

Abstract
Recent outbreaks of severe acute respiratory syndrome (SARS) have spurred intense research efforts around the world to deal with the serious threat to health posed by this novel coronavirus. A rapid, reliable diagnostic assay is needed for monitoring the spread of the disease. Here we report a method for eliminating false-negative results and increasing test sensitivity, based on the hypothesis that the message encoded by the nucleocapsid (N) gene is the most abundant during viral infection. Nasopharyngeal aspirates and stool samples were obtained from suspected SARS patients with major clinical symptoms and a significant history of close contact with infected patients. Total RNAs were extracted in a 96-well format, together with pig kidney epithelial (PK-15) cells as an internal control for extraction efficiency. PCR inhibitors were removed by ethanol precipitation, and a PCR for the pig β-actin gene was used as a positive control for all clinical samples. Samples were analyzed by a reverse transcriptase PCR assay. Northern blot analysis was performed to demonstrate differences in subgenomic transcripts of the virus, and a real-time quantitative PCR was employed to compare the sensitivities of two loci (1b and N). The detection rate of the assay reached 44.4% on day 9 after the onset of the disease. The diagnostic PCR amplifying the N gene gave an average of a 26.0% (6.3 to 60.0%) stronger intensity signal than that for the 1b gene. In conclusion, the nucleocapsid gene represents an additional sensitive molecular marker for the diagnosis of the SARS coronavirus and can be further adapted for use in a high-throughput platform assay.
Severe acute respiratory syndrome (SARS) is a recently emerged disease which causes patients to have various clinical symptoms, including fever (of 38°C or above for over 24 h), malaise, chills, headache, and body ache. Chest X-rays show changes that are compatible with pneumonia. Other symptoms include coughing, shortness of breath, and difficulty breathing (8). As of 1 July 2003, a total of 1,755 SARS cases and 298 SARS-related deaths were reported in Hong Kong, representing 21 and 37% of the global totals of SARS cases (8,437) and deaths (813), respectively (9). Serological testing is reliable as a retrospective diagnostic method, but antibody levels in the early phase postinfection may not be detectable. Since the disease is highly contagious and the diagnosis of the infection in the early phase of illness is important for patient care (11), it is important to develop a rapid and reliable molecular diagnostic assay for monitoring and controlling the disease.
Recently, a new strain of virus in the family Coronaviridae was discovered in SARS patients admitted to local hospitals (4), and its genome was completely sequenced a few weeks later (3, 7, 12). It is believed that this new virus causes SARS, and current diagnostic tests, including serological and molecular tests, are based on this virus. Reverse transcriptase PCR (RT-PCR) is employed for the rapid detection of virus in clinical samples. Primers amplifying regions within the RNA-dependent RNA polymerase-encoding sequence of the virus are being used for diagnosis in several of the World Health Organization network laboratories (in Germany, the United States, and Hong Kong). In Mouse hepatitis virus, a typical member of the genus Coronavirus, both the genomic RNA and mRNA transcripts are capped and possess common 3′ ends and common leader sequences on their 5′ ends. With this unique transcription strategy, it is expected that the copy numbers of different viral genes during the replication of the virus are different in the host cells (Fig. 1). The N gene that encodes the nucleocapsid protein has the most abundant copy number during virus replication, as all transcripts may carry the nucleotide sequence from the N gene, although they are not all in frame for translation of the gene product. Recently, Rota et al. (7) showed that when they used a digoxigenin-labeled riboprobe overlapping the 3′ untranslated region of the genome in a Northern blot analysis of the SARS-associated coronavirus (SARS-CoV), all transcripts showed a positive signal (7). Therefore, we hypothesized that an RT-PCR assay amplifying the 3′ region, including the N gene, of the viral genome would be more sensitive than amplifying the rest of the viral genome for diagnostic purposes. In this study, we report a new and improved RT-PCR assay for the detection of SARS-CoV in clinical samples.
FIG. 1.

Genome organization and transcription strategy of SARS-CoV HK-39. Genomic and mRNA transcripts are capped (black circles), carry leader sequences (vertical lines) at the 5′-proximal end, and are polyadenylated (A15). Arrows point to the positions of the intergenic sequence (CTAAACGAAC). After the release of the positive-sense genomic RNA into the cytoplasm of the host cell, the viral RNA-dependent RNA polymerase, encoded by ORF1a and -1b, is synthesized. It performs transcription of a full-length complementary (negative-sense) RNA, from which new genomic RNA, an overlapping set of subgenomic mRNA transcripts, and leader RNAs are synthesized. Note that all transcripts are preceded with common 5′ leader sequences and common 3′ ends. ORF1a and -1b, RNA-dependent RNA polymerase; S, the major peplomer glycoprotein; M, transmembrane glycoprotein; N, nucleocapsid protein; X1, X2, and X3, putative uncharacterized proteins.
MATERIALS AND METHODS
RNA isolation from clinical samples.
Clinical samples, including nasopharyngeal aspirates (NPAs) (n = 131) and stool specimens (n = 5), were provided by the Department of Microbiology, The University of Hong Kong (HKU). In addition, tracheal dispersion fluid and lung biopsies from index patient A, who was described previously (1), were also collected at three time points. Sample collection was conducted from 1 April to 28 April 2003 in local hospitals by a previously described procedure (6). Collected samples were stored at 4°C prior to RNA extraction, and viral RNAs were extracted within a few days of arrival. Briefly, total RNA extraction from clinical samples was performed with the SV Total RNA isolation system and the SV96 Total RNA isolation system (Promega, Madison, Wis.), with the following modifications to the manufacturer's protocol. Five hundred microliters of an NPA or stool sample in viral transport medium (containing, per liter, 2 g of sodium bicarbonate, 5 g of bovine serum albumin, 200 μg of vancomycin, 18 μg of amikacin, and 160 U of nystatin in Earle's balanced salt solution) was mixed with an equal volume of SV RNA lysis buffer containing 100 μl of pig kidney epithelial (PK-15) cells (ATCC CCL-33; 5.0 × 105 cell/ml) in complete minimum essential medium with Earle's salt (Invitrogen) as an internal control. The mixture was transferred to the wells of an SV96 binding plate or an individual binding tube for the SV RNA isolation system. After being washed with 500 μl of SV RNA wash solution prior to the elution step, the plate or tube was spun at 3,000 × g for 30 s to remove any residual wash solution. The RNA was then eluted with 50 μl of nuclease-free water and was collected in a clean 96-well PCR plate or 1.5-ml microcentrifuge tube by spinning of the plate or tube at 3,000 × g for 1 min. The eluted RNA was then concentrated by incubation on ice for 15 min in the presence of 5 μl of 3 M sodium acetate and 200 μl of 95% ethanol. After centrifugation at 3,000 × g at 4°C for 15 min, the RNA pellet was washed with 200 μl of 75% ethanol and dissolved with 12 μl of nuclease-free water. Extracted RNAs were immediately reverse transcribed to first-strand cDNAs.
First-strand cDNA synthesis and PCR.
Reverse transcription was performed with 200 U of Superscript II RT (Invitrogen) in a 20-μl reaction containing 0.15 μg of random hexamers, 1× RT buffer, 10 mM dithiothreitol, and 0.5 mM deoxynucleoside triphosphates. Reactions were incubated in a Peltier thermal cycler (MJ Research) for 50 min at 42°C, followed by 15 min at 70°C. Primers were designed according to the complete SARS-CoV genomic sequence of a local specimen, isolate HK-39, which was described previously (12) (accession no. AY278491). The forward primer (SRS251 [5′-GCAGTCAAGCCTCTTCTCG-3′], corresponding to nucleotides [nt] 28658 to 28676 of the HK-39 SARS-CoV genome) and reverse primer (SRS252 [5′-GCCTCAGCAGCAGATTTC-3′], corresponding to nt 28866 to 28883) amplified a 225-bp fragment from the region of the N gene that showed no homology to other coronaviruses. Primers amplifying the RNA-dependent RNA polymerase gene (1b gene) were used as parallel controls (coro3 [5′-TACACACCTCAGCGTTG-3′] and coro4 [5′-CACGAACGTGACGAAT-3′], corresponding to nt 18041 to 18057 and nt 18207 to 18222, respectively [Department of Microbiology, HKU]). Both amplicons were cloned into the pCR2.1 cloning vector. Serial dilutions of the plasmid, from 106 to 10−1 copies, were used to determine the dynamic range and optimal conditions for the PCRs. Another set of primers that amplified a 745-bp fragment from the pig β-actin gene was employed as an internal control for the diagnostic PCR assay (actin F [5′-TGAGACCTTCAACACGCC-3′] and actin R [5′-ATCTGCTGGAAGGTGGAC-3′]). A conventional PCR and gel electrophoresis were performed as a preliminary experiment. Briefly, 1 μl of cDNA from a clinical sample was amplified with 0.5 U of recombinant Taq DNA polymerase (Invitrogen Life Technologies) in a 25-μl reaction mixture containing 1× PCR buffer, 1.5 mM MgCl2, 0.1 mM deoxynucleoside triphosphates, and 0.5 pmol each of the forward and reverse primers. The reaction was incubated in a Peltier thermal cycler (MJ Research) for 3 min at 94°C, followed by 50 cycles of 94°C for 10 s, 56°C for 10 s, and 72°C for 10 s and a 10-min final extension step at 72°C. Amplicons were analyzed by 2% agarose gel electrophoresis. Quantitative real-time PCR using the Sybr Green fluorophore was performed for the diagnosis of clinical samples. In a 25-μl reaction, 1 μl of a cDNA template was mixed with 12.5 μl of 2× SYBR Green PCR master mix (Applied Biosystems) and 0.5 pmol each of the forward and reverse primers. The volume of the reaction was adjusted to 25 μl with distilled water. Reactions were performed with the iCycler iQ Real-Time PCR detection system (Bio-Rad) for 3 min at 94°C, followed by 50 cycles of 94°C for 10 s, 56°C for 10 s, and 72°C for 10 s. Fluorescence signals (FAM; excitation wavelength = 490 nm and emission wavelength = 530 nm) were collected at the end of each extension step during the PCR cycles. The threshold cycle (CT) of each sample was determined by the maximum curvature approach. Melting curve analysis was performed after a 10-min final extension. cDNAs from non-SARS patients, including patients suffering from adenovirus (n = 5), respiratory syncytial virus (n = 5), human metapneumovirus (n = 5), influenza A virus (n = 5), and influenza B virus (n = 5) infections, were used as negative controls for the assay.
Northern blot analysis.
SARS-CoV HK-39-infected Vero cells were provided by the Department of Microbiology, HKU, and the total RNA was extracted from cells by use of the TRIzol reagent (Invitrogen Life Technologies) according to the manufacturer's protocol. The total RNA (8 μg) was separated by electrophoresis in a 1% agarose gel containing 3.7% formaldehyde, transferred to a positively charged nylon membrane (Roche Diagnostic Corporation) by capillary blotting, and fixed by UV cross-linking. The cDNA synthesized with the same RNA sample was used as a template for probe synthesis. Four pairs of primers, amplifying fragments from the 1b gene (nt 18057 to 18222), the S gene (nt 21920 to 22107), the M gene (nt 26867 to 26996), and the N gene (nt 28658 to 28883), were used for probe synthesis (Table 1). Digoxigenin labeling of the probes and hybridization and detection of the bands were performed with the digoxigenin system, used according to the manufacturer's procedures (Roche Molecular Biochemicals). The signals were then analyzed by chemiluminescence.
TABLE 1.
DNA probes used for Northern blot analysis
| Probe | Region (nt) | Sequence |
|---|---|---|
| 1b | 18057-18222 | GATATAAAATTCAAGACTGAAGGATTATGTGTTGACATACCAGGCATACCAAAGGACATGACCTACCGTAGACTCATCTCTATGATGGGTTTCAAAATGAATTACCAAGTCAATGGTTACCCTAATATGTTTATCACCCGCGAAGAAGCTATTCGTCACGTTCGTG |
| S | 21920-22107 | CATGGGTACACAGACACATACTATGATATTCGATAATGCATTTAATTGCACTTTCGAGTACATATCTGATGCCTTTTCGCTTGATGTTTCAGAAAAGTCAGGTAATTTTAAACACTTACGAGAGTTTGTGTTTAAAAATAAAGATGGGTTTCTCTATGTTTATAAGGGCTATCAACCTATAGATGTAG |
| M | 26867-26996 | GCTGTGACATTAAGGACCTGCCAAAAGAGATCACTGTGGCTACATCACGAACGCTTTCTTATTACAAATTAGGAGCGTCGCAGCGTGTAGGCACTGATTCAGGTTTTGCTGCATACAACCGCTACCGTAT |
| N | 28658-28883 | GCAGTCAAGCCTCTTCTCGCTCCTCATCACGTAGTCGCGGTAATTCAAGAAATTCAACTCCTGGCAGCAGTAGGGGAAATTCTCCTGCTCGAATGGCTAGCGGAGGTGGTGAAACTGCCCTCGCGCTATTGCTGCTAGACAGATTG |
RESULTS AND DISCUSSION
A 96-well RT-PCR assay will provide a high-throughput platform for the monitoring and screening of suspected SARS patients and thus can be used to complement clinical diagnostic evaluations. To fulfill its diagnostic purpose, the assay should be reliable and its accuracy should be assured in order to prevent the occurrence of both false-negative and false-positive results. However, the accuracy of the test may be influenced by several factors. A common technical problem with PCRs is amplification failure due to the presence of PCR inhibitors. PCR inhibitors are commonly found in most biological samples containing heme compounds, including blood, aqueous and vitreous humors, heparin, EDTA, urine, and polyamines (2). For this study, NPA or stool samples were collected into transport medium to maintain the viability of the viral particles. Since inhibition of RT-PCRs was observed when total RNAs were used directly after extraction for first-strand cDNA synthesis without any treatment (25 of 27 samples) in a preliminary experiment (data not shown), total RNAs eluted from both an SV binding column and an SV96 binding plate were precipitated with 95% ethanol and 3 M sodium acetate and resuspended in 12 μl of nuclease-free water prior to first-strand cDNA synthesis. The results showed that amplification of the DNAs could be retained after this simple ethanol precipitation step. Positive results were obtained when we used either the SV or SV96 total RNA isolation system (data not shown). These results demonstrate that components either in the medium or in NPA or stool samples could affect the downstream process of the diagnostic test.
In addition, the current sample collection procedure involves dilution of the virus titer in the samples. This step compromises the sensitivity of the assay because the virus titer is generally low in nasal and throat swab specimens, especially during the early stage of infection (1). It has been suggested that the sensitivity of PCR tests for SARS-CoV depends on the quality of the specimen and the time of testing during the course of the illness (10). In order to increase the sensitivity of the test, we concentrated the total RNAs isolated from clinical samples prior to first-strand cDNA synthesis.
In order to avoid false-negative PCR results due to a failure during the process of RNA isolation and first-strand cDNA synthesis, total RNAs were extracted from clinical samples in parallel with viable PK-15 pig cells. Figure 2 shows the RT-PCR screening results for both NPA and stool clinical samples. Diagnostic PCRs were performed in parallel with β-actin as a PCR control. All clinical samples were positive by the β-actin PCR. These results indicate that RNAs and cDNAs were extracted and synthesized successfully from the samples in a single-step protocol. With this amplification control, total RNA isolation and cDNA synthesis from the samples can be ensured and false negatives can be eliminated. Moreover, the 96-well assay format developed for this study was adapted into a high-throughput screening protocol, with which we were able to obtain diagnostic results for >90 clinical samples in 3 h with one clinical technician, while the current protocol, for which samples are processed in individual tubes, can only handle about 30 to 50 samples per day per technician.
FIG. 2.

RT-PCR screening of clinical samples from suspected SARS patients. The upper bands in each row show a 745-bp DNA fragment amplified with actin F and actin R, while the lower bands are the amplicons by primers specific to the N gene of SARS-CoV (225 bp). cDNA samples synthesized from total RNAs extracted from NPA samples (A1 to H1) were used as templates in both N-gene-specific and β-actin-specific PCRs. The negative control (water) and positive control (cDNA from SARS-CoV-infected Vero cells) for the assay are indicated. Five-microliter samples of PCR products from two separate reactions, i.e., N-gene-specific PCR and β-actin-specific PCR, were mixed and loaded into the same well in a 2% agarose gel. M, 1 kb Plus molecular marker (Invitrogen).
Real-time quantitative PCR is more sensitive than conventional agarose gel electrophoresis-associated PCR (5) and was therefore employed for the purpose of SARS-CoV diagnosis. Positive signals were detected from 38 of 136 randomly selected clinical samples in both N-gene- and 1b-gene-specific PCRs. Among these 38 positive samples, 3 were stool samples (2.2%) and 35 were NPA samples (25.7%). The detection rates of the assay employing N-gene-specific RT-PCR at different time points are shown in Table 2. These 38 positive cases were confirmed by melting curve analysis of the PCR products. The specific Tms of the N gene and 1b gene PCR products (85.5 and 80.5°C, respectively) indicated that the target fragments were amplified in the reaction. The specificity of the assay was also validated with non-SARS patients samples, including samples from patients suffering from infections with adenovirus (n = 5), respiratory syncytial virus (n = 5), human metapneumovirus (n = 5), influenza A virus (n = 5), and influenza B virus (n = 5). All of these samples were negative by the RT-PCR assay (Fig. 3). These results indicate that the N-gene-specific RT-PCR assay is specific for SARS-CoV diagnosis.
TABLE 2.
Detection rates of N-gene-specific RT-PCR assay for SARS-CoV
| No. of days after onset of illness | No. of samples | No. of positive samples | Detection rate (%) |
|---|---|---|---|
| 1-2 | 15 | 2 | 13.3 |
| 3-4 | 17 | 4 | 23.5 |
| 5-6 | 15 | 4 | 26.7 |
| 7-8 | 13 | 5 | 38.5 |
| 9-10 | 9 | 4 | 44.4 |
| Negative control | 19 | 0 |
FIG. 3.

Specificity test for N-gene-specific PCR. An amplification plot of fluorescence intensity versus the number of PCR cycles is shown. Black lines show the dynamic range of N-gene-specific PCR with a serially diluted plasmid construct (from 101 to 106 copies). Results for NPA samples from non-SARS patients, including patients suffering from infections with adenovirus (n = 5), respiratory syncytial virus (n = 5), human metapneumovirus (n = 5), influenza A virus (n = 5), or influenza B virus (n = 5), are shown by gray lines. Triangles, SARS-CoV-positive NPA samples. NTC, no-template control. The x axis indicates the cycle number of the quantitative PCR, while the y axis represents the fluorescence intensity (FAM-490) over the background signal. The inset shows a melting curve analysis of the PCR products. Signals from positive (+ve) and negative (−ve) samples and from a no-template control are indicated.
In addition, we have also demonstrated that the N-gene-specific PCR is more sensitive than the 1b-gene-specific PCR. The amplification conditions for both PCR assays were first optimized with the plasmid constructs containing a 1:1 ratio of 1b and N gene fragments. The dynamic range of the N-gene-specific PCR was obtained (Fig. 4), and it was found to have lower CT values than that of the 1b-gene-specific PCR. This reveals that the N-gene-specific PCR can achieve a higher amplification efficiency than the 1b-gene-specific PCR when the same copy number is used as a template. On the other hand, PCRs with cDNAs from clinical samples or virus-infected Vero cells were performed next. Figure 5 shows the CT and half-maximal values of the fluorescence signals generated by N-gene- and 1b-gene-specific PCRs with NPAs, tracheal dispersion fluid, and lung biopsies collected from patient A at three different time points, as described previously (1). The results indicate that the fluorescence signals given by N-gene-specific PCRs are higher (26.0% average; range, 6.3 to 60%) than those by 1b-gene-specific PCRs for all positive samples. In addition, the CT values of N-gene-specific PCRs were lower (0.1 to 4.6 cycles) than those of 1b-gene-specific PCRs among most of the SARS-CoV-positive samples (Table 3). Statistic analysis indicated that the CT of the N-gene-specific PCR assay was significantly lower than that of the 1b-gene-specific assay (95% confidence interval, 0.74 to 2.23, by the F test). The stronger fluorescence signals and lower CT values of N-gene-specific PCRs provide a more sensitive diagnostic result for the assay.
FIG. 4.

Comparison of dynamic ranges of N-gene- and 1b-gene-specific PCRs. The dynamic ranges of both N-gene- and 1b-gene-specific PCRs were obtained with the same plasmid construct into which a 1:1 ratio of the corresponding amplicons were subcloned. The serially diluted plasmid, with copy numbers ranging from 10−1 to 105 copies, was used as a template for both PCRs. Triangles, N-gene-specific PCR; gray lines, 1b-gene-specific PCR. The inset shows CT values for a triplicate set of experiments of both PCRs with different copy numbers of the template. NTC, no-template control. The x axis indicates the cycle number of the quantitative PCR, while the y axis represents the fluorescence.
FIG. 5.

Amplification curve (A) and melting curve (B) of real-time quantitative PCR specific to 1b and N gene of SARS CoV. (A) Amplification plot of fluorescence intensity versus the number of PCR cycles. One-microliter samples of cDNAs from NPA, tracheal dispersion, and lung biopsy samples from patients with clinical symptoms were used as templates for PCRs. Fifty cycles of PCR were performed to achieve the saturation phase of the reaction. The x axis indicates the cycle number of the quantitative PCR, while the y axis represents the fluorescence intensity (FAM-490) above the background signal. The horizontal gray line indicates the threshold value calculated by the maximum curvature approach, and the baseline cycle CT was calculated automatically. The inset shows half-maximal fluorescence values (1/2max) and CT values of both PCRs with cDNAs from various tissues isolated from a key patient (patient A [1]) at three different time points. TW, tracheal wash; LW, lung wash. (B) Melting curves of PCR products. Melting curve analysis was performed after a 10-min further extension step. The Tm rose from 56 to 94°C for 76 0.5-s steps, while each set-point temperature was held for 7 s for data collection and analysis. The Tms of the 1b- and N-gene-specific PCR products were 80.5 and 85.5°C, respectively. The x axis indicates the temperature in degrees Celsius, while the y axis represents the fluorescence intensity (FAM-490) above the background signal. One microliter of water was used as a no-template control in the reaction.
TABLE 3.
Comparison of threshold cycles of 38 SARS-positive samples in N-gene- and 1b-gene-specific PCR assays
| Isolate |
Ct for PCR
|
ΔCta | |
|---|---|---|---|
| N | 1b | ||
| 56851 | 27.1 | 27.8 | 0.6 |
| 55751 | 27.6 | 27.7 | 0.1 |
| 62290 | 41.9 | 43.2 | 1.3 |
| 55531 | 41.0 | 43.1 | 2.1 |
| 55963 | 41.7 | 42.3 | 0.6 |
| 65733 | 43.6 | 44.9 | 1.3 |
| 34862 | 33.5 | 33.7 | 0.2 |
| 32814 | 38.6 | 40.8 | 2.2 |
| 33935 | 35.3 | 36.5 | 1.2 |
| 34861 | 31.4 | 31.7 | 0.3 |
| 34862 | 31.9 | 33.2 | 1.3 |
| 45971 | 43.6 | 48.2 | 4.6 |
| 45972 | 43.6 | 46.5 | 2.9 |
| 45973 | 42.1 | 43.2 | 1.1 |
| 69145 | 27.2 | 27.7 | 0.5 |
| 56386 | 32.8 | 33.8 | 1.0 |
| 55527 | 37.3 | 39.4 | 2.1 |
| 56851 | 23.9 | 26.1 | 2.2 |
| 69073 | 24.3 | 26.1 | 1.3 |
| 67423 | 28.7 | 29.4 | 0.7 |
| 67429 | 35.3 | 35.6 | 0.3 |
| 67438 | 28.4 | 27.5 | −0.9 |
| 68116 | 30.0 | 33.5 | 3.5 |
| 68118 | 36.7 | 37.7 | 1.0 |
| 68134 | 32.4 | 33.2 | 0.8 |
| 68184 | 30.6 | 32.4 | 1.8 |
| 68185 | 27.5 | 30.1 | 2.6 |
| 68187 | 40.3 | 41.5 | 1.2 |
| 68788 | 35.5 | 37.6 | 2.1 |
| 68791 | 34.8 | 35.3 | 0.5 |
| 68796 | 32.4 | 34.5 | 2.1 |
| 68798 | 28.8 | 28.4 | −0.4 |
| 68800 | 34.6 | 38.3 | 3.7 |
| 68801 | 31.9 | 32.8 | 0.9 |
| 70562 | 40.2 | 43.3 | 3.1 |
| 70589 | 35.5 | 38.2 | 2.7 |
| 70591 | 36.0 | 38.2 | 2.2 |
| 70059 | 41.4 | 43.1 | 1.7 |
ΔCt, Ctlb − CtN; average ΔCt, 1.49 ± 0.47 (95% confidence intervals, 0.74 to 2.23, by the F test).
Using cDNAs from SARS-CoV-infected Vero cells, we generated the amplification curves in Fig. 4, which show the differences between N-gene- and 1b-gene-specific PCRs. The CT values of the N-gene- and 1b-gene-specific PCRs were 35.3 and 37.8, respectively. This phenomenon probably had two main causes: (i) the expression level of the N gene was higher than that of the 1b gene and (ii) the copy number of the N gene was much higher than that of the 1b gene because each transcript preceded a copy of the N gene in SARS-CoV-infected cells. Northern blot analysis supported this hypothesis. When an N-gene-specific PCR product was used as a probe, at least five transcripts from the virus were hybridized and gave positive signals (Fig. 6). This result agrees with the finding that five subgenomic mRNAs were detected by Northern hybridization of RNAs from SARS-CoV-infected cells by use of a probe derived from the 3′ untranslated region (7). On the other hand, when the 1b-gene-specific PCR product was used as a probe, only two transcripts with a large molecular size were hybridized, demonstrating that the copy number of the N gene was much higher than that of the 1b gene during transcription and gene expression in host cells. The Northern hybridization result strongly supports the conclusion that PCR-amplified regions in the N gene of SARS-CoV are more sensitive than other regions as a target for diagnostic screening. Since the molecular characterization of the novel SARS-CoV is ongoing, the targeting of genomic segments of the virus for diagnostic applications is still unclear. The amplification of more than one genome region may increase the specificity of the test (11).
FIG. 6.
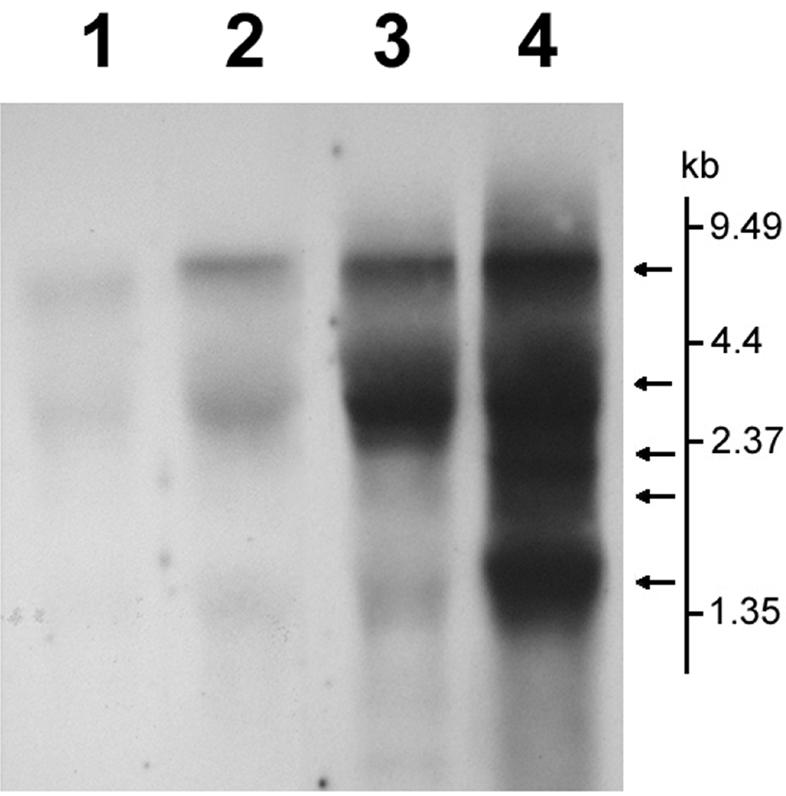
Northern blot analysis of SARS-CoV total RNA. The total RNA of SARS-CoV was extracted from SARS-CoV-infected Vero E6 cells. The RNA was separated in a 1% denaturing gel containing 6.29% formaldehyde. Afterwards, the RNA was transferred to a positively charged nylon membrane and hybridized with digoxigenin-labeled PCR fragments specific for the 1b, S, M, and N genes. Lane 1, 1b; lane 2, S; lane 3, M; lane 4, N. The vertical bar shows the molecular size reference. Arrows indicated the transcripts hybridized with the N probe. The signals were analyzed by chemiluminescence.
In conclusion, we have developed a new, sensitive, diagnostic RT-PCR test for the coronavirus associated with SARS. The assay provides a high-throughput, highly sensitive screening platform which enables us to test hundreds or thousands of suspected SARS cases each day in a single working line. The incorporation of PK-15 cells as an internal control in the assay and the use of the N gene as a diagnostic locus in addition to the 1b gene enhance the sensitivity and accuracy of the test. We are currently adapting the protocol for a 96-well single-step real-time quantitative PCR and sequencing format to shorten the time required for the test and to obtain information on genotypic variations of the virus.
Acknowledgments
This work was supported by Research Grant Council grant HKU 7553/03 M.
We thank C. C. Hon, Ken Y. C. Chow, Carol W. M. Chan, and Vince Y. Y. Li (Department of Zoology, HKU) for their efforts in the analysis of the genomic sequence of SARS-CoV strain HK-39. We thank Leo T. O. Lee (Department of Zoology, HKU) for his technical advice on real-time quantitative PCR. We also thank Leo L. M. Poon (Department of Microbiology, HKU) for providing us with clinical samples for this study.
REFERENCES
- 1.Drosten, C., S. Günther, W. Presier, S. Werf, H. Brodt, S. Becker, et al. 2003. Identification of a novel coronavirus in patients with severe acute respiratory syndrome. N. Engl. J. Med. 348:1967-1976. [DOI] [PubMed] [Google Scholar]
- 2.Fredricks, D., and D. Relman. 1998. Improved amplification of microbial DNA from blood cultures by removal of the PCR inhibitor sodium polyanetholesulfonate. J. Clin. Microbiol. 36:2810-2816. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Marra, M., S. Jones, C. Astell, R. Holt, A. Brooks-Wilson, et al. 2003. The complete genome sequence of SARS-associated coronavirus. Science 300:1377-1378. [DOI] [PubMed] [Google Scholar]
- 4.Peiris, J., S. Lai, L. Poon, Y. Guan, L. Yam, W. Lim, et al. 2003. Coronavirus as a possible cause of severe acute respiratory syndrome. Lancet 361:1319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Poon, L., K. Chan, O. Wong, W. Yam, K. Yuen, Y. Guan, D. Lo, and J. Peiris. 2003. Early diagnosis of SARS coronavirus infection by real time RT-PCR. J. Clin. Virol. 28:233-238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Poon, L., O. Wong, W. Luk, K. Yuen, J. Peiris, and Y. Guan. 2003. Rapid diagnosis of a coronavirus associated with severe acute respiratory syndrome (SARS). Clin. Chem. 49:953-955. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Rota, P., M. Oberste, S. Monroe, W. Nix, R. Campagnoli, J. Icenogle, et al. 2003. Characterization of a novel coronavirus associated with severe acute respiratory syndrome. Science 300:1394-1399. [DOI] [PubMed] [Google Scholar]
- 8.Tsang, K. W., P. L. Ho, G. C. Ooi, W. K. Yee, T. Wang, M. Chan-Yeung, W. K. Lam, W. H. Seto, L. Y. Yam, T. M. Cheung, P. C. Wong, B. Lam, M. S. Ip, J. Chan, K. Y. Yuen, and K. N. Lai. 2003. A cluster of cases of severe acute respiratory syndrome in Hong Kong. N. Engl. J. Med. 348:1977-1985. [DOI] [PubMed] [Google Scholar]
- 9.World Health Organization. 11. July 2003, posting date. Cumulative number of reported probable cases of severe acute respiratory syndrome (SARS) from 1 Nov 2002 to 11 July 2003, 17:00 GMT+2. [Online.] http://www.who.int/csr/sars/country/2003_07_11/en/.
- 10.World Health Organization. Use of laboratory methods for SARS diagnosis. [Online.] http://www.who.int/csr/sars/labmethods/en/.
- 11.Yam, W. C., K. H. Chan, L. L. M. Poon, Y. Guan, K. Y. Yuen, W. H. Seto, and J. S. M. Peiris. 2003. Evaluation of reverse transcription-PCR assays for rapid diagnosis of severe acute respiratory syndrome associated with a novel coronavirus. J. Clin. Microbiol. 41:4521-4524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Zeng, F., C. Chan, J. Chen, K. Chow, C. Hon, R. Hui, et al. 2003. The complete genome sequence of severe and acute respiratory syndrome coronavirus (SARS-CoV) strain HKU-39849 (HK39). Exp. Biol. Med. 288:866-873. [DOI] [PubMed] [Google Scholar]