

Abstract
Recently, whole genome sequencing approaches have pinpointed mutations in genes that were previously not associated with cancer. For Acute Myeloid Leukaemia (AML), and other myeloid disorders, these approaches revealed a high prevalence of mutations in genes encoding the chromosome cohesion complex, cohesin. Cohesin mutations represent a novel genetic pathway for AML, but how AML arises from these mutations is unknown. This review will explore the potential mechanisms by which cohesin mutations contribute to AML and other myeloid malignancies.
Keywords: Cohesin, Mutation, RUNX1, Myeloid, Leukemia, Transcription
Introduction
The development and pathology of acute myeloid leukemia (AML) can be caused by a number of genetic alterations, although the molecular basis of AML is not yet thoroughly understood. Chromosomal translocations and variations such as t(15;17), t(8:21), inv(16), t(9;21), t(9;11) are characteristic of AML, and suggest that genetic events play a key role in leukemogenesis [1]. However, nearly 50% of AML cases have a normal karyotype and lack major chromosome abnormalities. In an effort to elucidate the genetic basis of these cases, next-generation genome sequencing methods have been successfully used in recent years to identify many novel leukemogenic genes [2]. From these analyses, recurrent mutations in genes encoding subunits of the cohesin complex emerged in AML genomes. Several studies have now revealed that mutations in the cohesin complex are strongly associated with AML, and furthermore, that cohesin mutations are also found at high frequency in other related myeloid malignancies. Cohesin mutations could therefore represent a potential new molecular mechanism underpinning oncogenesis. Cohesin has multiple functions, including roles in cell division, nuclear architecture, DNA damage repair, development and transcription, and these functions have been the subject of several comprehensive recent reviews [3-9]. In this concise, focused review, we will discuss molecular functions of cohesin that have potential to influence the etiology and progression of AML and other myeloid malignancies.
Cohesin biology and cancer development
Cohesin is a large ring-shaped protein complex consisting of four major subunits: SMC1A, SMC3, RAD21 and STAG1/2 [7] (Figure 1). While best known for its role in mediating sister chromatid cohesion from S phase until M phase [7], cohesin also plays crucial roles in DNA damage repair and gene expression [10], human development [11] and cancer [12,13]. Cohesin mutations are found in several cancer types [14-16], however their contribution to oncogenesis is unclear with both overexpression and mutation of cohesin subunits being implicated in cancer. For example, overexpression of cohesin subunit RAD21 in breast cancer is associated with poor prognosis and resistance to chemotherapy [17]. Cohesin mutations must necessarily lead to reduced, but not absent function, since complete loss of cohesin function blocks mitosis and results in cell death [18,19]. Therefore, the cohesin mutations found in cancer are usually heterozygous or hypomorphic. The mechanisms by which cohesin mutations contribute to cancer probably involve multiple molecular pathways reflecting its non-mitotic molecular roles [12,13].
Figure 1.
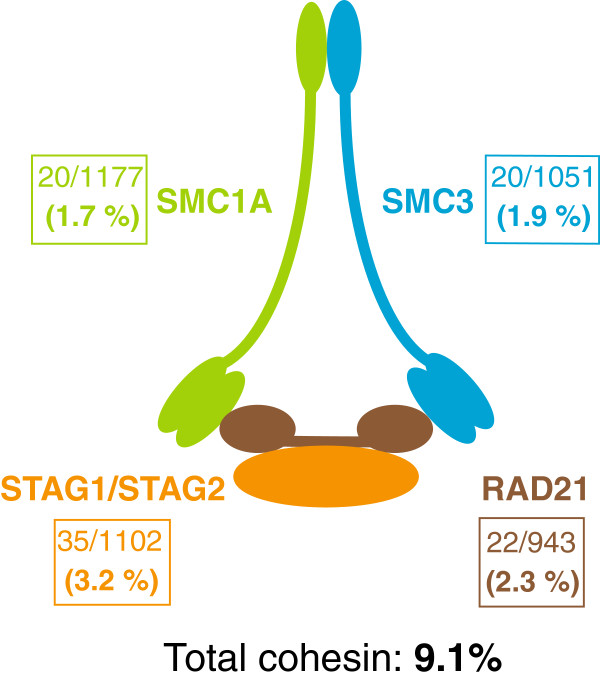
Frequency of cohesin mutations in AML. Cohesin is a multi-subunit protein complex that is involved in chromosome pairing, DNA repair and transcription regulation. Mutations within the individual protein components of cohesin occur at significant frequency in AML. Data from references 20–28 were combined to determine the mutation frequency (boxes) in each of the cohesin subunits (SMC1A, SMC3, STAG1/2, and RAD21). Details of mutations found in each study are presented in Table S1.
Cohesin mutations in myeloid malignancies
The association of cohesin mutations with myeloid malignancy is particularly striking. Data from the Cancer Genome Atlas Research Network (TCGA) revealed that a significant proportion of AMLs had mutations in subunits of cohesin [20]. Somatic variants in cohesin genes were identified in 26/200 cases of AML subjected to exome or whole genome sequencing [20,21]. Sequencing of a separate set of AML samples by Welch et al. identified cohesin mutations in 7/108 cases [21]. Cohesin mutations occurred primarily in French-American-British (FAB) M1 and M2 cases in the TCGA cohort, and Welch et al. found cohesin mutations exclusively in M1 cases [21]. The predominance of cohesin lesions in the most immature forms of AML suggests they were initiating events rather than passenger mutations [21]. Cohesin mutations co-occurred with NPM1, DNMT3A, TET2, or RUNX1 mutations in 17/19 cases [21], implying cooperation with other leukemogenic pathways. Mutations in cohesin genes represented one of just nine categories of mutations thought to actively contribute to leukemogenesis [20]. Our calculation of the rate of cohesin mutations in AML using the TCGA data [20,21] and other published studies to date [22-28] indicates that the total rate of cohesin mutation in AML is around 9% (Figure 1, Table 1). Further details of the contributing studies can be found in the accompanying supplementary table (see Additional file 1: Table S1).
Table 1.
Key findings from selected studies identifying cohesin mutations in myeloid malignancies
| Myeloid disorder(s) | Cohesin mutation rate | Rationale and key findings | Ref # |
|---|---|---|---|
| AML |
26/200 |
200 de novo AML samples were submitted to whole-genome/exome sequencing. The genes identified as recurrently mutated were grouped into nine functional categories important for AML: the cohesin complex was one such category. Cohesin complex mutations were mutually exclusive. |
[20] |
| (13%) | |||
| AML |
7/108 |
Whole-genome sequencing (WGS) of 24 normal-karyotype M1 and M3 AML samples. Cohesin genes were only mutated in M1 samples. Cohesin mutations were mutually exclusive and were not associated with chromosomal instability. |
[21] |
| (6.5%) | |||
| AML |
23/389 |
Targeted sequencing of cohesin genes in 389 AML samples. Cohesin mutations significantly co-occurred with NPM1 mutations. Allelic burden analysis suggested cohesin mutations occurred early in AML. |
[24] |
| (5.9%) | |||
| AML |
23/197 |
Targeted sequencing of 51 myeloid neoplasm candidate genes in 197 AML samples. Cohesin mutations were not associated with overall survival. |
[26] |
| (11.7%) | |||
| AML |
7/170 |
Targeted sequencing of AML candidate loci in 50 AML samples. RAD21 mutations were present in all AML subtypes and were significantly associated with RAS mutations. |
[27] |
| (4.1%) | |||
| AML |
12/158 |
WGS of eight MDS and subsequent secondary-AML patient genomes. Targeted sequencing of 94 MDS/AML candidate loci. Each clone contained at least one mutation that recurs in MDS/AML. STAG2 mutations significantly co-occurred with RUNX1 mutations. |
[28] |
| MDS |
(7.6%) |
||
| MDS |
Approximately 15% |
Targeted sequencing of 104 MDS/AML candidate genes in 944 MDS samples. 47 genes were recurrently mutated in MDS. 14 of these genes (including STAG2) could successfully predict survival-outcome risk groups. STAG2 and SMC1A mutations were significantly associated with adverse patient outcome. |
[29] |
| AML |
65/610 |
Targeted sequencing of cohesin complex genes was undertaken in a cohort of 610 samples from various myeloid neoplasms. The core components of cohesin were significantly mutated. Cohesin mutations were present in the major tumor population in 15/20 available samples, indicating that cohesin mutations often occur as early events in oncogenesis. |
[25] |
| MDS |
(10.7%) |
||
| CMML | |||
| CML | |||
| MPN | |||
| TAM |
39/86 |
WGS of the genomes of TAM, AMKL, and DS-AMKL patients. Progression to DS-AMKL required acquisition of further mutations, including RAD21, STAG2, NRAS, CTCF, EZH2, and TP53. |
[30] |
| AMKL |
(45.3%) | ||
| DS-AMKL | Cohesin mutations were present at a much higher rate in DS-AMKL than AMKL. Allelic burden analysis suggested that cohesin mutations occurred early in DS-AMKL. |
The emergence of cohesin mutations in AML prompted Thol et al.[24] to sequence cohesin complex genes in 389 AML samples, yielding a total of 23 mutations (5.9%). Mutations in cohesin subunits were mutually exclusive, and most mutations were found in karyotypically normal samples. A strong correlation was observed between mutations in cohesin and the known AML-associated gene nucleophosmin (NPM1), with NPM1-mutated patients twice as likely to also harbor a cohesin mutation compared with NPM1-normal. Cohesin mutation status was not prognostically informative, nor did it correlate with any differences in clinical features. Allelic burden analysis suggested that cohesin mutations occurred as an early event during leukemogenesis [24].
While most evidence for cohesin mutations in myeloid leukemia currently comes from AML, cohesin mutation is also implicated in related myeloid disorders. For example, Kon et al.[25] reported frequent mutations in cohesin components in a variety of myeloid neoplasms, including AML, myelodysplastic syndromes (MDS), chronic myelomonocytic leukemia (CMML), chronic myelogenous leukemia (CML) and classical myeloproliferative neoplasms (MPN). Deep sequencing revealed that the majority of cohesin mutations existed in the major tumor populations, indicating they arose early in neoplasia. Strikingly, despite cohesin’s known role in sister chromatid cohesion, myeloid malignancies with cohesin mutations were no more likely to be aneuploid than leukemias harboring other mutations [25]. Kon et al. conclude that, owing to their early origin and frequency in myeloid neoplasms, cohesin mutations actively contribute to leukemogenesis [25].
Further evidence of cohesin’s involvement in myeloid malignancies emerged from a recent study by Haferlach et al. showing that approximately 15% of patients with MDS harbor cohesin mutations [29]. The high proportion of cohesin mutations in MDS, combined with the fact that STAG2 and SMC1A mutations were significantly associated with poor survival outcome, strongly suggests that cohesin mutation is central to the development and prognosis of MDS [29].
Yoshida et al. identified a striking association of cohesin mutation with another myeloid dysplasia, DS-AMKL [30]. Down’s Syndrome (DS) patients can present with transient abnormal myelopoiesis (TAM) that is self-limiting in most cases. TAM is a myeloid proliferation resembling AML, and 10% of TAM progresses to non self-limiting acute megakaryoblastic leukemia (AMKL) in DS patients (DS-AMKL). Deep sequencing revealed that 53% of DS-AMKL samples had acquired cohesin mutations that were not found in somatic cells or the original TAM [30]. The high frequency of lesions in cohesin raises the strong possibility that cohesin mutation is instrumental to progression to DS-AMKL.
Despite the prevalence of cohesin mutations in myeloid dysplasia, the exact mechanism by which cohesin lesions contribute to cancer development is unclear. Accumulating evidence argues that cohesin mutation is an early event in myeloid oncogenesis. Welch et al. and TCGA showed that cohesin mutations mainly occur in the most immature AML subtypes [20,21]; clonal analysis by Kon et al.[25] and allelic burden analysis by Thol et al.[24] suggest that cohesin mutations occur as early events in leukemogenesis. What is the mechanism by which these mutations lead to cancer? In solid tumors with cohesin mutations, chromosome instability and aneuploidy have been suggested as the mechanisms by which cohesin mutation facilitates neoplasia [16,31,32], although other evidence argues against this idea [33]. For myeloid malignancies, a clear theme is emerging: heterozygous cohesin mutations do not cause chromosome instability [21,22,24,25,30] (for details, see Additional file 1: Table S1). This suggests that, at least in myeloid cancers, it is cohesin’s non-mitotic roles that contribute to oncogenesis.
Cohesin regulates gene transcription
Cohesin’s role in gene expression has been intensively investigated over the last 15 years [10]. Several examples of cohesin-dependent gene regulation have been found, including regulatory roles at developmental genes [5] and in stem cells [34]. One of the potential mechanisms by which cohesin regulates gene transcription is through mediating long-range communication events that form DNA loops, which regulate transcription [6]. Enhancers (which promote transcription) and insulators (which usually block transcription) are located in conserved regulatory elements (CREs) on chromosomes, and need not be close to the gene(s) they regulate. Cohesin is thought to physically connect distant CREs with gene promoters, in a cell type-specific manner, to modulate transcriptional outcomes [6] (Figure 2). Therefore, mutations in cohesin could impede cohesin binding to CREs, thereby altering their interaction with promoters, and subsequently gene activity. Similarly, mutations in the CREs that affect cohesin binding could alter transcription of the gene target(s) of that CRE.
Figure 2.
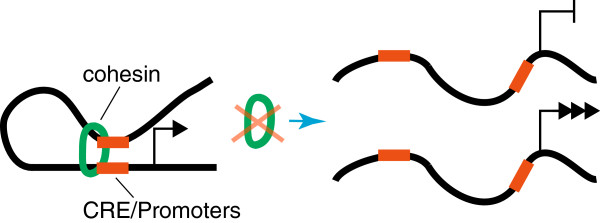
Cohesin regulates gene expression by controlling CRE-promoter interactions. CREs can regulate gene expression by physically contacting a promoter, but are often located at a distance (tens of kilobases and sometimes megabases) from the promoter. Cohesin is involved in the establishment and maintenance of CRE-promoter interactions and can thereby control gene expression. Loss of cohesin can lead to loss of CRE-promoter interactions, resulting in inappropriate gene repression, or gene activation.
In addition to connecting CREs with promoters, cohesin has an important role in organizing global genomic architecture. Cohesin binding of DNA together with CCCTC-binding factor (CTCF) helps to partition the genome into megabase-sized regions known as topologically associated domains (TADs) [35-37]. TADs are demarcated by boundaries that are characterized by the presence of cohesin and CTCF, housekeeping genes, tRNAs and short interspersed element (SINE) retrotransposons [35]. Within TADs are regions of local chromosome interactions, which allow CREs to come into physical proximity with gene promoters to modulate gene expression [38-40]. While TAD boundaries are conserved between cell types, the chromosome interactions within TADs vary, and provide a means for enabling cell type-specific transcription [35,38,40].
Although cohesin and CTCF frequently colocate on chromosomes [41-43], they appear to have distinct roles in genome architecture [40,44,45]. Cohesin influences gene expression by coordinating interactions between CREs and promoters within TADs [38,40], while CTCF is important for preventing interactions between TADs [40]. Cohesin deficiency reduces the number of chromosome interactions within TADs and leads to altered expression of many genes [38-40]: different genes to those dysregulated upon CTCF depletion [40]. Because of the cell type specificity of CRE-promoter interactions within TADs, cohesin deficiency could result in an abnormal transcriptional profile for a particular tissue type (Figure 3). Moreover, there are several genomic sites where cohesin binds exclusively of CTCF, in combination with tissue-specific transcription factors [44,45]. For example, in mouse primary liver cells and hepatocellular carcinoma cells (HepG2), CTCF-independent cohesin binding sites are associated with expression of liver-specific genes [44].
Figure 3.
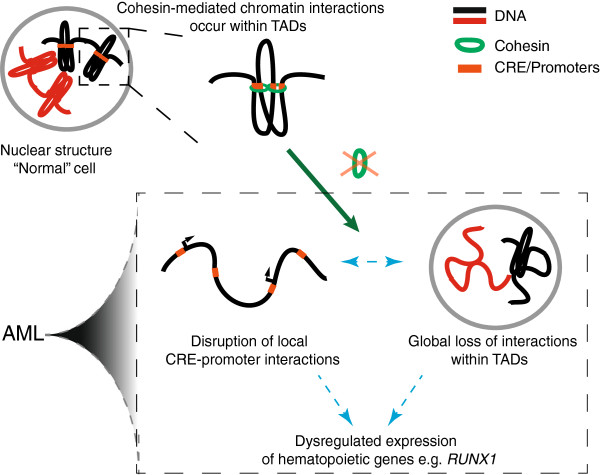
Model for cohesin’s role in AML and other myeloid malignancies. Cohesin has an important function in the nucleus: it mediates chromosome interactions within topologically associated domains (TADs). Within TADs, cohesin connects conserved regulatory elements (CREs) with promoters, thereby regulating gene transcription. When cohesin function is compromised by a heterozygous mutation, as in AML, this leads to loss of CRE-promoter communication at specific hematopoietic genes, such as RUNX1. The result is dysregulation of hematopoietic transcription programs, which could facilitate the development of AML. In addition, loss of tissue-specific sub-domain structures affects the global hematopoietic transcription program.
Strikingly, only a modest reduction in chromatin-bound cohesin is sufficient to cause changes in gene expression [46]. In human cells and mice, heterozygous mutations in the cohesin-loading factor NIPBL or in cohesin subunit SMC1A affect the expression of numerous genes [47,48]. In Drosophila, halving the gene dose of cohesin components robustly affects gene expression [49,50]. Therefore, leukemias with heterozygous cohesin mutations are also likely to be affected by the dysregulation of many genes.
Altered cohesin function has potential to perturb hematopoietic gene expression
A number of hematopoietic transcription factors are regulated by cohesin binding to CREs or promoters. For example, the hematopoietic transcription factor TAL1 is regulated at the transcriptional level by a chromatin hub containing cohesin and CTCF [51]. In further examples, the GATA2 gene contains an intronic +9.5 kb enhancer that is important for its expression [52], while the +85 ERG stem cell enhancer contains binding sites for a heptad of hematopoietic transcription factors and is thought to propagate a hematopoietic stem cell-like transcription profile [53]. Our survey of publically available ENCODE data revealed that cohesin binds both the GATA2 and ERG enhancers in hematopoietic cells (K562). Therefore, it is possible that cohesin mutation could alter the activity of these enhancers and their target genes in a leukemogenic setting.
RUNX1 transcription is altered by cohesin deficiency
The developmental transcription factor RUNX1 plays a particularly important role in myeloid malignancies. RUNX1 function is central to early myeloid differentiation and is absolutely required for definitive hematopoiesis [54,55]. RUNX1 is involved in chromosomal translocations, such as t(12;21) in acute lymphoblastic leukemia in childhood and t(8;21) in acute myeloid leukemia, and is also targeted by point mutations and deletions [56]. Leukemic alterations of RUNX1 lead to abnormal protein function and thus dysregulation of RUNX1 target genes. The importance of RUNX1 function in hematopoiesis and leukemia has generated great interest in determining the factors that regulate its expression.
It is interesting that DS-AMKL leukemias contain three copies of the RUNX1 gene (owing to trisomy 21), as well as having a remarkably high frequency of cohesin mutation (53%) [30]. Evocatively, data from zebrafish provided the first evidence that cohesin regulates tissue-specific Runx1 transcription. In developing zebrafish embryos, a null mutation in the rad21 subunit of cohesin blocked runx1 expression in hematopoietic mesoderm, but not in Rohon-Beard neurons [57]. That cohesin ablation affected hematopoietic progenitors, but not neurons, indicates that the transcriptional role of cohesin is tissue-specific in hematopoietic precursors.
In mouse, a CRE enhancer resides in an intron between the P1 (distal) and P2 (proximal) promoters of Runx1. This enhancer, termed +23 [58] or alternatively, +24 [59], is active only in precursors of hematopoietic stem cells where Runx1 is endogenously expressed [58,59]. Cohesin subunit Rad21 binds the Runx1 + 23/24 mouse enhancer region, which is also conserved in human [60]. ENCODE data from the leukemia K562 cell line indicates that the equivalent human CRE/enhancer also recruits cohesin subunits, together with CTCF [60].
In zebrafish, Marsman et al. showed that cohesin depletion altered the activity of intronic runx1 CREs [60]. Multiple binding sites were identified for cohesin and CTCF in the zebrafish runx1 gene, coinciding with active CREs in the intron between P1 and P2. Cohesin and CTCF determine the spatial distribution of runx1 transcripts in the zebrafish embryo at the onset of runx1 expression, likely by controlling intronic CRE activity and CRE-promoter interactions. CTCF appears to restrict the expression pattern of runx1, consistent with insulator activity, while cohesin is necessary for its expression in a specific subpopulation of hematopoietic progenitors [57,60].
Interestingly, Marsman et al. also showed that siRNA knock down of cohesin (but not CTCF) in HL-60 myelocytic leukemia cells enhanced RUNX1 transcription [60], indicating that cohesin’s transcriptional role is conserved in human cells. It is tempting to speculate that cohesin mutation leading to an increase in RUNX1 transcription might exacerbate myeloid malignancies that already have excess RUNX1; for example, DS-AMKL [30].
In summary, it appears that cohesin has a crucial role in cell type-specific regulation of Runx1, likely by mediating interactions between CREs and the promoters of Runx1. In support of this idea, ChIA-PET data generated in K562 cells using RNA polymerase II demonstrated that the two promoters of human RUNX1 are in physical proximity with each other, and with CREs in the intron between the two promoters [61]. It is not yet known whether these interactions regulate RUNX1, or whether they are cohesin-dependent. While formal proof of this kind of mechanism for cohesin regulation of Runx1 is still to come, the link between cohesin mutation and spatiotemporal Runx1 transcription may explain cohesin’s contribution to AML pathogenesis and other myeloid malignancies.
Conclusions
Mutations in cohesin comprise a novel genetic pathway significantly associated with the development of AML and related leukemias. While several types of cancer do have cohesin mutations, most cancers also harbor many additional mutations in multiple gene categories [15]. By contrast, AML genomes contain relatively fewer mutations than other cancer types, with only 23 genes significantly mutated [20]. Four of these genes correspond to cohesin subunits [15,20], indicating that cohesin mutations are particularly important to the progression of AML.
Why do myeloid disorders have a high prevalence of cohesin mutations in particular? The answer could reside in cohesin’s potential to mediate global transcriptional activity in a way that is also exquisitely cell type-specific.
Evidence that cohesin regulates cell type-specific global gene transcriptional programs, and in particular, expression of the AML-associated transcription factor, RUNX1, could explain why cohesin mutations are so prevalent in myeloproliferative disorders. Perhaps correct differentiation along the myeloid pathway relies on accurate expression of key genes (such as RUNX1) that can only respond to a full complement of cohesin. When cohesin function is impaired, differentiation of myeloid precursors might be prevented, facilitating dysplasia. These notions support previous hypotheses that cohesin is likely to play an important role in hematopoiesis [57,62].
Remarkably, cohesin binds to a majority of cell type-specific transcription factor binding sites, even when transcription factors themselves are evicted during mitosis [63]. In this manner, cohesin binding may ‘bookmark’ transcription factor binding sites to re-establish transcriptional programs after cell division [63], including sites for hematopoietic transcription factors.
Further research will be necessary to understand exactly how cohesin functions in normal and abnormal hematopoiesis, and how cohesin mutations cooperate with other genetic events to progress leukemia.
Abbreviations
AML: Acute myeloid leukemia; MDS: Myelodysplastic syndrome; CMML: Chronic myelomonocytic leukemia; CML: Chronic myelogenous leukemia; MPN: Myeloproliferative neoplasm; DS: Down’s Syndrome; TAM: Transient abnormal myelopoiesis; AMKL: Acute megakaryoblastic leukemia; CRE: Conserved regulatory element; CTCF: CCCTC-binding factor; TAD: Topologically associated domain; SINE: Short interspersed element.
Competing interests
The authors declare they have no competing interests.
Authors’ contributions
BL, JM, JO and JH wrote the article. BL researched the literature and prepared the tables. JO, JM, BL and JH prepared the figures. All authors read and approved the final manuscript.
Supplementary Material
Comprehensive summary of cohesin mutations in myeloid malignancies.
Contributor Information
Bryony Leeke, Email: bryony.leeke@otago.ac.nz.
Judith Marsman, Email: marju481@student.otago.ac.nz.
Justin M O’Sullivan, Email: justin.osullivan@auckland.ac.nz.
Julia A Horsfield, Email: julia.horsfield@otago.ac.nz.
Acknowledgements
Work in the authors’ laboratories was funded by the Royal Society of NZ Marsden Fund (11-UOO-027 to JAH; 10-UOA-023 to JOS), Leukemia & Blood Cancer NZ (JAH), Gravida Centre for Growth and Development (JAH/JOS) and the Maurice Wilkins Centre for Molecular Biodiscovery (JAH/JOS).
References
- Betz BL, Hess JL. Acute myeloid leukemia diagnosis in the 21st century. Arch Pathol Lab Med. 2010;134:1427–1433. doi: 10.5858/2010-0245-RA.1. [DOI] [PubMed] [Google Scholar]
- Sanders MA, Valk PJ. The evolving molecular genetic landscape in acute myeloid leukaemia. Curr Opin Hematol. 2013;20:79–85. doi: 10.1097/MOH.0b013e32835d821c. [DOI] [PubMed] [Google Scholar]
- Ball AR Jr, Chen YY, Yokomori K. Mechanisms of cohesin-mediated gene regulation and lessons learned from cohesinopathies. Biochim Biophys Acta. 2014;1839:191–202. doi: 10.1016/j.bbagrm.2013.11.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bose T, Gerton JL. Cohesinopathies, gene expression, and chromatin organization. J Cell Biol. 2010;189:201–210. doi: 10.1083/jcb.200912129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Horsfield JA, Print CG, Monnich M. Diverse developmental disorders from the one ring: distinct molecular pathways underlie the cohesinopathies. Front Genet. 2012;3:171. doi: 10.3389/fgene.2012.00171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Merkenschlager M, Odom DT. CTCF and cohesin: linking gene regulatory elements with their targets. Cell. 2013;152:1285–1297. doi: 10.1016/j.cell.2013.02.029. [DOI] [PubMed] [Google Scholar]
- Nasmyth K, Haering CH. Cohesin: its roles and mechanisms. Annu Rev Genet. 2009;43:525–558. doi: 10.1146/annurev-genet-102108-134233. [DOI] [PubMed] [Google Scholar]
- Skibbens RV, Colquhoun JM, Green MJ, Molnar CA, Sin DN, Sullivan BJ, Tanzosh EE. Cohesinopathies of a feather flock together. PLoS Genet. 2013;9:e1004036. doi: 10.1371/journal.pgen.1004036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu N, Yu H. The Smc complexes in DNA damage response. Cell Biosci. 2012;2:5. doi: 10.1186/2045-3701-2-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dorsett D, Strom L. The ancient and evolving roles of cohesin in gene expression and DNA repair. Curr Biol. 2012;22:R240–R250. doi: 10.1016/j.cub.2012.02.046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dorsett D, Krantz ID. On the molecular etiology of Cornelia de Lange syndrome. Ann N Y Acad Sci. 2009;1151:22–37. doi: 10.1111/j.1749-6632.2008.03450.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rhodes JM, McEwan M, Horsfield JA. Gene regulation by cohesin in cancer: is the ring an unexpected party to proliferation? Mol Cancer Res. 2011;9:1587–1607. doi: 10.1158/1541-7786.MCR-11-0382. [DOI] [PubMed] [Google Scholar]
- Xu H, Tomaszewski JM, McKay MJ. Can corruption of chromosome cohesion create a conduit to cancer? Nat Rev Cancer. 2011;11:199–210. doi: 10.1038/nrc3018. [DOI] [PubMed] [Google Scholar]
- Taylor CF, Platt FM, Hurst CD, Thygesen HH, Knowles MA. Frequent inactivating mutations of STAG2 in bladder cancer are associated with low tumour grade and stage and inversely related to chromosomal copy number changes. Hum Mol Genet. 2014;23:1964–1974. doi: 10.1093/hmg/ddt589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kandoth C, McLellan MD, Vandin F, Ye K, Niu B, Lu C, Xie M, Zhang Q, McMichael JF, Wyczalkowski MA, Leiserson MD, Miller CA, Welch JS, Walter MJ, Wendl MC, Ley TJ, Wilson RK, Raphael BJ, Ding L. Mutational landscape and significance across 12 major cancer types. Nature. 2013;502:333–339. doi: 10.1038/nature12634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barber TD, McManus K, Yuen KW, Reis M, Parmigiani G, Shen D, Barrett I, Nouhi Y, Spencer F, Markowitz S, Velculescu VE, Kinzler KW, Vogelstein B, Lengauer C, Hieter P. Chromatid cohesion defects may underlie chromosome instability in human colorectal cancers. Proc Natl Acad Sci U S A. 2008;105:3443–3448. doi: 10.1073/pnas.0712384105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu H, Yan M, Patra J, Natrajan R, Yan Y, Swagemakers S, Tomaszewski JM, Verschoor S, Millar EK, van der Spek P, Reis-Filho JS, Ramsay RG, O'Toole SA, McNeil CM, Sutherland RL, McKay MJ, Fox SB. Enhanced RAD21 cohesin expression confers poor prognosis and resistance to chemotherapy in high grade luminal, basal and HER2 breast cancers. Breast Cancer Res. 2011;13:R9. doi: 10.1186/bcr2814. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vass S, Cotterill S, Valdeolmillos AM, Barbero JL, Lin E, Warren WD, Heck MM. Depletion of Drad21/Scc1 in Drosophila cells leads to instability of the cohesin complex and disruption of mitotic progression. Curr Biol. 2003;13:208–218. doi: 10.1016/S0960-9822(03)00047-2. [DOI] [PubMed] [Google Scholar]
- Rollins RA, Korom M, Aulner N, Martens A, Dorsett D. Drosophila nipped-B protein supports sister chromatid cohesion and opposes the stromalin/Scc3 cohesion factor to facilitate long-range activation of the cut gene. Mol Cell Biol. 2004;24:3100–3111. doi: 10.1128/MCB.24.8.3100-3111.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cancer Genome Atlas Research N. Genomic and epigenomic landscapes of adult de novo acute myeloid leukemia. N Engl J Med. 2013;368:2059–2074. doi: 10.1056/NEJMoa1301689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Welch JS, Ley TJ, Link DC, Miller CA, Larson DE, Koboldt DC, Wartman LD, Lamprecht TL, Liu F, Xia J, Kandoth C, Fulton RS, McLellan MD, Dooling DJ, Wallis JW, Chen K, Harris CC, Schmidt HK, Kalicki-Veizer JM, Lu C, Zhang Q, Lin L, O'Laughlin MD, McMichael JF, Delehaunty KD, Fulton LA, Magrini VJ, McGrath SD, Demeter RT, Vickery TL. et al. The origin and evolution of mutations in acute myeloid leukemia. Cell. 2012;150:264–278. doi: 10.1016/j.cell.2012.06.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rocquain J, Gelsi-Boyer V, Adelaide J, Murati A, Carbuccia N, Vey N, Birnbaum D, Mozziconacci MJ, Chaffanet M. Alteration of cohesin genes in myeloid diseases. Am J Hematol. 2010;85:717–719. doi: 10.1002/ajh.21798. [DOI] [PubMed] [Google Scholar]
- Jan M, Snyder TM, Corces-Zimmerman MR, Vyas P, Weissman IL, Quake SR, Majeti R. Clonal evolution of preleukemic hematopoietic stem cells precedes human acute myeloid leukemia. Sci Transl Med. 2012;4:149. doi: 10.1126/scitranslmed.3004315. ra118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thol F, Bollin R, Gehlhaar M, Walter C, Dugas M, Suchanek KJ, Kirchner A, Huang L, Chaturvedi A, Wichmann M, Wiehlmann L, Shahswar R, Damm F, Gohring G, Schlegelberger B, Schlenk R, Dohner K, Dohner H, Krauter J, Ganser A, Heuser M. Mutations in the cohesin complex in acute myeloid leukemia: clinical and prognostic implications. Blood. 2014;123:914–920. doi: 10.1182/blood-2013-07-518746. [DOI] [PubMed] [Google Scholar]
- Kon A, Shih LY, Minamino M, Sanada M, Shiraishi Y, Nagata Y, Yoshida K, Okuno Y, Bando M, Nakato R, Ishikawa S, Sato-Otsubo A, Nagae G, Nishimoto A, Haferlach C, Nowak D, Sato Y, Alpermann T, Nagasaki M, Shimamura T, Tanaka H, Chiba K, Yamamoto R, Yamaguchi T, Otsu M, Obara N, Sakata-Yanagimoto M, Nakamaki T, Ishiyama K, Nolte F. et al. Recurrent mutations in multiple components of the cohesin complex in myeloid neoplasms. Nat Genet. 2013;45:1232–1237. doi: 10.1038/ng.2731. [DOI] [PubMed] [Google Scholar]
- Kihara R, Nagata Y, Kiyoi H, Kato T, Yamamoto E, Suzuki K, Chen F, Asou N, Ohtake S, Miyawaki S, Miyazaki Y, Sakura T, Ozawa Y, Usui N, Kanamori H, Kiguchi T, Imai K, Uike N, Kimura F, Kitamura K, Nakaseko C, Onizuka M, Takeshita A, Ishida F, Suzushima H, Kato Y, Miwa H, Shiraishi Y, Chiba K, Tanaka H, Comprehensive analysis of genetic alterations and their prognostic impacts in adult acute myeloid leukemia patients. Leukemia. 2014. doi:10.1038/leu.2014.55. [Epub ahead of print] [DOI] [PubMed]
- Dolnik A, Engelmann JC, Scharfenberger-Schmeer M, Mauch J, Kelkenberg-Schade S, Haldemann B, Fries T, Kronke J, Kuhn MW, Paschka P, Kayser S, Wolf S, Gaidzik VI, Schlenk RF, Rucker FG, Dohner H, Lottaz C, Dohner K, Bullinger L. Commonly altered genomic regions in acute myeloid leukemia are enriched for somatic mutations involved in chromatin remodeling and splicing. Blood. 2012;120:e83–e92. doi: 10.1182/blood-2011-12-401471. [DOI] [PubMed] [Google Scholar]
- Walter MJ, Shen D, Shao J, Ding L, White BS, Kandoth C, Miller CA, Niu B, McLellan MD, Dees ND, Fulton R, Elliot K, Heath S, Grillot M, Westervelt P, Link DC, DiPersio JF, Mardis E, Ley TJ, Wilson RK, Graubert TA. Clonal diversity of recurrently mutated genes in myelodysplastic syndromes. Leukemia. 2013;27:1275–1282. doi: 10.1038/leu.2013.58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haferlach T, Nagata Y, Grossmann V, Okuno Y, Bacher U, Nagae G, Schnittger S, Sanada M, Kon A, Alpermann T, Yoshida K, Roller A, Nadarajah N, Shiraishi Y, Shiozawa Y, Chiba K, Tanaka H, Koeffler HP, Klein HU, Dugas M, Aburatani H, Kohlmann A, Miyano S, Haferlach C, Kern W, Ogawa S. Landscape of genetic lesions in 944 patients with myelodysplastic syndromes. Leukemia. 2014;28:241–247. doi: 10.1038/leu.2013.336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoshida K, Toki T, Okuno Y, Kanezaki R, Shiraishi Y, Sato-Otsubo A, Sanada M, Park MJ, Terui K, Suzuki H, Kon A, Nagata Y, Sato Y, Wang R, Shiba N, Chiba K, Tanaka H, Hama A, Muramatsu H, Hasegawa D, Nakamura K, Kanegane H, Tsukamoto K, Adachi S, Kawakami K, Kato K, Nishimura R, Izraeli S, Hayashi Y, Miyano S. et al. The landscape of somatic mutations in Down syndrome-related myeloid disorders. Nat Genet. 2013;45:1293–1299. doi: 10.1038/ng.2759. [DOI] [PubMed] [Google Scholar]
- Solomon DA, Kim JS, Bondaruk J, Shariat SF, Wang ZF, Elkahloun AG, Ozawa T, Gerard J, Zhuang D, Zhang S, Navai N, Siefker-Radtke A, Phillips JJ, Robinson BD, Rubin MA, Volkmer B, Hautmann R, Kufer R, Hogendoorn PC, Netto G, Theodorescu D, James CD, Czerniak B, Miettinen M, Waldman T. Frequent truncating mutations of STAG2 in bladder cancer. Nat Genet. 2013;45:1428–1430. doi: 10.1038/ng.2800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guo G, Sun X, Chen C, Wu S, Huang P, Li Z, Dean M, Huang Y, Jia W, Zhou Q, Tang A, Yang Z, Li X, Song P, Zhao X, Ye R, Zhang S, Lin Z, Qi M, Wan S, Xie L, Fan F, Nickerson ML, Zou X, Hu X, Xing L, Lv Z, Mei H, Gao S, Liang C. et al. Whole-genome and whole-exome sequencing of bladder cancer identifies frequent alterations in genes involved in sister chromatid cohesion and segregation. Nat Genet. 2013;45:1459–1463. doi: 10.1038/ng.2798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Balbas-Martinez C, Sagrera A, Carrillo-de-Santa-Pau E, Earl J, Marquez M, Vazquez M, Lapi E, Castro-Giner F, Beltran S, Bayes M, Carrato A, Cigudosa JC, Dominguez O, Gut M, Herranz J, Juanpere N, Kogevinas M, Langa X, Lopez-Knowles E, Lorente JA, Lloreta J, Pisano DG, Richart L, Rico D, Salgado RN, Tardon A, Chanock S, Heath S, Valencia A, Losada A. et al. Recurrent inactivation of STAG2 in bladder cancer is not associated with aneuploidy. Nat Genet. 2013;45:1464–1469. doi: 10.1038/ng.2799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kagey MH, Newman JJ, Bilodeau S, Zhan Y, Orlando DA, van Berkum NL, Ebmeier CC, Goossens J, Rahl PB, Levine SS, Taatjes DJ, Dekker J, Young RA. Mediator and cohesin connect gene expression and chromatin architecture. Nature. 2010;467:430–435. doi: 10.1038/nature09380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dixon JR, Selvaraj S, Yue F, Kim A, Li Y, Shen Y, Hu M, Liu JS, Ren B. Topological domains in mammalian genomes identified by analysis of chromatin interactions. Nature. 2012;485:376–380. doi: 10.1038/nature11082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lieberman-Aiden E, van Berkum NL, Williams L, Imakaev M, Ragoczy T, Telling A, Amit I, Lajoie BR, Sabo PJ, Dorschner MO, Sandstrom R, Bernstein B, Bender MA, Groudine M, Gnirke A, Stamatoyannopoulos J, Mirny LA, Lander ES, Dekker J. Comprehensive mapping of long-range interactions reveals folding principles of the human genome. Science. 2009;326:289–293. doi: 10.1126/science.1181369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li Y, Huang W, Niu L, Umbach DM, Covo S, Li L. Characterization of constitutive CTCF/cohesin loci: a possible role in establishing topological domains in mammalian genomes. BMC Genomics. 2013;14:553. doi: 10.1186/1471-2164-14-553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seitan V, Faure A, Zhan Y, McCord R, Lajoie B, Ing-Simmons E, Lenhard B, Giorgetti L, Heard E, Fisher A, Flicek P, Dekker J, Merkenschlager M. Cohesin-based chromatin interactions enable regulated gene expression within pre-existing architectural compartments. Genome Res. 2013;23:2066–2077. doi: 10.1101/gr.161620.113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sofueva S, Yaffe E, Chan W-C, Georgopoulou D, Vietri Rudan M, Mira-Bontenbal H, Pollard SM, Schroth GP, Tanay A, Hadjur S. Cohesin-mediated interactions organize chromosomal domain architecture. EMBO J. 2013;32:3119–3129. doi: 10.1038/emboj.2013.237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zuin J, Dixon JR, van der Reijden MI, Ye Z, Kolovos P, Brouwer RW, van de Corput MP, van de Werken HJ, Knoch TA, van Ijcken WF, Grosveld FG, Ren B, Wendt KS. Cohesin and CTCF differentially affect chromatin architecture and gene expression in human cells. Proc Natl Acad Sci U S A. 2014;111:996–1001. doi: 10.1073/pnas.1317788111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stedman W, Kang H, Lin S, Kissil JL, Bartolomei MS, Lieberman PM. Cohesins localize with CTCF at the KSHV latency control region and at cellular c-myc and H19/Igf2 insulators. EMBO J. 2008;27:654–666. doi: 10.1038/emboj.2008.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wendt KS, Yoshida K, Itoh T, Bando M, Koch B, Schirghuber E, Tsutsumi S, Nagae G, Ishihara K, Mishiro T, Yahata K, Imamoto F, Aburatani H, Nakao M, Imamoto N, Maeshima K, Shirahige K, Peters JM. Cohesin mediates transcriptional insulation by CCCTC-binding factor. Nature. 2008;451:796–801. doi: 10.1038/nature06634. [DOI] [PubMed] [Google Scholar]
- Parelho V, Hadjur S, Spivakov M, Leleu M, Sauer S, Gregson HC, Jarmuz A, Canzonetta C, Webster Z, Nesterova T, Cobb BS, Yokomori K, Dillon N, Aragon L, Fisher AG, Merkenschlager M. Cohesins Functionally Associate with CTCF on Mammalian Chromosome Arms. Cell. 2008;132:422–433. doi: 10.1016/j.cell.2008.01.011. [DOI] [PubMed] [Google Scholar]
- Faure AJ, Schmidt D, Watt S, Schwalie PC, Wilson MD, Xu H, Ramsay RG, Odom DT, Flicek P. Cohesin regulates tissue-specific expression by stabilizing highly occupied cis-regulatory modules. Genome Res. 2012;22:2163–2175. doi: 10.1101/gr.136507.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmidt D, Schwalie P, Ross-Innes CS, Hurtado A, Brown G, Carroll J, Flicek P, Odom D. A CTCF-independent role for cohesin in tissue-specific transcription. Genome Res. 2010;20:578–588. doi: 10.1101/gr.100479.109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dorsett D, Merkenschlager M. Cohesin at active genes: a unifying theme for cohesin and gene expression from model organisms to humans. Curr Opin Cell Biol. 2013;25(3):327–333. doi: 10.1016/j.ceb.2013.02.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kawauchi S, Calof AL, Santos R, Lopez-Burks ME, Young CM, Hoang MP, Chua A, Lao T, Lechner MS, Daniel JA, Nussenzweig A, Kitzes L, Yokomori K, Hallgrimsson B, Lander AD. Multiple organ system defects and transcriptional dysregulation in the Nipbl(+/-) mouse, a model of Cornelia de Lange Syndrome. PLoS Genet. 2009;5:e1000650. doi: 10.1371/journal.pgen.1000650. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu J, Feldman R, Zhang Z, Deardorff MA, Haverfield EV, Kaur M, Li JR, Clark D, Kline AD, Waggoner DJ, Das S, Jackson LG, Krantz ID. SMC1A expression and mechanism of pathogenicity in probands with X-Linked Cornelia de Lange syndrome. Hum Mutat. 2009;30:1535–1542. doi: 10.1002/humu.21095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dorsett D, Eissenberg JC, Misulovin Z, Martens A, Redding B, McKim K. Effects of sister chromatid cohesion proteins on cut gene expression during wing development in Drosophila. Development. 2005;132:4743–4753. doi: 10.1242/dev.02064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schaaf CA, Misulovin Z, Sahota G, Siddiqui AM, Schwartz YB, Kahn TG, Pirrotta V, Gause M, Dorsett D. Regulation of the Drosophila Enhancer of split and invected-engrailed gene complexes by sister chromatid cohesion proteins. PLoS ONE. 2009;4:e6202. doi: 10.1371/journal.pone.0006202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou Y, Kurukuti S, Saffrey P, Vukovic M, Michie AM, Strogantsev R, West AG, Vetrie D. Chromatin looping defines expression of TAL1, its flanking genes and regulation in T-ALL. Blood. 2013;122:4199–4209. doi: 10.1182/blood-2013-02-483875. [DOI] [PubMed] [Google Scholar]
- Gao X, Johnson KD, Chang YI, Boyer ME, Dewey CN, Zhang J, Bresnick EH. Gata2 cis-element is required for hematopoietic stem cell generation in the mammalian embryo. J Exp Med. 2013;210:2833–2842. doi: 10.1084/jem.20130733. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Diffner E, Beck D, Gudgin E, Thoms JA, Knezevic K, Pridans C, Foster S, Goode D, Lim WK, Boelen L, Metzeler KH, Micklem G, Bohlander SK, Buske C, Burnett A, Ottersbach K, Vassiliou GS, Olivier J, Wong JW, Gottgens B, Huntly BJ, Pimanda JE. Activity of a heptad of transcription factors is associated with stem cell programs and clinical outcome in acute myeloid leukemia. Blood. 2013;121:2289–2300. doi: 10.1182/blood-2012-07-446120. [DOI] [PubMed] [Google Scholar]
- Wang Q, Stacy T, Binder M, Marin-Padilla M, Sharpe AH, Speck NA. Disruption of the Cbfa2 gene causes necrosis and hemorrhaging in the central nervous system and blocks definitive hematopoiesis. Proc Natl Acad Sci U S A. 1996;93:3444–3449. doi: 10.1073/pnas.93.8.3444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Okuda T, van Deursen J, Hiebert SW, Grosveld G, Downing JR. AML1, the target of multiple chromosomal translocations in human leukemia, is essential for normal fetal liver hematopoiesis. Cell. 1996;84:321–330. doi: 10.1016/S0092-8674(00)80986-1. [DOI] [PubMed] [Google Scholar]
- Schlegelberger B, Gohring G, Thol F, Heuser M. Update on cytogenetic and molecular changes in myelodysplastic syndromes. Leuk Lymphoma. 2012;53:525–536. doi: 10.3109/10428194.2011.618235. [DOI] [PubMed] [Google Scholar]
- Horsfield JA, Anagnostou SH, Hu JK, Cho KH, Geisler R, Lieschke G, Crosier KE, Crosier PS. Cohesin-dependent regulation of Runx genes. Development. 2007;134:2639–2649. doi: 10.1242/dev.002485. [DOI] [PubMed] [Google Scholar]
- Nottingham WT, Jarratt A, Burgess M, Speck CL, Cheng JF, Prabhakar S, Rubin EM, Li PS, Sloane-Stanley J, Kong ASJ, de Bruijn MF. Runx1-mediated hematopoietic stem-cell emergence is controlled by a Gata/Ets/SCL-regulated enhancer. Blood. 2007;110:4188–4197. doi: 10.1182/blood-2007-07-100883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ng CE, Yokomizo T, Yamashita N, Cirovic B, Jin H, Wen Z, Ito Y, Osato M. A Runx1 intronic enhancer marks hemogenic endothelial cells and hematopoietic stem cells. Stem Cells. 2010;28:1869–1881. doi: 10.1002/stem.507. [DOI] [PubMed] [Google Scholar]
- Marsman J, O’Neill AC, Kao BR, Rhodes JM, Meier M, Antony J, Monnich M, Horsfield JA. Cohesin and CTCF differentially regulate spatiotemporal runx1 expression during zebrafish development. Biochim Biophys Acta. 1839;2014:50–61. doi: 10.1016/j.bbagrm.2013.11.007. [DOI] [PubMed] [Google Scholar]
- Li G, Ruan X, Auerbach RK, Sandhu KS, Zheng M, Wang P, Poh HM, Goh Y, Lim J, Zhang J, Sim HS, Peh SQ, Mulawadi FH, Ong CT, Orlov YL, Hong S, Zhang Z, Landt S, Raha D, Euskirchen G, Wei CL, Ge W, Wang H, Davis C, Fisher-Aylor KI, Mortazavi A, Gerstein M, Gingeras T, Wold B, Sun Y. et al. Extensive promoter-centered chromatin interactions provide a topological basis for transcription regulation. Cell. 2012;148:84–98. doi: 10.1016/j.cell.2011.12.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Panigrahi AK, Pati D. Higher-order orchestration of hematopoiesis: is cohesin a new player? Exp Hematol. 2012;40:967–973. doi: 10.1016/j.exphem.2012.09.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yan J, Enge M, Whitington T, Dave K, Liu J, Sur I, Schmierer B, Jolma A, Kivioja T, Taipale M, Taipale J. Transcription factor binding in human cells occurs in dense clusters formed around cohesin anchor sites. Cell. 2013;154:801–813. doi: 10.1016/j.cell.2013.07.034. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Comprehensive summary of cohesin mutations in myeloid malignancies.