

Abstract
Neurofibrillary tangles (NFTs) are a pathological hallmark of Alzheimer's disease (AD); however, the relationship between NFTs and disease progression remains controversial. Analyses of tau animal models suggest that phenotypes coincide with accumulation of soluble aggregated tau species but not the accumulation of NFTs. The pathological role of prefilamentous tau aggregates, e.g., tau oligomeric intermediates, is poorly understood, in part because of methodological challenges. Here, we engineered a novel tau oligomer-specific antibody, T22, and used it to elucidate the temporal course and biochemical features of oligomers during NFT development in AD brain. We found that tau oligomers in human AD brain samples were 4-fold higher than those in the controls. We also revealed the role of oligomeric tau conformers in pretangles, neuritic plaques, and neuropil threads in the frontal cortex tissue from AD brains; this analysis uncovers a consistent code that governs tau oligomerization with regard to degree of neuronal cytopathology. These data are the first to characterize the role of tau oligomers in the natural history of NFTs, and they highlight the suitability of tau oligomers as therapeutic targets in AD and related tauopathies.—Lasagna-Reeves, C. A., Castillo-Carranza, D. L., Sengupta, U., Troncoso, J., Jackson, G. R., Kayed, R. Identification of oligomers at early stages of tau aggregation in Alzheimer's disease.
Keywords: neurodegeneration, conformational antibodies, tauopathies, protein misfolding
In Alzheimer's disease (AD) pathology, accumulation of the microtubule-associated protein tau takes place primarily in the neurons. Tuft-shaped astrocytes and coiled bodies of oligodendrocytes may be seen in AD, but their prevalence and density are linked to age (1, 2) rather than pathology. Tau accumulates in both the somatodendritic and axonal domains of neurons. Neuropil threads are related to the accumulation of tau in dendrites (3). The neuritic corona of the plaque is composed mainly of axonal processes enriched in tau (4). Tau also accumulates in the soma as neurofibrillary tangles (NFTs). Morphological stages during evolution of NFTs have been described (5–7). Hyman and colleagues (5) defined 3 stages of NFTs: pretangle phospho-tau aggregates (pre-NFTs), intraneuronal NFTs (iNFTs), and extraneuronal NFTs (eNFTs). Pretangles display granular cytoplasmic phospho-tau staining; the nucleus is detectable, and general morphology of the cell is normal. Intraneuronal NFTs contain aggregated filamentous structures within the cytoplasm; the nucleus is present but often eccentric or pyknotic. eNFTs show extracellular phospho-tau filaments in the shape of a neuronal cell body; however, these have no relation to the neuronal soma or nucleus, and they are also known as “ghost tangles.”
Tau undergoes many post-translational modifications, including glycosylation, ubiquitination, glycation, polyamination, nitrosylation, and truncation; the most important and disease-relevant tau post-translational modification is believed to be hyperphosphorylation, which can alter function and affect tau self-assembly, aggregation, and accumulation in NFTs (8, 9). All tau isoforms contain ≥30 phosphorylation sites (10, 11), most of which are believed to be in the dephosphorylated state in normal tau. Some degree of phosphorylation at many of these sites occurs in normal tau proteins; nevertheless, many of these sites are abnormally phosphorylated with respect to both location and quantity in NFTs (12, 13). Numerous tau kinases have been described, including MAPK (14), glycogen synthetase kinase-3β (15), MARK (16), cdk2, and cdk5 (17). In contrast, PP2A appears to be the principal tau phosphatase in vivo (18); PP1, PP2B, and PP2C are also capable of dephosphorylating tau in vitro (10, 19). Although tau hyperphosphorylation is confirmed in NFTs, its role in tau toxicity is not well understood. In Drosophila models, some investigators have reported that tau toxicity is not dependent on phosphorylation (20, 21); moreover, amyloid-β (Aβ) can induce tau fragmentation without any increase in full-length tau phosphorylation (22). Avila and colleagues (23) recently showed that tissue-nonspecific alkaline phosphatase dephosphorylates hyperphosphorylated tau protein once it is released on neuronal death and that only this dephosphorylated tau protein triggers further neuronal death.
Cell death and synaptic lesions occurred independently of NFT formation in mice expressing wild-type human tau (24, 25). Hippocampal synapse loss, impaired synaptic function, and microgliosis precede the formation of NFTs in the P301S mutant human tau transgenic mouse model (26), and similar results have been described in the Tau(RD)/δK280 mouse model (27), fly models (28), and zebrafish models (29). Tau oligomers were characterized in mice expressing P301L mutant human tau and a conditional model (rTg4510) expressing the same mutation. Surprisingly, accumulation of oligomeric tau, but not NFTs, correlated best with neuronal loss and behavioral deficits in these models. These findings suggest that the accumulation of tau oligomers, behavioral deficits, and neuronal loss precede the formation of NFTs (30, 31).
The published literature indicates that NFT formation alone is insufficient for neurodegeneration and suggests that soluble tau aggregates may be the most toxic and pathologically significant tau species (28, 30, 32–39). Tau oligomers are neurotoxic when applied extracellularly to cultured neuronal cells (40) and trigger increased intracellular calcium levels (41, 42). Moreover, we recently demonstrated that tau oligomers (but not fibrils) induce neurodegeneration and synaptic and mitochondrial dysfunction in vivo (43). However, only limited data are available regarding the presence of tau oligomers in patients with AD (44–46). Here, we investigate the role of tau oligomers in vivo using our novel tau oligomer specific antibody, T22. We were able to determine the presence of tau oligomers in AD brains, stage the appearance of oligomers immunohistochemically for the first time during NFT evolution, and evaluate the degree of phosphorylation and ubiquitination of these tau species. These data establish the pathological significance of tau oligomers in vivo and highlight their suitability as therapeutic targets for AD and other neurodegenerative tauopathies.
MATERIALS AND METHODS
Preparation of tau oligomers
Recombinant tau protein [tau-441 (2N4R) MW 45.9 kDa] was expressed and purified as described previously (47, 48). It was treated with 8 M urea to obtain monomeric tau; then it was dialyzed overnight against 1× PBS buffer (pH 7.4) and adjusted to 1 mg/ml with PBS, and aliquots of tau monomer (in PBS) were kept at −20°C. For preparation of oligomers, 300 μl of the tau stock (1 mg/ml) was added to 700 μl of 1× PBS, final concentration 0.3 mg/ml. Aβ42 oligomers (7 μl, 0.3 mg/ml) were added to the sample (seeds) and mixed by pipetting for 1 min. The sample was then incubated at room temperature for 1 h on an orbital shaker, and the resulting tau oligomers were used to seed a new patch of tau; this procedure was repeated three times to eliminate the residual Aβ seeds. The preparation and characterization of tau oligomers were performed as described previously (40, 43). Paired helical filament (PHF) tau fibrils from full-length recombinant tau protein were prepared using heparin according to well established protocols (47, 48).
T22 pAb production and characterization
The antigen (tau oligomers) was used to immunize two New Zealand White rabbits (Pacific Immunology Corp., Ramona, CA, USA) according to protocols approved by the Pacific Immunology institutional animal care and use committee and by the University of Texas Medical Branch. Each rabbit was immunized with 500 μl of antigen in complete Freund's adjuvant, followed by boosting twice at 4-wk intervals with 500 μl of antigen in incomplete Freund's adjuvant. The specificity of the novel antibody was determined by biochemical analysis (Western blot, dot blot, and ELISA) using well-characterized recombinant and synthetic samples.
Treatment of neuroblastoma cells with tau oligomers and T22
SH-SY5Y human neuroblastoma cells were maintained in DMEM (Life Technologies, Inc., Invitrogen, Carlsbad, CA, USA) supplemented with 10% FBS, glutamine (4 mM), penicillin (200 U/ml), streptomycin (200 μg/ml), and sodium pyruvate (1 μM). Cells were maintained at 37°C in 5% CO2. Cells (∼10,000/well) were plated in 96-well plates (Corning Glassworks, Corning, NY, USA) and grown overnight. Tau or Aβ and other amyloid species were added to the cells to a final concentration of 5 μM and incubated for 4 h. Cell viability was assessed spectrophotometrically using a 3-[4,5-dimethylthiazol-2-yl]-2,5-diphenyltetrazolium bromide (MTT)-based assay according to the manufacturer's specifications (Sigma-Aldrich, St. Louis, MO, USA).
Preparation of brain extracts
Postmortem brain tissues from subjects with AD and progressive supranuclear palsy (PSP) and control subjects were provided in the form of frozen blocks by the Institute for Brain Aging and Dementia (University of California–Irvine, Irvine, California, USA) and the Brain Resource Center at Johns Hopkins. AD cases consisted of pathologically severe AD, stage V–VI, (7). Each brain was homogenized in PBS with a protease inhibitor cocktail (catalog no. 11836145001; Roche Applied Science, Indianapolis, IN, USA), using a dilution of brain:PBS of 1:3 (w/v). Samples were then centrifuged at 10,000 rpm for 10 min at 4°C. The supernatants were portioned into aliquots, snap-frozen, and stored at −80°C until used (49).
Separation of soluble and insoluble tau
Separation of soluble and insoluble tau was performed in accordance with protocols developed by Lee et al. (50). In brief, each brain was homogenized in PBS with protease inhibitors (11836145001; Roche Applied Science) at a ratio of brain:PBS of 1:3 (w/v) and incubated 20 min at 4°C. Samples were then centrifuged for 20 min at 11,000 g at 4°C. The supernatant was collected and centrifuged at 100,000 g for 60 min at 4°C. Pellets from the first and second cold centrifugation were combined and resuspended in PHF extraction buffer at 1:10 (w/v). Samples were then centrifuged for 20 min at 15,000 g at 4°C. Sarcosyl was added to the supernatant to a final concentration of 1%, while stirring, for 1 h at room temperature. The samples were then centrifuged for 30 min at 100,000 g at 4°C. Finally, the pellet was resuspended in TBS buffer (1 ml of buffer for 25 g of starting material). For urea treatment, brain-derived PBS-soluble fraction samples were mixed with urea to reach a final concentration of 8 M, followed by incubation overnight at room temperature. The next day, samples were mixed with running buffer and loaded into the gel and analyzed by Western blot.
Western blot analysis
PBS-soluble fractions of brain extracts were run on bis-Tris SDS-PAGE gels and subsequently transferred onto nitrocellulose. After blocking overnight at 4°C with 10% nonfat dried milk, membranes were probed for 1 h at room temperature with anti-tau oligomer antibody T22 (1:1000), Tau-5 antibody (1:3000; Covance, Princeton, NJ, USA), or PHF-13 antibody (1:4000; Covance) diluted in 5% nonfat dried milk. T22 immunoreactivity was detected with horseradish peroxidase-conjugated anti-rabbit IgG (1:3000; The Jackson Laboratory, Bar Harbor, ME, USA); anti-mouse IgG (1:3000; The Jackson Laboratory) was used for Tau-5, and PHF-13. For signal detection, ECL Plus (Amersham-Pharmacia, Piscataway, NJ, USA) was used.
ELISA
For ELISA, plates were coated with 10 μl of the PBS-soluble fraction of brains using 0.1 M sodium bicarbonate (pH 9.6) as coating buffer, followed by incubation for 1 h at 37°C, washing 3 times with Tris-buffered saline with very low (0.01%) Tween 20 (TBS-T), and then blocking for 1 h at 37°C with 10% BSA (IgG-free TBS-T). The plates were then washed three times with TBS-T; T22 antibody (diluted 1:1000 in 5% nonfat milk in TBS-T) was added and allowed to react for 1 h at 37°C. The plates were then washed 3 times with TBS-T, and 100 μl of horseradish peroxidase-conjugated anti-rabbit IgG (diluted 1:10,000 in 5% nonfat milk in TBS-T; Promega, Madison, WI, USA) was added, followed by incubation for 1 h at 37°C. Finally, plates were washed 3 times with TBS-T and developed with 3,3,5,5-tetramethylbenzidine (TMB-1 component substrate) from KPL (Gaithersburg, MD, USA). The reaction was stopped with 100 μl of 1 M HCl, and samples were read at 450 nm.
Dot blot
Each sample (2 μl) was applied to a nitrocellulose membrane, blocked with 10% nonfat milk in TBS-T overnight at 4°C, washed 3 times for 5 min each with TBS-T, and incubated for 1 h at room temperature with the anti-tau oligomer antibody T22 (1:1000). The membranes were washed 3 times for 5 min each with TBS-T, incubated with horseradish peroxidase-conjugated anti-rabbit IgG (Promega) diluted 1:10,000 in 3% BSA/TBS-T, and incubated for 1 h at room temperature. The blots were washed 3 times with TBS-T and developed with a ECL chemiluminescence kit from Amersham-Pharmacia (RPN2132).
Immunohistochemistry (IHC)
IHC was performed on paraffin-embedded sections. In brief, sections (5 μm) were deparaffinized and rehydrated. Primary antibodies were detected with biotinylated goat anti-mouse IgG (1:2000; Jackson ImmunoResearch Laboratories Inc., West Grove, PA, USA) or biotinylated goat anti-rabbit IgG (1:1800; Jackson ImmunoResearch Laboratories) and visualized using an ABC reagent kit (Vector Laboratories, Burlingame, CA, USA), according to the manufacturer's recommendations. Bright-field images were acquired using a Nikon Eclipse 800 microscope, equipped with a Nikon DXM1200 color charge-coupled device (CCD) camera (Nikon Instruments Inc., Melville, NY, USA). Sections were counterstained with hematoxylin (Vector Laboratories) for nuclear staining. The following antibodies were used for immunostaining: rabbit anti-tau oligomers antibody T22 (1:700) and mouse anti-phospho tau (Ser202/Thr205) antibody AT8 (1:1000, Thermo Fisher Scientific, Waltham, MA, USA).
Immunofluorescence
Paraffin sections were deparaffinized, rehydrated, and washed in 0.01 M PBS 3 times for 5 min each time. After blocking in normal goat serum for 1 h, sections were incubated overnight with rabbit anti-tau oligomer antibody T22 (1:700). The next day, the sections were washed in PBS 3 times for 10 min each time and then incubated with goat anti-rabbit IgG Alexa Fluor 568 (1:700; Invitrogen) for 1 h. The sections were then washed 3 times for 10 min each time in PBS before incubation overnight with mouse AT8 (1:1000), anti-phospho tau (Thr231) PHF-6 (1:600, Covance), anti-ubiquitin antibody (1:50; Vector Laboratories), and anti-Von Willebrand factor antibody (1:200; Chemicon International Inc., Temecula, CA). The next day, the sections were washed in PBS 3 times for 10 min each before incubation with goat anti-IgG Alexa Fluor 488 (1:700; Invitrogen) for 1 h. Sections were washed and mounted in Vectashield mounting medium with DAPI (Vector Laboratories). The sections were examined using a Bio-Rad Radiance 2100 confocal system mounted on a Nikon Eclipse E800 microscope equipped with a CoolSnap-FX monochrome CCD camera (Photometrics, Tucson, AZ, USA) using a standard Nikon FITC, TX Red, and DAPI filter set for Alexa Fluor 488 and 568 and DAPI, respectively.
Confocal z stack
For confocal z-stack imaging, a Bio-Rad Radiance 2100 confocal system mounted on a Nikon Eclipse E800 microscope was used. To build the z-stack 3-dimensional (3D) construction, 27 confocal planes were captured. Each confocal plane had a thickness of 0.7 μm. The separation between planes was 0.2 μm.
Intensity correlation analysis (ICA)
ICA is based on the principle that for any set of values, the sum of the differences from the mean equals 0, i.e., ΣN (Ai − a) = 0, where a is the mean of the distribution with N values of Ai. In our case, N is the number of pixels, and Ai is the staining intensity for each pixel. It follows that for N pixels associated with 2 sets of random staining intensities (Ai and Bi), the sum of the product of their differences will also tend to 0; thus, ΣN (Ai − a) (Bi − b) ≈ 0. However, this is not the case if the two intensities are dependent (when the product tends to be a positive value) or if they are segregated (when the product tends to be a negative value). Thus, with dependent staining, ΣN (Ai − a) (Bi − b) > 0, whereas with segregated staining, ΣN (Ai − a) (Bi − b) < 0. We used this property to test for dependent or segregated staining between T22 and anti-ubiquitin antibody. This analysis was performed using ImageJ software (U.S. National Institutes of Health, Bethesda, MD, USA). For more details, see Li et al. (51).
Fluorescence resonance energy transfer (FRET)
For FRET experiments, immunostaining was observed using a Bio-Rad Radiance 2100 confocal system mounted on a Nikon Eclipse E800 microscope. The krypton-argon laser was used to excite Alexa Fluor 488 (excitation 488 nm) and Alexa Fluor 568 (excitation 569). The blue diode laser was used to excite DAPI (excitation 440). The energy transfer was detected as an increase in donor fluorescence after photobleaching of the acceptor fluorescence (i.e., donor dequenching; refs. 52–54). The FRET data are represented as an increase in donor fluorescence after photobleaching of the acceptor fluorescence. Four scans were saved from the confocal microscope for the FRET experiment. First, ubiquitin detected by Alexa Fluor 488 indirect immunostaining was excited with line 488. Second, T22 detected by Alexa Fluor 568 indirect immunostaining was excited with line 569. To perform the FRET experiment, an area was outlined on a T22-ubiquitin-immunoreactive cell, and this area was photobleached completely with 569-nm light, using 100% laser power. After thorough photobleaching to destroy the acceptor molecule, the area was rescanned with the 569 line, using 5% power to confirm that photobleaching was effective. Finally, the area was scanned after the photobleaching with the 488 line (∼5% power). FRET is detected as an increase in brightness of the Alexa Fluor 488 signal within the region where the acceptor dye, Alexa Fluor 568, was photobleached. ImageJ software was used to determine an increase in brightness after the photobleaching.
Analysis of tau pathology
Brain samples from 8 AD and 8 control cases were stained with T22 or AT8 and viewed with a Nikon AZ100M microscope. Ten visual fields in each case were randomly chosen, and the numbers of tau structures visible at ×20 were counted. Counts are displayed as the percentage of each specific structure in relation to the total number of structures counted (100%). For colocalization analysis, 16 AD cases were double-labeled with T22 and Tau-5. The percentage of tau structures that were double-labeled with both antibodies was compared with the percentage labeled by Tau-5 alone. Eight visual fields for each AD case were randomly chosen, and the numbers of structures stained by T22, Tau-5, or both, that were visible at ×20 were counted. For quantification analysis, the 16 cases were pooled together, and the results are displayed as the percentage of labeled structures that were either single- or double-labeled.
Statistical analysis
All statistical analyses were performed using OriginPro 8.0 software (OriginLab Corp., Northampton, MA, USA). The criterion for statistical significance was P < 0.01.
RESULTS
T22 antibody specifically recognizes tau oligomers
The specificity of T22 was examined by ELISA analysis, which shows the specificity of the antibody for tau oligomers (Fig. 1A); T22 does not show any significant reactivity for monomeric tau, Aβ oligomers, Aβ fibrils, α-synuclein oligomers, or α-synuclein fibrils (Fig. 1A). The signal produced by T22 for tau oligomers was comparable to the signal produced by Tau-5, which detects total tau. In the case of tau fibrils, it is possible to see some reactivity of T22 that could be produced by the presence of some residual oligomers in the preparation. To rule out any cross-reactivity of T22 with PHF tau fibrils, we performed dot blot analysis against tau monomers, tau oligomers, and PHF tau fibrils prepared by heparin-induced tau aggregation protocols (47, 48). In Supplemental Fig. S1A, it is clear that T22 is specific for the oligomeric forms of tau and does not detect tau monomer and PHF tau, indicating that the low reactivity observed for T22 with tau fibrils in the ELISA (Fig. 1A) was indeed due to the presence of some residual oligomers. Tau oligomers prepared in vitro are highly toxic in cell culture and in vivo (40, 43). T22 preincubated with tau oligomers neutralized their toxicity to neuroblastoma cells (Fig. 1B). However, when T22 was preincubated with Aβ oligomers, these aggregates remained toxic (Fig. 1B). Consistent with previous findings (40), tau fibrils and monomers were not toxic in cell culture, and incubation with T22 did not have any effects on these species (Fig. 1B). The results obtained by ELISA were confirmed by Western blot analysis using different samples prepared in vitro from tau and other amyloidogenic proteins (Supplemental Fig. S1A), which indicate that T22 is sequence conformation-dependent and oligomer-specific because T22 only recognizes tau oligomers and does not recognize tau fibrils or monomers nor does it recognize other protein oligomers or fibrils (e.g., Aβ, α-synuclein, or islet amyloid polypeptide; Supplemental Fig. S1A). It appears that T22 exhibits high affinity for tau oligomer dimers and trimers. We also noticed that T22 lost reactivity, and the signal appeared at a higher molecular level when tau oligomers were aged for 2 d. To determine T22 reactivity and specificity for tau oligomers formed in vivo, PBS-soluble fractions were prepared by homogenizing brain tissues from patients with PSP and performing an ELISA using T22; elevated levels of tau oligomers were observed in PSP brains in comparison with age-matched control brains. In addition, IHC was performed using T22, and we were able to detect tau oligomers in PSP brains (Supplemental Fig. S1B). The IHC confirmed that these oligomers are present in the pathological deposits of tau that were previously described in patients with PSP, such as coiled bodies and globose-type NFTs (55). In the frontal cortex from AD brains, Western blot analysis shows that T22 reacted with higher-molecular-mass tau but not with monomeric tau (Supplemental Fig. S1C). The bands detected appeared to be dimers, trimers, and tetramers. Tau-5, the linear epitope of which is in the proline-rich region (residues 210–230; ref. 56), reacted with monomeric and high-molecular-mass tau. The band detected at higher molecular mass (150 kDa) with Tau-5 in the age-matched control, which is not detected with T22, could be corresponding to tau multimers before adopting the oligomeric toxic conformation (Supplemental Fig. S1C). A Western blot with the same samples was also performed with the antibody PHF-13, which recognizes the phospho-epitope Ser396, an indicator of the late stage of tau aggregation and PHF formation. In Supplemental Fig. S1C, it is clear that T22 has weak reactivity with high-molecular-mass tau, which is recognized by PHF-13 with higher affinity, and most importantly Supplemental Fig. S1C shows that in some cases of AD PHF-13 reactivity is weak despite the presence of high amounts of tau oligomers detected by T22; this clearly demonstrates that T22 and PHF-13 recognize different forms of tau aggregates. It is also important to consider the fact that the molecular mass in Western blot analysis is not the best indicator of the level and nature of amyloid aggregates (57).
Figure 1.
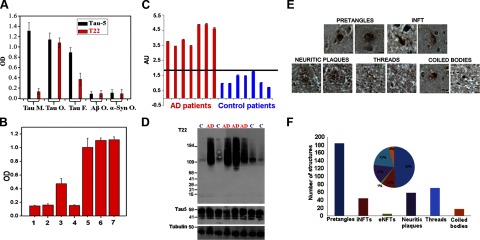
Specificity of T22 antibody for tau oligomers. A) ELISA analysis shows that T22 does not show any significant reactivity for monomeric tau, but some signal is observed in tau fibrils, possibly due to the cross-reactivity of the antibody or the presence of oligomers in the preparation. T22 does not recognize oligomers from other amyloidogenic proteins, such as Aβ or α-synuclein; Tau-5, which recognizes all forms of tau, was used as a positive control for the tau samples. B) T22 neutralizes tau oligomer toxicity when preincubated at an equimolar ratio, as assessed using the MTT toxicity assay and SY5Y human neuroblastoma cells. Tau oligomers (bar 1) and Aβ oligomers (bar 2) are highly toxic to neuroblastoma cells. T22 can neutralize the toxicity caused by tau oligomers (bar 3) but not Aβ oligomers (bar 4); tau fibrils were not toxic (bar 5). The same is true for monomeric tau (bar 6), as well as for the PBS control (bar 7). C–F) Antibody T22 preferentially recognizes tau oligomers in AD brain. C) ELISA analysis of PBS soluble fraction from AD and age-matched control brains. In all patients with AD, T22 signal corresponding to tau oligomers (red) was higher than that in age-matched controls (blue). Values were normalized with tubulin. AU, arbitrary units. D) Western blot of PBS-soluble fraction from AD and control frontal cortex. Top blot, T22; middle blot, Tau-5; bottom blot, tubulin loading controls. High-molecular-mass bands corresponding to tau oligomers detected by T22 are elevated in AD brain compared with control brain, in which the major T22 immunoreactive band probably corresponds to a dimer. T22 does not recognize monomeric tau; however, Tau-5-immunoreactive material corresponding to tau monomer is abundant in both AD and control samples (middle blot). E) Tau oligomer localization during the evolution of NFTs as assessed using T22. Photomicrographs of T22 staining developed using avidin-biotin complex with hematoxylin-counterstained paraffin sections. T22 labels pre-NFTs, iNFTs, neuritic coronae, threads, and coiled bodies. F) Quantitation of T22 immunoreactivity from a total of 377 structures; 184 of the lesions corresponded to pretangles, 71 to neuropil threads, and a lower quantity to other strutures: 44 iNFTs, 58 neuritic plaques, 17 coiled bodies, and 3 eNFTs. Inset: pie graph indicating percentage distribution of total T22 immunoreactivity among these various structures.
Elevated tau oligomers in AD frontal cortex
To examine the levels of tau oligomers in AD brain, PBS-soluble fractions were analyzed in triplicate by ELISA assays and compared with age-matched controls. Tau oligomers were elevated in AD brain compared with brain of age-matched controls (Fig. 1C); in some cases, tau oligomers in AD samples were 4-fold higher than in the control samples. Western blot analysis using T22 also showed elevated oligomers in AD brain compared with brain of controls (Fig. 1D). Under most conditions, T22 antibody does not detect monomeric tau; tau monomers are, however, recognized by Tau-5, with levels similar in patients with AD and controls (Fig. 1D). We followed well-established protocols to examine the distribution and solubility of tau oligomers (ref. 50; schematic representation in Supplemental Fig. S1D). By Western blot analysis, it was possible to see the tau oligomers in both sarcosyl-soluble and -insoluble fractions (Supplemental Fig. S1E). In the sarcosyl-soluble fraction, we mainly identified dimers and trimers; in the insoluble fraction, dimers, trimers, and tetramers were detected. After stripping, the same membranes were reprobed with Tau-5 (Supplemental Fig. S1F), revealing tau monomer in the soluble fraction and PHF tau in the insoluble fraction, both of which were undetectable using T22.
Tau oligomers are present in early stages of neuronal cytopathology in AD
To investigate the association of tau oligomers with AD cytopathology, 8 AD and 8 age-matched control brains were stained with T22. Photomicrographs showed T22 immunoreactivity in a variety of AD lesions, corresponding to pre-NFTs, iNFTs, neuritic coronae, threads, and coiled bodies (Fig. 1E). A total of 377 tau structures labeled with T22 were detected in 80 microscope fields (10 fields × 8 cases; Fig. 1E, F); only 16 structures were detected in the same number of fields in the age-matched controls. Specifically, 184 T22-positive structures (49%) corresponded to pretangles in AD, 71 (19%) to neuropil threads, 58 (15%) to neuritic coronae of plaques, 44 (12%) to iNFTs, 17 (4%) to coiled bodies, and 3 (1%) to eNFTs (Fig. 1F). All tau oligomers detected in age-matched controls were associated with pretangles (data not shown). The same AD and age-matched control samples were stained with AT8, which recognizes the double-phosphorylated epitope Ser202/Thr205 (58, 59) and mainly stains iNFTs and eNFTs (ref. 5 and data not shown). Using AT8, 79.46% of the tau structures detected corresponded to neuropil threads, 13.06% to eNFTs, 4% to iNFTs, and 2.58% to neuritic corona of the plaques; only 0.9% corresponded to pretangles (data not shown). These data suggest that our failure to detect eNFT with T22 is due to the absence of oligomers rather than variant pathology in our cases.
Immunofluorescence showed that tau oligomers represent a small fraction of total tau in the brain. To determine the tau oligomers as a percentage of total tau, double immunofluorescence was performed, using T22 and Tau-5 in 16 AD cases (Fig. 2A–C). For quantification, the results from 16 cases were pooled. On average, colocalization was identified in 16.36% of the structures stained; the majority (80.0%) were oligomer-free structures and showed immunoreactivity with Tau-5 alone. In addition, 3.60% of tau structures showed immunoreactivity with T22 alone; this could be due to the inaccessibility of the specific epitope for Tau-5 in these oligomers (Fig. 2D). These findings suggest that nearly 20% of total tau in AD brain frontal cortex corresponds to tau oligomers. Besides the presence of intracellular tau oligomers in neurons, extracellular oligomers were also observed in the perivascular space of AD frontal cortex (Fig. 2E–H). Qualitative IHC using T22 showed intense staining of vessel walls and around vessels in general (Fig. 2E–H). Tau oligomers were observed in perivascular spaces of veins and arterioles. The presence of punctuate tau-immunopositive deposits surrounding vessels has been observed in sporadic AD (60–62); however, these deposits have not been identified previously as oligomers. Double staining with von Willebrand factor antibody, which reacts specifically with endothelial cells (Fig. 2I), and T22 (Fig. 2J), confirmed the existence of extracellular tau oligomers surrounding and within blood vessels. Based on the finding of extracellular oligomers in the perivascular spaces of AD samples, it is tempting to speculate that this route might contribute to drainage of tau oligomers from the brain parenchyma and explain previous findings of elevated levels of extracellular tau in CSF in patients with AD and minimal cognitive impairment compared with controls (63–66).
Figure 2.
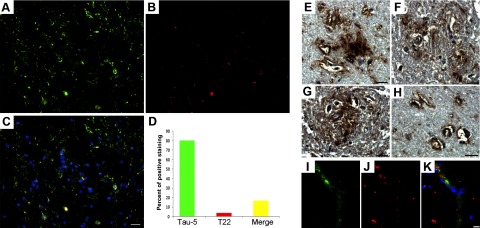
Tau oligomers constitute a small portion of total tau in AD frontal cortex. A–C) Representative immunofluorescent images of AD frontal cortex showing Tau-5 (A, green), T22 (B, red), and merge (C; also including DAPI, blue) confirm the presence of tau oligomers in situ. D) Histogram summarizing quantitation of 16 AD cases; only 20% of total aggregates corresponded to oligomers. Extracellular tau oligomers were also detected. E–H) Photomicrographs demonstrating diffuse extracellular tau oligomers (as detected by T22), including those in association with arterioles. Double immunofluorescence with von Willebrand factor antibody (I, green), T22 (J, red), and merge (K; also with DAPI, blue) suggested the presences of extracellular tau oligomers. Sections were stained with DAPI. Scale bar = 20 μm (A–C); 15 μm (E–K).
Phosphorylation states of tau oligomers in AD brains
Evidence suggests that phosphorylation of tau at Thr231 occurs early during tau aggregation and that it is essential to inhibit the ability of tau to bind and stabilize microtubules (67–69). Phosphorylated Thr231 antibody labels all stages of NFTs (5, 70). Using a commercially available antibody specific for this phospho-epitope, double immunofluorescence in combination with T22 was performed to determine the relationship of tau phosphorylation and oligomerization. Anti-phospho-tau Thr231 labeled the main stages of NFT formation (Fig. 3). Anti-phospho-tau Thr231 signal was present in the neuronal soma surrounding a nucleus with normal morphology, characteristic of pretangles (Fig. 3A, C). In later stages of NFT evolution (Fig. 3D, F), pThr231 immunoreactivity showed patterns including flame-shaped with dislocated or absent nuclei, corresponding to iNFTs and eNFTs, respectively. The staining pattern of T22 in some cases was inside of the neuronal soma around a healthy nucleus, confirming the presence of oligomers in pretangles (Fig. 3B, C). In other cases (Fig. 3E, F), tau oligomers were also present as flame shapes in cells with eccentric nuclei (iNFTs) but not in flame-shaped structures lacking nuclei, i.e., ghost tangles/eNFTs. Based on the stages of NFT evolution described by Iqbal and colleagues (71), we propose the following sequence of events that describe the interplay of phosphorylation and oligomerization. Tau phosphorylation at Thr231 corresponds to stage 0 (Fig. 3G). Aggregation is initiated, and tau forms oligomers phosphorylated at Thr231, corresponding to stage 1, or pretangles (Fig. 3H). Later, the neuron contains a mixture of tau oligomer, protofilaments, and filaments, corresponding to stage 2, or iNFTs (Fig. 3I). Finally, the neuron dies and an extraneuronal flame-shaped NFT pool remains, equivalent to stage 3, which is not recognized by T22 but is still detected by anti-phospho-tau Thr231 (Fig. 3J). This sequence (Fig. 3G–N) is supported by Western blot analysis, phosphorylated Thr231 in three AD cases and their age-matched controls, suggesting that tau phosphorylation at Thr231 occurs before the formation of oligomers (Fig. 3O).
Figure 3.

Phosphorylation of tau at Thr231 precedes the formation of tau oligomers. A–F) Double staining for anti-phospho Thr231 (A, D; green), T22 (B, E; red), and merge (C, F, plus DAPI in blue) demonstrate that tau oligomers are present at early and intermediate stages of aggregation and are preceded by phosphorylation at Thr231. G) Phospho-tau (Thr231) appears throughout the soma before its aggregation (stage 0). H) At early stages, the cells are positive for T22 and Thr231; nuclei and neuronal outlines appear normal (stage 1). I) In a more advanced stage, intraneuronal oligomers and phospho-tau aggregates colocalize, but the nucleus is eccentric (stage 2). J) Finally, at late stages, phospho-tau Thr231 signal remains, whereas T22 staining and nuclei are gone. K–N) Schematic diagram of tau oligomer formation and cell death, leading to the formation of a ghost tangle. Functional monomeric tau is phosphorylated in healthy neurons (K), then tau oligomerization occurs in the cytoplasm (L). Later, tau oligomers cause cell dysfunction and displacement of the nucleus to the side of the cell (M); these events finally lead to cell death and the formation of NFTs (N). O) Western blot analysis, using anti-phospho Thr231, indicated that tau is phosphorylated at this epitope in age-matched controls and detected high-molecular-mass bands in AD corresponding to tau filaments. Scale bars = 10 μm.
Phosphorylation at the epitope Ser202/Thr205 is regarded as a good marker for late-stage NFTs (5, 72). Hyman and colleagues (5) demonstrated that AT8 immunoreactivity is present primarily in eNFTs and in certain cases in iNFTs. These investigators also found that AT8 revealed dense neuropil thread staining. We double-stained frontal cortical sections with AT8 and T22 antibodies (Fig. 4). AT8 mainly stained eNFTs, whereas T22 labeled iNFTs but not eNFTs (Fig. 4A–F). T22 oligomeric staining preceded that of AT8; the staining pattern of AT8 focused exclusively on NFT evolution demonstrated that the 72.5% exhibited morphology corresponding to eNFTs, 22.3% to iNFTs, and only 5.2% to pretangles (Fig. 4K). In contrast, for T22, 79.5% showed pretangle morphology, 19% iNFTs, and 1.5% eNFTs (Fig. 4J). These results confirm that tau oligomers play a role in early stages during the natural history of NFTs.
Figure 4.
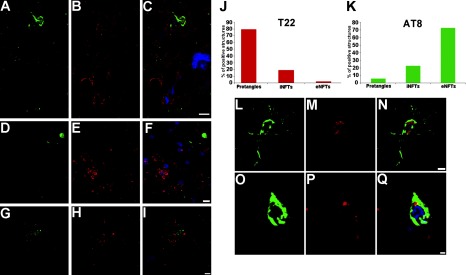
Tau oligomerization occurs before phosphorylation at Ser202/Thr205. A–I) Photomicrographs showing immunofluorescence signals for AT8 (A, D; green), T22 (B, E; red), and merge (C, F; also including DAPI, blue) localize tau oligomers in pretangles (F) and iNFTs (C), but not in eNFTs that are labeled with AT8 (C, F). In some instances, extracellular phosphorylated tau at Ser202/Thr205 (G, green) and extracellular oligomers (H, red) do not correlate (I, merge + DAPI). J, K) Distribution of oligomeric tau and phospho-Ser202/Thr205 tau among NFT species. J) Oligomeric species are distributed 79% in pretangles, 19% in iNFTs, and 2% in eNFTs. K) AT8 signal corresponded 72.4% to eNFTs, 22.3% to iNFTs, and only 5.21% to pretangles. L–Q) It is also possible to see some of AT8 and T22 signals in iNFTs, but in these cases the colocalization between AT8 (L, O; green) and T22 (M, P; red) is negligible (N, Q; merge + DAPI). Scale bars = 10 μm (A–I); 5 μm (L–N); 2 μm (O–Q).
In the frontal cortex regions of each AD brain analyzed, we observed areas that were exclusively stained for tau oligomers (Supplemental Fig. S2A–C) as well as some “transitional” areas that were labeled with T22 and AT8 but with little colocalization (Supplemental Fig. S2D–F). We also noted areas with abundant AT8 staining but lacking oligomers (Supplemental Fig. S2G–I). These data suggest that the presence of tau oligomers coincides with early AT8-positive NFT formation; tau phosphorylation at Ser202/Thr205 continues through late stages of aggregation, probably after or during the transition from the tau oligomers to protofilament and filament stages.
Time course of ubiquitination and tau oligomerization
We performed several experiments to determine the timing of tau ubiquitination relative to the formation of tau oligomers. First, standard double immunofluorescence was performed using anti-ubiquitin and T22. Tau oligomers at an early stage (pretangles) are not ubiquitinated (Fig. 5A–C). Ubiquitin immunoreactivity is abundant during later stages of oligomer aggregation (Fig. 5D–I) that correlate with iNFTs. Finally, at late stages of NFT evolution, after the transition from tau oligomers to filaments, eNFTs are still highly ubiquitinated (Fig. 5J–L). To ensure that the colocalization of signal in the immunofluorescence experiments was produced by the detection of the same complex (tau/ubiquitin) by both antibodies (T22 and anti-ubiquitin) and was not due to the overlapping of different focal planes, a 3D reconstruction was performed using confocal microscopy in brain sections stained with T22 and anti-ubiquitin antibodies. To build the 3D images, 27 confocal planes were captured, each with a thickness of 0.7 μm and a separation of 0.2 μm. The total constructed thickness was near 5.2 μm. The anti-ubiquitin signal is in the same confocal planes as the structures labeled with T22 (Supplemental Fig. S3A, B). We performed ICA to determine whether the staining intensities of ubiquitin and T22 vary in synchronicity. The results demonstrate positive synchrony between the signals produced by anti-ubiquitin and T22 antibodies, according to ICA (Supplemental Fig. S3D, F; represented by orange pseudocolor).
Figure 5.
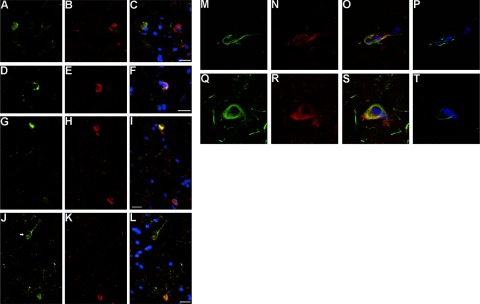
Ubiquitination of tau occurs at intermediate stages during NFT evolution, after appearance of oligomers. A–C) Immunofluorescence images of pretangles fail to show colocalization of anti-ubiquitin staining (A, green), T22 (B, red), and merge (C; with DAPI, blue). D–I) Tau oligomers (E, H; red) are highly ubiquitinated (D, G; green) at intermediate stages. F, I) Merged images plus DAPI. J–L) Structures labeled with anti-ubiquitin (J, green; arrow) that resemble eNFTs are not detected with T22 (K, red), suggesting that ubiquitination continues after the transition of oligomer to fibril. L) Merged image + DAPI. M–T) Ubiquitinated tau oligomers positive for FRET are present in iNFTs. Laser scanning confocal images depicting anti-ubiquitin staining (M, Q; green), T22 signal (N, R; red), and merged images plus DAPI (O, S); colocalization of anti-ubiquitin and tau oligomers is seen only in iNFTs (O, S). Increased donor fluorescence (ubiquitin, Alexa Fluor 488) is observed only in iNFTs, showing the presence of FRET exclusively in these structures (P, T). Scale bars = 10 μm.
To further define and characterize the colocalization between ubiquitin and tau oligomers, we performed FRET. These data confirmed the proximity of ubiquitin and tau oligomers, with a distance of <124 Å (Foster radius 62 Å) between the two molecules (Fig. 5M–T and Supplemental Fig. S4). After complete photobleaching of acceptor molecules (tau oligomers, Alexa Fluor 568), the donor energy (ubiquitin, Alexa Fluor 488) increased, suggesting an energy transfer from donor to acceptor and, therefore, proximity between ubiquitin and tau oligomers. All of the NFTs positive for FRET were iNFTs. The presence of FRET interactions in iNFTs demonstrates that colocalization of ubiquitin and tau oligomers is not due to the presence of other ubiquitinated proteins or tau species surrounding tau oligomers. Together, these data demonstrate that tau oligomers are initially not ubiquitinated, but at later stages, possibly just before becoming filaments, these oligomers tend to become ubiquitinated.
DISCUSSION
The amyloid cascade hypothesis (73, 74) holds that Aβ accumulation precedes tau pathology; however, tau pathology has been described in the absence of Aβ (75). Recently, tau pathology was reported in the entorhinal-hippocampal region preceding the onset of Aβ pathology; moreover, the surprising discovery of tau pathology in children and young adults in the absence of Aβ accumulation (76) calls into question the sequential ordering of events posited by the amyloid cascade. The sequence of pathological events in the amyloid cascade was necessarily based on scoring of NFT pathology, given a historical lack of tools to detect tau oligomers immunohistochemically. The novel natural history of tau oligomers during NFT evolution defined here suggests the need for more detailed evaluation of the relationship between amyloid and tau, in keeping with recent commentaries (77, 78).
The significance of soluble amyloid oligomers and their critical role in neurodegeneration has become more accepted for multiple neurodegenerative diseases, including AD (32, 57, 79–83). The aggregation pathways through which the natively unfolded tau protein adopts higher order structures and fibrillizes have important implications for clarifying the pathogenesis of tauopathies, such as AD, and may lead to the identification of potential drug targets. Details pertaining to the aggregation of soluble and insoluble tau remain unclear. A large body of evidence suggests that tau oligomers, rather than fibrillar aggregates, may be cytotoxic (28, 30, 32–39, 46). A recent study shows the presence of tau oligomers in cell culture and animals overexpressing tau (44), but the authors were unable to verify the specific sizes of oligomeric tau species accumulated in AD brains. Moreover, a similar tau oligomer species was detected in AD brains (46), which the authors described as a tau dimer of similar size and appearance on a Western blot to the species we described here.
Our Western blot assays of AD brain homogenates indicate reactivity of T22 for a subset of tau bands also labeled by Tau-5. A band just below 190 kDa, which could be a dimer or trimer, was elevated in all of our AD cases. Monomeric tau proteins normally associate with each other, forming dimers, followed by a change in conformation that generates larger tau oligomers (40). This result confirms the relevance of tau dimers in the aggregation process, which has already been shown biochemically by others using transgenic mouse models and AD brain (44, 46). Because of technical difficulties in extracting stable aggregates of tau from human brain under the conditions necessary to preserve these conformations, it is impossible to determine whether these bands represent different isoforms of tau or whether they reflect a hyperphosphorylation state of the aggregates. Our Western blot samples were run on the gel immediately after addition of the sample buffer, without boiling; nonetheless we usually visualize oligomers as a smeared band in the gel, which is characteristic of oligomeric species and bears similarities to other amyloidogenic proteins. When brain samples are treated with 8 M urea, all proteins become denatured, including tau oligomers, and only the monomeric isomers can be detected using Tau-5. No bands were detected with T22 (data not shown). This experiment confirms that denaturating agents affect the aggregation state of the protein, disrupting the oligomeric conformation of tau. This appears to be a roadblock with current techniques designed to identify isoforms or post-translational modifications in tau, because T22 is unable to detect these structures under extreme denaturing conditions. The high specificity of T22 in vivo allowed us to analyze the levels of tau oligomers in patients with AD and their age-matched controls using ELISA and determine, by IHC, the relationship of oligomeric tau to total tau more accurately.
For many years, the biochemical characterization of tau aggregation and PHF formation involved well-standardized protocols based on the use of sarcosyl (50, 84). Using this method, we identified tau oligomers in two fractions, suggesting that there could be at least two distinct types of tau oligomers: sarkosyl-soluble tau and sarkosyl-insoluble tau. Soluble tau oligomers appear to be mainly dimers/trimers, and, in the case of insoluble tau oligomers, the structures go from dimers to form tetramers and larger structures. It is well known that the size of amyloid oligomers is not a reliable indicator of their conformation and aggregation state (57, 85–87).
Phosphorylation of Thr231 occurs early during evolution of NFTs. Phosphorylation at Thr231 inhibits the binding of tau to microtubules (67, 69), and this phospho-epitope is present in all types of NFTs (5, 88). We observed that the phospho-epitope Thr231 is present in pretangles, iNFTs, and eNFTs and that this phosphorylation precedes tau oligomerization. Phosphorylation at Ser202 and Thr205 has been related to late phospho-tau changes (5). Using the commercially available antibody AT8, we confirmed the specificity of T22 for tau oligomers detected only in pretangles and iNFTs. There is generally poor colocalization of T22 and AT8 (Fig. 4L–Q), in keeping with the hypothesis that tau oligomers are converted to protofilaments and filaments before phosphorylation at Ser202/Thr205. Another possibility could be that the phosphor-epitope Ser202/Thr205 is not accessible when tau adapts the oligomeric conformation, thus making it impossible to detect it with AT8. Other studies conclude that the phosphorylation at Ser202/Thr205 occurs at early stages of pathology (89, 90), but we have to keep in mind that in our study we did not perform any antigen-retrieval protocol to preserve the oligomeric conformation of tau and its reactivity with T22. Even though these results suggest that tau undergoes different modification, which may be associated with its adaptation of different conformations, at this time it is hard to establish the temporal relationship between phosphorylation and the formation of tau oligomers based on the end-stage pathology. In AD frontal cortex, we detect areas exclusively positive for T22, others exclusively positive for AT8, and a third subset that seems to represent the transition of oligomers to NFTs. It is intriguing to consider these data in light of novel studies suggesting that aggregated tau can transmit a misfolded state from one cell to another and indicating that tau oligomers could play a key role in this process (91–93). In AD, NFTs appear first in the hippocampus, the basal nucleus of Meynert, and the brainstem (7, 94–96) and then subsequently spread to other brain regions, including the neocortex, in a stereotypical pattern. However, at the cellular level, it is unclear how tau pathology spreads, and further work is necessary to define this process. Nevertheless, our results provide strong evidence that tau oligomers progress in an orderly sequence in AD brain.
The ubiquitin-proteasome system degrades misfolded, unassembled, or damaged proteins that could otherwise form potentially toxic aggregates (97). Studies have indicated that tau ubiquitination may take place at a rather early stage in tau aggregation (98, 99). Here, we demonstrate that at early stages (pretangles), tau oligomers are not ubiquitinated, but that these oligomers become ubiquitinated in iNFTs. Tau ubiquitination continues during the transition phase from oligomers to filaments (eNFTs). Having an excess of abnormally phosphorylated soluble tau in tauopathies may exceed the capacity of the proteasome, with excess ubiquitinated phospho-tau shifted into stable aggregates. Taking into consideration these published data and our results, we suggest that tau oligomers are highly ubiquitinated for proteasomal degradation, but at later stages these ubiquitinated oligomers inhibit the proteasome. It is also possible that the excess of these oligomers may exceed the capacity of the proteasome, thereby shifting excess ubiquitinated tau into more stable and less toxic aggregates.
We have demonstrated the specificity of the novel antibody T22 for tau oligomers in vivo. This unique tool has provided us with the exceptional opportunity to use tau oligomers as a reliable biomarker to differentiate patients with AD from age-matched controls and to begin to analyze the chronological relationship between tau oligomerization and its post-translational modifications, such as phosphorylation and ubiquitination (Fig. 6). Our data validate tau oligomers as a relevant factor in AD and present T22 as an invaluable tool to answer unresolved questions regarding the relation between tau aggregation, amyloid, and disease progression.
Figure 6.

Schematic diagram of NFT life cycle and the role of oligomers in NFT formation and disease progression. 1) Under normal conditions, tau participates in the association and dissociation of microtubules, conferring dynamics to the system. 2) Post-translational modifications of tau, e.g., phosphorylation, can dissociate tau from microtubules. This dissociation produces an increase in the cytosolic concentration of the protein that exceeds the minimal tau concentration necessary to support conformational changes and leads to an electrostatic modification in the molecule that enables it to form a side chain-side chain interaction, culminating in the formation of a tau-tau dimer. Once these dimers are formed and adopt a stable structure, they can begin a process of nucleation, forming oligomers. 3) Subsequently, tau continues the aggregation process and undergoes additional post-translational modifications, such as phosphorylation and ubiquitination. These oligomers (in 2 and 3) are highly toxic and can induce memory deficits in mice and seed the aggregation of monomeric unmodified tau. Then tau oligomers form tau filaments, termed PHFs; these tau structures undergo new modifications and form NFTs. 4) Finally, after cell death, NFTs maintain their flame shape and form “ghost tangles” or eNFTs. Dashed arrows indicate that extracellular tau oligomers can induce the aggregation of monomeric tau from healthy neurons.
Supplementary Material
Acknowledgments
The authors thank Dr. Bridget E. Hawkins for her suggestions and critical reading of the manuscript. The authors are grateful to Prof. Carl Cotman [University of California–Irvine (UCI), Irvine, CA, USA] and the UCI–Alzheimer's Disease Research Center brain bank for providing brain samples, Dr. Marcos Guerrero-Munoz for his help in the purification and characterization of tau oligomers, and Dr. Adriana Paulucci for her help in using the confocal and image analyses.
This work was supported by the Cullen Family Trust for Health Care, the Alzheimer's Drug Discovery Foundation, and the Mitchell Center for Neurodegenerative Diseases. R.K. is the founder of ConImm, Inc. and has patent applications on the compositions and methods related to tau oligomers.
This article includes supplemental data. Please visit http://www.fasebj.org to obtain this information.
- 3D
- 3-dimensional
- Aβ
- amyloid-β
- AD
- Alzheimer's disease
- CCD
- charge-coupled device
- eNFT
- extraneuronal neurofibrillary tangle
- FRET
- fluorescence resonance energy transfer
- ICA
- intensity correlation analysis
- IHC
- immunohistochemistry
- iNFT
- intraneuronal neurofibrillary tangle
- MTT
- 3-[4,5-dimethylthiazol-2-yl]-2,5-diphenyltetrazolium bromide
- NFT
- neurofibrillary tangle
- PHF
- paired helical filament
- pre-NFT
- pre-neurofibrillary tangle phospho-tau aggregate
- PSP
- progressive supranuclear palsy
- TBS-T
- Tris-buffered saline with Tween 20.
REFERENCES
- 1. Schultz C., Ghebremedhin E., Del Tredici K., Rub U., Braak H. (2004) High prevalence of thorn-shaped astrocytes in the aged human medial temporal lobe. Neurobiol. Aging 25, 397–405 [DOI] [PubMed] [Google Scholar]
- 2. Umahara T., Tsuchiya K., Ikeda K., Kanaya K., Iwamoto T., Takasaki M., Mukai K., Shibata N., Kato S. (2002) Demonstration and distribution of tau-positive glial coiled body-like structures in white matter and white matter threads in early onset Alzheimer's disease. Neuropathology 22, 9–12 [DOI] [PubMed] [Google Scholar]
- 3. Braak H., Braak E. (1988) Neuropil threads occur in dendrites of tangle-bearing nerve cells. Neuropathol. Appl. Neurobiol. 14, 39–44 [DOI] [PubMed] [Google Scholar]
- 4. Adalbert R., Nogradi A., Babetto E., Janeckova L., Walker S. A., Kerschensteiner M., Misgeld T., Coleman M. P. (2009) Severely dystrophic axons at amyloid plaques remain continuous and connected to viable cell bodies. Brain 132, 402–416 [DOI] [PubMed] [Google Scholar]
- 5. Augustinack J. C., Schneider A., Mandelkow E. M., Hyman B. T. (2002) Specific tau phosphorylation sites correlate with severity of neuronal cytopathology in Alzheimer's disease. Acta Neuropathol. 103, 26–35 [DOI] [PubMed] [Google Scholar]
- 6. Arnold S. E., Hyman B. T., Flory J., Damasio A. R., Van Hoesen G. W. (1991) The topographical and neuroanatomical distribution of neurofibrillary tangles and neuritic plaques in the cerebral cortex of patients with Alzheimer's disease. Cereb. Cortex 1, 103–116 [DOI] [PubMed] [Google Scholar]
- 7. Braak H., Braak E. (1991) Neuropathological staging of Alzheimer-related changes. Acta Neuropathol. 82, 239–259 [DOI] [PubMed] [Google Scholar]
- 8. Alonso A. C., Li B., Grundke-Iqbal I., Iqbal K. (2008) Mechanism of tau-induced neurodegeneration in Alzheimer disease and related tauopathies. Curr. Alzheimer. Res. 5, 375–384 [DOI] [PubMed] [Google Scholar]
- 9. Lee V. M., Goedert M., Trojanowski J. Q. (2001) Neurodegenerative tauopathies. Annu. Rev. Neurosci. 24, 1121–1159 [DOI] [PubMed] [Google Scholar]
- 10. Buee L., Bussiere T., Buee-Scherrer V., Delacourte A., Hof P. R. (2000) Tau protein isoforms, phosphorylation and role in neurodegenerative disorders. Brain Res. Brain Res. Rev. 33, 95–130 [DOI] [PubMed] [Google Scholar]
- 11. Goedert M., Spillantini M. G., Cairns N. J., Crowther R. A. (1992) Tau proteins of Alzheimer paired helical filaments: abnormal phosphorylation of all six brain isoforms. Neuron 8, 159–168 [DOI] [PubMed] [Google Scholar]
- 12. Matsuo E. S., Shin R. W., Billingsley M. L., Van deVoorde A., O'Connor M., Trojanowski J. Q., Lee V. M. (1994) Biopsy-derived adult human brain tau is phosphorylated at many of the same sites as Alzheimer's disease paired helical filament tau. Neuron 13, 989–1002 [DOI] [PubMed] [Google Scholar]
- 13. Morishima-Kawashima M., Hasegawa M., Takio K., Suzuki M., Yoshida H., Watanabe A., Titani K., Ihara Y. (1995) Hyperphosphorylation of tau in PHF. Neurobiol. Aging 16, 365–371; discussion 371–380 [DOI] [PubMed] [Google Scholar]
- 14. Drewes G., Lichtenberg-Kraag B., Doring F., Mandelkow E. M., Biernat J., Goris J., Doree M., Mandelkow E. (1992) Mitogen activated protein (MAP) kinase transforms tau protein into an Alzheimer-like state. EMBO J. 11, 2131–2138 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Hanger D. P., Hughes K., Woodgett J. R., Brion J. P., Anderton B. H. (1992) Glycogen synthase kinase-3 induces Alzheimer's disease-like phosphorylation of tau: generation of paired helical filament epitopes and neuronal localisation of the kinase. Neurosci. Lett. 147, 58–62 [DOI] [PubMed] [Google Scholar]
- 16. Drewes G., Trinczek B., Illenberger S., Biernat J., Schmitt-Ulms G., Meyer H. E., Mandelkow E. M., Mandelkow E. (1995) Microtubule-associated protein/microtubule affinity-regulating kinase (p110mark). A novel protein kinase that regulates tau-microtubule interactions and dynamic instability by phosphorylation at the Alzheimer-specific site serine 262. J. Biol. Chem. 270, 7679–7688 [DOI] [PubMed] [Google Scholar]
- 17. Baumann K., Mandelkow E. M., Biernat J., Piwnica-Worms H., Mandelkow E. (1993) Abnormal Alzheimer-like phosphorylation of tau-protein by cyclin-dependent kinases cdk2 and cdk5. FEBS Lett. 336, 417–424 [DOI] [PubMed] [Google Scholar]
- 18. Goedert M., Jakes R., Qi Z., Wang J. H., Cohen P. (1995) Protein phosphatase 2A is the major enzyme in brain that dephosphorylates tau protein phosphorylated by proline-directed protein kinases or cyclic AMP-dependent protein kinase. J. Neurochem. 65, 2804–2807 [DOI] [PubMed] [Google Scholar]
- 19. Johnson G. V., Stoothoff W. H. (2004) Tau phosphorylation in neuronal cell function and dysfunction. J. Cell Sci. 117, 5721–5729 [DOI] [PubMed] [Google Scholar]
- 20. Chatterjee S., Sang T. K., Lawless G. M., Jackson G. R. (2009) Dissociation of tau toxicity and phosphorylation: role of GSK-3β, MARK and Cdk5 in a Drosophila model. Hum. Mol. Genet. 18, 164–177 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Talmat-Amar Y., Arribat Y., Redt-Clouet C., Feuillette S., Bouge A. L., Lecourtois M., Parmentier M. L. (2011) Important neuronal toxicity of microtubule-bound Tau in vivo in Drosophila. Hum. Mol. Genet. 20, 3738–3745 [DOI] [PubMed] [Google Scholar]
- 22. Reifert J., Hartung-Cranston D., Feinstein S. C. (2011) Amyloid β-mediated cell death of cultured hippocampal neurons reveals extensive tau fragmentation without increased full-length Tau phosphorylation. J. Biol. Chem. 286, 20797–20811 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Diaz-Hernandez M., Gomez-Ramos A., Rubio A., Gomez-Villafuertes R., Naranjo J. R., Miras-Portugal M. T., Avila J. (2010) Tissue-nonspecific alkaline phosphatase promotes the neurotoxicity effect of extracellular tau. J. Biol. Chem. 285, 32539–32548 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Andorfer C., Acker C. M., Kress Y., Hof P. R., Duff K., Davies P. (2005) Cell-cycle reentry and cell death in transgenic mice expressing nonmutant human tau isoforms. J. Neurosci. 25, 5446–5454 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Polydoro M., Acker C. M., Duff K., Castillo P. E., Davies P. (2009) Age-dependent impairment of cognitive and synaptic function in the htau mouse model of tau pathology. J. Neurosci. 29, 10741–10749 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Yoshiyama Y., Higuchi M., Zhang B., Huang S. M., Iwata N., Saido T. C., Maeda J., Suhara T., Trojanowski J. Q., Lee V. M. (2007) Synapse loss and microglial activation precede tangles in a P301S tauopathy mouse model. Neuron 53, 337–351 [DOI] [PubMed] [Google Scholar]
- 27. Mocanu M. M., Nissen A., Eckermann K., Khlistunova I., Biernat J., Drexler D., Petrova O., Schonig K., Bujard H., Mandelkow E., Zhou L., Rune G., Mandelkow E. M. (2008) The potential for β-structure in the repeat domain of tau protein determines aggregation, synaptic decay, neuronal loss, and coassembly with endogenous Tau in inducible mouse models of tauopathy. J. Neurosci. 28, 737–748 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Wittmann C. W., Wszolek M. F., Shulman J. M., Salvaterra P. M., Lewis J., Hutton M., Feany M. B. (2001) Tauopathy in Drosophila: neurodegeneration without neurofibrillary tangles. Science 293, 711–714 [DOI] [PubMed] [Google Scholar]
- 29. Paquet D., Bhat R., Sydow A., Mandelkow E. M., Berg S., Hellberg S., Falting J., Distel M., Koster R. W., Schmid B., Haass C. (2009) A zebrafish model of tauopathy allows in vivo imaging of neuronal cell death and drug evaluation. J. Clin. Invest. 119, 1382–1395 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Berger Z., Roder H., Hanna A., Carlson A., Rangachari V., Yue M., Wszolek Z., Ashe K., Knight J., Dickson D., Andorfer C., Rosenberry T. L., Lewis J., Hutton M., Janus C. (2007) Accumulation of pathological tau species and memory loss in a conditional model of tauopathy. J. Neurosci. 27, 3650–3662 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Spires T. L., Orne J. D., SantaCruz K., Pitstick R., Carlson G. A., Ashe K. H., Hyman B. T. (2006) Region-specific dissociation of neuronal loss and neurofibrillary pathology in a mouse model of tauopathy. Am. J. Pathol. 168, 1598–1607 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Brunden K. R., Trojanowski J. Q., Lee V. M. (2008) Evidence that non-fibrillar tau causes pathology linked to neurodegeneration and behavioral impairments. J. Alzheimers Dis. 14, 393–399 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Marx J. (2007) Alzheimer's disease. A new take on tau. Science 316, 1416–1417 [DOI] [PubMed] [Google Scholar]
- 34. Meraz-Rios M. A., Lira-De Leon K. I., Campos-Pena V., De Anda-Hernandez M. A., Mena-Lopez R. (2010) Tau oligomers and aggregation in Alzheimer's disease. J. Neurochem. 112, 1353–1367 [DOI] [PubMed] [Google Scholar]
- 35. Kayed R., Jackson G. R. (2009) Prefilament tau species as potential targets for immunotherapy for Alzheimer disease and related disorders. Curr. Opin. Immunol. 21, 359–363 [DOI] [PubMed] [Google Scholar]
- 36. Santacruz K., Lewis J., Spires T., Paulson J., Kotilinek L., Ingelsson M., Guimaraes A., DeTure M., Ramsden M., McGowan E., Forster C., Yue M., Orne J., Janus C., Mariash A., Kuskowski M., Hyman B., Hutton M., Ashe K. H. (2005) Tau suppression in a neurodegenerative mouse model improves memory function. Science 309, 476–481 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Sydow A., Van der Jeugd A., Zheng F., Ahmed T., Balschun D., Petrova O., Drexler D., Zhou L., Rune G., Mandelkow E., D'Hooge R., Alzheimer C., Mandelkow E. M. (2011) Tau-induced defects in synaptic plasticity, learning, and memory are reversible in transgenic mice after switching off the toxic Tau mutant. J. Neurosci. 31, 2511–2525 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Morsch R., Simon W., Coleman P. D. (1999) Neurons may live for decades with neurofibrillary tangles. J. Neuropathol. Exp. Neurol. 58, 188–197 [DOI] [PubMed] [Google Scholar]
- 39. Spires-Jones T. L., Kopeikina K. J., Koffie R. M., de Calignon A., Hyman B. T. (2011) Are tangles as toxic as they look? J. Mol. Neurosci. 45, 438–444 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Lasagna-Reeves C. A., Castillo-Carranza D. L., Guerrero-Muoz M. J., Jackson G. R., Kayed R. (2010) Preparation and characterization of neurotoxic tau oligomers. Biochemistry 49, 10039–10041 [DOI] [PubMed] [Google Scholar]
- 41. Gomez-Ramos A., Diaz-Hernandez M., Cuadros R., Hernandez F., Avila J. (2006) Extracellular tau is toxic to neuronal cells. FEBS Lett. 580, 4842–4850 [DOI] [PubMed] [Google Scholar]
- 42. Gomez-Ramos A., Diaz-Hernandez M., Rubio A., Miras-Portugal M. T., Avila J. (2008) Extracellular tau promotes intracellular calcium increase through M1 and M3 muscarinic receptors in neuronal cells. Mol. Cell. Neurosci. 37, 673–681 [DOI] [PubMed] [Google Scholar]
- 43. Lasagna-Reeves C. A., Castillo-Carranza D. L., Sengupta U., Clos A. L., Jackson G. R., Kayed R. (2011) Tau oligomers impair memory and induce synaptic and mitochondrial dysfunction in wild-type mice. Mol. Neurodegener. 6, 39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Sahara N., Maeda S., Murayama M., Suzuki T., Dohmae N., Yen S. H., Takashima A. (2007) Assembly of two distinct dimers and higher-order oligomers from full-length tau. Eur. J. Neurosci. 25, 3020–3029 [DOI] [PubMed] [Google Scholar]
- 45. Maeda S., Sahara N., Saito Y., Murayama S., Ikai A., Takashima A. (2006) Increased levels of granular tau oligomers: an early sign of brain aging and Alzheimer's disease. Neurosci. Res. 54, 197–201 [DOI] [PubMed] [Google Scholar]
- 46. Patterson K. R., Remmers C., Fu Y., Brooker S., Kanaan N. M., Vana L., Ward S., Reyes J. F., Philibert K., Glucksman M. J., Binder L. I. (2011) Characterization of prefibrillar tau oligomers in vitro and in Alzheimers disease. J. Biol. Chem. 286, 23063–23076 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Margittai M., Langen R. (2004) Template-assisted filament growth by parallel stacking of tau. Proc. Natl. Acad. Sci. U. S. A. 101, 10278–10283 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Margittai M., Langen R. (2006) Side chain-dependent stacking modulates tau filament structure. J. Biol. Chem. 281, 37820–37827 [DOI] [PubMed] [Google Scholar]
- 49. Lasagna-Reeves C. A., Glabe C. G., Kayed R. (2011) Amyloid-β annular protofibrils evade fibrillar fate in Alzheimer's disease brain. J. Biol. Chem. 286, 22122–22130 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Lee V. M., Wang J., Trojanowski J. Q. (1999) Purification of paired helical filament tau and normal tau from human brain tissue. Methods Enzymol. 309, 81–89 [DOI] [PubMed] [Google Scholar]
- 51. Li Q., Lau A., Morris T. J., Guo L., Fordyce C. B., Stanley E. F. (2004) A syntaxin 1, Gαo, and N-type calcium channel complex at a presynaptic nerve terminal: analysis by quantitative immunocolocalization. J. Neurosci. 24, 4070–4081 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Sharma N., McLean P. J., Kawamata H., Irizarry M. C., Hyman B. T. (2001) α-Synuclein has an altered conformation and shows a tight intermolecular interaction with ubiquitin in Lewy bodies. Acta Neuropathol. 102, 329–334 [DOI] [PubMed] [Google Scholar]
- 53. Knowles R. B., Chin J., Ruff C. T., Hyman B. T. (1999) Demonstration by fluorescence resonance energy transfer of a close association between activated MAP kinase and neurofibrillary tangles: implications for MAP kinase activation in Alzheimer disease. J. Neuropathol. Exp. Neurol. 58, 1090–1098 [DOI] [PubMed] [Google Scholar]
- 54. Kinoshita A., Whelan C. M., Smith C. J., Mikhailenko I., Rebeck G. W., Strickland D. K., Hyman B. T. (2001) Demonstration by fluorescence resonance energy transfer of two sites of interaction between the low-density lipoprotein receptor-related protein and the amyloid precursor protein: role of the intracellular adapter protein Fe65. J. Neurosci. 21, 8354–8361 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Yoshida M. (2006) Cellular tau pathology and immunohistochemical study of tau isoforms in sporadic tauopathies. Neuropathology 26, 457–470 [DOI] [PubMed] [Google Scholar]
- 56. Carmel G., Mager E. M., Binder L. I., Kuret J. (1996) The structural basis of monoclonal antibody Alz50's selectivity for Alzheimer's disease pathology. J. Biol. Chem. 271, 32789–32795 [DOI] [PubMed] [Google Scholar]
- 57. Glabe C. G. (2008) Structural classification of toxic amyloid oligomers. J. Biol. Chem. 283, 29639–29643 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Biernat J., Mandelkow E. M. (1999) The development of cell processes induced by tau protein requires phosphorylation of serine 262 and 356 in the repeat domain and is inhibited by phosphorylation in the proline-rich domains. Mol. Biol. Cell 10, 727–740 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Goedert M., Jakes R., Vanmechelen E. (1995) Monoclonal antibody AT8 recognises tau protein phosphorylated at both serine 202 and threonine 205. Neurosci. Lett. 189, 167–169 [DOI] [PubMed] [Google Scholar]
- 60. Vidal R., Calero M., Piccardo P., Farlow M. R., Unverzagt F. W., Mendez E., Jimenez-Huete A., Beavis R., Gallo G., Gomez-Tortosa E., Ghiso J., Hyman B. T., Frangione B., Ghetti B. (2000) Senile dementia associated with amyloid beta protein angiopathy and tau perivascular pathology but not neuritic plaques in patients homozygous for the APOE-ε4 allele. Acta Neuropathol. 100, 1–12 [DOI] [PubMed] [Google Scholar]
- 61. Hernandez F., Avila J. (2011) Intra and extracellular protein interactions with tau. Curr. Alzheimer. Res. 7, 670–676 [DOI] [PubMed] [Google Scholar]
- 62. Avila J. (2011) Intracellular and extracellular tau. Front. Neurosci. 4, 49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Andreasen N., Minthon L., Vanmechelen E., Vanderstichele H., Davidsson P., Winblad B., Blennow K. (1999) Cerebrospinal fluid tau and Aβ42 as predictors of development of Alzheimer's disease in patients with mild cognitive impairment. Neurosci. Lett. 273, 5–8 [DOI] [PubMed] [Google Scholar]
- 64. Galasko D., Clark C., Chang L., Miller B., Green R. C., Motter R., Seubert P. (1997) Assessment of CSF levels of tau protein in mildly demented patients with Alzheimer's disease. Neurology 48, 632–635 [DOI] [PubMed] [Google Scholar]
- 65. Blom E. S., Giedraitis V., Zetterberg H., Fukumoto H., Blennow K., Hyman B. T., Irizarry M. C., Wahlund L. O., Lannfelt L., Ingelsson M. (2009) Rapid progression from mild cognitive impairment to alzheimer's disease in subjects with elevated levels of tau in cerebrospinal fluid and the APOEε4/ε4 Genotype. Dement. Geriatr. Cogn. Disord. 27, 458–464 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Shaw L. M., Vanderstichele H., Knapik-Czajka M., Clark C. M., Aisen P. S., Petersen R. C., Blennow K., Soares H., Simon A., Lewczuk P., Dean R., Siemers E., Potter W., Lee V. M., Trojanowski J. Q. (2009) Cerebrospinal fluid biomarker signature in Alzheimer's disease neuroimaging initiative subjects. Ann. Neurol. 65, 403–413 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Sengupta A., Kabat J., Novak M., Wu Q., Grundke-Iqbal I., Iqbal K. (1998) Phosphorylation of tau at both Thr 231 and Ser 262 is required for maximal inhibition of its binding to microtubules. Arch. Biochem. Biophys. 357, 299–309 [DOI] [PubMed] [Google Scholar]
- 68. Cho J. H., Johnson G. V. (2004) Primed phosphorylation of tau at Thr231 by glycogen synthase kinase 3β (GSK3β) plays a critical role in regulating tau's ability to bind and stabilize microtubules. J. Neurochem. 88, 349–358 [DOI] [PubMed] [Google Scholar]
- 69. Lin Y. T., Cheng J. T., Liang L. C., Ko C. Y., Lo Y. K., Lu P. J. (2007) The binding and phosphorylation of Thr231 is critical for Tau's hyperphosphorylation and functional regulation by glycogen synthase kinase 3β. J. Neurochem. 103, 802–813 [DOI] [PubMed] [Google Scholar]
- 70. Jicha G. A., Lane E., Vincent I., Otvos L., Jr., Hoffmann R., Davies P. (1997) A conformation- and phosphorylation-dependent antibody recognizing the paired helical filaments of Alzheimer's disease. J. Neurochem. 69, 2087–2095 [DOI] [PubMed] [Google Scholar]
- 71. Bancher C., Brunner C., Lassmann H., Budka H., Jellinger K., Wiche G., Seitelberger F., Grundke-Iqbal I., Iqbal K., Wisniewski H. M. (1989) Accumulation of abnormally phosphorylated tau precedes the formation of neurofibrillary tangles in Alzheimer's disease. Brain Res. 477, 90–99 [DOI] [PubMed] [Google Scholar]
- 72. Uchihara T., Nakamura A., Yamazaki M., Mori O. (2001) Evolution from pretangle neurons to neurofibrillary tangles monitored by thiazin red combined with Gallyas method and double immunofluorescence. Acta Neuropathol. 101, 535–539 [DOI] [PubMed] [Google Scholar]
- 73. Hardy J., Selkoe D. J. (2002) The amyloid hypothesis of Alzheimer's disease: progress and problems on the road to therapeutics. Science 297, 353–356 [DOI] [PubMed] [Google Scholar]
- 74. Selkoe D. J. (1994) Alzheimer's disease: a central role for amyloid. J. Neuropathol. Exp. Neurol. 53, 438–447 [DOI] [PubMed] [Google Scholar]
- 75. Braak H., Braak E. (1997) Frequency of stages of Alzheimer-related lesions in different age categories. Neurobiol. Aging 18, 351–357 [DOI] [PubMed] [Google Scholar]
- 76. Braak H., Del Tredici K. (2011) The pathological process underlying Alzheimer's disease in individuals under thirty. Acta Neuropathol. 121, 171–181 [DOI] [PubMed] [Google Scholar]
- 77. Small S. A., Duff K. (2008) Linking Aβ and tau in late-onset Alzheimer's disease: a dual pathway hypothesis. Neuron 60, 534–542 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78. Duyckaerts C. (2011) Tau pathology in children and young adults: can you still be unconditionally baptist? Acta Neuropathol. 121, 145–147 [DOI] [PubMed] [Google Scholar]
- 79. Haass C., Selkoe D. J. (2007) Soluble protein oligomers in neurodegeneration: lessons from the Alzheimer's amyloid beta-peptide. Nat. Rev. Mol. Cell Biol. 8, 101–112 [DOI] [PubMed] [Google Scholar]
- 80. Caughey B., Baron G. S., Chesebro B., Jeffrey M. (2009) Getting a grip on prions: oligomers, amyloids, and pathological membrane interactions. Annu. Rev. Biochem. 78, 177–204 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81. Glabe C. G. (2006) Common mechanisms of amyloid oligomer pathogenesis in degenerative disease. Neurobiol. Aging 27, 570–575 [DOI] [PubMed] [Google Scholar]
- 82. Oddo S., Vasilevko V., Caccamo A., Kitazawa M., Cribbs D. H., LaFerla F. M. (2006) Reduction of soluble Aβ and tau, but not soluble Aβ alone, ameliorates cognitive decline in transgenic mice with plaques and tangles. J. Biol. Chem. 281, 39413–39423 [DOI] [PubMed] [Google Scholar]
- 83. Lesne S., Koh M. T., Kotilinek L., Kayed R., Glabe C. G., Yang A., Gallagher M., Ashe K. H. (2006) A specific amyloid-β protein assembly in the brain impairs memory. Nature 440, 352–357 [DOI] [PubMed] [Google Scholar]
- 84. Rostagno A., Ghiso J. (2009) Isolation and biochemical characterization of amyloid plaques and paired helical filaments. Curr. Protoc. Cell Biol. Chapter 3, Unit 3.33, 1–33 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85. Campioni S., Mannini B., Zampagni M., Pensalfini A., Parrini C., Evangelisti E., Relini A., Stefani M., Dobson C. M., Cecchi C., Chiti F. (2011) A causative link between the structure of aberrant protein oligomers and their toxicity. Nat. Chem. Biol. 6, 140–147 [DOI] [PubMed] [Google Scholar]
- 86. Kayed R., Canto I., Breydo L., Rasool S., Lukacsovich T., Wu J., Albay R., 3rd, Pensalfini A., Yeung S., Head E., Marsh J. L., Glabe C. (2010) Conformation dependent monoclonal antibodies distinguish different replicating strains or conformers of prefibrillar Aβ oligomers. Mol. Neurodegener. 5, 57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87. Kayed R., Head E., Sarsoza F., Saing T., Cotman C. W., Necula M., Margol L., Wu J., Breydo L., Thompson J. L., Rasool S., Gurlo T., Butler P., Glabe C. G. (2007) Fibril specific, conformation dependent antibodies recognize a generic epitope common to amyloid fibrils and fibrillar oligomers that is absent in prefibrillar oligomers. Mol. Neurodegener. 2, 18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88. Arriagada P. V., Growdon J. H., Hedley-Whyte E. T., Hyman B. T. (1992) Neurofibrillary tangles but not senile plaques parallel duration and severity of Alzheimer's disease. Neurology 42, 631–639 [DOI] [PubMed] [Google Scholar]
- 89. Braak E., Braak H., Mandelkow E. M. (1994) A sequence of cytoskeleton changes related to the formation of neurofibrillary tangles and neuropil threads. Acta Neuropathol. 87, 554–567 [DOI] [PubMed] [Google Scholar]
- 90. Kimura T., Ono T., Takamatsu J., Yamamoto H., Ikegami K., Kondo A., Hasegawa M., Ihara Y., Miyamoto E., Miyakawa T. (1996) Sequential changes of tau-site-specific phosphorylation during development of paired helical filaments. Dementia 7, 177–181 [DOI] [PubMed] [Google Scholar]
- 91. Clavaguera F., Bolmont T., Crowther R. A., Abramowski D., Frank S., Probst A., Fraser G., Stalder A. K., Beibel M., Staufenbiel M., Jucker M., Goedert M., Tolnay M. (2009) Transmission and spreading of tauopathy in transgenic mouse brain. Nat. Cell Biol. 11, 909–913 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92. Frost B., Diamond M. I. (2010) Prion-like mechanisms in neurodegenerative diseases. Nat. Rev. Neurosci. 11, 155–159 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93. Brundin P., Melki R., Kopito R. (2010) Prion-like transmission of protein aggregates in neurodegenerative diseases. Nat. Rev. Mol. Cell Biol. 11, 301–307 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94. Pearson R. C., Esiri M. M., Hiorns R. W., Wilcock G. K., Powell T. P. (1985) Anatomical correlates of the distribution of the pathological changes in the neocortex in Alzheimer disease. Proc. Natl. Acad. Sci. U. S. A. 82, 4531–4534 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95. Delacourte A., Sergeant N., Wattez A., Maurage C. A., Lebert F., Pasquier F., David J. P. (2002) Tau aggregation in the hippocampal formation: an ageing or a pathological process? Exp. Gerontol. 37, 1291–1296 [DOI] [PubMed] [Google Scholar]
- 96. Lace G., Savva G. M., Forster G., de Silva R., Brayne C., Matthews F. E., Barclay J. J., Dakin L., Ince P. G., Wharton S. B. (2009) Hippocampal tau pathology is related to neuroanatomical connections: an ageing population-based study. Brain 132, 1324–1334 [DOI] [PubMed] [Google Scholar]
- 97. Ciechanover A., Orian A., Schwartz A. L. (2000) Ubiquitin-mediated proteolysis: biological regulation via destruction. Bioessays 22, 442–451 [DOI] [PubMed] [Google Scholar]
- 98. Dickey C. A., Yue M., Lin W. L., Dickson D. W., Dunmore J. H., Lee W. C., Zehr C., West G., Cao S., Clark A. M., Caldwell G. A., Caldwell K. A., Eckman C., Patterson C., Hutton M., Petrucelli L. (2006) Deletion of the ubiquitin ligase CHIP leads to the accumulation, but not the aggregation, of both endogenous phospho- and caspase-3-cleaved tau species. J. Neurosci. 26, 6985–6996 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99. Cripps D., Thomas S. N., Jeng Y., Yang F., Davies P., Yang A. J. (2006) Alzheimer disease-specific conformation of hyperphosphorylated paired helical filament-Tau is polyubiquitinated through Lys-48, Lys-11, and Lys-6 ubiquitin conjugation. J. Biol. Chem. 281, 10825–10838 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.