

Abstract
One of the factors determining a high degree of heterogeneity in the HIV population is recombination-based variation, which leads to the emergence of the virus variants with a mosaic genome. An example is CRF63_02A1, an HIV-1 variant currently spreading in the Siberian region of Russia. To prove that this HIV-1 variant is a new circulating recombinant form that had emerged as a result of repeated recombination between CRF02_AG and subtype A, we have isolated seven full-length HIV genomes and theoretically analyzed them, that is, reconstructed the phylogenetic relationships, determined recombination breakpoints and regions, and compared them with the regions known for CRF02_AG.
Introduction
Human immunodeficiency virus has one of the highest evolutionary rates among the viruses, which is determined by specific features of its replication. Recombination is among these specific features; it allows for the emergence of the virus variants with the genome composed of genomic regions of various HIV genetic strains (subtypes). According to accepted international HIV nomenclature,1 such recombinant viruses with limited spreading (one individual or one epidemic chain) are referred to as unique recombinant forms (URFs), while wider spread viruses displaying a stable genetic structure are referred to as circulating recombinant forms (CRFs). Currently, more than 60 CRFs are known. In the majority of cases, recombination involves the main nine HIV-1 subtypes of group M, which gives rise to the primary circulating recombinant forms. However, as early as 2002, a recombinant HIV-1 form with the genome carrying certain regions of CRF01_AE and other virus subtypes was described.2 In this case, we can discuss the next step in the evolution of this virus, namely, the emergence of second-generation recombinants produced via recombination between a CRF and major HIV subtypes. Taking into account the fact that recombination during virus replication leads to replacement of mutation clusters rather than the emergence of individual mutations, the effect of such substitutions on the function of a protein may correspondingly play a key role and requires further studies.
The initially existing regional specificity in the circulation of HIV genetic variants has recently undergone considerable changes. Ever increasing population migration enhances transfer of endemic HIV subtypes to new regions, growth in HIV abundance, and coexistence of several HIV variants in the same area. In turn, these events enhance recombination between circulating HIV variants, thereby creating a favorable situation for a stable increase in the genetic diversity of spreading virus variants.3–5
Recently, the HIV epidemic in the Russian Federation has displayed a stable trend of deterioration unlike that in the majority of developed countries. According to the official data, over 720,000 HIV-infected persons were recorded in this country as of the beginning of 2013.6 The situation in Russia is also complicated by a significant increase in labor migrants, including those from regions with a high rate of HIV as well as virus subtypes untypical of the Russian Federation, such as Ukraine, Uzbekistan, Tajikistan, and other countries of Central Asia.7,8 Until now, the epidemiological situation in most Russian regions has been characterized by the prevalence of HIV-1 subtype A (over 90%) and the stable circulation of subtype B (4–6%).9,10 The other genetic variants of the virus either remain at a low level of abundance in local epidemics or are sporadically recorded without further spreading.11 However, genetic monitoring of the HIV variants circulating in the Siberian region has shown that recombinant HIV CRF02_AG variants, not characteristic of this area, have become considerably more abundant in Novosibirsk.
In 2011, a complete genome of one HIV isolate (10.RU.6637) was determined; this isolate belongs to the most abundant cluster of “Siberian” CRF02_AG variants. Analysis of the 10.RU.6637 genome has demonstrated that the virus in question is a new recombinant form of CRF63_02A1, resulting from a repeated recombination between CRF02_AG and HIV-1 subtype A.12 In 2011–2012, we isolated HIV-1 CRF63_02A1 variants, which fell into the same cluster with the “Siberian” variants in the phylogenetic tree constructed based on the pol gene and recovered in Chechnya, Rostov-on-Don, Novokuznetsk, and Kemerovo. Our phylogenetic analysis of nucleotide sequences of the 02_AG variants deposited with the GenBank by other research teams has shown that infections with the HIV-1 CRF63_02A1 recombinant form have also been recorded in the city of Blagoveshchensk (Kazakhstan) and in Kyrgyzstan.13
To study the structure of the complete HIV-1 genomes, we have selected 13 blood plasma samples of patients living in different regions of Russia and countries of Central Asia and infected with subtype A (three samples) as well as with recombinant HIV-1 CRF63_02A1 variants (seven samples) and CRF02_AG (three samples) variants.
Materials and Methods
RNA was extracted from the blood plasma; the virus-specific fragments were isolated and sequenced as earlier described.12 For theoretical analysis, reference nucleotide sequences of the main HIV-1 subtypes and the recombinant forms of 02_AG and 01_AE were selected using the Los Alamos HIV Sequence Database (www.hiv.lanl.gov/). Multiple alignment of the nucleotide sequences was constructed with the Muscle and T-Coffee programs and edited with BioEdit. The phylogenetic trees were built using PhyML v. 3.0 and Mega 5; the optimal model for calculating evolutionary distances was selected with FindModel (www.hiv.lanl.gov/content/sequence/findmodel/). Phylogenetic analysis was conducted using two methods—distant neighbor joining (NJ) and maximum likelihood (ML). The statistical significance of phylogenetic tree topologies was estimated using bootstrap analysis; the nucleotide sequence of HIV-1 subtype O (GenBank accession number AJ302646) was used as an outgroup. Two searching approaches, implemented in the programs jpHMM14 and Recco,15 were utilized to detect the possible recombination events between different subtypes.
Results and Discussion
Theoretical analysis involved the 13 full-genome HIV-1 sequences determined in this work and the earlier sequenced HIV-1 10.RU.6637 (JN230353) (Table 1). In total, 11 determined nucleotide sequences of recombinant form AG and three complete genomes of Russian HIV-1 subtype A isolates were used for phylogenetic analysis. The ML phylogenetic tree for the genomes studied in this work and selected HIV-1 reference sequences are shown in Fig. 1. The phylogenetic branch of HIV-1 CRF02_AG may be distinctly divided into two subbranches. One subbranch (Fig. 1, I) contains all the HIV-1 isolates characteristic of African countries as well as the HIV-1 strain IbNG, reference for the circulating recombinant form CRF02_AG. The other subbranch (Fig. 1, II) contains all the HIV-1 genomic sequences determined in this work and one virus isolate from Uzbekistan. Such a topology of the studied genomes with a bootstrap value exceeding 97% is also observed in the NJ tree (data not shown). In turn, two HIV-1 clusters are distinguishable in subbranch II. The first cluster contains an Uzbekistan HIV-1 sequence (GenBank accession number AY829214), which clusters together with the studied genomes from Novosibirsk (10.RU.6509), Uzbekistan (11.RU.6939), and an isolate from Tajikistan (11.RU.6900).
Table 1.
Epidemiological Data for the Patients Whose HIV-1 Isolates Were Used for Near Full-Length Sequencing and Analysis
| No. | HIV-1 genome code | Region of infection | Date of blood sampling | HIV-1 subtype |
|---|---|---|---|---|
| 1 | 10.RU6637 | Russia (Novosibirsk) | September 07, 2010 | 02_AG/A |
| 2 | 10.RU6649 | Russia (Novosibirsk) | September 07, 2010 | 02_AG/A |
| 3 | 11.RU.18n | Russia (Novosibirsk) | April 01, 2011 | 02_AG/A |
| 4 | 10.RU.6829 | Russia (Novosibirsk) | October 19, 2010 | 02_AG/A |
| 5 | 10.RU.5983 | Russia (Novosibirsk) | May 28, 2010 | 02_AG/A |
| 6 | 09.RU.4829 | Russia (Novosibirsk) | December 08, 2009 | 02_AG/A |
| 7 | 10.RU.6366 | Russia (Novosibirsk) | August 01, 2010 | 02_AG/A |
| 8 | 12.RU.15r | Russia (Rostov) | March 01, 2012 | 02_AG/A |
| 9 | 11.RU.6939 | Uzbekistan (Tashkent) | August 23, 2011 | 02_AG |
| 10 | 11.RU.6900 | Tajikistan | July 22, 2011 | 02_AG |
| 11 | 10.RU.6509 | Russia (Novosibirsk) | August 11, 2010 | 02_AG |
| 12 | 10.RU.6792 | Russia (Novosibirsk) | September 21, 2010 | A |
| 13 | 10.RU.6617 | Russia (Samara) | September 06, 10 | A |
| 14 | 11.RU.6950 | Russia (Novorossiysk) | August 16, 2011 | A |
FIG. 1.

Maximum likelihood (ML) phylogenetic tree of HIV-1 nucleotide sequences (the 14 Russian isolates studied in this work are boldfaced).
The second cluster contains an earlier described isolate, 10.RU.6637 (GenBank accession number JN230353) and the seven studied HIV-1 02_AG genomes, namely, five HIV-1 isolates recovered from Novosibirsk inhabitants, one from Rostov, and one from Novokuznetsk. The genomes of the recombinant HIV-1 02_AG form were additionally partitioned into two groups according to the clustering of the phylogenetic tree: one group contained the genomes identical to those from Central Asian countries (designated as AG1) and the other contained the genomes from Russian cities (group AG2). The genetic distances within the groups of HIV-1 genetic variants and between these groups were determined for the recombinant HIV-1 02_AG variants and subtype A. Analysis of these genetic distances within group AG1 has shown that they are comparable to the distances for subtype A; as for the second group, AG2, these distances appear to be 2-fold shorter. Another specific feature is the distance between HIV-1 subtype A and group AG2. This distance appears shorter (dA – AG2=0.1449) than that between subtype A and group AG1 (dA – AG1=0.1848), thereby favoring the hypothesis on recombination between the viruses of subtype A and the CRF02_AG of Asian type (see Table 2).
Table 2.
Evolutionary Distances Between the HIV-1 Genomes Belonging to Different Genetic Clusters
| A | AG1 | AG2 | |
|---|---|---|---|
| A | 0.0533 | 0.1848 | 0.1449 |
| AG1 | 0.0519 | 0.0699 | |
| AG2 | 0.0222 |
To prove that the 11 studied Russian recombinant genomes of HIV-1 02_AG had different structures, it was necessary to determine the recombination breakpoints for each genome and to compare them with the structure of a known genome, the classical HIV-1 CRF02_AG. Several methods allow this task to be implemented. To increase the reliability of the results, it is better to use several programs. The first step here was determination of the recombination breakpoints in the analyzed genomes in the presence of the sequences of HIV-1 subtypes A and G. The program jpHMM demonstrated differences in the detected recombination breakpoints from the standard HIV-1 CRF02_AG breakpoints for eight genomes belonging to the Russian cluster (Fig. 2).
FIG. 2.

Scheme for arrangement of recombination regions in the genomes belonging to (I, upper scheme) the Central Asian cluster of HIV-1 recombinant form CRF02_AG and (II, lower scheme) and the Novosibirsk cluster of HIV-1 02_AG.
Three HIV genomes (10.RU.6366, 12.RU.15r, and 10.RU.6829) “lacked” the recombination region homologous to subtype G localized to between recombination breakpoints 3 and 4. When involving in the analysis not only the exact values for recombination breakpoints, but also the information about recombination intervals and an uncertain region, the region in these genomes localized to between recombination breakpoints 3 and 4 appears to be the uncertain region, that is, the region where the a posteriori probability value for the predicted subtype is lower than the threshold value.
The accuracy for the detection of recombination breakpoints in the HIV genome provided by jpHMM may vary for different regions. Analysis of standard deviations has shown that these values are minimal and do not exceed 2 for breakpoints 1, 4, 6, 8, and 10 and are maximal for breakpoints 5 and 7. For the remaining three recombination breakpoints, the fluctuations are caused by solitary insignificant deviations in individual viral genomes (Fig. 3).
FIG. 3.

Assessment of standard deviations for 10 recombination breakpoints in the analyzed HIV-1 genomes determined with jpHMM. For convenience, the minimal value for the HIV-1 genomes was subtracted from each breakpoint.
The data on standard deviations for 10 recombination breakpoints correlate with the jpHMM-computed lengths for the recombination intervals (Kendall's rank coefficient, 0.966 and Spearman's rank coefficient, 0.987), which allows for the estimation of prediction accuracy for different positions of recombination breakpoints. The reason for such a variation in deviations may be associated with the specific features of recombining regions. The program Recco was selected as an alternative to jpHMM in performing recombination analysis. The jpHMM data for recombination breakpoints were confirmed by the Recco program. However, the recombination region absent in three of the HIV genomes studied according to jpHMM analysis appeared present in these genomes when using Recco computations.
According to the hypothesis on recombination between HIV-1 subtype A and CRF02_AG,12 it was necessary to confirm the presence of both virus ancestors in the genome structure of the new HIV-1 recombinant form. For this purpose, the classical HIV-1 CRF02_AG sequences were included into the multiple alignment. Subsequent analysis has shown that the eight examined HIV-1 02_AG genomes contain two to five inserts homologous to subtype A, while the remaining genome part of these eight HIV variants retained the homology to the recombinant HIV-1 form CRF02_AG. Noteworthy is the absence of some recombination regions for part of the genomes. Figure 4 shows the arrangement of recombination regions.
FIG. 4.

Scheme for arrangement of the recombination regions in the analyzed HIV-1 02_AG genomes: (A) recombination regions of the new recombinant form HIV-1 CRF63_02A1 and (B) recombination regions of the HIV-1 CRF02_AG.
The genome of 10.RU.6649 contains the least number of recombination regions, displaying only five of 11 regions; two regions with the homology to subtype A and one region similar to the HIV-1 AG sequence are absent. Five HIV genomes lack the region localized between breakpoints 4 and 5, i.e., region V (Fig. 4A), homologous to the HIV-1 variant CRF02_AG. The 12.RU.15r lacks region X, which is similar to subtype A. As in the case with region IV in the analysis of recombination breakpoints without involving the HIV-1 recombinant form CRF02_AG in the multiple alignment, all these “lost” regions are ambiguity areas, where statistical significance for a correct attribution to a particular HIV-1 subtype is below the corresponding allowed level. Similar to the first case, the absence of recombination regions for some genomes is not validated by the Recco program.
As the third approach confirming a unique structure of this new recombinant form, phylogenetic trees were constructed for some recombination regions of HIV genomes. When determining the coordinates of viral genomic regions for construction of phylogenetic trees, only the alignment positions that according to both programs (jpHMM and Recco) were not the uncertain region and fell beyond recombination intervals were taken into account. First, it was necessary to comprehensively study the genomic regions in HIV-1 variants that displayed homology to HIV-1 subtype A according to these programs, namely, regions III, V, VII, X, and XIII (Fig. 4). In addition, the question concerning the recombination regions absent in some genomes still remains open. These regions are region IV for three genomes of the analyzed virus variants, which displays homology to subtype G, and region VI for five HIV-1 genomes, which in the new circulating recombinant HIV-1 from CRF02_AG/A displays homology to HIV-1 CRF02_AG in the majority of analyzed genomes. The unrooted trees are shown in Fig. 5.
FIG. 5.

Phylogenetic trees for HIV-1 genomic regions belonging to different subtypes: region III, region IV, region V, region VI, region VII, region X, and region XIII. A black circle encloses the CRF63_02A1 genomes. The samples used included major subtypes A, B, and G as well as HIV-1 CRF02_AG sequences.
According to the constructed trees, five regions homologous to subtype A are present in all eight recombinant HIV-1 genomes for which additional recombination is assumed, including the earlier studied isolate 10.RU.6637, while the entire remaining genome part of the studied isolates retains homology to HIV-1 CRF02_AG. The trees for the questionable HIV-1 genomic regions, which according to previous results lacked some recombination regions, refuted this fact. Unfortunately, the small size of regions VII and X makes it difficult to establish a reliable conclusion concerning their presence in the genomes; however, all eight genomes cluster with Russian HIV-1 subtype A in the phylogenetic trees constructed for the genetic sequences of these regions.
These results of recombination analysis suggest that the eight recombinant HIV-1 genomes of Russian origin (10.RU.6637, 10.RU.6649, 11.RU.18n, 10.RU.6829, 10.RU.5983, 09.RU.4829, 10.RU.6366, and 12.RU.15r) display a genetic structure distinct from that of HIV-1 CRF02_AG, which appears as the presence of additional regions with a close similarity to HIV-1 subtype A. In total, five such regions have been found. Thus, the structure of the new recombinant form that emerged via recombination between HIV-1 subtype A and CRF02_AG may be represented as shown in Fig. 6.
FIG. 6.
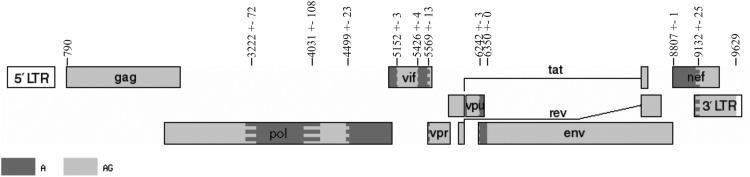
Scheme for the arrangement of recombination regions of the new circulating recombinant form HIV-1 CRF63_02A1.
Accession Numbers
The sequences described in this article were submitted to the GenBank Nucleotide Sequence Database under accession numbers JX500694–JX500706.
Acknowledgment
The work was partially funded by the Ministry of Education and Science of the Russian Federation under the program for support of leading scientific schools (grant NSh-2996.2012.4).
Author Disclosure Statement
No competing financial interests exist.
References
- 1.Robertson DL, Anderson JA, Bradac JA, et al. : HIV-1 nomenclature proposal. Science 2000;288:55–56 [DOI] [PubMed] [Google Scholar]
- 2.Wilbe K, Casper C, Albert J, and Leitner T: Identification of two CRF11-cpx genomes and two preliminary representatives of a new circulating recombinant form (CRF13-cpx) of HIV type 1 in Cameroon. AIDS Res Hum Retroviruses 2002;18:849–856 [DOI] [PubMed] [Google Scholar]
- 3.Parczewski M, Leszczyszyn-Pynka M, Bander D, Urbanska A, and Boroń-Kaczmarska A: HIV-1 subtype D infections among caucasians from northwestern Poland—phylogenetic and clinical analysis. PLoS One 2012;7: e31674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Galimand J, Frange P, Rouzioux C, Deveau C, Avettand-Fenoël V, Ghosn J, Lascoux C, Goujard C, Meyer L, and Chaix ML: Evidence of HIV type 1 complex and second generation recombinant strains among patients infected in 1997–2007 in France: ANRS CO06 PRIMO Cohort. AIDS Res Hum Retroviruses 2010;26:645–651 [DOI] [PubMed] [Google Scholar]
- 5.Thomson MM. and Nájera R: Increasing HIV-1 genetic diversity in Europe. J Infect Dis 2007;196:1120–1124 [DOI] [PubMed] [Google Scholar]
- 6.Joint United Nations Programme on HIV/AIDS: UNAIDS report on the global AIDS epidemic 2012/UNAIDS, 2012
- 7.Saad MD, Aliev Q, Botros BAM, et al. : Genetic forms of HIV type 1 in the former Soviet Union dominate the epidemic in Azerbaijan. AIDS Res Human Retroviruses 2006;22:796–800 [DOI] [PubMed] [Google Scholar]
- 8.Carr J, Nadai Y, Eyzaguirre L, et al. : Outbreak of a West African recombinant of HIV-1 in Tashkent, Uzbekistan. J Acquir Immune Defic Syndr 2005;39:570–575 [PubMed] [Google Scholar]
- 9.Bobkov AF, Kazennova EV, Selimova LM, et al. : Temporal trends in the HIV-1 epidemic in Russia: Predominance of subtype A. J Med Virol 2004;74:191–196 [DOI] [PubMed] [Google Scholar]
- 10.Thomson MM, Vázquez de Parga E, Vinogradova A, et al. : New insights into the origin of the HIV type 1 subtype A epidemic in former Soviet Union countries derived from sequence analyses of preepidemically transmitted viruses. AIDS Res Hum Retroviruses 2007;23:1599–1604 [DOI] [PubMed] [Google Scholar]
- 11.Roudinskii NI, Sukhanova AL, Kazennova EV, et al. : Diversity of human immunodeficiency virus type 1 subtype A and CRF03_AB protease in Eastern Europe: Selection of the V77I variant and its rapid spread in injecting drug user populations. J Virol 2004;78:11276–11287 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Baryshev PB, Bogachev VV, and Gashnikova NM: Genetic characterization of an isolate of HIV type 1 AG recombinant form circulating in Siberia, Russia. Arch Virol 2012;157:2335–2341 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Laga VIu, Kazennova EV, Vasil'ev AV, Lapovok IA, Ismailova A, Beĭsheeva N, Asybalieva N, and Bobkova MR: Molecular-genetic characterization of the HIV-1 variants abundant in Kirghizia. Vopr Virusol 2012;57:26–32 [PubMed] [Google Scholar]
- 14.Schultz A-K, Zhang M, Bulla I, Leitner T, Korber B, Morgenstern B, and Stanke M: jpHMM: Improving the reliability of recombination prediction in HIV-1. Nucleic Acids Res 2009;37:W647–W651 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Maydt J. and Lengauer T: Recco: Recombination analysis using cost optimization. Bioinformatics 2006;22:1064–1071 [DOI] [PubMed] [Google Scholar]