

Abstract
Obscurins (∼70–-870 kDa), encoded by the single OBSCN gene, are cytoskeletal proteins originally identified in striated muscles with structural and regulatory roles. Recently, analysis of 13,023 genes in breast and colorectal cancers identified OBSCN as one of the most frequently mutated genes, implicating it in cancer formation. Herein we studied the expression profile of obscurins in breast, colon, and skin cancer cell lines and their involvement in cell survival. Immunoblot analysis demonstrated significant reduction of obscurin proteins in cancer cells, resulting from decreased mRNA levels and/or the presence of mutant transcripts. In normal epithelium, obscurins localize in cytoplasmic puncta, the cell membrane, and the nucleus. Accordingly, subcellular fractionation demonstrated the presence of 2 novel nuclear isoforms of ∼110 and ∼120 kDa. Nontumorigenic MCF10A breast epithelial cells stably transduced with shRNAs targeting giant obscurins exhibited increased viability (∼30%) and reduced apoptosis (∼20%) following exposure to the DNA-damaging agent etoposide. Quantitative RT-PCR further indicated that the antiapoptotic genes BAG4 and HAX1 were up-regulated (1.5- and 1.4-fold, respectively), whereas initiator caspase-9 and death caspase-3 transcripts were down-regulated (0.8- and 0.6-fold, respectively). Our findings are the first to pinpoint critical roles for obscurins in cancer development by contributing to the regulation of cell survival.—Perry, N. A., Shriver, M., Mameza, M. G., Grabias, B., Balzer, E., Kontrogianni-Konstantopoulos, A. Loss of giant obscurins promotes breast epithelial cell survival through apoptotic resistance.
Keywords: OBSCN gene, TP53 gene, MCF10, MCF7, MDA-MB-231
Accumulation of genetic alterations in oncogenes and tumor suppressor genes underlies the development and progression of cancer (1–3). Recently, a systematic analysis of 13,023 genes in breast and colorectal cancers identified 189 “candidate cancer genes” displaying somatic mutations at high frequency (4). OBSCN, which was originally believed to code for the muscle-specific obscurin proteins (5, 6), was identified as one of the 189 candidate cancer genes. Of those, only TP53 and OBSCN were common to both breast and colorectal cancers. While the presence of multiple mutations in the TP53 gene in both tumor types was unsurprising (7), the identification of somatic mutations in the OBSCN gene was unexpected and intriguing. Following these initial studies, Balakrishnan et al. (8) evaluated the mutational profiles of 19 candidate genes in 3 highly aggressive and lethal cancers: glioblastoma, melanoma, and pancreatic ductal adenocarcinoma. Their analysis of the OBSCN gene identified an additional somatic missense mutation in melanoma as well as a germline variant in glioblastoma, previously reported as a somatic missense mutation (4). Consistent with these findings, using whole human genome arrays combined with bioinformatics analysis, Price et al. (9) reported a single set of genes, consisting of OBSCN and C9orf65, a homologue of the Drosophila PRUNE2 gene, as a reliable 2-gene expression classifier that distinguishes between gastrointestinal stromal tumors (GISTs) and leiomyosarcoma (LMS) tumors. Taken together, these studies provided evidence of a potential role of the OBSCN gene in cancer predisposition and formation that was previously unsuspected.
The prototypical obscurin, obscurin-A, was identified in 2001 as the third giant protein, along with titin and nebulin, contributing to the structural organization and contractile activity of vertebrate striated muscles (10). The OBSCN gene spans 150 kb on human chromosome 1q42 and undergoes complex alternative splicing, giving rise to at least 4 isoforms (5, 6). Obscurin-A has a molecular mass of ∼720 kDa and a modular architecture composed of tandem adhesion and signaling motifs (10, 11). The NH2-terminal half of the molecule contains repetitive immunoglobulin (Ig) and fibronectin-III (FN-III) domains, while its COOH-terminal portion mainly consists of signaling domains, including an isoleucine- and glutamine-containing calmodulin-binding domain motif, a src-homology-3 domain, and tandem Rho-guanine nucleotide exchange factor (Rho-GEF) and pleckstrin homology motifs, interspersed by unique, nonmodular sequences. In addition to obscurin-A, OBSCN gives rise to another giant form of the protein, obscurin-B (5, 6). Obscurin-B has a molecular mass of ∼870 kDa and also contains 2 putative serine/threonine kinase domains (SKI and SKII), which replace the nonmodular COOH terminus of obscurin-A. Notably, the 2 serine/threonine kinases may be expressed independently as smaller forms, containing one (∼70 kDa) or both (∼160 kDa) kinase domains (12).
Obscurins interact with diverse protein partners located in distinct subcellular compartments within muscle cells, including cytoskeletal components (13–17), internal membrane proteins (18, 19), and signaling molecules (20–22). More important, gain- or loss-of-function approaches using cell culture systems and animal models have clearly demonstrated that obscurins play pivotal roles in the assembly, maintenance, and contractile activity of striated muscle cells, through their dynamic interactions with other muscle proteins (23–29).
In the current study, we sought to investigate the expression profile of obscurins in normal and cancer epithelial cells and their involvement in the regulation of cell survival. Obscurins are virtually absent from breast, skin, and colon cancer cells, but abundantly expressed in their normal counterparts, where they exhibit cytosolic, membrane, and nuclear distributions. Using multiple means, we demonstrate that down-regulation of the giant obscurins in MCF10A breast epithelial cells results in significantly enhanced viability and reduced apoptosis on exposure to the DNA-damaging agent etoposide. Our studies are the first to document important and novel functions of the OBSCN gene that extend beyond the organization and activity of muscle cells, and to suggest that obscurins may contribute to tumor suppression, a property that is lost in cancer cells due to their reduced or altered expression.
MATERIALS AND METHODS
Unless otherwise noted, all chemicals were purchased from Sigma-Aldrich (St. Louis, MO, USA).
Human cell lines and culturing conditions
The following human cell lines were purchased from American Type Culture Collection (ATCC; Manassas, VA, USA) and maintained at 37°C, 5% CO2: MCF10A (nontumorigenic breast epithelium), MCF7 (breast adenocarcinoma), MDA-MB-231 (breast adenocarcinoma), FHC (normal fetal colon epithelium), and LS174T (colorectal adenocarcinoma). SCC13 (skin squamous carcinoma) and primary cultures of normal skin keratinocytes were kindly provided by Dr. Gautam Adhikary and Dr. Richard L. Eckert (Department of Biochemistry and Molecular Biology, University of Maryland School of Medicine). Cell lines were cultured according to established protocols from ATCC. SCC13 cells were cultured in DMEM medium containing 10% FBS, and primary keratinocytes were isolated and maintained as described previously (30).
Generation of protein lysates and Western blotting
Cell lysates were prepared in radio immunoprecipitation assay (RIPA) buffer supplemented with protease inhibitors (Roche, Mannheim, Germany). Lysates were electrophoresed on SDS-NuPAGE gel (Invitrogen, Carlsbad, CA, USA) and transferred to nitrocellulose membranes. Primary antibodies used in this study include homemade obscurin-COOH (300 ng/ml; ref. 19), obscurin-Rho-GEF (300 ng/ml; ref. (15), and obscurin-kinase (150 ng/ml; ref. 31), as well as commercial antibodies to β-actin (Santa Cruz Biotechnology, Santa Cruz, CA, USA), lamin-B (Thermo-Fisher, Waltham, MA), glyceraldehyde-3-phosphate dehydrogenase (GAPDH; Applied Biosystems, Carlsbad, CA), CoxII (Molecular Probes, Carlsbad, CA, USA), BAG-4 (Epitomics, Burlingame, CA, USA), HAX-1 (Becton Dickinson, Franklin Lakes, NJ, USA), poly-ADP-ribose-polymerase (PARP) 1/2 (Santa Cruz Biotechnology), and caspases 3 and 9 (Cell Signaling, Danvers, MA, USA). Alkaline phosphatase-conjugated anti-mouse or anti-rabbit IgG (1:4,000; Jackson ImmunoResearch, West Grove, PA, USA) were used, and immunoreactive bands were visualized with a chemiluminescence detection kit (Applied Biosystems).
RNA isolation, cDNA synthesis, and PCR amplification
Total RNA was isolated from cultured cells with the TRIzol reagent (Invitrogen) and reverse transcribed using the SuperScript III First-Strand Synthesis Kit (Invitrogen). PCR amplification reactions were performed with the High Fidelity Platinum PCR SuperMix (Invitrogen), using the following primer sets: sense primer a, 5′-ATGGATCAGCCACAGTTC-3′, and antisense primer a′, 5′-AGCGCACTTGCCTCGCCGAT-3′, amplified the region between bp 45 and 550; sense primer b, 5′-GGCTACCACGTGCTGACCCT-3′, and antisense primer b′, 5′-ACCCTCTGTGAACTTGGC-3′, amplified the region between bp 9219 and 10,409; sense primer c, 5′-CCGCTTCCGTGTGGCAGCTGT-3′, and antisense primer c′, 5′-TGACCCACTGACCACCA-3′, amplified the region between bp 13,821 and 15,017; sense primer d, 5′-GGCCTGCCTAAGGTGGAG-3′, and antisense primer d′, 5′-GTGCCCGTCACGCGGCAGGCCAG-3′, amplified the region between bp 16,446 and 18,164; sense primer e, 5′-GCTGGTGAACCGGCTGGGCTCCGCGC-3′, and antisense primer e′, 5′-TCAGGGCTGTGCCGCCTCATCCCCATC-3′ amplified the region between bp 18,593 and 19,907; sense primer e and antisense primer e″: 5′-GGTGGAGAACGCCATGGCTG-3′ amplified the region between bp 18,593 and 19,800; and sense primer f, 5′-CCCTACAGCAGCCCC-3′, in combination with antisense primer f′, 5′-GCGCACCTGGGCCAG-3′, amplified the region between bp 22,950 and 23,950. All amplicons were subcloned into the StrataClone PCR cloning vector pSC-A (Stratagene, Santa Clara, CA, USA) and sequenced.
Inhibition of lysosomal degradation
MCF7 cells were seeded in 6-well plates and grown to ∼70% confluency. Cells were treated with 100 μM chloroquine diphosphate or vehicle control and collected at various time points, harvested in RIPA buffer, and immunoblotted, as above.
Nuclear isolation
Cells grown on 150-mm culture dishes were washed in PBS and harvested in ice-cold homogenization buffer containing 10 mM HEPES (pH 7.9), 1.5 mM MgCl2, 10 mM KCl, and 0.5 mM DTT, supplemented with protease and phosphatase inhibitors. Homogenates were incubated on ice for 5 min in the presence of 0.3% Nonidet P-40. Sucrose was added to a final concentration of 0.5 M, and lysates were loaded on top of a 1.6 M sucrose cushion and fractionated at 30,000 g. Proteins in the cytosolic (supernatant) fraction were extracted by adding 1% Nonidet P-40, 0.5% sodium deoxycholate, and 0.1% SDS on ice for 3 h. Isolated nuclei (pellet) were resuspended in RIPA buffer, and proteins were extracted on ice for 3 h with occasional vortexing. Nuclear and cytosolic fractions were subsequently centrifuged at 16,100 g for 30 min at 4°C, and the supernatants were used for immunoblotting, as described above.
Immunofluorescence combined with confocal microscopy
Cells were fixed with 2% paraformaldehyde, permeabilized with 0.1% Triton X-100 in PBS containing 1 mg/ml BSA (PBS/BSA), and blocked with 5% normal goat serum. Cells were subsequently incubated with primary antibodies overnight, including rabbit polyclonal obscurin-COOH (3 μg/ml; ref. 19) and obscurin-kinase (3 μg/ml; ref. 31), guinea pig obscurin-Rho-GEF (3 μg/ml; ref. 15), and mouse monoclonal obscurin-NH2 (3 μg/ml; ref. 11), as well as antigiantin (Abcam). Alexa Fluor-568 goat anti-rabbit IgG, Alexa Fluor-488 goat anti-mouse IgG, or Cy5 goat anti-guinea pig IgG (Molecular Probes) served as secondary antibodies. Cells were analyzed using a LSM510 confocal microscope (Carl Zeiss AG, Jena, Germany) at ×63 view.
Cell transfection and generation of stable cell clones
A set of 4 short hairpin RNA (shRNA) constructs that specifically target the human OBSCN gene along with a set of 2 control (scrambled) shRNAs were commercially obtained (Origene Technologies, Rockville, MD, USA). Transfections of MCF10A cells were conducted using Lipofectamine 2000 (Invitrogen), and 2 μg/ml puromycin was added to the medium to select for stably transfected cells 3 d post-transfection. Stable clones expressing control shRNA-1, obscurin shRNA-2, obscurin shRNA-3, obscurin shRNA-4, or obscurin shRNA-5 plasmids were isolated and propagated in complete medium in the presence of 1.5 μg/ml puromycin. All experiments were performed with multiple clonal isolates from each stable cell line expressing control or obscurin shRNA. Similar results were obtained with all obscurin shRNAs tested.
Cell viability assay
MCF10A cells stably expressing control or obscurin shRNAs were seeded in quadruplicate in 96-well plates at a density of 5000 cells/well, treated with 50, 100, and 150 μM etoposide or dimethyl sulfoxide (DMSO; vehicle) for 48 h and immediately assayed or allowed to recover in etoposide-free medium for 72 h. Activated 2,3-bis-(2-methoxy-4-nitro-5-sulfophenyl)-2H-tetrazolium-5-carboxanilide (XTT) was added for 3 h, and absorbance was obtained at 450 nm. Viability of etoposide-treated samples was determined relative to DMSO vehicle control.
Anchorage-independent growth using soft agar assay
SeaPlaque agarose (Cambrex, East Rutherford, NJ, USA) was diluted to 0.8% in the appropriate culture medium containing 150 μM etoposide or DMSO vehicle, and 2 ml was added to each well of a 6-well plate. After cooling, 2 ml of 0.5% agarose containing 7500 cells/ml was added to each well and allowed to cool before plates were placed in a 37°, 5% CO2 incubator. Culture medium containing vehicle or etoposide was added gently on top of the agarose layers 24 h later and changed daily. Colony growth continued for 21 d, at which point colonies were stained with crystal violet and blindly counted per 10 fields. MCF7 cells were used as positive control for colony growth.
Cell cycle analysis
MCF10A cells expressing control or obscurin shRNAs were treated with 150 μM etoposide for 48 h. Adherent and floating cells were collected and fixed in 75% ethanol at −20°C overnight, treated with 1 mg/ml RNase, and resuspended in 20 μg/ml propidium iodide. Analysis was performed on a FACScan flow cytometer (BD Biosciences, San Jose, CA, USA), and data were analyzed with FlowJo software (TreeStar, Ashland, OR, USA) using the Watson algorithm.
Mitotic index determination
Stably transduced MCF10A cells with control or obscurin shRNAs were grown to ∼50% confluency and treated with 150 μM etoposide or DMSO for 48 h, then 500 ng/ml of nocodazole was added during the last 10 h to trap cells in mitosis. Floating and adherent cells were collected and fixed at −20°C with 75% ethanol overnight. Fixed cells were washed with PBS, followed by Stain Buffer (Dulbecco's PBS with 2% FBS and 0.09% NaN3; Becton Dickinson), and incubated for 20 min with phospho-serine-28-Histone-H3 (pS28-H3) antibody conjugated with Alexa-647 (Becton Dickinson). Following washing with Stain Buffer, cells were treated with RNase (1 mg/ml) and propidium iodide (10 μg/ml). G0/G1, S, and G2 stages of the cell cycle were determined based on DNA content measured with propidium iodide, and mitotic cells were identified as G2 DNA content cells with high levels of pS28-H3 staining. Cells were collected on a FACSCanto 2-laser, digital cytometer (BD Biosciences), and data were analyzed using the FlowJo software (TreeStar).
DNA laddering assay
DNA laddering was assessed by 1% agarose gel electrophoresis. Genomic DNA was extracted with the DNAeasy Tissue kit (Qiagen, Hilden, Germany) from parental MCF10A cells and stable cell clones expressing control or obscurin shRNAs, exposed to 150 μM etoposide for 48 h.
Flow cytometry and apoptosis
MCF10A cells stably expressing control or obscurin shRNAs were plated in 6-well plates and grown to 70% confluence. Cells were treated with 150 μM of etoposide or DMSO vehicle control in complete medium for 48 h. Adherent cells were detached, washed twice with PBS, and stained for apoptotic markers as described in the PE Annexin V Apoptosis Detection Kit I (BD Biosciences). Fluorescence-activated flow cytometry (FACS) analysis of ∼10,000 cells/trial was completed using a Becton Dickinson FACScan instrument, and early apoptotic cells were defined as those positive for annexin V staining and negative for 7-AAD dye incorporation.
cDNA microarrays and GO analysis
cDNA microarray experiments were performed in triplicate as described previously (32–34). Using the Trizol reagent (Invitrogen) coupled with RNeasy columns (Qiagen), total RNA was isolated from MCF10A cells stably expressing control or obscurin shRNA, following treatment with 150 μM etoposide for 48 h. RNA (10 μg) was then reverse transcribed using random hexamers and aminoallyl-dUTP using the Superscript III kit (Invitrogen). cDNA from control and obscurin shRNA-expressing cells was coupled to NHS Cy3 and Cy5, respectively, mixed, and hybridized to microarray slides printed with a set of 19,596 unique human RNAs (Agilent, Santa Clara, CA, USA). Expression levels from individual genes were determined from the scanned microarray slides using Genepix Pro (Molecular Devices, Sunnyvale, CA, USA), and the ratio of median channel intensities of the data was normalized to 1. Comparisons between the expression levels of genes in control and obscurin-knockdown cells were performed using the unpaired Student's t test and considered to be statistically significant at values of P < 0.05. Further microarray data analysis involved only statistically significant genes. The Institute for Genome Research Multiexperiment Viewer software (35, 36) and the annotations publicly available from the Gene Ontology Database (37) were used for classification of differentially regulated genes in specific biological processes. The complete results of the cDNA microarray analysis are available at the U.S. National Center for Biotechnology Information (NCBI) Gene Expression Omnibus, accession number GSE35494 (http://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE35494).
Quantitative RT-PCR
Total RNA was isolated from MCF10A cells stably transfected with control and obscurin shRNAs using the Trizol reagent following treatment with 150 μM etoposide, and reversed transcribed into cDNA using the iScript cDNA Synthesis kit (Bio-Rad, Hercules, CA, USA). cDNA was used in quantitative RT-PCR studies with the iQ SYBR Green kit (Bio-Rad) and MyiQ Real Time PCR Detection System (Bio-Rad). Primer sets that specifically amplified portions of GAPDH (reference gene), caspase-3, caspase-9, BAG-4, and HAX-1 variant-1 were generated according to the recommendations of the Real Time PCR Application Guide (Bio-Rad) and included the following: for GAPDH, sense primer, 5′-TGATGACATCAAGAAGGTGGTGAAG-3′, and antisense primer, 5′-TCCTTGGAGGCCATGTGGGCCAT-3′; for caspase-3, sense primer, 5′-GAGGCGGTTGTAGAAGAGTTTCGTG-3′, and antisense primer, 5′-CGCTAACTCCTCACGGCCT-3′; for caspase-9, sense primer, 5′-CGCCATGGACGAAGCGGAT-3′, and antisense primer, 5′-CCGCTGGATGTCCTCGATCA-3′; for BAG-4, sense primer, 5′-AGACCACCTGGCTGGGAGAA-3′, and antisense primer, 5′-ATGGTGGCTGCTCCTGGTG-3′; and for HAX-1 variant-1, sense primer, 5′-CGGAGCCACAGAGATCCCTT-3′, and antisense primer, 5′-GGCTGAAGCTGAAGCCGAAG-3′. PCR products were verified by 1% agarose gel electrophoresis and sequencing. Experimental efficiencies were calculated according to methods described by the manufacturer (Bio-Rad) and ranged between 90 and 100% for BAG-4, HAX-1, and GAPDH and 98–107% for caspase-3 and caspase-9. Data are reported as an average cycle threshold (Ct) value and as fold difference between control and obscurin shRNA-expressing cells, calculated using the 2−ΔΔCt method.
Reproducibility and statistics
All experiments were performed in triplicate, a minimum of 3 times. Data are presented as mean values of independent measurements, and statistical significance was assessed using Student's t test (P<0.005).
RESULTS
Obscurins exhibit reduced expression in epithelial cancers
Sequencing analysis identified OBSCN as one of the most prevalently mutated genes in several types of epithelial cancers (4). Given that OBSCN was originally considered muscle specific, this finding raised a number of questions about its expression profile and roles in normal and cancer epithelial cells. To this end, we examined the expression levels of the obscurin proteins and transcripts using immunoblotting and reverse transcription-polymerase chain reaction (RT-PCR), respectively.
Using an antibody against the COOH terminus (19) of giant obscurins A and B (Fig. 1A), we detected 5 major immunoreactive bands in normal breast (MCF10A), skin (primary keratinocytes), and colon (FHC) epithelial cells of ∼720 and ∼870 kDa, corresponding to obscurins A and B, respectively, and of ∼80, ∼110, and ∼160 kDa (Fig. 1B), which may represent smaller obscurin isoforms of yet unknown identity (5, 6, 11). Interestingly, obscurin proteins were markedly reduced (>70%) in breast (MCF7 and MDA-MB-231), skin (SCC13), and colon (LS174T) cancer cell lines, with giant obscurins being virtually absent in all cancer cell lines tested, with the exception of MDA-MB-231 (Fig. 1B).
Figure 1.

Obscurins are abundantly expressed in normal epithelium but nearly absent from cancer epithelium. A) Domain architecture of the giant obscurin isoforms A and B; the locations of epitopes used for generation of different obscurin antibodies and of primer sets used for amplification of select obscurin fragments are also depicted. B) Immunoblots for obscurins in normal (MCF10A, primary keratinocytes and FHC) and tumor breast (MCF7 and MDA-MB-231), skin (SCC13), and colon (LS174T) cell lines. Equal loading of homogenates was ensured by measuring protein concentration and probing for β-actin. Reduced expression to virtual absence of the giant obscurins A and B (arrows) was observed in all cancer cell lines tested, relative to their normal counterparts; moreover, decreased expression of the smaller forms of obscurin was detected, albeit at variable extents. C) Semiquantitative RT-PCR was employed to compare the expression levels of obscurin transcripts between normal and cancer cell lines, using primer sets aa′, bb′, cc′, dd′, ee′, ee′, and ff′, spanning the entire length of the longest obscurin mRNA, as shown in A. Use of equivalent amounts of cDNA was achieved by measuring absorbance and tested by amplifying a portion of β-actin. Obscurin transcripts were readily detected in all normal cell lines. However, a notable decrease in their amounts was observed in all cancer cell lines examined, with the exception of MCF7 cells, which exhibited a modest up-regulation of the obscurin transcripts. D) Treatment of MCF7 cells with 100 μM of chloroquine diphosphate, an inhibitor of lysosomal acidification and degradation, restored the levels of giant obscurins A and B at detectable levels. GAPDH was used as loading control.
To examine whether the decrease in the amounts of obscurins at the protein level corresponded to a similar reduction at the transcript level, we performed a series of semiquantitative RT-PCR assays, using primer sets (aa′–ff′) that spanned the entire length of the obscurin mRNA (Fig. 1A). Obscurin amplicons of the expected sizes were detected in all normal epithelial cell lines examined (Fig. 1C). However, a significant reduction in their amounts was observed in all cancer cell lines, compared to their normal counterparts, with the exception of MCF7 cells, in which a marked increase was apparent (Fig. 1C).
The simultaneous reduction of obscurins at the protein level and increase at the mRNA level in MCF7 cells suggested that obscurin transcripts may carry mutations that lead to the generation of altered or truncated forms of the protein that are unstable and thus susceptible to degradation. To test this possibility, we sequenced select segments of the 21- to 24-kb obscurin transcripts expressed in MCF7 and MCF10A cells, focusing on signaling domains and flanking Ig motifs present in the COOH terminus (Fig. 1A; fragments obtained with primer sets cc′, dd′, ee′, ee″, and ff′ were analyzed by sequencing). Our findings are shown in Supplemental Table S1 and include verification of 7 nucleotide changes that were previously identified (38), as well as documentation of 12 novel mutations present in MCF7, but not MCF10A, cells. Of those 12 mutations, 3 are synonymous, 7 are missense, 1 is nonsense, and another is an insertion of 38 bp that causes frame shift and premature termination. Consistent with the presence of mutations in the obscurin transcripts that may alter the expression profile and/or stability of the resulting proteins, treatment of MCF7 cells with chloroquine, an inhibitor of lysosomal acidification and degradation, restored the amounts of giant obscurins A and B to detectable levels (Fig. 1D), while treatment with the proteasomal inhibitor MG132 did not have any effect (data not shown).
Taken together, these results demonstrate the presence of different obscurin isoforms in normal epithelial cells, and their marked reduction or loss in their malignant counterparts. The decreased levels of obscurin proteins in cancer epithelium may result from reduced amounts of (e.g., MDA-MB-231, SCC13, and LS174T) or mutant (e.g., MCF7) obscurin transcripts.
Obscurins are present in diverse locations in normal breast epithelium
To examine the subcellular distribution of obscurins in nontumorigenic epithelium and gain insights about their activities, we focused our subsequent studies in the MCF10A breast epithelial cell line. Using a panel of antibodies targeted to different domains along the length of the obscurin proteins (Fig. 1A), we found that they are present in diverse subcellular compartments (Fig. 2A–D). Interestingly, all 4 antibodies, directed to the extreme NH2 terminus (Fig. 2A), the Rho-GEF domain (Fig. 2B), the COOH terminus (Fig. 2C), or the Ig domain preceding SKI (Fig. 2D; we refer to this antibody as kinase antibody, as it specifically recognizes obscurin isoforms that contain SKI or SKI and SKII), exhibited similar staining patterns, including perinuclear and cytosolic puncta (arrows), membrane accumulation (arrowheads), especially at sites of cell-cell contact, and nuclear localization (asterisks). To learn whether the perinuclear puncta codistribute with markers present in internal membrane systems, we performed a series of colocalization studies using the COOH-antibody in combination with markers to endosomes, endoplasmic reticulum, mitochondria, and Golgi. Only a coincident distribution with the Golgi marker giantin was readily detected (Fig. 2E–E″). Conversely, examination of the distribution of obscurins in MCF7 and MDA-MB-231 cells confirmed our immunoblot analysis, indicating that their expression levels are considerably reduced, and revealed a weak and diffuse cytosolic staining pattern, but no discernible structures (data not shown).
Figure 2.
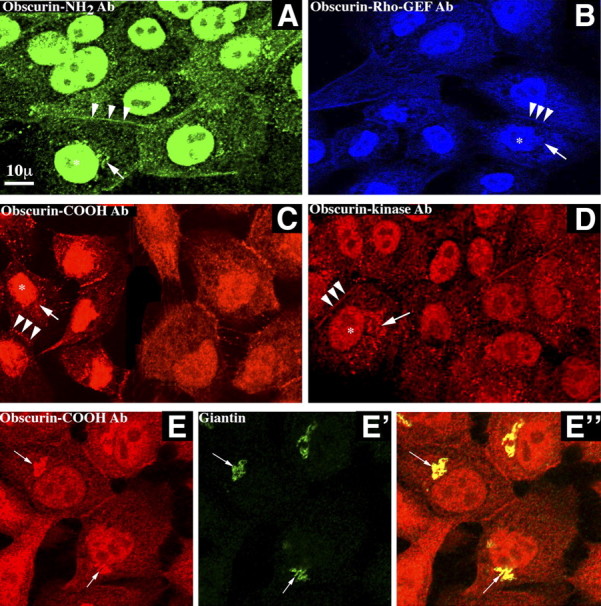
Obscurins localize to several subcellular compartments in normal breast epithelial cells. A–D) We used 4 different antibodies: obscurin NH2-terminal (A), obscurin Rho-GEF (B), obscurin COOH-terminal (C), and obscurin-kinase (D) to examine the distribution of obscurins in normal MCF10A breast epithelial cells. Antibodies in A–C recognize both giant obscurins A and B, while the kinase antibody in D recognizes obscurin-B as well as the smaller kinase-containing isoforms (see Fig. 1A for location of respective epitopes). Notably, all 4 antibodies exhibited similar staining patterns, indicating that obscurins are present at the cell membrane, and specifically at cell-cell junctions (arrowheads), the nucleus (asterisks), and in perinuclear/cytoplasmic puncta (arrows). E–E″) Perinuclear puncta of obscurins (E) colocalize with giantin (E′), a Golgi marker protein, as seen in the overlay image (E″). Scale bar = 10 μm.
Novel obscurin isoforms selectively concentrate in the nucleus of MCF10A cells
Our immunofluorescent data clearly demonstrated that some obscurins are present in the nucleus of MCF10A cells. To confirm this finding and identify the isoforms that may reside in the nucleus, we performed subcellular fractionation to isolate nuclear and cytosolic fractions from MCF10A cells, which we subsequently used in immunoblot analysis (Fig. 3). Using 3 different obscurin antibodies (Fig. 1A) directed against the Rho-GEF domain (Fig. 3A), the COOH terminus (Fig. 3B), and the Ig domain preceding SKI (Fig. 3C), we identified at least 2 distinct obscurin isoforms that were selectively enriched in the nuclear fraction of MCF10A cells. These novel isoforms exhibited molecular masses of ∼110 and ∼120 kDa. The ∼110-kDa isoform was detected by both the Rho-GEF (Fig. 3A, asterisk) and COOH terminus (Fig. 3B, asterisk) antibodies, whereas the ∼120-kDa isoform was detected only by the kinase antibody (Fig. 3C, arrowhead). Although the precise molecular identity of these novel isoforms is unknown, the ∼110-kDa isoform appears to contain COOH-terminal epitopes present in both obscurin A and B, while the ∼120-kDa isoform carries COOH-terminal epitopes present only in obscurin B (Fig. 1A).
Figure 3.

Novel isoforms of obscurin are present in the nucleus of breast epithelial cells. A–C) MCF10A cells were fractionated into nuclear and cytosolic extracts and immunoblotted with antibodies to the Rho-GEF domain (A), COOH-terminal region (B), and kinase domains (C). Giant obscurins A (∼720 kDa) and B (∼870 kDa) were entirely absent from the nuclear fraction, while 2 novel isoforms of ∼110 kDa (asterisk, containing the Rho-GEF and COOH-terminal epitopes) and ∼120 kDa (arrowhead, containing the kinase epitopes) were selectively enriched in the nuclear fraction. D) Purity of the obtained fractions was assessed by blotting for nuclear (top panel, lamin-B), mitochondrial (middle panel, CoxII) and cytosolic (bottom panel, GAPDH) markers. Neither CoxII nor GAPDH was detected in the nuclear fraction, while only trace amounts of lamin B were found in the cytosolic extract.
Down-regulation of giant obscurins A and B enhances survival and reduces apoptosis in MCF10A cells treated with the DNA-damaging agent etoposide
The apparent reduction or absence of obscurins A and B in epithelial cancer cell lines prompted us to examine their roles in cell survival and apoptosis. To this end, we generated stable MCF10A cell lines expressing shRNA plasmids targeting obscurins A and B. All shRNAs directed against obscurins (i.e., shRNA-2 targeting Ig24, shRNA-3 targeting Ig51, shRNA-4 targeting Ig9, and shRNA-5 targeting Ig45) efficiently reduced the amounts of giant obscurins, while control (scramble) shRNA-1 had no effect (Fig. 4A and Supplemental Fig. S1A).
Figure 4.

Knockdown of the giant obscurin isoforms increases survival of MCF10A cells following exposure to the DNA damaging agent, etoposide. A) MCF10A cells were stably transfected with shRNA-2, targeting Ig24, or control (scramble) shRNA-1. shRNA-2 is able to effectively down-regulate the expression of giant obscurins A and B; GAPDH serves as loading control. B) MCF10A cells expressing control shRNA-1 or obscurin shRNA-2 were seeded at a density of 5000 cells/well of 96-well plates, and treated with 50, 100, and 150 μM etoposide or DMSO vehicle control for 48 h. Viability was assessed using the XTT assay, which detects metabolic activity. Blanked absorbance values of vehicle control cells were normalized to 1, and viability of etoposide treated cells was expressed as a percentage of that of vehicle control cells. Etoposide treatments of 100 and 150 μM resulted in statistically significant differences in viability between obscurin-deficient and control cells. Error bars = sd. *P < 0.005; t test. C) Phase-contrast images of shRNA-1 and shRNA-2 stable clones were obtained under a ×10 objective in the absence or presence of etoposide. Major morphological disruptions and cell death are apparent in control shRNA-1 cells on etoposide treatment, while obscurin shRNA-2-expressing cells are not affected as dramatically.
To examine whether down-regulation or absence of the giant obscurins provides the cells with a survival advantage under stress conditions, we subjected MCF10A cells transduced with control shRNA-1 or obscurin shRNA-2 to DMSO vehicle or etoposide (50–150 μM) treatment for 48 h and used the XTT assay to determine cell viability (Fig. 4B). Stable MCF10A clones expressing obscurin shRNA-2 showed significantly higher viability compared to clones expressing control shRNA-1, which depended on the etoposide dose and ranged between ∼15% at 50 μM, ∼20% at 100 μM, and ∼30% at 150 μM. Consistent with these findings, phase contrast images of shRNA-1- and shRNA-2-treated cells indicated that the former exhibited dramatic morphological disruptions, ultimately leading to increased cell death on etoposide treatment (Fig. 4C). To further demonstrate the validity of our findings, we performed XTT viability assays using MCF10A cells stably transfected with obscurin shRNAs 3-5. In all cases, down-regulation of giant obscurins provided cells with a similar survival advantage on etoposide exposure (Supplemental Fig. S1B, C), compared to control cells.
The ability of cells to proliferate in anchorage independent conditions, in the absence of extracellular matrix and cell-cell contact derived cues, is a classic marker of tumorigenicity. Given the enhanced viability that obscurin-deficient MCF10A cells demonstrated when adhered to a hard substratum, we next examined their ability to survive and grow in anchorage-independent conditions (Fig. 5). While MCF10A cells do not generally form colonies in soft agar, down-regulation of giant obscurins was sufficient to allow the growth of small, yet discernible colonies in the absence or presence of 150 μM etoposide (Fig. 5A, bottom panels); to the contrary, the foci observed in control cells were invariably single cells (Fig. 5A, top panels). Consistent with this, obscurin shRNA-2 treated cells formed twice as many colonies in soft agar compared to control shRNA-1-transduced cells (Fig. 5B).
Figure 5.

Down-regulation of giant obscurins A and B allows MCF10A cells to survive and grow in anchorage-independent conditions. A) Equal numbers of MCF10A cells expressing control shRNA-1 or obscurin shRNA-2 were plated in 0.5% agarose in the presence of DMSO vehicle control or 150 μM etoposide, allowed to grow for 21 d, and stained with crystal violet. Representative images (×4 view) of cells expressing control shRNA-1 (top panels) or obscurin shRNA-2 (bottom panels) without and with etoposide are shown. B) After crystal violet staining, the numbers of foci per 10 fields of view were blindly counted for each experimental condition. Error bars = sd. *P < 0.005; t test.
Etoposide is a known carcinogen that induces DNA damage resulting in cell cycle arrest at the G1 checkpoint in cells expressing wild-type p53 (39, 40), such as MCF10A. We therefore next examined whether down-regulation of giant obscurins altered the cell cycle of MCF10A cells on treatment with etoposide. To this end, we exposed shRNA-1- and shRNA-2-expressing cells to 150 μM etoposide for 48 h (Fig. 6A), stained them with propidium iodide, and processed them for FACS analysis. Evaluation of their DNA content revealed no significant differences in the cell cycle distribution between control and obscurin-knockdown cells (Fig. 6B), indicating that loss of obscurins did not allow MCF10A cells to bypass the G1 DNA-damage checkpoint. Similarly, when tested for the accumulation of mitotic cells in the presence of nocodazole, both control and obscurin-null cells were effectively arrested in the presence of etoposide, with no cells entering mitosis (Fig. 6C, D). Consequently, the increased survival we observed in obscurin-knockdown cells in the XTT assays was not mediated by their increased proliferative capacity. However, we did note a modest, yet reproducible, decrease in the sub-G1 population in cells transduced with obscurin shRNA-2 (Fig. 6B), suggesting that a lower percentage of obscurin-deficient cells died in the presence of etoposide, compared to control shRNA-1-expressing cells. We therefore hypothesized that the increased viability of MCF10A cells that lack the giant isoforms of obscurin was due to enhanced apoptotic resistance.
Figure 6.

Knockdown of giant obscurins does not allow MCF10A cells to escape DNA damage-induced cell cycle arrest. A) MCF10A cells expressing control shRNA-1 (gray) or obscurin shRNA-2 (black) were treated with 150 μM etoposide, and DNA content was analyzed 48 h later. DNA content of n indicates cells in G1, and 2n indicates cells in G2/M. B) Percentage of cells from A in each cell cycle phase. No statistically significant difference (P>0.005; t test) was observed. C, D) Etoposide-treated control shRNA-1-expressing (C) and obscurin shRNA-2-expressing cells (D) cells were also treated with nocodazole, and the number of mitotic cells was counted; no mitotic cells were detected in either cell population.
To test this hypothesis, shRNA-1- and shRNA-2-transduced clones were treated with 150 μM etoposide for 48 h, and their apoptotic levels were evaluated by measuring externalization of phosphatidylserine by FACS analysis. A set of representative images is included in Fig. 7A, and bar graphs generated from averaging 3 independent experiments are shown in Fig. 7B. MCF10A cells expressing obscurin shRNA-2 exhibited significantly diminished levels of apoptosis compared to cells expressing control shRNA-1 (Fig. 7B; ∼10% and 30%, respectively). Notably, obscurin-deficient MCF10A cells showed no difference in their apoptotic levels in the absence or presence of etoposide (Fig. 7B; ∼8% and ∼10%, respectively), whereas shRNA-1-expressing cells exhibited increased apoptotic levels in response to etoposide (Fig. 7B; ∼4 to ∼30%). DNA laddering further confirmed these results (Fig. 7C), demonstrating that shRNA-1-transduced cells exhibited significant genomic DNA cleavage following etoposide treatment, while shRNA-2-expressing cells showed a DNA pattern similar to untreated parental MCF10A cells.
Figure 7.

MCF10A obscurin-null cells resist apoptotic death following exposure to etoposide. A) Representative images from FACS analysis of MCF10A cells expressing control shRNA-1 or obscurin shRNA-2 cells following DMSO treatment or treatment with 150 μM etoposide for 48 h. Early apoptotic cells, present in the bottom right quadrant, were defined as those that stained positive for annexin V (indicating localization of phosphatidylserine to the outer leaflet of the cell membrane), but negative for 7-AAD (a DNA dye that indicates catastrophic damage to the plasma membrane that occurs during late apoptosis and necrosis). B) Comparison of the percentages of early apoptotic shRNA-1- and shRNA-2-expressing cells after treatment with DMSO or etoposide. MCF10A cells transduced with shRNA-2 exhibit lower apoptotic levels (∼10%) compared to cells transduced with control shRNA-2 (∼30%), after exposure to etoposide. Moreover, shRNA-1-expressing cells show a statistically significant increase in apoptosis following exposure to etoposide (∼26%), whereas shRNA-2-expressing cells do not (∼2%). Error bars = sd. *P < 0.005; t test. C) Genomic DNA was isolated from MCF10A parental, control shRNA-1, and obscurin shRNA-2-expressing cells following DMSO or etoposide exposure. A DNA laddering assay further confirmed the reduced susceptibility to apoptosis exhibited by the obscurin-deficient MCF10A cells, compared to controls.
Select antiapoptotic and proapoptotic genes are differentially regulated in obscurin-deficient MCF10A cells
The increased apoptotic resistance that MCF10A cells exhibited following down-regulation of obscurins prompted us to identify signaling pathways that may mediate this effect. To this end, we analyzed the transcriptome of obscurin-deficient MCF10A cells following exposure to 150 μM etoposide for 48 h compared to control cells using cDNA microarrays coupled with Gene Ontogeny profiling. The complete set of results has been deposited at the NCBI GEO Repository. Notably, a number of genes involved in apoptosis were differentially regulated on obscurin knockdown, including the antiapoptotic genes BCL2, BAG4, HAX1, and BAG3, as well as the executioner CASP3. We focused our analysis on these genes and used quantitative real-time PCR (qRT-PCR) to validate the observed expression changes. Determination of the Ct values of the above genes (Fig. 8A) revealed significant up-regulation of BAG4 (∼1.5-fold) and HAX1 variant-1 (∼1.4-fold) transcripts (Fig. 8B), but not of BCL2 and BAG3 (data not shown). Conversely, the transcript level of caspase-3 was significantly diminished, exhibiting a ∼0.6-fold reduction. We also tested for changes in the expression of CASP9, which activates CASP3 in the intrinsic apoptotic cascade, and found a modest yet significant reduction of ∼0.8-fold on abrogation of giant obscurins.
Figure 8.

Down-regulation of obscurins A and B results in altered expression of pro- and antiapoptotic genes. We used quantitative RT-PCR to determine the relative expression levels of BAG4, HAX1, CASP3, and CASP9. A) Number of PCR cycles required to reach threshold fluorescence intensity for each tested gene in both control shRNA-1 and obscurin shRNA-2 expressing cells. The reported Ct value for each gene was normalized to that of GAPDH, which served as reference gene. Error bars = sd. *P < 0.005; t test. B) Fold change of the expression levels of each transcript between shRNA-1- and shRNA-2-transduced cells, subjected to etoposide treatment, was calculated with the 2−ΔΔCt method.
To further confirm these findings, we used immunoblot analysis to examine the expression levels of the respective proteins (Supplemental Fig. S2). Consistent with our qRT-PCR data, the amounts of BAG4 protein were significantly elevated in shRNA-2 compared to shRNA-1-transduced cells; however, we were unable to detect an increase in the levels of the HAX1 variant-1 protein (see Discussion). Moreover, the levels of cleaved caspases 9 and 3 (i.e., active forms; Supplemental Fig. S2, arrowheads) were lower in shRNA-2 compared to shRNA-1-transduced cells. Consistent with this, poly-ADP-ribose-polymerase (Supplemental Fig. S2, PARP, arrow), a substrate of caspase-3, was cleaved to a lesser extent in shRNA-2 compared to shRNA-1 stable clones (Supplemental Fig. S2, double arrow), further validating that obscurin deficiency reduces susceptibility to apoptosis.
DISCUSSION
In the present study, we examined the expression profile and properties of giant obscurins A and B in normal and cancer mammary epithelium. To date, the giant forms of obscurin have been studied extensively in striated muscles, where they have been implicated in myofibril assembly and stabilization as well as in anchoring the contractile cytoskeleton to the surrounding sarcoplasmic reticulum membranes (for review, see ref. 11). Consistent with this, a single missense mutation in Ig48 has been causally linked to the development of hypertrophic cardiomyopathy in humans (41). Despite obscurins' critical roles in normal muscle function, little is known regarding their presence and roles in other tissues. Herein we report the expression of multiple obscurin isoforms in normal epithelial cells, contrary to the widely held notion that they are selectively present in striated muscles. More important, obscurins are virtually lost on malignant transformation of epithelial cells. Consistent with this, down-regulation of giant obscurins A and B provides a survival advantage to normal breast epithelial cells subjected to stress, by reducing their susceptibility to apoptosis. Based on the above evidence, we propose that obscurins may act as tumor suppressors in normal breast epithelium.
Our studies demonstrated the presence of minute amounts of obscurins in all cancer cell lines tested, compared to their nonmalignant counterparts, which may be attributed to the concomitantly reduced levels of their respective transcripts. Interestingly, MCF7 breast cancer cells contained increased amounts of obscurin transcripts compared to nontumorigenic MCF10A cells, despite the virtual absence of the encoded proteins. To explain this paradoxical observation, we hypothesized that the obscurin transcripts present in MCF7 cells may carry mutations that render the resulting proteins unstable and prone to rapid degradation. We therefore sequenced select portions of the obscurin transcripts present in MCF7 cells and compared them to those obtained from MCF10A cells. We confirmed the presence of several nucleotide changes that were previously reported in the NCBI dbSNP database (42) and further identified 9 novel mutations that were exclusively present in cDNA prepared from MCF7 cells, which resulted in truncated or mutant forms of the giant obscurins (Supplemental Table S1). These observations, along with our experimental findings indicating that loss of giant obscurins promotes cell survival, suggest that some of these polymorphisms may be driver, rather than passenger, mutations causally associated with cancer development (43).
Previous reports have indicated that select forms of obscurins are present in the sarcoplasm, the sarcolemma and transiently the nucleus of striated muscle cells (ref. 44 and our unpublished observations). Consistent with this, our current studies revealed that obscurins are present in cytoplasmic puncta, the cell membrane, and the nucleus in normal breast epithelial cells. Further examination of the presence of obscurins in cytoplasmic puncta indicated their preferential accumulation at the Golgi apparatus. Although obscurins have been previously reported to directly associate with components of internal membrane systems (18, 19), their presence in the Golgi membranes of breast epithelial cells is novel. Moreover, the prominent nuclear localization of obscurins was also surprising. Subcellular fractionation demonstrated that the giant obscurin isoforms were absent from the nucleus; however, a ∼110-kDa protein containing the Rho-GEF and COOH-terminal epitopes and a ∼120-kDa protein that includes at least one kinase domain (SKI) were exclusively found in the nuclear fraction. Both the ∼110- and ∼120-kDa isoforms are markedly reduced in cancer cell lines compared to their normal counterparts (Fig. 1B and our unpublished observations), suggesting that small obscurin isoforms containing the Rho-GEF or kinase domains may participate in the regulation of transcription or the progression of the cell cycle via novel signaling pathways. Consequently, their absence may contribute to the dysregulation of these processes in cancer cells. Notably, no obscurin isoform has been predicted to contain a nuclear localization signal, and thus a binding partner remains to be found that shuttles the small obscurins into the nucleus. One possible candidate is Ran binding protein-9, a ubiquitously expressed protein, which is a known binding partner of the Rho-GEF domain of obscurin and is localized to the nucleus of newly formed myotubes (15). This scenario, which could account for nuclear import of the ∼110-kDa isoform, is currently under investigation in our laboratory.
Down-regulation of the giant obscurin isoforms in normal breast epithelial cells provided them with a survival advantage, following exposure to the DNA-damaging agent etoposide, even in anchorage-independent conditions. Given that loss of giant obscurins neither accelerated the cell cycle nor enhanced the proliferative capacity of MCF10A cells following exposure to stress, we surmised that the observed increase in viability was attributed to their ability to resist apoptosis. Resistance to apoptosis is a distinguishing trait that a cell acquires in the course of becoming tumorigenic (2). Given that apoptosis fails to be activated in the absence of obscurins, we propose that obscurins may be acting as tumor suppressors. Many tumor suppressors, for instance, p53, exert their effects at least in part by activation of apoptosis in damaged cells (45). However, tumors often bear mutations in multiple genes (46, 47); therefore in the absence of additional activating mutations to oncogenes that cause increased proliferation, loss of tumor suppressor genes may not result in uncontrolled growth. Similarly, up-regulation of antiapoptotic proteins may not enhance proliferation induced by a potent oncogene (48). This situation is consistent with the loss of giant obscurins in MCF10A cells, which are not cotransformed by a strong oncogene; on transfection with an shRNA directed to giant obscurins, cells do not proliferate uncontrollably or bypass cell-cycle checkpoints, but instead persist in a premalignant state due to their enhanced ability to resist apoptosis. Consistent with this, cDNA array experiments coupled with qRT-PCR analysis demonstrated that select pro- and antiapoptotic genes were differentially regulated in response to down-regulation of giant obscurins A and B. In particular, the transcript levels of caspase-9 and -3, whose abrogation or loss has been intimately associated with cancer development (49, 50), were significantly reduced on obscurin knockdown. Moreover, the amounts of cleaved (active) caspase-9 and -3 peptides were diminished in obscurin-deficient MCF10A cells. Conversely the transcript and protein levels of the antiapoptotic gene BAG4 were notably up-regulated. Interestingly, overexpression of BAG4 has been shown to induce malignant transformation of MCF10A cells (51), while increased amounts of BAG4 protein have been detected in pancreatic cancer (52). Although the transcript levels of the antiapoptotic HAX1 variant-1 were also increased, we failed to detect a concomitant increase at the protein level. This may be due to the lack of specific antibodies to variant-1 and to the fact that the HAX1 gene undergoes extensive splicing giving rise to multiple isoforms with similar molecular masses (53), but opposing roles in regulating apoptosis (our unpublished observations). Notably, increased levels of select HAX1 transcripts and proteins have been correlated with advanced stages of breast cancer (54). Thus, our findings strongly suggest that in normal breast epithelium, obscurins may belong to the vast network of proteins that control programmed cell death and survival.
In summary, the present study has unraveled novel roles for the giant obscurin proteins in the development and progression of cancer. Our findings are consistent with obscurins acting as tumor suppressors in normal breast epithelial cells; when obscurins are lost, cells exhibit enhanced survival and decreased apoptosis in response to DNA damage. Further investigations will focus on elucidation of the molecular mechanisms through which obscurins exert their tumor-suppressing activities.
Supplementary Material
This article includes supplemental data. Please visit http://www.fasebj.org to obtain this information.
Acknowledgments
The authors thank Dr. Konstantinos Konstantopoulos (Department of Chemical and Biomolecular Engineering, Johns Hopkins University) and Drs. Michele I. Vitolo and Stuart S. Martin (Department of Physiology, University of Maryland) for helpful discussions and detailed protocols, Drs. Gautam Adhikary and Richard L. Eckert (Department of Biochemistry and Molecular Biology, University of Maryland School of Medicine) for providing primary cultures of keratinocytes, and Ms. Li-Yen Hu for contributing to the analysis of the mutation data. Flow cytometry analyses were performed at the Greenebaum Cancer Center Shared Flow Cytometry Facility (University of Maryland).
This work was supported by a pilot grant from the Johns Hopkins Physical Sciences-Oncology Center, the Johns Hopkins Institute for Nanobiotechnology (U54CA143868), to A.K.-K.
This article includes supplemental data. Please visit http://www.fasebj.org to obtain this information.
- DMSO
- dimethyl sulfoxide
- FACS
- fluorescence-activated flow cytometry
- FN-III
- fibronectin III
- GAPDH
- glyceraldehyde-3-phosphate dehydrogenase
- GIST
- gastrointestinal stromal tumor
- Ig
- immunoglobulin
- LMS
- leiomyosarcoma
- PARP
- poly-ADP-ribose-polymerase
- qRT-PCR
- quantitative real-time polymerase chain reaction
- Rho-GEF
- Rho-guanine exchange factor
- RIPA
- radioimmunoprecipitation assay
- shRNA
- short hairpin RNA
- SKI
- serine/threonine kinase domain I
- SKII
- serine/threonine kinase domain II
- XTT
- 2,3-bis-(2-methoxy-4-nitro-5-sulfophenyl)-2H-tetrazolium-5-carboxanilide
REFERENCES
- 1.Vogelstein B., Kinzler K. W. (2004) Cancer genes and the pathways they control. Nat. Med. , 789–799 [DOI] [PubMed] [Google Scholar]
- 2.Hanahan D., Weinberg R. A. (2000) The hallmarks of cancer. Cell , 57–70 [DOI] [PubMed] [Google Scholar]
- 3.Futreal P. A., Kasprzyk A., Birney E., Mullikin J. C., Wooster R., Stratton M. R. (2001) Cancer and genomics. Nature , 850–852 [DOI] [PubMed] [Google Scholar]
- 4.Sjoblom T., Jones S., Wood L. D., Parsons D. W., Lin J., Barber T. D., Mandelker D., Leary R. J., Ptak J., Silliman N., Szabo S., Buckhaults P., Farrell C., Meeh P., Markowitz S. D., Willis J., Dawson D., J., Willson K. V., Gazdar A. F., Hartigan J., Wu L., Liu C., Parmigiani G., Park B. H., Bachman K. E., Papadopoulos N., Vogelstein B., Kinzler K. W., Velculescu V. E. (2006) The consensus coding sequences of human breast and colorectal cancers. Science , 268–274 [DOI] [PubMed] [Google Scholar]
- 5.Russell M. W., Raeker M. O., Korytkowski K. A., Sonneman K. J. (2002) Identification, tissue expression and chromosomal localization of human obscurin-MLCK, a member of the titin and Dbl families of myosin light chain kinases. Gene , 237–246 [DOI] [PubMed] [Google Scholar]
- 6.Fukuzawa A., Idowu S., Gautel M. (2005) Complete human gene structure of obscurin: implications for isoform generation by differential splicing. J. Muscle Res. Cell Motility , 427–434 [DOI] [PubMed] [Google Scholar]
- 7.Vogelstein B., Lane D., Levine A. J. (2000) Surfing the p53 network. Nature , 307–310 [DOI] [PubMed] [Google Scholar]
- 8.Balakrishnan A., Bleeker F. E., Lamba S., Rodolfo M., Daniotti M., Scarpa A., van Tilborg A. A., Leenstra S., Zanon C., Bardelli A. (2007) Novel somatic and germline mutations in cancer candidate genes in glioblastoma, melanoma, and pancreatic carcinoma. Cancer Res. , 3545–3550 [DOI] [PubMed] [Google Scholar]
- 9.Price N. D., Trent J., El-Naggar A. K., Cogdell D., Taylor E., Hunt K. K., Pollock R. E., Hood L., Shmulevich I., Zhang W. (2007) Highly accurate two-gene classifier for differentiating gastrointestinal stromal tumors and leiomyosarcomas. Proc. Natl. Acad. Sci. U. S. A. , 3414–3419 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Kontrogianni-Konstantopoulos A., Ackermann M. A., Bowman A. L., Yap S. V., Bloch R. J. (2009) Muscle giants: molecular scaffolds in sarcomerogenesis. Physiol. Rev. , 1217–1267 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Kontrogianni-Konstantopoulos A., Bloch R. J. (2005) Obscurin: a multitasking muscle giant. J. Muscle Res. Cell Motil. , 419–426 [DOI] [PubMed] [Google Scholar]
- 12.Borisov A. B., Raeker M. O., Russell M. W. (2008) Developmental expression and differential cellular localization of obscurin and obscurin-associated kinase in cardiac muscle cells. J. Cell. Biochem. , 1621–1635 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Ackermann M. A., Hu L. Y. R., Bowman A. L., Bloch R. J., Kontrogianni-Konstantopoulos A. (2009) Obscurin interacts with a novel isoform of MyBP-C slow at the periphery of the sarcomeric M-band and regulates thick filament assembly. Mol. Biol. Cell , 2963–2978 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Bang M. L., Centner T., Fornoff F., Geach A. J., Gotthardt M., McNabb M., Witt C. C., Labeit D., Gregorio C. C., Granzier H., Labeit S. (2001) The complete gene sequence of titin, expression of an unusual approximate to 700-kDa titin isoform, and its interaction with obscurin identify a novel Z-line to I-band linking system. Circ. Res. , 1065–1072 [DOI] [PubMed] [Google Scholar]
- 15.Bowman A. L., Catino D. H., Strong J. C., Randall W. R., Kontrogianni-Konstantopoulos A., Bloch R. J. (2008) The Rho-guanine nucleotide exchange factor domain of obscurin regulates assembly of titin at the Z-disk through interactions with Ran binding protein 9. Mol. Biol. Cell , 3782–3792 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Fukuzawa A., Lange S., Holt M., Vihola A., Carmignac V., Ferreiro A., Udd B., Gautel M. (2008) Interactions with titin and myomesin target obscurin and obscurin-like 1 to the M-band: implications for hereditary myopathies. J. Cell Sci. , 1841–1851 [DOI] [PubMed] [Google Scholar]
- 17.Young P., Ehler E., Gautel M. (2001) Obscurin, a giant sarcomeric Rho guanine nucleotide exchange factor protein involved in sarcomere assembly. J. Cell Biol. , 123–136 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Bagnato P., Barone V., Giacomello E., Rossi D., Sorrentino V. (2003) Binding of an ankyrin-1 isoform to obscurin suggests a molecular link between the sarcoplasmic reticulum and myofibrils in striated muscles. J. Cell Biol. , 245–253 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Kontrogianni-Konstantopoulos A., Jones E. M., van Rossum D. B., Bloch R. J. (2003) Obscurin is a ligand for small ankyrin 1 in skeletal muscle. Mol. Biol. Cell , 1138–1148 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Coisy-Quivy M., Touzet O., Bourret A., Hipskind R. A., Mercier J., Fort P., Philips A. (2009) TC10 controls human myofibril organization and is activated by the sarcomeric RhoGEF obscurin. J. Cell Sci. , 947–956 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Ford-Speelman D. L., Roche J. A., Bowman A. L., Bloch R. J. (2009) The Rho-guanine nucleotide exchange factor domain of obscurin activates RhoA signaling in skeletal muscle. Mol. Biol. Cell , 3905–3917 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Qadota H., McGaha L. A., Mercer K. B., Stark T. J., Ferrara T. M., Benian G. M. (2008) A novel protein phosphatase is a binding partner for the protein kinase domains of UNC-89 (obscurin) in Caenorhabditis elegans. Mol. Biol. Cell , 2424–2432 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Borisov A., Kontrogianni-Konstantopoulos A., Bloch R. J., Westfall M. W., Russell M. W. (2003) Functional role of obscurin in assembly and remodeling of the contractile system. Circulation , 245–253. [Google Scholar]
- 24.Borisov A. B., Kontrogianni-Konstantopoulos A., Bloch R. J., Westfall M. V., Russell M. W. (2004) Dynamics of obscurin localization during differentiation and remodeling of cardiac myocytes: obscurin as an integrator of myofibrillar structure. J. Histochem. Cytochem. , 1117–1127 [DOI] [PubMed] [Google Scholar]
- 25.Kontrogianni-Konstantopoulos A., Catino D. H., Strong J. C., Randall W. R., Bloch R. J. (2004) Obscurin regulates the organization of myosin into A bands. Am. J. Physiol. Cell Physiol. , C209–C217 [DOI] [PubMed] [Google Scholar]
- 26.Kontrogianni-Konstantopoulos A., Catino D. H., Strong J. C., Sutter S., Borisov A. B., Pumplin D. W., Russell M. W., Bloch R. J. (2006) Obscurin modulates the assembly and organization of sarcomeres and the sarcoplasmic reticulum. FASEB J. , 2102–2111 [DOI] [PubMed] [Google Scholar]
- 27.Lange S., Ouyang K., Meyer G., Cui L., Cheng H., Lieber R. L., Chen J. (2009) Obscurin determines the architecture of the longitudinal sarcoplasmic reticulum. J. Cell Sci. , 2640–2650 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Raeker M. O., Bieniek A. N., Ryan A. S., Tsai H. J., Zahn K. M., Russell M. W. (2010) Targeted deletion of the zebrafish obscurin A RhoGEF domain affects heart, skeletal muscle and brain development. Dev. Biol. , 432–443 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Raeker M. O., Su F. Y., Geisler S. B., Borisov A. B., Kontrogianni-Konstantopoulos A., Lyons S. E., Russell M. W. (2006) Obscurin is required for the lateral alignment of striated myofibrils in zebrafish. Dev. Dyn. , 2018–2029 [DOI] [PubMed] [Google Scholar]
- 30.Yap S. V., Koontz J. M., Kontrogianni-Konstantopoulos A. (2011) HAX-1: a family of apoptotic regulators in health and disease. J. Cell. Physiol. , 2752–2761 [DOI] [PubMed] [Google Scholar]
- 31.Hu L.-Y. R., Valenti J., Kontrogianni-Konstantopoulos A. (2010) Localization of obscurin kinases in the extracellular matrix of striated muscle cells. Mol. Biol. Cell (Suppl.), 988383 (abst.) [Google Scholar]
- 32.Abulencia J. P., Gaspard R., Healy Z. R., Gaarde W. A., Quackenbush J., Konstantopoulos K. (2003) Shear-induced cyclooxygenase-2 via a JNK2/c-Jun-dependent pathway regulates prostaglandin receptor expression in chondrocytic cells. J. Biol. Chem. , 28388–28394 [DOI] [PubMed] [Google Scholar]
- 33.Healy Z. R., Lee N. H., Gao X., Goldring M. B., Talalay P., Kensler T. W., Konstantopoulos K. (2005) Divergent responses of chondrocytes and endothelial cells to shear stress: cross-talk among COX-2, the phase 2 response, and apoptosis. Proc. Natl. Acad. Sci. U. S. A. , 14010–14015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Zhu F., Wang P., Kontrogianni-Konstantopoulos A., Konstantopoulos K.. Prostaglandin (PG)D(2) and 15-deoxy-Delta(12,14)-PGJ(2), but not PGE(2), mediate shear-induced chondrocyte apoptosis via protein kinase A-dependent regulation of polo-like kinases. Cell Death Differ. , 1325–1334 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Saeed A. I., Bhagabati N. K., Braisted J. C., Liang W., Sharov V., Howe E. A., Li J., Thiagarajan M., White J. A., Quackenbush J. (2006) TM4 microarray software suite. Methods Enzymol. , 134–193 [DOI] [PubMed] [Google Scholar]
- 36.Saeed A. I., Sharov V., White J., Li J., Liang W., Bhagabati N., Braisted J., Klapa M., Currier T., Thiagarajan M., Sturn A., Snuffin M., Rezantsev A., Popov D., Ryltsov A., Kostukovich E., Borisovsky I., Liu Z., Vinsavich A., Trush V., Quackenbush J. (2003) TM4: a free, open-source system for microarray data management and analysis. BioTechniques , 374–378 [DOI] [PubMed] [Google Scholar]
- 37.Ashburner M., Ball C. A., Blake J. A., Botstein D., Butler H., Cherry J. M., Davis A. P., Dolinski K., Dwight S. S., Eppig J. T., Harris M. A., Hill D. P., Issel-Tarver L., Kasarskis A., Lewis S., Matese J. C., Richardson J. E., Ringwald M., Rubin G. M., Sherlock G. (2000) Gene ontology: tool for the unification of biology. The Gene Ontology Consortium. Nat. Genet. , 25–29 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Sherry S. T., Ward M. H., Sirotkin K. (1999) dbSNP: database for single nucleotide polymorphisms and other classes of minor genetic variation. Genome Res. , 677–679 [PubMed] [Google Scholar]
- 39.Fan S., el-Deiry W. S., Bae I., Freeman J., Jondle D., Bhatia K., Fornace A. J., Magrath I., Kohn K. W., O'Connor P. M. (1994) p53 gene mutations are associated with decreased sensitivity of human lymphoma cells to DNA damaging agents. Cancer Res. , 5824–5830 [PubMed] [Google Scholar]
- 40.Gudas J. M., Nguyen H., Li T., Sadzewicz L., Robey R., Wosikowski K., Cowan K. H. (1996) Drug-resistant breast cancer cells frequently retain expression of a functional wild-type p53 protein. Carcinogenesis , 1417–1427 [DOI] [PubMed] [Google Scholar]
- 41.Arimura T., Matsumoto Y., Okazaki O., Hayashi T., Takahashi M., Inagaki N., Hinohara K., Ashizawa N., Yano K., Kimura A. (2007) Structural analysis of obscurin gene in hypertrophic cardiomyopathy. Biochem. Biophys. Res. Commun. , 281–287 [DOI] [PubMed] [Google Scholar]
- 42.Sherry S. T., Ward M. H., Kholodov M., Baker J., Phan L., Smigielski E. M., Sirotkin K. (2001) dbSNP: the NCBI database of genetic variation. Nucleic Acids Res. , 308–311 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Stratton M. R., Campbell P. J., Futreal P. A. (2009) The cancer genome. Nature , 719–724 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Bowman A. L., Kontrogianni-Konstantopoulos A., Hirsch S. S., Geisler S. B., Gonzalez-Serratos H., Russell M. W., Bloch R. J. (2007) Different obscurin isoforms localize to distinct sites at sarcomeres. FEBS Lett. , 1549–1554 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Sherr C. J. (2004) Principles of tumor suppression. Cell , 235–246 [DOI] [PubMed] [Google Scholar]
- 46.Hahn W. C., Counter C. M., Lundberg A. S., Beijersbergen R. L., Brooks M. W., Weinberg R. A. (1999) Creation of human tumour cells with defined genetic elements. Nature , 464–468 [DOI] [PubMed] [Google Scholar]
- 47.Viney J. L. (1995) Transgenic and gene knockout mice in cancer. Res. Cancer Metastasis Rev. , 77–90 [DOI] [PubMed] [Google Scholar]
- 48.Strasser A., Harris A. W., Bath M. L., Cory S. (1990) Novel primitive lymphoid tumours induced in transgenic mice by cooperation between myc and bcl-2. Nature , 331–333 [DOI] [PubMed] [Google Scholar]
- 49.Devarajan E., Sahin A. A., Chen J. S., Krishnamurthy R. R., Aggarwal N., Brun A.-M., Sapino A., Zhang F., Sharma D., Yang X.-H., Tora A. D., Mehta K. (2002) Down-regulation of caspase 3 in breast cancer: a possible mechanism for chemoresistance. Oncogene , 8843–8851 [DOI] [PubMed] [Google Scholar]
- 50.Olsson M., Zhivotovsky B. (2011) Caspases and cancer. Cell Death Differ. , 1441–1449 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Yang Z. Q., Streicher K. L., Ray M. E., Abrams J., Ethier S. P. (2006) Multiple interacting oncogenes on the 8p11-p12 amplicon in human breast cancer. Cancer Res. , 11632–11643 [DOI] [PubMed] [Google Scholar]
- 52.Ozawa F., Friess H., Zimmermann A., Kleeff J., Buchler M. W. (2000) Enhanced expression of silencer of death domains (SODD/BAG-4) in pancreatic cancer. Biochem. Biophys. Res. Commun. , 988383 (abst.) [DOI] [PubMed] [Google Scholar]
- 53.Fadeel B., Grzybowska E. (2009) HAX-1: a multifunctional protein with emerging roles in human disease. Biochim. Biophys. Acta , 1139–1148 [DOI] [PubMed] [Google Scholar]
- 54.Trebinska A., Rembiszewska A., Ciosek K., Ptaszynski K., Rowinski S., Kupryjanczyk J., Siedlecki J. A., Grzybowska E. A. (2010) HAX-1 overexpression, splicing and cellular localization in tumors. BMC Cancer , 76. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.