

Abstract
The most common disease-causing mutation in the cystic fibrosis transmembrane conductance regulator (CFTR) gene is the out-of-frame deletion of 3 nucleotides (CTT). This mutation leads to the loss of phenylalanine-508 (ΔF508) and a silent codon change (SCC) for isoleucine-507 (I507-ATC→ATT). ΔF508 CFTR is misfolded and degraded by endoplasmic reticulum-associated degradation (ERAD). We have demonstrated that the I507-ATC→ATT SCC alters ΔF508 CFTR mRNA structure and translation dynamics. By comparing the biochemical and functional properties of the I507-ATT and I507-ATC ΔF508 CFTR, we establish that the I507-ATC→ATT SCC contributes to the cotranslational misfolding, ERAD, and to the functional defects associated with ΔF508 CFTR. We demonstrate that the I507-ATC ΔF508 CFTR is less susceptible to the ER quality-control machinery during translation than the I507-ATT, although 27°C correction is necessary for sufficient cell-surface expression. Whole-cell patch-clamp recordings indicate sustained, thermally stable cAMP-activated Cl− transport through I507-ATC and unstable function of the I507-ATT ΔF508 CFTR. Single-channel recordings reveal improved gating properties of the I507-ATC compared to I507-ATT ΔF508 CFTR (NPo=0.45±0.037 vs. NPo=0.09±0.002; P<0.001). Our results signify the role of the I507-ATC→ATT SCC in the ΔF508 CFTR defects and support the importance of synonymous codon choices in determining the function of gene products.—Lazrak, A., Fu, L., Bali, V., Bartoszewski, R., Rab, A., Havasi, V., Keiles, S., Kappes, J., Kumar, R., Lefkowitz, E., Sorscher, E. J., Matalon, S., Collawn, J. F., Bebok, Z. The silent codon change I507-ATC→ATT contributes to the severity of the ΔF508 CFTR.
Keywords: ABC protein, channel gating, patch-clamp, single-nucleotide polymorphism, sSNP, cotranslational folding
Synonymous single-nucleotide polymorphisms (sSNPs) have been shown to affect mRNA and protein conformation or alter the expression levels and function of gene products (1–6). As an example, a naturally occurring sSNP leads to the synthesis of an ABC transporter (ABCB1 or P-glycoprotein) with altered transport properties (2). Studies on catechol-O-methyltransferase (COMT) polymorphisms (1, 7–9) demonstrated that an sSNP near the translation initiation site alters the translation efficiency via entropy-driven changes in mRNA structure (6).
Our studies have focused on the most common mutation in the human cystic fibrosis transmembrane conductance regulator (CFTR) gene, ΔF508 CFTR (10, 11). The CFTR protein is a cyclic adenosine monophosphate (cAMP)-activated chloride (Cl−) channel (12), predominantly expressed in epithelial cells that line the luminal surface of the secretory glands of the respiratory and gastrointestinal tracts (13). Deletion of 3 nucleotides from CFTR that comprise either the last 2 nucleotides (TC) of the isoleucine 507 codon (I507-ATC) and the first nucleotide (T) of F508 (F508 TTT), or alternatively the last nucleotide (C) of I507-ATC and the first 2 nucleotides (TT) of F508 TTT, results in the loss of F508 (ΔF508) and a silent codon change (SCC; ATC→ATT) for I507 (Fig. 1; 10, 11, 14, 15). The resulting gene product is referred to as ΔF508 CFTR. The ΔF508 CFTR protein is misfolded and subjected to endoplasmic reticulum-associated degradation (ERAD; refs. 16–19). Notably, when ΔF508 CFTR is rescued from ERAD by culturing cells at 27°C, it retains partial cAMP-activated Cl− channel function (20, 21). However, the protein stability, functional half-life, and channel properties of the rescued ΔF508 CFTR are severely compromised (22, 23).
Figure 1.
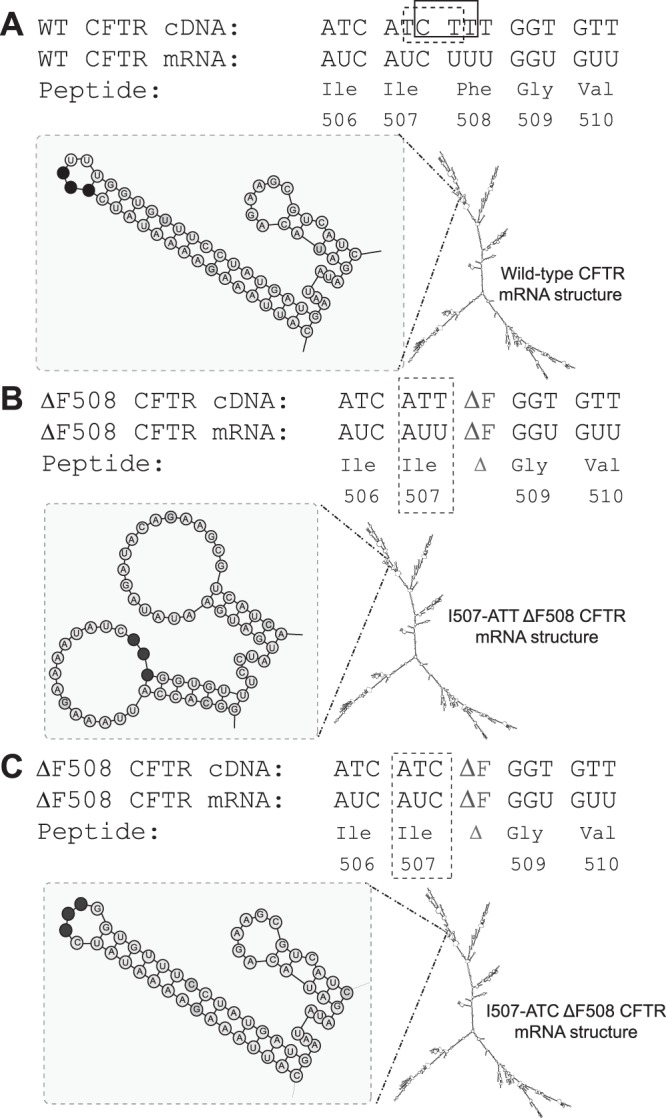
CFTR cDNA, mRNA, and protein sequences in the vicinity of the ΔF508 mutation depicting predicted mRNA structural elements. Deletion of 3 nucleotides, CTT (rectangle) or TCT (dashed line), results in the loss of phenylalanine at the 508 position of the CFTR protein and an SCC (ATC to ATT) for isoleucine 507. The mRNA secondary structural predictions are shown under each sequence. The predicted structures in the vicinity of the mutation were confirmed through biochemical analysis. A) Wild-type CFTR. B) I507-ATT ΔF508 CFTR. C) I507-ATC ΔF508 CFTR. Structural elements in the mutation region (left), the 3 nucleotides (ATC or ATT) corresponding to I507 are shown as black circles.
While the folding, trafficking, and functional properties of the ΔF508 CFTR protein have been extensively studied (for review see ref. 24), the possibility that the I507-ATC→ATT substitution contributes to the severity of the mutation has not been examined. Because of the generally assumed insignificance of synonymous codon alterations, the functional defect associated with this mutation has been attributed to the loss of the phenylalanine (F) residue. However, we have demonstrated that the ATC→ATT SCC in I507 of the human ΔF508 CFTR alters the structure of the mRNA in the vicinity of the mutation and alters the translational dynamics of ΔF508 CFTR compared to the wild-type protein (3). Notably, changing the I507-ATT codon back to ATC in ΔF508 CFTR eliminated the mRNA secondary structure changes and the translation rate differences observed between the wild-type and I507-ATT ΔF508 CFTR mRNAs (3).
Thus, the studies on P-glycoprotein, COMT, and CFTR strongly suggest that understanding how SCCs contribute to the structure and function of the gene product may provide valuable information regarding the significance of codon redundancy. To elucidate the role of the I507-ATC→ATT SCC in the severity of the ΔF508 CFTR mutation, we performed a series of biochemical and electrophysiological assays on cell clones stably expressing the I507-ATT or I507-ATC-ΔF508 CFTR. Our results support the concept that silent codon variations can have dramatic and dynamic effects on protein structure and function, and therefore should always be considered in determining the structure and function of other gene products.
MATERIALS AND METHODS
Cell lines
Human embryonic kidney 293 (HEK293) cells expressing wild-type, I507-ATT ΔF508, and I507-ATC ΔF508 CFTR were developed, cloned, characterized, and maintained as described previously (3). HEK293 cells without CFTR expression were used as control in some experiments. To rescue the mutant proteins from ERAD, cells were cultured at 27°C for 24 h prior to the start of the experiments as specified.
CFTR mRNA level measurements
CFTR mRNA levels were measured by qRT-PCR as described previously (25, 26). Total RNA was isolated using the RNeasy mini kit (Qiagen, Valencia, CA, USA). Quantitative real-time PCR was performed using the ABI StepOnePlus sequence detection system (Applied Biosystems, Foster City, CA, USA). Total CFTR mRNA levels were evaluated using Assay-On-Demand primer mix (assay ID: Hs00357011_m1). Glyceraldehyde 3-phosphate dehydrogenase (GAPDH; assay ID: Hs99999905_m1) mRNA levels were studied in parallel as internal control. Results were expressed as CFTR mRNA levels relative to GAPDH mRNA (mean±sd). Primer efficiencies for GAPDH and CFTR assays were 101 and 95%, respectively.
CFTR protein measurements
CFTR protein levels were assayed by Western blot according to our previously described protocol (25, 27). Briefly, cells were lysed in radioimmunoprecipitation assay (RIPA) buffer [50 mM Tris-HCl, pH 8.0; 1% Nonidet P-40; 0.5% deoxycholate; 0.1% SDS; 150 mM NaCl; and Complete Protease Inhibitor (Roche, Indianapolis, IN, USA)]. Proteins were separated by SDS/PAGE and transferred onto PVDF membranes (Bio-Rad, Hercules, CA, USA). For the detection of CFTR fragments, proteins were lysed in a buffer containing 1% (v/v) Nonidet P40, 50 mM Tris/HCl (pH 7.5), 150 mM NaCl, and protease inhibitor cocktail (Roche). Before electrophoresis, lysates were incubated with modified Laemmli sample buffer containing 105 mM DTT as described previously (28). Proteins were separated on 8–16% gradient gels (Invitrogen). Full-length CFTR and CFTR fragments were detected with a monoclonal antibody (MM13-4, 1:500 dilution; Millipore, Billerica, MA, USA), recognizing the N-terminal tail of CFTR. Horseradish peroxidase (HRP)-labeled secondary antibodies and Super Signal West Pico chemiluminescence substrate (Pierce, Rockford, IL, USA) were used for visualization of protein bands. Densitometry was performed using ImageJ software (U.S. National Institutes of Health, Bethesda, MD, USA).
Cell surface biotinylation and CFTR cell surface half-life measurements
Cell surface CFTR half-lives were determined as described previously (29). Cells were treated with 0.2 mg/ml cycloheximide for the time periods specified. Cell surface proteins were labeled with biotin LC hydrazide, the cells were lysed in RIPA buffer (50 mM Tris-HCl, pH 8.0; 1% Nonidet P-40; 0.5% deoxycholate; 0.1% SDS; 150 mM NaCl; and Complete Protease Inhibitor). CFTR was immunoprecipitated with 24-1 monoclonal antibody raised against the C-terminal tail of CFTR. Proteins were separated by SDS-PAGE, and biotinylated CFTR was detected with HRP-labeled avidin. Chemiluminescence was induced with West Pico Super Signal peroxide solution. Densitometry was performed using ImageJ software.
Metabolic pulse-chase assay
CFTR stability and processing efficiencies were analyzed as described previously (30, 31), with specific conditions listed below. Cells were methionine starved for 20 min using methionine- and cysteine-free medium, followed by pulse labeling with 100 μCi/ml of 35S-methionine (EXPRE35S35S Protein Labeling Mix, Perkin Elmer, Santa Clara, CA, USA) for 20 min. Radioactively labeled proteins were chased using regular growth medium for the time periods specified. Cells were lysed in RIPA buffer (50 mM Tris-HCl, pH 8.0; 1% Nonidet P-40; 0.5% deoxycholate; 0.1% SDS; 150 mM NaCl; and Complete Protease Inhibitor) and CFTR was immunoprecipitated with a monoclonal antibody recognizing the C-terminal tail of CFTR (31). Densitometry was performed using ImageJ software.
Measurement of whole-cell currents in HEK293 cells expressing wild type CFTR, I507-ATT ΔF508 CFTR, and I507-ATC ΔF508 CFTR
All electrophysiology measurements were performed at room temperature (20–22°C; ref.32). HEK293 clones stably expressing wild-type, I507-ATT, or I507-ATC ΔF508 CFTR were seeded onto coverslips and 24 h prior to any measurements were placed into a 27°C incubator with 95% air/5% CO2. For measurements of whole-cell currents, coverslips containing HEK293 cells were placed onto a recording chamber mounted on the stage of an Olympus IMT-2 inverted fluorescent microscope (Olympus, Tokyo, Japan). Cells were perfused with an external solution (ES) containing 145 mM CsCl, 2 mM MgCl2, 2 mM CaCl2, 5.5 mM glucose, and 10 mM HEPES (pH 7.4; 1 N NaOH). Patch pipettes were filled with the internal solution (IS) containing 135 mM CsCl, 10 mM KCl, 2 mM MgCl2, 0.1 mM EGTA, 5.5 mM glucose, 0.200 mM Na2ATP, and 10 mM HEPES (pH 7.2). Pipette resistance was 3–5 MΩ when filled with the IS solution. The pipette-offset potential was corrected just prior to the gigaseal formation. Series resistance and transient capacitance were compensated using the patch-clamp amplifier (Axopatch 200B, Axon; Molecular Devices, LLC, Sunnyvale, CA, USA). Whole-cell seal resistance values were ≥10 GΩ, and capacitances were ∼20 pF. The holding potential was 0 mV during whole-cell measurements. Whole-cell current-voltage (I-V) relationships were elicited by a step-pulse protocol from −80 to +80 mV in 20-mV increments for 1 s duration, using the Clampex program (PCLAMP; Molecular Devices). After baseline I-V relationships were obtained, cells were perfused with ES containing 10 μM forskolin and 100 μM 3-isobutyl-1-methylxanthine (IBMX) to increase intracellular cAMP levels, and I-V measurements were repeated 5–7 min later. No recordings were performed unless the stability of the whole-cell configuration (seal) was confirmed. To document the extent to which the cAMP activated currents were through CFTR, we added 50 μM GlyH-101 (Millipore, Billerica, MA, USA) or 50 μM CFTRinh-172 (Sigma-Aldrich, St. Louis, MO, USA) into the perfusion solution and repeated the I-V measurements. Averages were calculated at 300 ms when the currents reached a steady state. The average values shown in the I-V curves were the mean values of the currents between 600 and 900 ms for each step. We observed that cell size did not correlate with currents; therefore, we opted to analyze cells with similar morphology and size. In additional experiments, the time course of CFTR activation by forskolin + IBMX was assessed by applying a voltage pulse protocol that hyperpolarized the holding membrane potential from 0 to −60 mV, then depolarized it from 0 to +60 mV, returning to holding potential (0 mV) while recording current. Each step lasted 2.58 s. Time-course studies were performed for the maximum of 60 min.
CFTR single-channel activity measurements in the cell-attached mode, patch-clamp technique
Single-channel activity of CFTR channels was recorded in cell-attached mode of the patch-clamp technique (33, 34). Recordings were performed only from gigaseals with resistance of >10 GΩ. Cells were perfused with a solution containing 145 mM KCl, 10 mM NaCl, 2 mM MgCl2, and 1 M HEPES (pH 7.4; 1 N KOH). Because of the high K+ and low Na+ concentrations in the bathing solution, cell membrane and patch potential were depolarized to ∼0 mV. The pipette solution had the same composition as the bath solution. During all measurements, the patch potential was held at 50 or 100 mV using the patch amplifier (Axopatch 200B; Molecular Devices). The holding potential (Vh) across the patch was calculated using the following formula: Vh = Vcell − Vp; since Vcell = 0, Vh = − Vp, where Vcell is the cell membrane resting potential, and Vp is the applied voltage through the pipette, as described previously (34). After stable recordings were obtained, cells were perfused with the same bath solution containing 10 μM forskolin and 100 μM IBMX. Data were analyzed with PClamp software. Single-channel recordings were analyzed using Clampfit software (Molecular Devices; refs. 34, 35). The product of open probability × number of channels (NPo), represents the proportion of the time during which activated channels remain open and was calculated as described previously (35, 36).
Codon analysis
CFTR protein and cDNA sequences from 43 different organisms were downloaded from UniProt (http://www.uniprot.org) and EMBL (http://www.embl.org) [PMID:22102590, PMID:15608199]. The sequence analysis package MEGA [PMID:21546353] was used to construct a codon-aligned multiple sequence alignment of all cDNA sequences. Custom scripts were used to calculate codon usage bias for each aligned amino acid in the multiple sequence alignment covering the various functional domains of the CFTR protein.
RESULTS
Not every codon change alters CFTR mRNA structure
Studies on synonymous codons have to be performed in a controlled environment to ensure that differences among the single-nucleotide variants can be attributed to the SCC rather than other variances that may be present in the expression vectors. The intracellular trafficking and functional properties of the CFTR proteins expressed from the wild-type, I507-ATT, or I507-ATC ΔF508 CFTR were investigated in HEK293 cell clones stably expressing these variants (3). The wild-type CFTR expression vector (pcDNA3.1.WT-CFTR) we used for the construction of the I507-ATT and I507 ATC ΔF508 CFTR variants contains a CFTR cDNA insert with 3 sSNPs (TGAAAAT→CGAGAAC) at nucleotide positions 1880–1886, which were introduced at the original construction of the expression vector for efficient plasmid propagation (ref. 12 and U.S. patent 5863770; http://www.patentgenius.com/patent/5863770). This finding indicates that the choice of codon in this region was important to propagate the plasmid in bacteria prior to mammalian gene delivery. Also, this finding is consistent with the idea of codon optimization as a tool to achieve high plasmid yield (37). The TGAAAAT→CGAGAAC silent substitutions must be present in most CFTR expression vectors constructed from the original plasmid described by Rommens et al. (12). Significantly, mammalian CFTR expression vectors containing these silent nucleotide variations have been applied to study CFTR expression, trafficking, and function by numerous investigators worldwide.
To ensure that in our studies the only difference among the constructs is represented by the C→T alteration at the third nucleotide of the I507 codon, we obtained full-length sequences of the expression vectors (Heflin Center of Genetics Core Facility, University of Alabama at Birmingham) and then aligned those with the GenBank (NM_000492) sequence of the human CFTR cDNA. In addition to the 3-nt deletion in ΔF508 CFTR and the T→C alteration in the I507-ATC ΔF508 CFTR (Fig. 1), we identified 2 G→A nonsynonymous codon alterations compared to the GenBank sequences (nt 2490; V470M and nt 4423; V1475M). This naturally occurring G→A (nt 2490) polymorphism results in a valine-to-methionine change (V470M; ref. 38) and is represented as ∼60% methionine (M) and 40% valine (V) in >30,000 patient samples tested at Ambry Genetics. However, in agreement with the one common ancestry theory of the ΔF508 mutation (39), the amino acid residue at the 470 position of CFTR in ΔF508 patients is always methionine, as analyzed by Ambry Genetics from >500 patient samples. The nt 4423 V1475M polymorphism has not been reported previously and was found originally in the pcDNA3.1 wild-type CFTR expression vector constructed by Moyer et al. (40). Notably, these polymorphisms are present in all CFTR constructs expressed in our model cell clones (wild-type, I507-ATT, and I507-ATC ΔF508 CFTR) and the only sequence difference between the I507-ATT and I507-ATC ΔF508 CFTR vectors is the T or C in the third position of I507 codon.
To investigate the possibility that the above noted nucleotide variations may affect the secondary structure of the CFTR mRNA, we analyzed the predicted mRNA secondary structures of the full-length mRNA sequences of the inserts for wild type, I507-ATC and I507-ATT ΔF508 CFTR using Mfold (41–44). The results indicate that the predicted secondary structures of the wild-type, I507-ATT and I507-ATC ΔF508 CFTR mRNAs transcribed from our vectors are identical with the predicted mRNA structures of the GenBank sequences (NM_000492; ref. 3). More notably, the only significant difference between the structures is localized to the ΔF508 mutation region and is caused by the C→T alteration (Fig. 1). Specifically, neither the silent nucleotide changes that were introduced to improve plasmid propagation nor the two nonsynonymous (V→M) variations alter the structure of the CFTR mRNA. This observation emphasizes the prospective significance of codon selection with respect to mRNA secondary structural formation in specific regions. In the case of CFTR, deletion of 3 nucleotides (CTT; ΔF508 CFTR), and the I507-ATC→ATT SCC in ΔF508 CFTR demonstrate a significant effect on the mRNA secondary structure in the region of mutation, while the other substitutions do not.
Differences in the biochemical properties of the I507-ATT and I507-ATC ΔF508 CFTR proteins
To identify differences in the stability, folding and function of the I507-ATT-ΔF508 and I507-ATC ΔF508 CFTR encoded proteins, we used HEK293 cell clones stably expressing I507-ATT or I507-ATC ΔF508 (3). We applied a classic strategy that rescues ΔF508 CFTR from ERAD, based on the temperature sensitivity of the native, I507-ATT ΔF508 CFTR (21). ΔF508 CFTR rescue was accomplished by culturing the cells at 27°C for 24 h. Cells grown under physiological conditions (37°C) were tested as a control. Culturing cells at low temperature reduced CFTR mRNA levels in both cell lines, but I507-ATC ΔF508 CFTR mRNA levels were 1.54 ± 0.175 (n=6) higher than I507-ATT ΔF508 CFTR mRNA levels under both conditions (37 and 27°C; Fig. 2A). In contrast to the minor mRNA level differences (1.5-fold), the full-length protein levels of the I507-ATC ΔF508 CFTR were ∼5-fold higher than the I507-ATT (37°C; Fig. 2B). This represents ∼2.5-fold higher I507-ATC ΔF508 CFTR band B levels (37°C) when normalized to mRNA levels (Fig. 2C). Low-temperature (27°C) culture of cells resulted in the rescue (appearance of band C, the post-Golgi form of CFTR) of both variants from ERAD (Fig. 2D).
Figure 2.

Biochemical properties of the I507-ATT and I507-ATC ΔF508 CFTR. A) CFTR mRNA levels were measured by qRT-PCR from cells cultured at physiological temperature (37°C) and following a low-temperature (27°C) culture for 24 h (n=6). GAPDH mRNA levels were assessed as an internal control. CFTR mRNA levels are plotted relative to GAPDH under each condition. I507-ATC ΔF508 CFTR levels were ∼50% higher under both conditions than I507-ATT. *P < 0.05. B) CFTR protein levels in cells expressing I507-ATT or I507-ATC ΔF508 CFTR were compared using Western blot. β-Actin served as a loading control. Cell lysates from wild-type CFTR-expressing cells were studied for comparison. Band B represents the core-glycosylated, ER form of CFTR; band C is the fully glycosylated, post-Golgi form. C) I507-ATT and I507-ATC ΔF508 CFTR protein levels, measured by densitometry of Western blots. MM13-4 monoclonal antibody was used to detect CFTR. Results are plotted relative to mRNA levels; n = 6. *P < 0.01. D) Western blot studies to compare the effects of 24 h of low temperature (27°C) culture on I507-ATT and I507-ATC ΔF508 CFTR levels and processing. Considering the higher mRNA levels in I507-ATC cells (A), the loading of total proteins was adjusted accordingly. I507-ATT (50 μg) and I507-ATC (35 μg) total cellular proteins were loaded, and the differences are represented by the lower intensity of β-actin bands in I507-ATC ΔF508 cells. Note higher levels of I507-ATC ΔF508 CFTR band B. Representative gels are shown; n = 4.
Because the significantly higher I507-ATC ΔF508 CFTR protein levels compared to I507-ATT ΔF508 were not attributable to mRNA differences (Fig. 2), and based on our previous results (3), we hypothesized that they might result from changes in translation dynamics leading to altered recognition of the I507-ATC ΔF508 CFTR by the ER quality control machinery. We based our hypothesis on previous reports indicating that CFTR folds cotranslationally (45) and ΔF508 CFTR might be selected for ERAD before completion of the polypeptide chain (46, 47). To test whether changes in ERAD contribute to higher I507-ATC ΔF508 CFTR protein levels, we measured the effects of ALLN, a cell permeable protease inhibitor with efficient proteasome inhibitory effect on the levels of the ΔF508 CFTR variants under standard culture conditions (37°C). Without ALLN, we could detect only minimal amounts of the full-length (band B) I507-ATT ΔF508 CFTR by Western blotting if cells were cultured at 37°C. In contrast, a more pronounced band of I507-ATC ΔF508 was present under the same conditions (Fig. 3A). In the presence of proteasome inhibition, a > 8-fold increase in I507-ATT ΔF508 CFTR band B was found, compared with only a 2-fold increase in I507-ATC ΔF508 CFTR levels (Fig. 3A, right panel). These results indicate that most I507-ATT ΔF508 CFTR is degraded cotranslationally as described previously (46, 47). Therefore, these data support our hypothesis that the cotranslational ER membrane insertion of the I507-ATC ΔF508 CFTR is more efficient than the I507-ATT ΔF508 and results in higher levels of I507-ATC ΔF508 CFTR band B.
Figure 3.

Differences in the folding and processing of I507-ATT and I507-ATC ΔF508 CFTR. A) Proteasome inhibitor sensitivity differences between the CFTR variants. We compared CFTR protein levels prior to proteasome inhibition (ALLN −), and at 4 or 8 h following ALLN treatment (100 μM) (ALLN +). β-Actin levels are shown as a loading control. Loading: 50 μg of total cellular proteins under each condition. Top panel: representative gels. Bottom panel: relative increase in band B (ER-form of CFTR); densities are shown for each variant as a result of 4 h ALLN treatment (n=3). *P < 0.01. B) N-terminal fragment pattern differences between the I507-ATT and I507-ATC ΔF508 CFTR. Total cellular proteins (5 μg) were loaded from HEK293 cells expressing wild-type, I507-ATT, I507-ATC ΔF508 CFTR, or without CFTR expression (no CFTR). Representative gels and densitometry results of the lanes are shown for each (n=4). β-Actin: loading control. Arrows indicate a band only present in wild-type and I507-ATC ΔF508 CFTR-expressing cell lysates. C) Metabolic pulse-chase experiments were performed to measure the stability of the ER resident variant proteins following a 20-min pulse with 35S methionine/cysteine protein labeling mix. Decay of radioactive proteins was followed after addition of regular medium. Top panel: representative gels. Bottom panel: CFTR band B stability and maturation efficiency are plotted relative to band B densities at T0 (100%); n = 4. D) Cell turnover of the low-temperature-rescued CFTR variants was measured following cyclohexamide block and cell surface biotinylation as described in Materials and Methods. Top panel: representative gels. Band C represents biotinylated, cell surface CFTR following cyclohexamide treatment for the indicated periods of time at 37°C. Bottom panel: half-lives of the I507-ATT ΔF508 and I507-ATC ΔF508 CFTR (n=4). No significant difference was found between the cell surface stability of the proteins (NS, n=4).
Considering the differences in translation dynamics among the wild-type, I507-ATT, and I507-ATC ΔF508 CFTR (3) and the higher levels of full-length I507-ATC ΔF508 CFTR compared to the I507-ATT variant (Fig. 2), we hypothesized that the folding of the I507-ATC ΔF508 CFTR differs from the I507-ATT and therefore, it is less accessible to ERAD during synthesis. To test this, we analyzed the natural fragmentation of the CFTR variants using a monoclonal antibody (MM13-4) that recognizes the N-terminal tail of CFTR (Fig. 3B). Monoclonal antibodies have been used to identify differences between the fragmentation of wild-type and ΔF508 CFTR previously (28). The MM13-4 antibody recognized multiple bands of different molecular masses in both the I507-ATT and I507-ATC ΔF508 CFTR expressing cell lysates and identified a similar fragment in wild-type CFTR expressing cell lysates as described by Tosoni et al. (28). Notably, the fragment patterns of the I507-ATT and I507-ATC ΔF508 CFTR were markedly different. The results of proteasome sensitivity and fragmentation studies support our hypothesis that the choice of synonymous codon for I507 contributes to the cotranslational folding of ΔF508 CFTR and that I507-ATC can be distinguished from I507-ATT CFTR.
To analyze the post-translational processing of the full-length I507-ATT and I507-ATC ΔF508 CFTR proteins, we performed metabolic pulse-chase experiments (Fig. 3C). We used a monoclonal antibody (24-1) recognizing the C-terminal tail of CFTR for immunoprecipitation to identify only full-length CFTR. Consistent with the results of the in vitro translation studies (3) and Western blot experiments (Figs. 2 and 3), the radioactively labeled band B of the I507-ATC ΔF508 CFTR was significantly stronger than the I507-ATT ΔF508 CFTR. When we followed the decay of the labeled proteins, the I507-ATC ΔF508 encoded protein was more stable during the early period of the chase. As anticipated, based on the loss of phenylalanine in both constructs, by the end of the chase (6 h), the majority of both variants were degraded. We also observed a minor (5–8% of total labeled band B; n=4) maturation efficiency of the I507-ATC ΔF508 CFTR (band C) (Fig. 3C). We did not observe post-ER forms (band C) of the I507-ATT ΔF508 CFTR. The minimum I507-ATC ΔF508 CFTR maturation efficiency implied that in order to identify possible functional differences between the variants, both had to be rescued from ERAD. Thus, we chose low-temperature growth conditions (27°C) to rescue and determine the functional properties of the ΔF508 CFTR variants.
Prior to electrophysiological analysis, we compared the cell surface expression and stability of the low-temperature-rescued I507-ATT and I507-ATC encoded ΔF508 CFTR proteins (Fig. 3D). These experiments were based on our previous results indicating that the protein half-life of the low-temperature-rescued I507-ATT ΔF508 CFTR was significantly longer than the functional half-life when cells were returned to 37°C culture conditions (22). To test for differences in the stability of rescued I507-ATT vs. I507-ATC ΔF508 CFTR, we cultured the cells expressing the constructs at 27°C for 24 h and then returned them to 37°C for the time periods specified (Fig. 3D). Cell surface CFTR was detected using a biotinylation assay (22, 27, 30) and surface half-life was monitored during a cycloheximide (CHX) blockade as described previously (29). The results demonstrate that the cell surface protein half-lives of the low-temperature-rescued I507-ATT ΔF508 and I507-ATC ΔF508 CFTR were identical (∼2 h) when the cells were replaced to 37°C. These results are consistent with our previous findings testing the cell surface stability of I507-ATT ΔF508 CFTR (22, 30). Furthermore, these results suggest that once the translation of the I507-ATT and I507-ATC ΔF508 CFTR variants is completed, the stability of the protein is determined by the loss of F508. Therefore, the minimum post-ER trafficking (5–8%) of the I507-ATC ΔF508 CFTR may be the result of higher full-length protein levels. However, the similar protein stability does not exclude the possibility of functional differences between the variants.
Enhanced thermal stability of the I507-ATC ΔF508 CFTR
Previous studies have indicated that the functional half-life of low-temperature-rescued ΔF508 CFTR is shorter than its protein half-life would predict (22, 48, 49). To test whether this was true to both I507-ATT ΔF508 and I507-ATC ΔF508 CFTR, we performed whole-cell patch clamp studies. Wild-type CFTR-expressing cells, cultured under physiological conditions (37°C), were studied as control. I507-ATT ΔF508 and I507-ATC ΔF508 CFTR-expressing cells were studied following low-temperature correction at 27°C for 24 h.
The characteristics of whole cell currents recorded from wild-type, I508-ATT, and I507-ATC ΔF508 CFTR are shown in Fig. 4. To demonstrate that the forskolin+IBMX-activated whole-cell currents were CFTR-mediated, we measured the average I-V relationships for each CFTR variant ∼5 min after activation with forskolin + IBMX, when the currents reached maximum value, and following perfusion with two different CFTR inhibitors GlyH-101 (32) and CFTRinh-172 (50, 51). We studied cells of similar size and morphology and the mean capacitances (pF) ± 1 sem (number of cells) were as follows: wild-type CFTR = 21.2 ± 1.3 (n=20); I507-ATT ΔF508 CFTR = 20.4 ± 1.2 (n=20); I507-ATC ΔF508 CFTR = 20.7 ± 1.2 (n=20). Because cells with similar size and morphology were chosen for the studies and the mean capacitances were very similar, normalization of the currents to cell size was not necessary. The average I-V curves (mean± se, n=7–8) were close to linear, in the voltage range tested, with slight inward rectification as described elsewhere (51–53). A complete inhibition of the cAMP-activated currents was found both by GlyH-101 (50 μM) or CFTRinh-172 (50 μM) (Fig. 4A).
Figure 4.

Whole-cell currents of wild-type, I507-ATT, and I507-ATC ΔF508 CFTR-expressing cells prior to and following cAMP activation. Whole-cell patch-clamp studies were performed as described in Materials and Methods. Wild-type CFTR-expressing HEK293 cells cultured under physiological conditions (37°C). I507-ATT ΔF508 and I507-ATC ΔF508 CFTR-expressing cells were cultured at 27°C for 24 h prior to measurements, which were performed at room temperature. After formation of seals, currents were activated by perfusing the cells with forskolin (10 μM) + IBMX (100 μM), and I-V relationships were measured. Cells were then perfused with solutions containing forskolin (10 μM) + IBMX (100 μM) and either GlyH101 (50 μM) or CFTRinh-172 (50 μM), and I-V relationships were repeated at ∼5 min, when the currents reached maximum. A) Average I-V relationships of the cyclic-AMP-dependent currents in wild type, I507-ATT, and I507-ATC ΔF508 CFTR-expressing HEK293 cells. Mean ± sem I-V relationships (n=8) prior to (control) and following forskolin + IBMX induction and after deactivation with GlyH-101 (n=8) or CFTRinh-172 (n=7). The I-V curves are linear (53), with minimal inward rectifications at large positive voltages, as reported previously (52). B–E) Time-course characteristics of cAMP-activated whole-cell currents recorded from wild-type, I507-ATT, and I507-ATC ΔF508 CFTR-expressing cells. Forskolin + IBMX-activated currents (I, pA) were recorded using the whole-cell patch-clamp technique at room temperature, as described in Materials and Methods. Wild-type CFTR-expressing cells were cultured under standard conditions; I507-ATT and I507-ATC ΔF508 CFTR-expressing cells were tested immediately after a low-temperature (24 h, 27°C) rescue. B) Summary of whole-cell recordings from HEK293 cells expressing wild-type CFTR (GlyH-101, n=8; and CFTRinh-172 deactivation, n=7). C, D) Summary of whole-cell recordings from HEK293 cells expressing I507-ATT (C; n=11) or I507-ATC (D; n=9) ΔF508 CFTR. Properties of forskolin + IBMX-activated currents are plotted as means ± sem. E) Similar magnitude tracings from I507-ATT and I507-ATC ΔF508 CFTR-expressing cells are plotted in the same graph. CFTR channel activity spontaneously declined to the baseline after 20 min of maximum activation in cells expressing I507-ATT ΔF507 CFTR (open squares). Low-temperature-rescued I507-ATC ΔF508 CFTR channels (solid circles) sustained their currents for 40 min after reaching the maximum at 10 min following activation and were blocked by GlyH-101. F) Cell surface expression of I507-ATT and I507-ATC ΔF508 CFTR. Cell surface CFTR levels were measured following cell surface biotinylation at 3 different time points (0, 10, and 40 min) following forskolin + IBMX activation. Cell surface CFTR levels increased in both cell lines on activation of the channels. Representative gels are shown at top (band C); β-actin was detected to ensure equal loading. Bar graphs compare band C densities at different time points (0, 10, and 40 min) following channel activation (n=4). Bar graphs indicate that although I507-ATT ΔF508 CFTR cell surface levels increased, the channel activity declined. *P < 0.01.
In additional experiments, we investigated the stability of the forskolin + IBMX activated whole-cell currents in wild type, I507-ATT ΔF508, and I507-ATC ΔF508 CFTR-expressing cells by measuring whole cell currents while changing the membrane potential from 0 to −60 or 0 to 60 mV as described in Materials and Methods. The forskolin+IBMX activated whole-cell currents of cells with wild-type CFTR remained stable or decreased slightly during the recording period (40 min) and addition of either GlyH101 or CFTRinh-172 resulted in a quick and complete blockade of the channels (Fig. 4B). In this first set of experiments, we studied I507-ATT and I507-ATC ΔF508 CFTR-expressing cells immediately after a 24 h rescue at 27°C. When the whole-cell current recordings were performed with cells expressing I507-ATT ΔF508 CFTR, the cAMP-activated currents began to decline spontaneously, 10 min after perfusion with IBMX + forskolin and returned to baseline in 20 min (Fig. 4C). In contrast, perusion of cells expressing I507-ATC ΔF508 CFTR with forskolin + IBMX resulted in stable, wild-type CFTR-like currents for a 40-min period; at this time perfusion with GlyH-101 decreased the currents to baseline levels (Fig. 4C). Notably, the average whole-cell currents recorded from I507-ATT ΔF508 CFTR-expressing cells were lower (∼750 pA) than those of I507-ATC ΔF508 CFTR (∼1500 pA) and wild-type CFTR (∼2500 pA) (Fig. 4B–D).
To test whether the cAMP activated current stability differences between the I507-ATT and I507-ATC variants were the result of lower total currents recorded from I507-ATT ΔF508 CFTR-expressing cells (∼750 pA), we selected similar magnitude recordings from I507-ATT and I507-ATC ΔF508 CFTR-expressing cells and tested the stability of the currents over time (Fig. 4E). The results indicate that the spontaneous decline of whole-cell currents in I507-ATT ΔF508 CFTR-expressing cells is not the result of lower whole-cell channel activity.
Because previous studies indicated that cAMP activation enhances the levels of CFTR in the plasma membrane (54), we also compared the cell surface levels of the ΔF508 CFTR variants following cAMP activation. Addition of forskolin + IBMX resulted in increased cell surface CFTR levels for both variants. More notably, the cell surface levels of the I507-ATT ΔF508 and I507-ATC ΔF508 proteins were similar 40 min following addition of forskolin + IBMX (Fig. 4F). As noted in Fig. 4C, at that time, currents from the I507-ATT ΔF508 CFTR-expressing cells had declined to baseline levels. These results suggest that the declining currents seen in cells expressing the I507-ATT ΔF508 variant were due to the deactivation of CFTR rather than reduced surface protein levels (Fig. 4B–F).
To further analyze the differences in the thermal stability of the I507-ATT-ΔF508 and I507-ATC-ΔF508 CFTR following low-temperature rescue, we replaced the cells to 37°C for 30 min prior to patch-clamp studies (which were performed at room temperature). Small (∼250 pA) and transient currents were recorded from I507-ATT ΔF508 CFTR-expressing cells (Fig. 5A). However, considerable higher currents were seen across 507-ATC-ΔF508 CFTR cell, which remained stable over a 40-min period; again, these currents were completely inhibited by GlyH-101 (Fig. 5B). Notably, cell surface biotinylation revealed similar levels of mature CFTR proteins at the cell surface during the time course of this experiment (Fig. 5A, B; inlays).
Figure 5.

Functional stabilities of the low-temperature-rescued I507-ATT and I507-ATC ΔF508 CFTR at 37°C. A) Summary of whole-cell patch-clamp recordings and cell surface CFTR measurements in I507-ATT ΔF508 CFTR cells following 24 h rescue at 27°C followed by 30 min culture at 37°C. Only small (∼250 pA maximum current) and transient currents were recorded (means±sem, n=11), while cell-surface CFTR levels increased during the time course of the activation (0, 10, 40 min) by forskolin (F) + IBMX (inset, band C). B) Summary of whole-cell patch-clamp recordings from cells expressing I507-ATC ΔF508 CFTR after 30 min at 37°C. Activation of CFTR in cells expressing the I507-ATC variant resulted in sustained currents of ∼1500 pA (means±sem, n=9), and the protein was present on the cell surface (inset). C) After 4 h of culture at 37°C, we could not activate I507-ATT ΔF508 CFTR (data not shown), and the currents recorded from the I507-ATC ΔF508 CFTR were transient (means±sem, n=5). D) Cell-surface protein levels of the I507-ATT and I507-ATC ΔF508 CFTR variants were similar following 4 h culture at 37°C and represented ∼15% of the low-temperature-rescued protein measured immediately after low-temperature culture (band C). β-Actin is shown as a loading control n = 4.
The next set of experiments was performed following a 4 h incubation of the low-temperature-corrected I507-ATT and I507-ATC ΔF508 CFTR-expressing cells at 37°C. We chose this time point based on our previous studies showing a total loss of I507-ATT ΔF508 CFTR activity after 4 h (22). Indeed, we could not record forskolin + IBMX-activated currents in I507-ATT ΔF508 CFTR-expressing cells after 4 h. In contrast, currents were recorded across the membranes of cells expressing I507-ATC ΔF508 CFTR, although they decayed with time (Fig. 5C). When we measured the cell surface protein levels of the two variants after the 4 h culture at physiological temperature (37°C), we did not observe significant differences as only a small portion (∼15%) of the rescued proteins remained on the cell surface in both cell lines (Fig. 5D).
Single-channel recordings indicate differences between the channel properties of the I507-ATT ΔF508 and I507-ATC ΔF508 CFTR
Previous studies indicated that the ΔF508 CFTR folding defect led to impaired channel gating (20, 55). Further, while low-temperature culture improved the processing of the mutant, it did not restore physiological channel function (21). Based on the observed differences among the ΔF508 CFTR variants that involve translational dynamics, ER stability (3), proteasome sensitivity (Fig. 3), and functional stability (Figs. 4 and 5), we tested whether these alterations resulted in functional correction of the I507-ATC ΔF508 CFTR channels. To address this, we measured CFTR single-channel activities of cells patched in the cell attached mode as we did for epithelial sodium channels (56) following a 24 h rescue of I507-ATT and I507-ATC ΔF508 CFTR at 27°C. We used wild-type CFTR-expressing cells as a control (Fig. 6A, D). As shown in Fig. 6A–C, addition of forskolin + IBMX into the bath solution 3 min prior to recordings activated the channels. The time course of forskolin activation for wild-type CFTR is shown in Fig. 6D. Mean values for NPo of forskolin + IBMX channels are shown in Fig. 6E. In agreement with previous reports (20), the NPo of wild-type CFTR was 0.55 ± 0.064 (n=6), and the NPo of the I507-ATT ΔF508 CFTR was 0.09 ± 0.002 (n=4). Notably, the gating of the I507-ATC ΔF508 CFTR differed significantly from the I507-ATT ΔF508 CFTR and was similar to wild-type CFTR, with an NPo of 0.45 ± 0.037 (n=6). The conductances of single channels [mean ± sem values (pS); n=number of patches] were as follows: wild-type CFTR = 6.1 ± 0.14 (n=9); I507-ATT ΔF508 CFTR = 6.26 ± 0.07 (n=9); I507-ATC ΔF508 CFTR = 6.5 ± 0.1 (n=9). These results imply that the I507-ATC→ATT codon alteration is responsible for the gating defect associated with the ΔF508 CFTR mutation.
Figure 6.

Single-channel properties of wild-type and low-temperature-rescued I507-ATT and I507-ATC ΔF508 CFTR. ATP-dependent, forskolin + IBMX-activated conformational changes in the CFTR protein result in the opening and closing of the channel. Channel gating (closed↔open transition) was calculated following addition of cAMP agonists (10 μM forskolin + 100 μM IBMX) to activate the channel by phosphorylation. A–C) Representative tracings of single channel activities for wild-type (A), I507-ATT (B), and I507-ATC (C) ΔF508 CFTR. After formation of the seals, CFTR was activated by forskolin + IBMX 3 min prior to recordings. D) Time course of wild-type CFTR activation following addition of forskolin. E) Open probability represents the proportion of the time during which activated channels remain open. NPo (number of single channels times their open probabilities) in cell attached patches from HEK293 cells expressing wild type, I507-ATT, and I507-ATC ΔF508 CFTR were 0.55 ± 0.064, 0.09 ± 0.002, and 0.45 ± 0.037, respectively (X±sem; n≥ 5). *P < 0.001.
DISCUSSION
Significance of analyzing silent nucleotide alterations
A large number of human disorders have now been linked to synonymous mutations (for review see ref. 57). More specifically, a study that analyzed >20,000 disease and SNP correlations indicated that non-synonymous SNPs (nsSNPs) and sSNPs shared similar probability for disease association (58). Because silent SNPs may lead to the synthesis of a protein product with the same amino acid sequence but different structural and functional properties (2, 3, 59, 60), silent SNPs should no longer be overlooked and should be taken into account during treatment development programs. However, not every silent nucleotide change alters protein function (2, 61) and the molecular mechanisms by which sSNPs contribute to disease pathogenesis are diverse and poorly understood (62). Therefore, investigating the consequences of specific sSNPs, such as the subject of our studies (I507-ATC→ATT), will provide important information regarding the cellular mechanisms by which sSNPs affect the function of a gene product. Even more significantly, these studies will provide valuable clues for understanding the obvious need for codon redundancy.
Codon-usage bias for Phe508-TTT in the NBD domains of CFTR
Here we demonstrate that the I507-ATC→ATT SCC in the human ΔF508 CFTR contributes to the function of the gene product. As illustrated in Fig. 1, individuals with I507-ATC -ΔF508 CFTR would exist when the same deletion occurred if F508 was encoded by its synonym TTC. However, in agreement with the common ancestry theory of the ΔF508 mutation, as this mutation has been found in association exclusively with one marker haplotype 23-31-13 (39), it is unlikely that patients with I507-ATC ΔF508 CFTR variant exist. Indeed, analysis of >30,000 patient samples sequenced at AmbryGenetics revealed no I507-ATC ΔF508 CFTR. More surprisingly, analysis of all non-ΔF508 sequences from 30,000 cohorts indicated no F508-TTC either, suggesting a codon usage bias for F-TTT in this particular location. The majority of these patients are of Caucasian descent, mixed gender and all have symptoms consistent with CF.
Considering the lack of F508-TTC in the population tested for cystic fibrosis (CF) symptoms, we analyzed the codon usage for both isoleucine (ATC, ATT, ATA) and phenylalanine (TTT and TTC) in all domains of CFTR (Fig. 7). The rationale for this is that the choice of codon can affect gene expression and cellular functions, as it influences diverse processes, including mRNA structure, translation, and protein folding (63). Interestingly, analysis of codon usage reveled a strong bias toward F-TTT (0.89 TTT vs. 0.11 TTC) in the NBD1 and NBD2 domains of the human CFTR, while in the majority of the CFTR coding regions, both synonyms appear with similar frequency (0.55 TTT vs. 0.45 TTC) (Fig. 7B). Considering the low number of phenylalanine codons (four) in the NBD1-encoding domain of the human CFTR, we examined the phenylalanine codons in the NBD1 domains of CFTR from 43 organisms. Our analysis revealed that the F508-TTT codon was used conservatively in 42 of the 43 organisms and the codon bias for F-TTT in the NBD domains is present across the 43 organisms (Fig. 7C and Table 1). These results, together with the variable codon occurrence in other regions of CFTR, are consistent with the idea that F-TTT for F508 may have a significant role in determining the structure and/or function of CFTR not only in humans, but also in other organisms.
Figure 7.
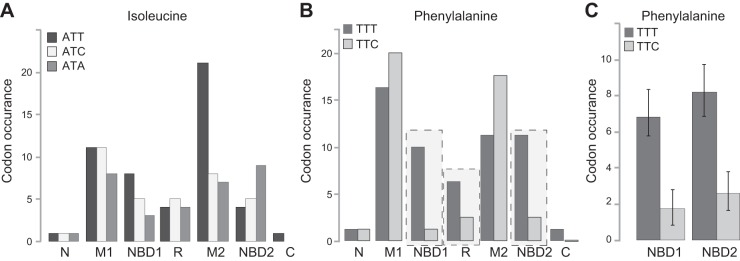
Phenylalanine-TTT codon usage bias in the NBD domains of CFTR. A) Isoleucine codon usage in different human CFTR domains. B) Phenylalanine codon usage in different human CFTR domains. N, N-terminal tail; M1, membrane spanning domain 1 with extra- and intracellular loops; NBD1, nucleotide binding domain 1 (E449–G646); R, regulatory domain (C647–G836); M2, membrane-spanning domain 2 with extra- and intracellular loops; NBD2, nucleotide binding domain 2 (A1225–P1443); C, C-terminal tail. A strong codon usage bias was found for phenylalanine in the NBD and R domains of the human CFTR (rectangles). C) Phenylalanine codon usage in the NBD1 and NBD2 domains of CFTR in 43 different organisms (n=43, P<0.001). Strong codon usage bias for TTT was found in the NBD domains of CFTR in all 43 organisms tested. See Table 1 for domain sequences.
Table 1.
Alignment of the CFTR NBD1 domain cDNA sequences from 43 organisms
| Accession no. | Organism scientific name | Codon-based multiple sequence alignment (partial sequence) |
|---|---|---|
| AAA35680 | Homo sapiens (human) | ACC ATT AAA GAA AAT ATC ATC TTT GGT GTT TCC TAT GAT GAA |
| ABC87455 | Gorilla gorilla (gorilla) | ACC ATT AAA GAA AAT ATC ATC TTT GGT GTT TCC TAT GAT GAA |
| AAR16253 | Pan troglodytes (chimpanzee) | ACC ATT AAA GAA AAT ATC ATC TTT GGT GTT TCC TAT GAT GAA |
| AAC14011 | Macaca mulatta (Rhesus monkey) | ACC ATT AAA GAA AAT ATC ATC TTT GGT GTT TCC TAT GAT GAA |
| ABC87491 | Chlorocebus aethiops (monkey | ACC ATT AAA GAA AAT ATC ATC TTT GGT GTT TCC TAT GAT GAA |
| ABI75275 | Ateles geoffroyi (monkey) | ACC ATT AAA GAA AAT ATC ATC TTT GGT GTT TCT TAT GAT GAA |
| ABJ08890 | Aotus nancymaae (monkey) | ACC ATT AAA GAA AAT ATA ATC TTT GGT GTT TCC TAT GAT GAA |
| ABI75310 | Salmiri boliviensis (monkey) | ACC ATT AAA GAA AAT ATC ATC TTT GGT GTT TCC TAT GAT GAA |
| ABC87466 | Pongo abelii (Sumatran orangutan) | ACC ATT AAA GAA AAT ATC ATC TTT GGT GTT TCC TAT GAT GAA |
| ABJ08867 | Nomascus leucogenys (gibbon) | ACC ATT AAA GAA AAT ATC ATC TTT GGT GTT TCC TAT GAT GAA |
| AAA30772 | Bos taurus (cattle) | ACC ATT AAA GAT AAC ATC ATC TTT GGT GTT TCC TAT GAT GAA |
| ABB89806 | Eqqus caballus (horse) | ACC ATT AAA GAA AAT ATC ATC TTT GGT GTT TCC TAC GAT GAG |
| AAA68600 | Ovis aries (sheep) | ACC ATT AAA GAT AAC ATC ATC TTT GGT GTT TCC TAT GAT GAA |
| AAS98211 | Sus scrofa (pig) | ACC ATT AAA GAA AAC ATC ATC TTT GGT GTT TCC TAT GAT GAG |
| BAK78871 | Cavia porcellus (pig) | ACC ATC AAA GAA AAC ATC ATC TTT GGT GTT TCC TAT GAT GAG |
| AAV40962 | Canis lupus (dog) | ACC ATT AAA GAA AAC ATC ATC TTT GGT GTT TCC TAT GAT GAG |
| ABG66652 | Loxodonta africana (elephant) | ACC ATT AAA GAA AAC ATC ATC TTT GGT GTT TCC TAT GAT GAG |
| AAR16315 | Rattus norvegicus (rat) | ACT ATC AAA GAA AAT ATC ATC TTT GGT GTT TCC TAT GAT GAG |
| AAA37417 | Mus musculus (mouse) | ACT ATC AAA GAA AAT ATC ATC TTT GGT GTT TCC TAT GAT GAG |
| AAC48608 | Oryctolagus cuniculus (rabbit) | ACC ATT AAA GAA AAC ATC ATC TTT GGT GTT TCC TAT GAT GAG |
| AAD46905 | Macaca fuscata (macaque) | ACC ATT AAA GAA AAT ATC ATC TTT GGT GTT TCC TAT GAT GAA |
| AAD46907 | Macaca nemestrina (macaque) | ACC ATT AAA GAA AAT ATC ATC TTT GGT GTT TCC TAT GAT GAA |
| AAD46904 | Papio anubis (olive baboon) | ACC ATT AAA GAA AAT ATC ATC TTT GGT GTT TCC TAT GAT GAA |
| AAF80467 | Macaca fascicularis (macaque) | ACC ATT AAA GAA AAT ATC ATC TTT GGT GTT TCC TAT GAT GAA |
| AAX13970 | Trichosurus vulpecula (brushtail) | ACT ATT AAA GAA AAT ATT ATT TTT GGT GTT TCT TAT GAT GAA |
| AAY88985 | Atelerix albiventris (hedgehog) | ACC ATA AAA GAA AAT ATC ATC TTT GGT GTT TCC TAT GAT GAG |
| ABA90397 | Callithrix jacchus (marmoset) | ACC ATT AAA GAA AAT ATC ATC TTT GGT GTT TCC TAT GAT GAA |
| ABA90408 | Otolemur garnetti (galago) | ACC ATT AAA GAA AAC ATC ATC TTT GGT GTT TCC TAT GAT GAG |
| ABB89784 | Carollia perspicillata (bat) | ACC ATT AAG GAA AAC ATC ATC TTT GGT GTT TGC TAT GAT GAG |
| ABC87480 | Rhinolophus ferrumequinum (bat) | ACC ATT AAA GAA AAC ATC ATC TTT GGT GTT TCC TAT GAT GAG |
| ABB89795 | Callicebus moloch (red-bellied titi) | ACC ATT AAA GAA AAT ATC ATC TTT GGT GTT TCC TAT GAT GAA |
| ABB89826 | Microcebus murinus (lemur) | ACC ATT AAA GAA AAT ATC ATC TTT GGC GTT TCC TAT GAT GAA |
| ABB89835 | Didelphis virginiana (opossum) | ACT ATT AAA GAA AAC ATC ATT TTT GGT GTT TCT TAT GAT GAA |
| ABI75286 | Muntiacus muntjak vaginalis | ACC ATT AAA GAT AAC ATC ATC TTT GGT GTT TCC TAT GAT GAA |
| ABI93634 | Dasypus novemcinctus (armadillo) | ACC ATT AAA GAA AAC ATC ATC TTT GGT GTT TGT TAT GAT GAG |
| ABI93660 | Mustela putorius (ferret) | ACC ATC AAA GAA AAC ATC ATC TTT GGT GTT TCT TAT GAT GAG |
| ABI93680 | Ornithorhynchus anatinus (platypus) | ACC ATC AAG GAG AAC ATC GTC TTC GGG GTT TCT TTC GAT GAG |
| ABJ08857 | Colobus guereza (guereza) | ACC ATT AAA GAA AAT ATC ATC TTT GGT GTT TCC TAT GAT GAA |
| ABJ08877 | Muntiacus reevesi (muntjak) | ACC ATT AAA GAT AAC ATC ATC TTT GGT GTT TCC TAT GAT GAA |
| CAA46348 | Xenopus laevis (frog) | ACC ATC AAA GAG AAT ATA GTA TTT GGC GTT TCC TAT GAT CAG |
| ABK34432 | Gallus gallus (chicken) | ACA ATC AAA GAA AAC ATC ATT TTT GGT GTA TCT TAT GAC GAA |
| AAA49616 | Squalus acanthias (dogfish) | ACA ATC AAG GAC AAC ATC ATA TTT GGA CTT TCA TAT GAT GAG |
| CAK10920 | Danio rerio (zebrafish) | ACC ATC CGA GAC AAC ATC CTC TTT GGC CTG ACC TAC GAC GAG |
See Fig. 7. Sequence accession number and organism for each of the 43 CFTR cDNA sequences analyzed for codon usage are listed. Table includes the partial multiple sequence alignment that corresponds to region 501–514 of the human CFTR protein sequence; 39 of 43 sequences were from mammals and are listed on top.
One explanation for the F-TTT codon bias in the human CFTR NBD domains and 42 other organisms might be its evolutionary importance. It was proposed 25 yr ago that codon preference contributes to gene expression levels (64), and today it is evident that codon usage bias is necessary for translational regulation and also has a strong effect on bacterial selection (63). Similar functions of codon usage bias are likely to exist in mammals, and it will be of interest to elucidate the potential role of codon preference in both the regulation of gene expression and the function of resulting gene products. Therefore, our results may provide important background information for studies analyzing codon usage bias in other genes.
Based on the lack of CF patients with I507-ATC ΔF508 CFTR and the absence of F508-TTC in the tested population, one may question the biological significance of our results. However, it is important that comparing the native I507-ATT to the I507-ATC variant allowed us to demonstrate the deleterious effect of this single nucleotide change in ΔF508 patients. This is significant since, despite large-scale efforts, the progress toward understanding the ΔF508 dysfunction remains slow. The studies presented herein demonstrate that one not-so-silent nucleotide change (I507-ATC→ATT) can affect the function of the gene product significantly, while others, such as the changes that were introduced for efficient plasmid propagation, remain silent. Furthermore, it is obvious from our studies that detailed sequence analysis of constructs that are used to study CFTR biogenesis and function will be necessary to draw conclusion about the maturation, degradation, and function of the gene product.
Effects of the silent (I507-ATC→ATT) codon change on ΔF508 CFTR biogenesis and degradation
Earlier studies analyzing the process that directs misfolded ΔF508 CFTR to the proteasome have demonstrated that ΔF508 CFTR is co- and post-translationally ubiquitinated both in chaperone-dependent and chaperone-independent fashion, followed by retrotranslocation from the ER to the proteasome (46, 47, 65). Our observation that the I507-ATC ΔF508 CFTR is less accessible to the ER quality control system than the I507-ATT ΔF508 CFTR fits this paradigm. Specifically, higher levels of full-length I507-ATC ΔF508 CFTR are consistent with the idea that less incompletely translated protein is extracted from the ER prior to completion of the peptide chain. This hypothesis is also supported by the observed differences in CFTR fragment patterns between the I507-ATT and I507-ATC ΔF508 CFTR using a monoclonal antibody against the N-terminal tail of the protein.
The reduced accumulation of I507-ATC ΔF508 CFTR in the presence of proteasome inhibition also points to the differences in the folding state and consequent recognition by the ER quality control of the variants. Furthermore, the results of metabolic pulse-chase studies demonstrate that, while the I507-ATC ΔF508 CFTR is more stable during the early chase periods, most of the core glycosylated (band B) degrades by 4 h, supporting the idea of less cotranslational degradation compared to the I507-ATT ΔF508 CFTR. Two important questions arise from these results: Why does a more properly folded protein gets degraded by ERAD? Does any of the I507-ATC ΔF508 CFTR that escaped early ERAD traffic to the cell membrane under physiological conditions?
To address these questions, it must first be noted that the I507-ATC-encoded protein lacks F508, and a number of studies have analyzed the consequences of the phenylalanine deletion on various steps of ΔF508 CFTR folding (24, 66, 67) and degradation (46, 65, 68, 69). Thomas and colleagues (66, 67) applied a series of misfolding and suppressor mutations in the nucleotide binding and transmembrane domains of CFTR to evaluate their effects on the folding and maturation of the protein. The conclusion from these studies is that the ΔF508 deletion carries multiple consequences regarding both intradomain and interdomain interactions that are critical for protein folding during the later steps of translation. Furthermore, the studies analyzing co- and post-translational ubiquitination and deubiquitination of CFTR (46, 65, 68, 69) suggest that changes in folding dynamics can alter the levels of the full-length protein in the ER, as supported by our results. The results presented in Fig. 3 also suggest that, while the single-nucleotide change alters the cotranslational folding of the protein resulting in differences during cotranslational degradation, the stability of the full-length, low-temperature-rescued cell surface proteins is identical. Therefore, it is evident that the major determinant of rescued ΔF508 CFTR cell surface stability is the folding defect caused by the loss of F508, as has been established by Lukacs and colleagues (23, 70, 71). Our results do not imply that the silent I507-ATC alteration overcomes the complete folding defect caused by the deletion of phenylalanine, rather suggest that it improves protein structure leading to more stable function.
ΔF508 CFTR dysfunction and the role of the I507-ATT sSNP
Dysfunction of the rescued ΔF508 CFTR has been extensively analyzed (for review see ref. 24), but not all the functional defects, including the thermal instability of the rescued protein (49) are fully understood. Therefore, the results of our electrophysiological studies are significant and establish that both the thermal instability and the channel gating defects of the ΔF508 CFTR can be suppressed by expressing the I507-ATC variant. Most significantly, the I507-ATC variant exhibits channel properties similar to wild-type CFTR. These results support our original hypothesis that the mRNA structure and the translation dynamics modify the folding state of the mutant CFTR. Our findings are also consistent with previously reported sSNP caused abnormalities in P-glycoprotein, another ABC-transporter with structural similarity to CFTR. These studies demonstrated that one specific sSNP (ATC to ATT) in exon 26 of the 209-kb-long gene resulted in altered ligand binding and transport properties of the transporter, and these differences were attributable to altered protein conformation (2). However, the multidrug resistance gene 1 (MDR1) gene variant did not contain a primary mutation confounding protein misfolding and ERAD. Our studies clearly demonstrate that the I507-ATT exacerbates the protein-folding defect of ΔF508 CFTR, and contributes to the severity of the mutation by altering channel properties.
The most striking differences between the I507-ATT- and ATC-encoded ΔF508 CFTR proteins are represented by thermal stability and channel-gating properties at the cell surface. Thermal instability of rescued human ΔF508 CFTR has been described by multiple laboratories (49, 72, 73). A conclusion from these studies was that cellular manipulations that rescue the native (I507-ATT) ΔF508 CFTR may not correct all folding defects (73). However, to properly interpret SCC-induced alterations in mRNA, protein conformation and effects on both thermal stability and channel-gating properties, it is necessary to consider fundamental aspects of ΔF508 CFTR function and cell surface turnover. CFTR is an ATP-gated ion channel, and opening of the pore is facilitated by the phosphorylation-dependent dimerization of the NBD domains. This is associated with conformational changes in the membrane spanning segments (74, 75). In addition, both cell surface inactivation and degradation of the native ΔF508 CFTR are known consequences of thermal unfolding (23, 71). Based on these observations, our results are consistent with a model in which I507-ATC-induced mRNA structural alterations lead to changes in protein folding and a product with enhanced stability.
Most notably, our single-channel measurements provide evidence that the I507-ATC codon change corrects the most significant functional ΔF508 CFTR defect. Single-channel tracings and the calculated NPo values are consistent with physiological NBD domain dimerization and/or conformation-dependent associations between the NBDs and transmembrane segments to maintain pore opening. In support of this hypothesis, previous studies have demonstrated that stabilization of ΔF508 NBD1 by second-site mutation improves channel gating (76, 77). A similar conclusion was reached by recent studies that applied human-mouse CFTR chimeras and identified regions that partially rescue and alter CFTR gating defects (78).
In summary, we identified that a previously unappreciated variation (I507ATC→ATT) in the human ΔF508 CFTR contributes to the severity of the most frequent mutation that leads to CF. Our previous findings regarding the mRNA structure differences and translation dynamic alterations with the experiments presented herein are consistent with the hypothesis that the choice of synonymous codons in certain genetic regions contributes to the expression and function of the gene product by altering mRNA and protein structure of the gene product. Further, we provide compelling evidence for the significance of silent nucleotide variations in the regulation of gene expression.
Acknowledgments
This publication was made possible by U.S. National Institutes of Health grants HL076587 (Z.B.), DK060065 (J.F.C.), 5U01ES015676 and 5R01HL031197 (A.L. and S.M.), GM095639 (J.C.K.), University of Alabama at Birmingham Center for Clinical and Translational Science UL1TR000165 (National Center for Advancing Translational Sciences and National Center for Research Resources; R.K. and E.L.), P30 DK07248Z, and North American Cystic Fibrosis Foundation grants Sorsch05xx0, CFF R464 (E.J.S.), and CFF DELUCA03G0 (J.C.K).
The authors thank Ms. Nancy Brissie for her outstanding technical support in organizing, maintaining, and preparing the cell clones used in these studies.
Footnotes
- cAMP
- cyclic adenosine monophosphate
- CF
- cystic fibrosis
- CFTR
- cystic fibrosis transmembrane conductance regulator
- COMT
- catechol-O-methyltransferase
- ERAD
- endoplasmic reticulum-associated degradation
- ES
- external solution
- F
- phenylalanine
- GAPDH
- glyceraldehyde 3-phosphate dehydrogenase
- HEK293
- human embryonic kidney 293
- HRP
- horseradish peroxidase
- I
- isoleucine
- IBMX
- 3-isobutyl-1-methylxanthine
- IS
- internal solution
- nsSNP
- non-synonymous single nucleotide polymorphism
- RIPA
- radioimmunoprecipitation assay
- SCC
- silent codon change
- SNP
- single nucleotide polymorphism
- sSNP
- synonymous single-nucleotide polymorphism
REFERENCES
- 1. Nackley A. G., Shabalina S. A., Tchivileva I. E., Satterfield K., Korchynskyi O., Makarov S. S., Maixner W., Diatchenko L. (2006) Human catechol-O-methyltransferase haplotypes modulate protein expression by altering mRNA secondary structure. Science 314, 1930–1933 [DOI] [PubMed] [Google Scholar]
- 2. Kimchi-Sarfaty C., Oh J. M., Kim I. W., Sauna Z. E., Calcagno A. M., Ambudkar S. V., Gottesman M. M. (2007) A “silent” polymorphism in the MDR1 gene changes substrate specificity. Science 315, 525–528 [DOI] [PubMed] [Google Scholar]
- 3. Bartoszewski R. A., Jablonsky M., Bartoszewska S., Stevenson L., Dai Q., Kappes J., Collawn J. F., Bebok Z. (2010) A synonymous single nucleotide polymorphism in DeltaF508 CFTR alters the secondary structure of the mRNA and the expression of the mutant protein. J. Biol. Chem. 285, 28741–28748 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Tsai C. J., Sauna Z. E., Kimchi-Sarfaty C., Ambudkar S. V., Gottesman M. M., Nussinov R. (2008) Synonymous mutations and ribosome stalling can lead to altered folding pathways and distinct minima. J. Mol. Biol. 383, 281–291 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Tsao D., Diatchenko L., Dokholyan N. V. (2011) Structural mechanism of S-adenosyl methionine binding to catechol O-methyltransferase. PLoS One 6, e24287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Tsao D., Shabalina S. A., Gauthier J., Dokholyan N. V., Diatchenko L. (2011) Disruptive mRNA folding increases translational efficiency of catechol-O-methyltransferase variant. Nucleic Acids Res. 39, 6201–6212 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Diatchenko L., Anderson A. D., Slade G. D., Fillingim R. B., Shabalina S. A., Higgins T. J., Sama S., Belfer I., Goldman D., Max M. B., Weir B. S., Maixner W. (2006) Three major haplotypes of the beta2 adrenergic receptor define psychological profile, blood pressure, and the risk for development of a common musculoskeletal pain disorder. Am. J. Med. Genet. B Neuropsych. Genet. 141B, 449–462 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Diatchenko L., Nackley A. G., Slade G. D., Bhalang K., Belfer I., Max M. B., Goldman D., Maixner W. (2006) Catechol-O-methyltransferase gene polymorphisms are associated with multiple pain-evoking stimuli. Pain 125, 216–224 [DOI] [PubMed] [Google Scholar]
- 9. Nackley A. G., Shabalina S. A., Lambert J. E., Conrad M. S., Gibson D. G., Spiridonov A. N., Satterfield S. K., Diatchenko L. (2009) Low enzymatic activity haplotypes of the human catechol-O-methyltransferase gene: enrichment for marker SNPs. PLoS One 4, e5237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Riordan J. R., Rommens J. M., Kerem B., Alon N., Rozmahel R., Grzelczak Z., Zielenski J., Lok S., Plavsic N., Chou J. L., Drumm M. L., Iannuzzi M. C., Collins F. C., Tsui L. C.1989) Identification of the cystic fibrosis gene: cloning and characterization of complementary DNA. Science 245, 1066–1073 [DOI] [PubMed] [Google Scholar]
- 11. Zielenski J., Rozmahel R., Bozon D., Kerem B., Grzelczak Z., Riordan J. R., Rommens J., Tsui L. C. (1991) Genomic DNA sequence of the cystic fibrosis transmembrane conductance regulator (CFTR) gene. Genomics 10, 214–228 [DOI] [PubMed] [Google Scholar]
- 12. Rommens J. M., Dho S., Bear C. E., Kartner N., Kennedy D., Riordan J. R., Tsui L. C., Foskett J. K. (1991) cAMP-inducible chloride conductance in mouse fibroblast lines stably expressing the human cystic fibrosis transmembrane conductance regulator. Proc. Natl. Acad. Sci. U. S. A. 88, 7500–7504 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Sheppard D. N., Welsh M. J. (1999) Structure and function of the CFTR chloride channel. Physiol. Rev. 79, S23–45 [DOI] [PubMed] [Google Scholar]
- 14. Kerem B., Rommens J. M., Buchanan J. A., Markiewicz D., Cox T. K., Chakravarti A., Buchwald M., Tsui L. C. (1989) Identification of the cystic fibrosis gene: genetic analysis. Science 245, 1073–1080 [DOI] [PubMed] [Google Scholar]
- 15. Rommens J. M., Iannuzzi M. C., Kerem B., Drumm M. L., Melmer G., Dean M., Rozmahel R., Cole J. L., Kennedy D., Hidaka N., Zsiga M., Buchwald M., Riordan J. R., Tsui L. C., Collins F.S.1989) Identification of the cystic fibrosis gene: chromosome walking and jumping. Science 245, 1059–1065 [DOI] [PubMed] [Google Scholar]
- 16. Jensen T. J., Loo M. A., Pind S., Williams D. B., Goldberg A. L., Riordan J. R. (1995) Multiple proteolytic systems, including the proteasome, contribute to CFTR processing. Cell 83, 129–135 [DOI] [PubMed] [Google Scholar]
- 17. Ward C. L., Omura S., Kopito R. R. (1995) Degradation of CFTR by the ubiquitin-proteasome pathway. Cell 83, 121–127 [DOI] [PubMed] [Google Scholar]
- 18. Brodsky J. L., Wojcikiewicz R. J. (2009) Substrate-specific mediators of ER associated degradation (ERAD). Curr. Opin. Cell Biol. 21, 516–521 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Rotin D., Staub O. (2011) Role of the ubiquitin system in regulating ion transport. Pflügers Arch. 461, 1–21 [DOI] [PubMed] [Google Scholar]
- 20. Dalemans W., Barbry P., Champigny G., Jallat S., Dott K., Dreyer D., Crystal R. G., Pavirani A., Lecocq J. P., Lazdunski M. (1991) Altered chloride ion channel kinetics associated with the delta F508 cystic fibrosis mutation. Nature 354, 526–528 [DOI] [PubMed] [Google Scholar]
- 21. Denning G. M., Anderson M. P., Amara J. F., Marshall J., Smith A. E., Welsh M. J. (1992) Processing of mutant cystic fibrosis transmembrane conductance regulator is temperature-sensitive. Nature 358, 761–764 [DOI] [PubMed] [Google Scholar]
- 22. Jurkuvenaite A., Chen L., Bartoszewski R., Goldstein R., Bebok Z., Matalon S., Collawn J. F. (2010) Functional stability of rescued delta F508 cystic fibrosis transmembrane conductance regulator in airway epithelial cells. Am. J. Respir. Cell Mol. Biol. 42, 363–372 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Sharma M., Benharouga M., Hu W., Lukacs G. L. (2001) Conformational and temperature-sensitive stability defects of the delta F508 cystic fibrosis transmembrane conductance regulator in post-endoplasmic reticulum compartments. J. Biol. Chem. 276, 8942–8950 [DOI] [PubMed] [Google Scholar]
- 24. Lukacs G. L., Verkman A. S. (2012) CFTR: folding, misfolding and correcting the DeltaF508 conformational defect. Trends Mol. Med. 18, 81–91 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Bartoszewski R., Brewer J. W., Rab A., Crossman D. K., Bartoszewska S., Kapoor N., Fuller C., Collawn J. F., Bebok Z. (2011) The unfolded protein response (UPR)-activated transcription factor X-box-binding protein 1 (XBP1) induces microRNA-346 expression that targets the human antigen peptide transporter 1 (TAP1) mRNA and governs immune regulatory genes. J. Biol. Chem. 286, 41862–41870 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Bartoszewski R., Rab A., Twitty G., Stevenson L., Fortenberry J., Piotrowski A., Dumanski J. P., Bebok Z. (2008) The mechanism of cystic fibrosis transmembrane conductance regulator transcriptional repression during the unfolded protein response. J. Biol. Chem. 283, 12154–12165 [DOI] [PubMed] [Google Scholar]
- 27. Fu L., Rab A., Tang L. P., Rowe S. M., Bebok Z., Collawn J. F. (2012) Dab2 is a key regulator of endocytosis and post-endocytic trafficking of the cystic fibrosis transmembrane conductance regulator. Biochem. J. 441, 633–643 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Tosoni K., Stobbart M., Cassidy D. M., Venerando A., Pagano M. A., Luz S., Amaral M. D., Kunzelmann K., Pinna L. A., Farinha C. M., Mehta A. (2013) CFTR mutations altering CFTR fragmentation. Biochem. J. 449, 295–305 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Loo T. W., Bartlett M. C., Clarke D. M. (2011) Benzbromarone stabilizes DeltaF508 CFTR at the cell surface. Biochemistry 50, 4393–4395 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Varga K., Goldstein R. F., Jurkuvenaite A., Chen L., Matalon S., Sorscher E. J., Bebok Z., Collawn J. F. (2008) Enhanced cell-surface stability of rescued DeltaF508 cystic fibrosis transmembrane conductance regulator (CFTR) by pharmacological chaperones. Biochem. J. 410, 555–564 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Varga K., Jurkuvenaite A., Wakefield J., Hong J. S., Guimbellot J. S., Venglarik C. J., Niraj A., Mazur M., Sorscher E. J., Collawn J. F., Bebok Z. (2004) Efficient intracellular processing of the endogenous cystic fibrosis transmembrane conductance regulator in epithelial cell lines. J. Biol. Chem. 279, 22578–22584 [DOI] [PubMed] [Google Scholar]
- 32. Londino J. D., Lazrak A., Jurkuvenaite A., Collawn J. F., Noah J. W., Matalon S. (2013) Influenza matrix protein 2 alters CFTR expression and function through its ion channel activity. Am. J. Physiol. Lung Cell. Mol. Physiol. 304, L582–L592 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Hamill O. P., Marty A., Neher E., Sakmann B., Sigworth F. J. (1981) Improved patch-clamp techniques for high-resolution current recording from cells and cell-free membrane patches. Pflügers Arch. 391, 85–100 [DOI] [PubMed] [Google Scholar]
- 34. Lazrak A., Iles K. E., Liu G., Noah D. L., Noah J. W., Matalon S. (2009) Influenza virus M2 protein inhibits epithelial sodium channels by increasing reactive oxygen species. FASEB J. 23, 3829–3842 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Lazrak A., Matalon S. (2003) cAMP-induced changes of apical membrane potentials of confluent H441 monolayers. Am. J. Physiol. Lung Cell. Mol. Physiol. 285, L443–L450 [DOI] [PubMed] [Google Scholar]
- 36. Lazrak A., Chen L., Jurkuvenaite A., Doran S. F., Liu G., Li Q., Lancaster J. R., Jr., Matalon S. (2012) Regulation of alveolar epithelial Na+ channels by ERK1/2 in chlorine-breathing mice. Am. J. Respir. Cell Mol. Biol. 46, 342–354 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Makrides S. C. (1996) Strategies for achieving high-level expression of genes in Escherichia coli. Microbiol. Rev. 60, 512–538 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Cuppens H., Lin W., Jaspers M., Costes B., Teng H., Vankeerberghen A., Jorissen M., Droogmans G., Reynaert I., Goossens M., Nilius B., Cassiman J. J. (1998) Polyvariant mutant cystic fibrosis transmembrane conductance regulator genes. The polymorphic (Tg)m locus explains the partial penetrance of the T5 polymorphism as a disease mutation. J. Clin. Invest. 101, 487–496 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Morral N., Bertranpetit J., Estivill X., Nunes V., Casals T., Gimenez J., Reis A., Varon-Mateeva R., Macek M., Jr., Kalaydjieva L., Angelicheva D., Dancheva R., Romeo G., Russo M. P., Garnerone S., Restagno G., Ferrari M., Magnani C., Claustres M., Desgeorges M., Schwartz M., Schwarz M., Dallapiccola B., Novelli G., Ferec C., de Arce M., Nemeti M., Kere J., Anvret M., Dahl N., Kadasi L. (1994) The origin of the major cystic fibrosis mutation (delta F508) in European populations. Nat. Genet. 7, 169–175 [DOI] [PubMed] [Google Scholar]
- 40. Moyer B. D., Loffing J., Schwiebert E. M., Loffing-Cueni D., Halpin P. A., Karlson K. H., Ismailov I. I., Guggino W. B., Langford G. M., Stanton B. A. (1998) Membrane trafficking of the cystic fibrosis gene product, cystic fibrosis transmembrane conductance regulator, tagged with green fluorescent protein in madin-darby canine kidney cells. J. Biol. Chem. 273, 21759–21768 [DOI] [PubMed] [Google Scholar]
- 41. Mathews D. H., Turner D. H., Zuker M. (2007) RNA secondary structure prediction. In Current Protocols in Nucleic Acid Chemistry (Beaucage S. I., ed) pp. 11.2.1–11.2.17, John Wiley & Sons; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Zuker M. (2000) Calculating nucleic acid secondary structure. Curr. Op. Struct. Biol. 10, 303–310 [DOI] [PubMed] [Google Scholar]
- 43. Zuker M., Jacobson A. B. (1998) Using reliability information to annotate RNA secondary structures. RNA 4, 669–679 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Zuker M., Jacobson A. B. (1995) “Well-determined” regions in RNA secondary structure prediction: analysis of small subunit ribosomal RNA. Nucleic Acids Res. 23, 2791–2798 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Kleizen B., van Vlijmen T., de Jonge H. R., Braakman I. (2005) Folding of CFTR is predominantly cotranslational. Mol. Cell 20, 277–287 [DOI] [PubMed] [Google Scholar]
- 46. Meacham G. C., Patterson C., Zhang W., Younger J. M., Cyr D. M. (2001) The Hsc70 co-chaperone CHIP targets immature CFTR for proteasomal degradation. Nat. Cell Biol. 3, 100–105 [DOI] [PubMed] [Google Scholar]
- 47. Sato S., Ward C. L., Kopito R. R. (1998) Cotranslational ubiquitination of cystic fibrosis transmembrane conductance regulator in vitro. J. Biol. Chem. 273, 7189–7192 [DOI] [PubMed] [Google Scholar]
- 48. Heda G. D., Tanwani M., Marino C. R. (2001) The Delta F508 mutation shortens the biochemical half-life of plasma membrane CFTR in polarized epithelial cells. Am. J. Physiol. Cell Physiol. 280, C166–C174 [DOI] [PubMed] [Google Scholar]
- 49. Wang W., Okeyo G. O., Tao B., Hong J. S., Kirk K. L. (2011) Thermally unstable gating of the most common cystic fibrosis mutant channel (DeltaF508): “rescue” by suppressor mutations in nucleotide binding domain 1 and by constitutive mutations in the cytosolic loops. J. Biol. Chem. 286, 41937–41948 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Ma T., Thiagarajah J. R., Yang H., Sonawane N. D., Folli C., Galietta L. J., Verkman A. S. (2002) Thiazolidinone CFTR inhibitor identified by high-throughput screening blocks cholera toxin-induced intestinal fluid secretion. J. Clin. Invest. 110, 1651–1658 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Muanprasat C., Sonawane N. D., Salinas D., Taddei A., Galietta L. J., Verkman A. S. (2004) Discovery of glycine hydrazide pore-occluding CFTR inhibitors: mechanism, structure-activity analysis, and in vivo efficacy. J. Gen. Physiol. 124, 125–137 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Cai Z., Scott-Ward T. S., Sheppard D. N. (2003) Voltage-dependent gating of the cystic fibrosis transmembrane conductance regulator Cl- channel. J. Gen. Physiol. 122, 605–620 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Gray M. A., Plant S., Argent B. E. (1993) cAMP-regulated whole cell chloride currents in pancreatic duct cells. Am. J. Physiol. 264, C591–602 [DOI] [PubMed] [Google Scholar]
- 54. Howard M., Jiang X., Stolz D. B., Hill W. G., Johnson J. A., Watkins S. C., Frizzell R. A., Bruton C. M., Robbins P. D., Weisz O. A. (2000) Forskolin-induced apical membrane insertion of virally expressed, epitope-tagged CFTR in polarized MDCK cells. Am. J. Physiol. Cell Physiol. 279, C375–C382 [DOI] [PubMed] [Google Scholar]
- 55. Cui L., Aleksandrov L., Hou Y. X., Gentzsch M., Chen J. H., Riordan J. R., Aleksandrov A. A. (2006) The role of cystic fibrosis transmembrane conductance regulator phenylalanine 508 side chain in ion channel gating. J. Physiol. 572, 347–358 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Lazrak A., Jurkuvenaite A., Chen L., Keeling K. M., Collawn J. F., Bedwell D. M., Matalon S. (2011) Enhancement of alveolar epithelial sodium channel activity with decreased cystic fibrosis transmembrane conductance regulator expression in mouse lung. Am. J. Physiol. Lung Cell. Mol. Physiol. 301, L557–L567 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Sauna Z. E., Kimchi-Sarfaty C. (2011) Understanding the contribution of synonymous mutations to human disease. Nat. Rev. Genet. 12, 683–691 [DOI] [PubMed] [Google Scholar]
- 58. Chen R., Davydov E. V., Sirota M., Butte A. J. (2010) Non-synonymous and synonymous coding SNPs show similar likelihood and effect size of human disease association. PLoS One 5, e13574. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Diatchenko L., Nackley A. G., Slade G. D., Fillingim R. B., Maixner W. (2006) Idiopathic pain disorders–pathways of vulnerability. Pain 123, 226–230 [DOI] [PubMed] [Google Scholar]
- 60. Diatchenko L., Slade G. D., Nackley A. G., Bhalang K., Sigurdsson A., Belfer I., Goldman D., Xu K., Shabalina S. A., Shagin D., Max M. B., Makarov S. S., Maixner W. (2005) Genetic basis for individual variations in pain perception and the development of a chronic pain condition. Hum. Mol. Genet. 14, 135–143 [DOI] [PubMed] [Google Scholar]
- 61. Tseng S. C., Kimchi-Sarfaty C. (2011) SNPs in ADAMTS13. Pharmacogenomics 12, 1147–1160 [DOI] [PubMed] [Google Scholar]
- 62. Plotkin J. B., Kudla G. (2011) Synonymous but not the same: the causes and consequences of codon bias. Nat. Rev. Genet. 12, 32–42 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Sharp P. M., Emery L. R., Zeng K. (2010) Forces that influence the evolution of codon bias. Philo. Trans. R. Soc. Lond. B 365, 1203–1212 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Sharp P. M., Li W. H. (1987) The codon Adaptation Index–a measure of directional synonymous codon usage bias, and its potential applications. Nucleic Acids Res. 15, 1281–1295 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Younger J. M., Chen L., Ren H. Y., Rosser M. F., Turnbull E. L., Fan C. Y., Patterson C., Cyr D. M. (2006) Sequential quality-control checkpoints triage misfolded cystic fibrosis transmembrane conductance regulator. Cell 126, 571–582 [DOI] [PubMed] [Google Scholar]
- 66. Thibodeau P. H., Richardson J. M., 3rd, Wang W., Millen L., Watson J., Mendoza J. L., Du K., Fischman S., Senderowitz H., Lukacs G. L., Kirk K., Thomas P. J. (2010) The cystic fibrosis-causing mutation deltaF508 affects multiple steps in cystic fibrosis transmembrane conductance regulator biogenesis. J. Biol. Chem. 285, 35825–35835 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Thibodeau P. H., Brautigam C. A., Machius M., Thomas P. J. (2005) Side chain and backbone contributions of Phe508 to CFTR folding. Nat. Struct. Mol. Biol. 12, 10–16 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Morito D., Hirao K., Oda Y., Hosokawa N., Tokunaga F., Cyr D. M., Tanaka K., Iwai K., Nagata K. (2008) Gp78 cooperates with RMA1 in endoplasmic reticulum-associated degradation of CFTRDeltaF508. Mol. Biol. Cell 19, 1328–1336 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Henderson M. J., Vij N., Zeitlin P. L. (2010) Ubiquitin C-terminal hydrolase-L1 protects cystic fibrosis transmembrane conductance regulator from early stages of proteasomal degradation. J. Biol. Chem. 285, 11314–11325 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Okiyoneda T., Barriere H., Bagdany M., Rabeh W. M., Du K., Hohfeld J., Young J. C., Lukacs G. L. (2010) Peripheral protein quality control removes unfolded CFTR from the plasma membrane. Science 329, 805–810 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Sharma M., Pampinella F., Nemes C., Benharouga M., So J., Du K., Bache K. G., Papsin B., Zerangue N., Stenmark H., Lukacs G. L. (2004) Misfolding diverts CFTR from recycling to degradation: quality control at early endosomes. J. Cell Biol. 164, 923–933 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Liu X., O'Donnell N., Landstrom A., Skach W. R., Dawson D. C. (2012) Thermal instability of DeltaF508 cystic fibrosis transmembrane conductance regulator (CFTR) channel function: protection by single suppressor mutations and inhibiting channel activity. Biochemistry 51, 5113–5124 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Aleksandrov A. A., Kota P., Cui L., Jensen T., Alekseev A. E., Reyes S., He L., Gentzsch M., Aleksandrov L. A., Dokholyan N. V., Riordan J. R. (2012) Allosteric modulation balances thermodynamic stability and restores function of DeltaF508 CFTR. J. Mol. Biol. 419, 41–60 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Vergani P., Lockless S. W., Nairn A. C., Gadsby D. C. (2005) CFTR channel opening by ATP-driven tight dimerization of its nucleotide-binding domains. Nature 433, 876–880 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Kirk K. L., Wang W. (2011) A unified view of cystic fibrosis transmembrane conductance regulator (CFTR) gating: combining the allosterism of a ligand-gated channel with the enzymatic activity of an ATP-binding cassette (ABC) transporter. J. Biol. Chem. 286, 12813–12819 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. Jih K. Y., Li M., Hwang T. C., Bompadre S. G. (2011) The most common cystic fibrosis-associated mutation destabilizes the dimeric state of the nucleotide-binding domains of CFTR. J. Physiol. 589, 2719–2731 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77. Roxo-Rosa M., Xu Z., Schmidt A., Neto M., Cai Z., Soares C. M., Sheppard D. N., Amaral M. D. (2006) Revertant mutants G550E and 4RK rescue cystic fibrosis mutants in the first nucleotide-binding domain of CFTR by different mechanisms. Proc. Natl. Acad. Sci. U. S. A. 103, 17891–17896 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78. Dong Q., Ostedgaard L. S., Rogers C., Vermeer D. W., Zhang Y., Welsh M. J. (2012) Human-mouse cystic fibrosis transmembrane conductance regulator (CFTR) chimeras identify regions that partially rescue CFTR-DeltaF508 processing and alter its gating defect. Proc. Natl. Acad. Sci. U. S. A. 109, 917–922 [DOI] [PMC free article] [PubMed] [Google Scholar]