the full DNA-binding site on a protein is DOT's exhaustive search and the ability to keep a large number of the most favorable dockings. The DOT potentials capture the electrostatic properties of dsDNA and DNA-binding proteins sufficiently well to localize the majority of favorable DNA placements to DNA-binding regions and indicate the orientation of the bound DNA axis over those regions. In all cases, the distribution of at least the most favorable 1,000 DNA placements followed that of the top 30. Even though both the protein and the DNA were represented as rigid molecules, evidence for global conformational changes of the DNA, such as bending or kinking, was found in these large clusters. These ensembles therefore provide a template that could be used to derive the bound DNA conformation from, for example, a library of dsDNA structures created by systematically adjusting conformational parameters91 or the conic section methods described by Banitt and Wolfson.92 The UNG study suggests that the rigid-body ensembles can also indicate binding sites for regions of ssDNA, though not provide details on the interactions. The large clusters of DNA placements are in sharp contrast to results from protein-protein docking. A cluster corresponding to the correct complex may be found, but the problem is distinguishing this cluster from other clusters that predict very different interactions.
The rigid-body docking of DOT is less successful at predicting the contacts of the DNA with the protein. A mixture of major and minor groove binding over specific protein features was found for B-DNA docking to the unbound structures of both FadR and UNG. One B-DNA cluster among the most favorable configurations correctly aligned the correct DNA sequence with FadR side chains involved in recognition. However, shifts along the DNA axis by one or more base pairs gave placements with very similar energies. Knowledge of critical residues and the DNA recognition motif, or use of symmetry in the case of dimeric proteins such as FadR, can help to select the correct cluster. These configurations would provide templates for more detailed studies that allow flexibility in both components.
As we found in protein-protein docking,17 using the fullest possible models is critical for good docking results, especially for unbound molecules. Interacting partners may influence DNA specificity93, 94 or the orientation of DNA at the binding site. For example, represention of FadR as a monomer gave significantly poorer alignment of docked DNA with the known DNA orientation.33 A second example was the linker histone. Docking DNA fragments to the linker histone identified the three DNA-binding surfaces, but, in the full nucleosome model, the orientation of the DNA at each surface is different.
We made use of one advantage that protein-DNA docking has over protein-protein docking: the rigidity of dsDNA allows good models to be built based on sequence alone. We have found that 8 to 12-bp dsDNA fragments with a canonical B-DNA structure work well as the moving molecule in DOT. These fragments are short enough to accommodate the varied topography of protein surfaces, but long enough to mimic the elongated distribution of negative charge. In contrast, a peptide structure based on sequence alone would have a poor chance of representing the properties of the same peptide as part of the three-dimensional structure of a protein. In DOT, a DNA fragment that represents a much longer DNA substrate is best assigned as the moving molecule, which is described by atomic point charges that are uniform throughout the molecule. If DNA is the stationary molecule, its electrostatic potential, calculated by Poisson-Boltzmann methods, becomes strongly modulated around the ends of the DNA due to solvent effects.33 With this treatment, only the four central base pairs of a 12-bp dsDNA fragment show the full negative potential. Here, DNA was the moving molecule in all cases except that of the linker histone-nucleosome complex in which the stationary molecule was the nucleosome. We added sufficiently long DNA segments to ensure a good electrostatics model for the nucleosome dyad and linker DNA.
The application of computational docking to protein-DNA interactions may have different goals than for protein-protein interactions. A crystallographic structure of a stable protein-protein complex is very likely to be a good representation of the interaction. A good test of the computational method is reproduction of this complex from unbound structures, where each structure contains the complete interacting surface. In contrast, the short DNA fragments typically found in protein-DNA crystallographic structures can, at best, represent only part of the full DNA-binding site, as in UNG. A crystallographer's goal is to design a DNA substrate that successfully cocrystallizes and occupies a critical region, such as the catalytic site of a protein. But the crystal environment can influence where and how the DNA fragment binds within this large site. One example is hairpin DNA structures that were designed with an overhang to fit into the nuclease site of the DNA repair protein Mre11. Although Mre11-DNA complexes formed crystals of good resolution, the DNA fragment does not occupy the nuclease site.95 It is unclear what part of the biological interaction the bound fragment mimics or if it even represents a key contact. In such intricate systems, which may bind multiple DNA segments, we do not have a model for the DNA substrate comparable to an unbound protein structure. As we have shown for UNG, a complete search using a linear, B-form DNA model can map out a full DNA-binding site, indicate the orientation of DNA within the site, and aid the design of experiments that test specific structural hypotheses suggested by the computational results. Thus, for protein-DNA interactions, computational docking can not only help to identify protein-DNA interactions in the absence of a crystal structure, but also expand structural understanding beyond known crystallographic structures.
Acknowledgments
This paper is dedicated to the memory of Professor Virgil L. Woods, Jr. (1948-2012), who developed the hydrogen/deuterium-exchange mass spectrometry technology used in this work.
This work was supported by the National Science Foundation (DBI 99-04559), the National Institutes of Health (GM070996, GM46312, AI081982, RR029388), and the University of California, San Diego, Center for AIDS Research (CFAR, National Institutes of Health grant P30 AI036214).
Literature Cited
- 1.Tan S, Hunziker Y, Pellegrini L, Richmond TJ. Crystallization of the yeast MATα2/MCM1/DNA ternary complex: general methods and principles for protein/DNA cocrystallization. J Mol Biol. 2000;297:947–959. doi: 10.1006/jmbi.2000.3606. [DOI] [PubMed] [Google Scholar]
- 2.Janin J, Henrick K, Moult J, Ten Eyck L, Sternberg MJE, Vajda S, Vakser I, Wodak SJ. CAPRI: a Critical Assessment of PRedicted Interactions. Proteins. 2003;52:2–9. doi: 10.1002/prot.10381. [DOI] [PubMed] [Google Scholar]
- 3.Méndez R, Leplae R, De Maria L, Wodak SJ. Assessment of blind predictions of protein-protein interactions: current status of docking methods. Proteins. 2003;52:51–67. doi: 10.1002/prot.10393. [DOI] [PubMed] [Google Scholar]
- 4.Méndez R, Leplae R, Lensink MF, Wodak SJ. Assessment of CAPRI predictions in rounds 3-5 shows progress in docking procedures. Proteins. 2005;60:150–169. doi: 10.1002/prot.20551. [DOI] [PubMed] [Google Scholar]
- 5.Giudice E, Lavery R. Simulations of nucleic acids and their complexes. Acc Chem Res. 2002;35:350–357. doi: 10.1021/ar010023y. [DOI] [PubMed] [Google Scholar]
- 6.van Dijk M, van Dijk ADJ, Hsu V, Boelens R, Bonvin AMJJ. Information-driven protein-DNA docking using HADDOCK: it is a matter of flexibility. Nucl Acids Res. 2006;34:3317–3325. doi: 10.1093/nar/gkl412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.van Dijk M, Bonvin AMJJ. Pushing the limits of what is achievable in protein-DNA docking: benchmarking HADDOCK's performance. Nucl Acids Res. 2010;38:5634–5647. doi: 10.1093/nar/gkq222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Knegtel RMA, Antoon J, Rullmann C, Boelens R, Kaptein R. MONTY: a Monte Carlo approach to protein-DNA recognition. J Mol Biol. 1994;235:318–324. doi: 10.1016/s0022-2836(05)80035-x. [DOI] [PubMed] [Google Scholar]
- 9.Knegtel RMA, Boelens R, Kaptein R. Monte Carlo docking of protein-DNA complexes: Incorporation of DNA flexibility and experimental data. Protein Eng. 1994;7:761–767. doi: 10.1093/protein/7.6.761. [DOI] [PubMed] [Google Scholar]
- 10.Tzou WS, Hwang MJ. Modeling helix-turn-helix protein-induced DNA bending with knowledge-based distance restraints. Biophys J. 1999;77:1191–1205. doi: 10.1016/S0006-3495(99)76971-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Sandmann C, Cordes F, Saenger W. Structure model of a complex between the factor for inversion stimulation (FIS) and DNA: modeling protein-DNA complexes with dyad symmetry and known protein structures. Proteins. 1996;25:486–500. doi: 10.1002/prot.8. [DOI] [PubMed] [Google Scholar]
- 12.Moreira IS, Fernandes PA, Ramos MJ. Protein-protein docking dealing with the unknown. J Comput Chem. 2010;31:317–342. doi: 10.1002/jcc.21276. [DOI] [PubMed] [Google Scholar]
- 13.Katchalski-Katzir E, Shariv I, Eisenstein M, Friesem AA, Aflalo C, Vakser IA. Molecular surface recognition: determination of geometric fit between proteins and their ligands by correlation techniques. Proc Natl Acad Sci USA. 1992;89:2195–2199. doi: 10.1073/pnas.89.6.2195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Vakser IA, Aflalo C. Hydrophobic docking: a proposed enhancement to molecular recognition techniques. Proteins. 1994;20:320–329. doi: 10.1002/prot.340200405. [DOI] [PubMed] [Google Scholar]
- 15.Ten Eyck LF, Mandell JG, Roberts VA, Pique ME. In: Hayes A, Simmons M, editors. Surveying molecular interactions with DOT; Proceedings of the 1995 ACM/IEEE Supercomputing Conference; San Diego. Los Alamitos, CA: IEEE Computer Society Press; 1995. www.sdsc.edu/CCMS/Papers/DOT_sc95.html. [Google Scholar]
- 16.Mandell JG, Roberts VA, Pique ME, Kotlovyi V, Mitchell JC, Nelson E, Tsilgeny I, Ten Eyck LF. Protein docking using continuum electrostatics and geometric fit. Prot Eng. 2001;14(2):105–113. doi: 10.1093/protein/14.2.105. [DOI] [PubMed] [Google Scholar]
- 17.Roberts VA, Thompson EE, Pique ME, Perez MS, Ten Eyck LF. DOT2: macromolecular docking with improved biophysical models. J Comput Chem. 2013;34:1743–1758. doi: 10.1002/jcc.23304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Heifetz A, Katchalski-Katzir E, Eisenstein M. Electrostatics in protein-protein docking. Protein Sci. 2002;11:571–587. doi: 10.1110/ps.26002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Gabb HA, Jackson RM, Sternberg MJE. Modelling protein docking using shape complementarity, electrostatics and biochemical information. J Mol Biol. 1997;272:106–120. doi: 10.1006/jmbi.1997.1203. [DOI] [PubMed] [Google Scholar]
- 20.Vakser IA. Evaluation of GRAMM low-resolution docking methodology on the hemagglutinin-antibody. Proteins. 1997;1:226–230. [PubMed] [Google Scholar]
- 21.Chen R, Li L, Weng Z. ZDOCK: an initial-stage protein-docking algorithm. Proteins. 2003;52:80–87. doi: 10.1002/prot.10389. [DOI] [PubMed] [Google Scholar]
- 22.Kozakov D, Brenke R, Comeau SR, Vajda S. PIPER: an FFT-based protein docking program with pairwise potentials. Proteins. 2006;65:392–406. doi: 10.1002/prot.21117. [DOI] [PubMed] [Google Scholar]
- 23.Li L, Guo D, Huang Y, Liu S, Xiao Y. ASPDock: protein-protein docking algorithm using atomic solvation parameters model. BMC Bioinformatics. 2011;12:36. doi: 10.1186/1471-2105-12-36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Bajaj C, Chowdhury R, Siddavanahalli V. F2Dock: Fast Fourier protein-protein docking. IEEE/ACM Trans Comput Biol Bioinform. 8:45–58. 2011. doi: 10.1109/TCBB.2009.57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Ritchie DW, Kemp GJL. Protein docking using spherical polar Fourier correlations. Proteins. 2000;39:178–194. [PubMed] [Google Scholar]
- 26.Garzon JI, Lopéz-Blanco JR, Pons C, Kovacs J, Abagyan R, Fernandez-Recio J, Chacon P. FRODOCK: a new approach for fast rotational protein-protein docking. Bioinformatics. 25:2544–2551. 2009. doi: 10.1093/bioinformatics/btp447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Duhovny D, Nussinov R, Wolfson HJ. Efficient unbound docking of rigid molecules. In: Guigo R, Gusfield D, editors. WABI, Proceedings of the 2nd Workshop on Algorithms in Bioinformatics. Springer; Berlin: 2002. pp. 185–200. Lecture Notes in Computer Science. [Google Scholar]
- 28.Mashiach E, Schneidman-Duhovny D, Peri A, Shavit Y, Nussinov R, Wolfson HJ. An integrated suite of fast docking algorithms. Proteins. 78:3197–3204. 2010. doi: 10.1002/prot.22790. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Aloy P, Moont G, Gabb HA, Querol E, Aviles FX, Sternberg MJE. Modelling repressor proteins docking to DNA. Proteins. 33:535–549. 1998. [PubMed] [Google Scholar]
- 30.De Luca L, Pedretti A, Vistoli G, Barreca ML, Villa L, Monforte P, Chimirri A. Analysis of the full-length integrase-DNA complex by a modified approach for DNA docking. Biochem Biophys Res Commun. 2003;310:1083–1088. doi: 10.1016/j.bbrc.2003.09.120. [DOI] [PubMed] [Google Scholar]
- 31.Hopfner KP, Karcher A, Craig L, Woo TT, Carney JP, Tainer JA. Structural biochemistry and interaction architecture of the DNA double-strand break repair Mre11 nuclease and Rad50-ATPase. Cell. 2001;105:473–485. doi: 10.1016/s0092-8674(01)00335-x. [DOI] [PubMed] [Google Scholar]
- 32.Adesokan AA, Roberts VA, Lee KW, Lins RD, Briggs JM. Prediction of HIV-1 integrase/viral DNA interactions in the catalytic domain by fast molecular docking. J Med Chem. 2004;47:821–828. doi: 10.1021/jm0301890. [DOI] [PubMed] [Google Scholar]
- 33.Roberts VA, Case DA, Tsui V. Predicting interactions of winged-helix transcription factors with DNA. Proteins. 2004;57:172–187. doi: 10.1002/prot.20193. [DOI] [PubMed] [Google Scholar]
- 34.Zhu HM, Chen WZ, Wang CX. Docking dinucleotides to HIV-1 integrase carboxyl-terminal domain to find possible DNA binding sites. Bioorg Med Chem Lett. 2005;15:475–477. doi: 10.1016/j.bmcl.2004.10.003. [DOI] [PubMed] [Google Scholar]
- 35.Fan L, Roberts VA. Complex of linker histone H5 with the nucleosome and its implications for chromatin packing. Proc Natl Acad Sci USA. 2006;103:8384–8389. doi: 10.1073/pnas.0508951103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Fanelli F, Ferrari S. Prediction of MEF2A-DNA interface by rigid body docking: a tool for fast estimation of protein mutational effects on DNA binding. J Struct Biol. 2006;153:278–283. doi: 10.1016/j.jsb.2005.12.002. [DOI] [PubMed] [Google Scholar]
- 37.Fan L, Fuss JO, Cheng QJ, Arvai AS, Hammel M, Roberts VA, Cooper PK, Tainer JA. XPD helicase structures and activities: insights into the cancer and aging phenotypes from XPD mutations. Cell. 2008;133:789–800. doi: 10.1016/j.cell.2008.04.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Halperin I, Ma B, Wolfson H, Nussinov R. Principles of docking: an overview of search algorithms and a guide to scoring functions. Proteins. 2002;47:409–443. doi: 10.1002/prot.10115. [DOI] [PubMed] [Google Scholar]
- 39.Lins RD, Briggs JM, Straatsma TP, Carlson HA, Greenwald J, Choe S, McCammon JA. Molecular dynamics studies on the HIV-1 integrase catalytic domain. Biophys J. 1999;76:2999–3011. doi: 10.1016/s0006-3495(99)77453-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Lins RD, Adesokan AA, Soares TA, Briggs JM. Investigations on human immunodeficiency virus type 1 integrase/DNA binding interactions via molecular dynamics and electrostatic calculations. Pharmacol Ther. 2000;85:123–131. doi: 10.1016/s0163-7258(99)00059-5. [DOI] [PubMed] [Google Scholar]
- 41.Luger K, Mader AW, Richmond RK, Saftent DF, Richmond TJ. Crystal structure of the nucleosome core particle at 2.8 Å resolution. Nature. 1997;389:251–260. doi: 10.1038/38444. [DOI] [PubMed] [Google Scholar]
- 42.Ramakrishnan V, Finch JT, Graziano V, Lee PL, Sweet RM. Crystal structure of globular domain of histone H5 and its implications for nucleosome binding. Nature. 1993;362:219–223. doi: 10.1038/362219a0. [DOI] [PubMed] [Google Scholar]
- 43.Macke T, Case DA. Modeling unusual nucleic acid structures. In: Leontes NB, Santa Lucia J Jr, editors. Molecular Modeling of Nucleic Acids. Vol. 682. American Chemical Society; Washington, DC: 1998. pp. 379–393. [Google Scholar]
- 44.Roberts VA, Pique ME, Hsu S, Li S, Slupphaug G, Rambo RP, Jamison J, Liu T, Lee JH, Tainer JA, Ten Eyck LF, Woods VL., Jr Combining H/D exchange mass spectroscopy and computational docking reveals extended DNA-binding surface on uracil-DNA-glycosylase. Nucl Acids Res. 2012;40(13):6070–6081. doi: 10.1093/nar/gks291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Tsui V, Case DA. Molecular dynamics simulations of nucleic acids with a generalized Born solvation model. J Am Chem Soc. 2000;122:2489–2498. [Google Scholar]
- 46.Gilson MK, Davis ME, Luty BA, McCammon JA. Computation of electrostatic forces on solvated molecules using the Poisson-Boltzmann equation. J Phys Chem. 1993;97:3591–3600. doi: 10.1063/1.1924448. [DOI] [PubMed] [Google Scholar]
- 47.Baker NA, Sept D, Joseph S, Holst JJ, McCammon JA. Electrostatics of nanosystems: application to microtubules and the ribosome. Proc Natl Acad Sci USA. 2001;98:10037–10041. doi: 10.1073/pnas.181342398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Roberts VA, Pique ME. Definition of the interaction domain for cytochrome c on cy-tochrome c oxidase. III. Prediction of the docked complex by a complete, systematic search. J Biol Chem. 1999;274:38051–38060. doi: 10.1074/jbc.274.53.38051. [DOI] [PubMed] [Google Scholar]
- 49.DiRusso CC, Black PN, Weimar JD. Molecular inroads into the regulation and metabolism of fatty acids, lessons from bacteria. Prog Lipid Res. 1999;38:129–197. doi: 10.1016/s0163-7827(98)00022-8. [DOI] [PubMed] [Google Scholar]
- 50.DiRusso CC, Heimert TL, Metzger AK. Characterization of FadR, a global transcriptional regulator of fatty acid metabolism in Escherichia coli. J Biol Chem. 1992;267:8685–8691. [PubMed] [Google Scholar]
- 51.van Aalten DMF, DiRusso CC, Knudsen J, Wierenga RK. Crystal structure of FadR, a fatty acid-responsive transcription factor with a novel acyl coenzyme A-binding fold. EMBO J. 2000;19:5167–5177. doi: 10.1093/emboj/19.19.5167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Xu Y, Heath RJ, Li Z, Rock CO, White SW. The FadR-DNA complex. J Biol Chem. 2001;276:17373–17379. doi: 10.1074/jbc.M100195200. [DOI] [PubMed] [Google Scholar]
- 53.van Aalten DMF, DiRusso CC, Knudsen J. The structural basis of acyl coenzyme A-dependent regulation of the transcription factor FadR. EMBO J. 2001;20:2041–2050. doi: 10.1093/emboj/20.8.2041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Kalodimos CG, Biris N, Bonvin AMJJ, Levandoski MM, Guennuegues M, Boelens R, Kaptein R. Structure and flexibility adaptation in nonspecific and specific protein-DNA complexes. Science. 2004;305:386–389. doi: 10.1126/science.1097064. [DOI] [PubMed] [Google Scholar]
- 55.Krokan HE, Standal R, Slupphaug G. DNA glycosylases in the base excision repair of DNA. Biochem J. 1997;325:1–16. doi: 10.1042/bj3250001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Friedman JI, Stivers JT. Detection of damaged DNA bases by DNA glycosylase enzymes. Biochemistry. 2010;49:4957–4967. doi: 10.1021/bi100593a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Slupphaug G, Mol CD, Kavli B, Arvai AS, Krokan HE, Tainer JA. A nucleotide-flipping mechanism from the structure of human uracil-DNA glycosylase bound to DNA. Nature. 1996;384:87–92. doi: 10.1038/384087a0. [DOI] [PubMed] [Google Scholar]
- 58.Parikh SS, Mol CD, Slupphaug G, Bharati S, Krokan HE, Tainer JA. Base excision repair initiation revealed by crystal structures and binding kinetics of human uracil-DNA glycosylase with DNA. EMBO J. 1998;17:5214–5226. doi: 10.1093/emboj/17.17.5214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Parikh SS, Walcher G, Jones GD, Slupphaug G, Krokan HE, Blackburn GM, Tainer JA. Uracil-DNA glycosylase–DNA substrate and product structures: conformational strain promotes catalytic efficiency by coupled stereoelectronic effects. Proc Natl Acad Sci USA. 2000;97:5083–5088. doi: 10.1073/pnas.97.10.5083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Parker JB, Bianchet MA, Krosky DJ, Friedman JI, Amzel LM, Stivers JT. Enzymatic capture of an extrahelical thymine in the search for uracil in DNA. Nature. 2007;449:433–437. doi: 10.1038/nature06131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Sperry JB, Shi X, Rempel DL, Nishimura Y, Akashi S, Gross ML. A mass spectrometric approach to the study of DNA-binding proteins: interaction of human TRF2 with telomeric DNA. Biochemistry. 2008;47:1797–1807. doi: 10.1021/bi702037p. [DOI] [PubMed] [Google Scholar]
- 62.Black BE, Brock MA, Bedard S, Woods VL, Jr, Cleveland DW. An epigenetic mark generated by the incorporation of CENP-A into centromeric nucleosomes. Proc Natl Acad Sci USA. 2007;104:5008–5013. doi: 10.1073/pnas.0700390104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Nagelhus TA, Haug T, Singh KK, Keshav KF, Skorpen F, Otterlei M, Bharati S, Lindmo T, Benichou S, Benarous R, Krokan HE. A sequence in the N-terminal region of human uracil-DNA glycosylase with homology to XPA interacts with the C-terminal part of the 34 kDa subunit of replication protein A. J Biol Chem. 1997;272:6561–6566. doi: 10.1074/jbc.272.10.6561. [DOI] [PubMed] [Google Scholar]
- 64.Otterlei M, Warbrick E, Nagelhus TA, Haug T, Slupphaug G, Akbari M, Aas PA, Steinsbekk K, Bakke O, Krokan HE. Post-replicative base excision repair in replication foci. EMBO J. 1999;18:3834–3844. doi: 10.1093/emboj/18.13.3834. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Mer G, Bochkarev A, Gupta R, Bochkareva E, Frappier L, Ingles CJ, Edwards AM, Chazin WJ. Structural basis for the recognition of DNA repair proteins UNG2, XPA, and RAD52 by replication factor RPA. Cell. 2000;103:449–458. doi: 10.1016/s0092-8674(00)00136-7. [DOI] [PubMed] [Google Scholar]
- 66.Kosaka H, Hoseki J, Nakagawa N, Kuramitsu S, Masui R. Crystal structure of family 5 uracil-DNA glycosylase bound to DNA. J Mol Biol. 2007;373:839–850. doi: 10.1016/j.jmb.2007.08.022. [DOI] [PubMed] [Google Scholar]
- 67.Slupphaug G, Eftedal I, Kavli B, Bharati S, Helle NM, Haug T, Levine DW, Krokan HE. Properties of a recombinant human uracil-DNA glycosylase from the UNG gene and evidence that UNG encodes the major uracil-DNA glycosylase. Biochemistry. 1995;34:128–138. doi: 10.1021/bi00001a016. [DOI] [PubMed] [Google Scholar]
- 68.Vassylyev DG, Morikawa K. Precluding uracil from DNA. Structure. 1996;4:1381–1385. doi: 10.1016/s0969-2126(96)00145-1. [DOI] [PubMed] [Google Scholar]
- 69.Cao C, Jiang YL, Stivers JT, Song F. Dynamic opening of DNA during the enzymatic search for a damaged base. Nat Struct Mol Biol. 2004;11:1230–1236. doi: 10.1038/nsmb864. [DOI] [PubMed] [Google Scholar]
- 70.Jaskolski M, Alexandratos JN, Bujacz G, Wlodawer A. Piecing together the structure of retroviral integrase, an important target in AIDS therapy. FEBS J. 2009;276:2926–2946. doi: 10.1111/j.1742-4658.2009.07009.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Hare S, Gupta SS, Valkov E, Engelman A, Cherepanov P. Retroviral intasome assembly and inhibition of DNA strand transfer. Nature. 2010;464:232–236. doi: 10.1038/nature08784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Maertens GN, Hare S, Cherepanov P. The mechanism of retroviral integration from X-ray structures of its key intermediates. Nature. 2010;468:326–330. doi: 10.1038/nature09517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Jenkins TM, Esposito D, Engelman A, Craigie R. Critical contacts between HIV-1 integrase and viral DNA identified by structure-based analysis and photo-crosslinking. EMBO J. 1997;16:6849–6859. doi: 10.1093/emboj/16.22.6849. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Vignali M, Workman JL. Location and function of linker histones. Nature Struct Biol. 1998;5:1025–1028. doi: 10.1038/4133. [DOI] [PubMed] [Google Scholar]
- 75.Duggan MM, Thomas JO. Two DNA-binding sites on the globular domain of histone H5 are required for binding to both bulk and 5 S reconstituted nucleosomes. J Mol Biol. 2000;304:21–33. doi: 10.1006/jmbi.2000.4205. [DOI] [PubMed] [Google Scholar]
- 76.Sun J-M, Ali Z, Lurz R, Ruiz-Carrillo A. Replacement of histone H1 by H5 in vivo does not change the nucleosome repeat length of chromatin but increases its stability. EMBO J. 1990;9:1651–1658. doi: 10.1002/j.1460-2075.1990.tb08285.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Simpson RT. Structure of the chromatosome, a chromatin particle containing 160 base pairs of DNA and all the histones. Biochemistry. 1978;17:5524–5531. doi: 10.1021/bi00618a030. [DOI] [PubMed] [Google Scholar]
- 78.Staynov DZ, Crane-Robinson C. Footprinting of linker histones H5 and H1 on the nucleosome. EMBO J. 1988;7:3685–3691. doi: 10.1002/j.1460-2075.1988.tb03250.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Zhou YB, Gerchman SE, Ramakrishnan V, Travers A, Muyldermans S. Position and orientation of the globular domain of linker histone H5 on the nucleosome. Nature. 1998;395:402–405. doi: 10.1038/26521. [DOI] [PubMed] [Google Scholar]
- 80.Hayes JJ, Pruss D, Wolff AP. Contacts of the globular domain of histone H5 and core histones with DNA in a “chromatosome”. Proc Natl Acad Sci USA. 1994;91:7817–7821. doi: 10.1073/pnas.91.16.7817. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Thomas JO, Wilson CM. Selective radiolabeling and identification of a strong nucleosome binding site on the globular domain of histone H5. EMBO J. 1986;5:3531–3537. doi: 10.1002/j.1460-2075.1986.tb04679.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Buckle RS, Maman JD, Allan J. Site-directed mutagenesis studies on the binding of the globular domain of linker histone H5 to the nucleosome. J Mol Biol. 1992;223:651–659. doi: 10.1016/0022-2836(92)90981-o. [DOI] [PubMed] [Google Scholar]
- 83.Mirzabekov AD, Pruss DV, Ebralidse KK. Chromatin superstructure-dependent crosslinking with DNA of the histone H5 residues Thr1, His25, and His62. J Mol Biol. 1989;211:479–491. doi: 10.1016/0022-2836(90)90366-T. [DOI] [PubMed] [Google Scholar]
- 84.Schalch T, Duda S, Sargent DF, Richmond TJ. X-ray structure of a tetranucleosome and its implications for the chromatin fibre. Nature. 2005;436:138–141. doi: 10.1038/nature03686. [DOI] [PubMed] [Google Scholar]
- 85.Crane-Robinson C, Ptitsyn OB. Binding of the globular domain of linker histones H5/H1 to the nucleosome: a hypothesis. Prot Eng. 1989;2:577–582. doi: 10.1093/protein/2.8.577. [DOI] [PubMed] [Google Scholar]
- 86.Syed SH, Goutte-Gattat D, Becker NB, Meyer S, Shukla MS, Hayes JJ, Everaers R, Angelov D, Bednar J, Dimitrov S. Single-base resolution mapping of H1-nucleosome interactions and 3D organization of the nucleosome. Proc Natl Acad Sci USA. 2010;107:9620–9625. doi: 10.1073/pnas.1000309107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Meyer S, Becker NB, Syed SH, Goutte-Gattat D, Shukla MS, Hayes JJ, Angelov D, Bednar J, Dimitrov S, Everaers R. From crystal and NMR structures, footprints and cryo-electron-micrographs to large and soft structures: nanoscale modeling of the nucleosomal stem. Nucl Acids Res. 2011;39:9139–9154. doi: 10.1093/nar/gkr573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Nadassy K, Wodak SJ, Janin J. Structural features of protein-nucleic acid recognition sites. Biochemistry. 1999;38:1999–2017. doi: 10.1021/bi982362d. [DOI] [PubMed] [Google Scholar]
- 89.Jones S, van Heyningen P, Berman HM, Thornton JM. Protein-DNA interactions: a structural analysis. J Mol Biol. 1999;287:877–896. doi: 10.1006/jmbi.1999.2659. [DOI] [PubMed] [Google Scholar]
- 90.Jones S, Shanahan HP, Berman HM, Thornton JM. Using electrostatic potentials to predict DNA-binding sites on DNA-binding proteins. Nucl Acids Res. 2003;31:7189–7198. doi: 10.1093/nar/gkg922. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.van Dijk M, Bonvin AMJJ. 3D-DART: a DNA structure modelling server. Nucl Acids Res. 2009;37:W235–W239. doi: 10.1093/nar/gkp287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Banitt I, Wolfson HJ. ParaDock: a flexible non-specific DNA-rigid protein docking algorithm. Nucl Acids Res. 2011;39:e135. doi: 10.1093/nar/gkr620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Pan Y, Tsai CJ, Ma B, Nussinov R. How do transcription factors select specific binding sites in the genome? Nature Struct Mol Biol. 2009;16:1118–1120. doi: 10.1038/nsmb1109-1118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Pan Y, Tsai CJ, Ma B, Nussinov R. Mechanisms of transcription factor selectivity. Trends Genet. 2010;26:75–83. doi: 10.1016/j.tig.2009.12.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Williams RS, Moncalian G, Williams JS, Yamada Y, Limbo O, Shin DS, Groocock LM, Cahill D, Hitomi C, Guenther G, Moiani D, Carney JP, Russell P, Tainer JA. Mre11 dimers coordinate DNA end bridging and nuclease processing in double-strand-break repair. Cell. 2008;135:97–109. doi: 10.1016/j.cell.2008.08.017. [DOI] [PMC free article] [PubMed] [Google Scholar]