

Abstract
The CXCR4/SDF1 axis participates in various cellular processes, including cell migration, which is essential for skeletal muscle repair. Although increasing evidence has confirmed the role of CXCR4/SDF1 in embryonic muscle development, the function of this pathway during adult myogenesis remains to be fully elucidated. In addition, a role for CXCR4 signaling in muscle maintenance and repair has only recently emerged. Here, we have demonstrated that CXCR4 and stromal cell-derived factor-1 (SDF1) are up-regulated in injured muscle, suggesting their involvement in the repair process. In addition, we found that notexin-damaged muscles showed delayed muscle regeneration on treatment with CXCR4 agonist (AMD3100). Accordingly, small-interfering RNA-mediated silencing of SDF1 or CXCR4 in injured muscles impaired muscle regeneration, whereas the addition of SDF1 ligand accelerated repair. Furthermore, we identified that CXCR4/SDF1-regulated muscle repair was dependent on matrix metalloproteinase-10 (MMP-10) activity. Thus, our findings support a model in which MMP-10 activity modulates CXCR4/SDF1 signaling, which is essential for efficient skeletal muscle regeneration.
Introduction
Skeletal muscle possesses remarkable regenerative potential, which relies on the activation of adult muscle stem cells known as satellite cells [1,2]. These cells usually remain in a quiescent state within their niche beneath the basal lamina of myofibers [3]. However, in response to injury or stress, satellite cells become activated, proliferate, differentiate, and fuse with each other (or with existing myofibers) to generate new muscle [4]. In order to restore normal tissue architecture, satellite cells from distant fibers should cross the basal membranes of their resident myofibers and migrate toward damaged sites [1,2,5]. These key processes, which are required for efficient muscle repair, are consistent with the notion that satellite cells have the ability to respond to chemokines and actively express/secrete matrix metalloproteinases (MMPs).
Chemokines constitute a family of structurally related low-molecular-weight cytokines that play fundamental roles in cellular trafficking and migration [6]. Stromal cell-derived factor-1 (SDF1) belongs to the CXC chemokine subfamily and was identified as a pre-B cell stimulatory factor [7]. SDF1 is known to be a chemoattractant for stem/progenitor cells [8,9] and mainly functions as a ligand for the G protein-coupled receptor, CXCR4 [10]. Notably, SDF1-mediated CXCR4 activation is crucial in the regulation of various biological processes involving cell motility, chemotactic response, cell adhesion, gene transcription, cell proliferation, and survival [11–15]. In addition, SDF1 seems to play a beneficial role in tissue repair after myocardial infarction [16–18], vascular disease [19,20], and fracture healing [21]; however, it may also contribute to cancer progression [22,23]. The discovery of SDF1 and CXCR4 expression in skeletal muscle has led to the study of their potential roles in embryonic myogenesis [24]. In fact, muscle progenitor cells, which express CXCR4, have been shown to migrate toward SDF1 during the generation of new myofibers in limb muscles [25]. Accordingly, null mice for CXCR4 have a reduced number of satellite cells, leading to muscular deficiencies in the embryo [26]. However, the function of the CXCR4/SDF1 axis during adult myogenesis remains to be fully investigated [27–30], and its role in muscle repair has only recently begun to be characterized [31].
Myogenic cells produce and secrete MMPs, a family of zinc endopeptidases that degrade components of the extracellular matrix (ECM) and participate in diverse cellular processes [32–34]. In addition, the CXCR4/SDF1 signaling axis and MMPs have been shown to participate in some of the same cellular processes, including migration of muscle-derived cells during muscle repair [5,35,36]. This might suggest that these two systems functionally interact. In this regard, MMP-10 (stromelysin-2), which belongs to the stromelysin family of MMPs, might be of primary interest. Although MMP-10 has been shown to be involved in various pathologies, including cancer and vascular disease [37–39], we recently described its pivotal role in muscle maintenance and repair. In fact, MMP-10 deficiency delays muscle repair after injury and deteriorates the dystrophic phenotype of mdx mice. Furthermore, the administration of recombinant human MMP-10 (rhMMP-10) improves muscular regeneration after damage [40].
In the present study, we demonstrated that the CXCR4/SDF1 axis is required for efficient muscle repair. Accordingly, we observed that the CXCR4 agonist, AMD3100, delayed skeletal muscle regeneration. In addition, small-interfering RNA (siRNA)-mediated silencing of SDF1 or CXCR4 in injured muscle impaired muscle regeneration, whereas the addition of SDF1 ligand accelerated the repair process. In addition, our in vitro data revealed that the CXCR4/SDF1 axis functions mechanistically at the level of satellite cell-derived myoblasts, which are directly responsible for muscle repair. Furthermore, we identified that MMP-10 might mediate the effect of CXCR4/SDF1 on injured muscle, supporting the existence of a functional interaction between these systems. Collectively, our findings reveal a new area of investigation, which could contribute to the development of novel therapies for muscle injury and/or dystrophic diseases.
Materials and Methods
Mice
Wild-type (WT; C57BL/6J) mice were purchased from Harlan Iberica. MMP-10 knockout (KO) mice were provided by Dr. W.C. Parks from the University of Washington. Genotypes for MMP-KO mice were determined through PCR analysis of mouse ear DNA using specific primers for MMP-10 (sense 1, 5′-TGTGTAGTGCCTACACTAAGCCA-3′; sense 2, 5′-TGCCTCGTCCTGCAGTTCATTC-3′; antisense, 5′-TAAGGGTGTGAGTCTTCATGGAT-3′). All animals were used at 8 weeks of age.
Model of muscle injury and in vivo treatments
Muscle damage was induced by an intramuscular injection of 10 μL (10 μg/mL) of Notechis scutatus scutatus notexin (Latoxan) into the left tibialis anterior (TA) muscle, leaving the right TA as a control [phosphate-buffered saline (PBS)]. In some experimental conditions, notexin was delivered to both TA muscles. After 24-h notexin treatments, we intramuscularly injected SDF1 (100 ng/mL; R&D), AMD3100 (10 μM; Sigma), or rhMMP-10 (100 ng/mL; R&D) into TA muscles as indicated, leaving contra-lateral muscles as controls.
A pair of specific siRNA sequences was pooled for each target, including SDF1 (siSDF1), CXCR4 (siCXCR4), and MMP-10 (siMMP-10) (Stealth siRNA; Invitrogen). These siRNAs were prepared in atelocollagen (Koken) according to the manufacturer's instructions and injected intramuscularly into notexin-treated left TA, whereas right injured TA was injected with empty atelocollagen as a control. Control siRNA (siControl) was also prepared and delivered under the same conditions (Invitrogen). The following specific siRNA sequences were used: SDF1, 5′-AGUGUGCAUUGACCCGAAAtt-3′ and 5′-UUUCGGGUCAAUGCACACUtg-3′; CXCR4, 5′-GGUACUUUGGGAAAUUUUUtt-3′ and 5′-AAAAAUUUCCCAAAGUACCag-3′; MMP-10, 5′-GACAGAUAACAGAUGAUUUtt-3′ and 5′-CGGAGACUUUUACCUUUUtt-3′.
Mice were sacrificed at 7 and 14 days after treatment by cervical dislocation and their TA muscles were removed.
Isolation and culturing of satellite cells
WT mice were killed by cervical dislocation, and the extensor digitorum longus (EDL) muscles were carefully removed. Myofibers were then isolated from EDL muscles as previously described (40). For suspension culture, myofibers were incubated in DMEM with 10% horse serum (PAA Laboratories), 0.5% chick embryo extract (ICN Flow), 4 mM l-glutamine, and 1% penicillin–streptomycin (37°C in 5% CO2). For adherent cultures, myofibers were grown in Lab-Tek® eight-well chamber slides (Nunc), which were coated with 1 mg/mL Matrigel (Collaborative Research, Inc.) and incubated at 37°C in 5% CO2. The satellite cells were then passaged and re-plated at a high density in order to examine myogenic differentiation and fusion. In addition, the medium was replaced with DMEM containing 2% horse serum for myogenic differentiation. In some cases, BrdU (10 μM) was added to the cell culture medium. When required, some muscle-derived cells as well as the media secreted by them were collected at different time points to examine different stages of differentiation. To this, we collected cells and media at 3, 6, and 9 days after re-plating satellite cells to obtain satellite cell-derived myoblasts, myoblasts that began to fuse into myotubes and myotubes with the capacity to spontaneously contract (contractile myotubes), respectively.
In vitro treatments
Satellite cell-derived myoblasts were grown to 30% confluency in six-well plates and transfected with siRNA. A second transfection was performed 24 h later. The siRNA duplexes were diluted in OptiMEM (Invitrogen) at 20 pmol/well and incubated with Lipofectamine 2000 (Invitrogen) in OptiMEM, according to the manufacturer's instructions. For myogenic differentiation, cells were transferred to eight-well chamber slides (Lab-Tek) immediately after the second transfection and maintained in differentiation medium (DMEM plus 2% horse serum).
Mouse SDF1 cDNA was cloned into pcDNA3.1/V5 plasmid (Invitrogen; K4800-01) and transfected into the COS-1 cell line (ATCC; CLR-1650) using Fugene-6 (Roche). Transfected cells were selected with geneticin (50 ng/mL; Invitrogen) and plated in six-well plates (100,000 cells/well) a day before isolating myofibers. Some COS-1 cells were transfected with the empty pcDNA plasmid, serving as control cells. The secretion of SDF1 into the media (10 ng/mL) was confirmed using an SDF1 ELISA Kit (Roche). Single EDL myofibers, along with their associated satellite cells, were cocultured with SDF1-transfected COS-1 cells in transwell plates (0.4 μm pore size; Corning) for 72 h. Some of them underwent a 24-h pretreatment with AMD3100 (10 μM). In addition, SDF1 (R&D) was sometimes added to the media of plated satellite cells or suspended single myofibers (10 ng/mL).
Immunostaining
Skeletal muscles were frozen in liquid nitrogen-cooled isopentane and serial 9-μm cryosections were collected at 200-μm intervals through the entire muscle. Sections were blocked with 20% goat serum and immunostained with mouse anti-embryonic myosin heavy chain (eMyHC) (1:100; DSHB), anti-Pax7 (1:100; DSHB), and anti-Ki67 (1:100; NeoMarkers). For all nuclear staining, 4,6-diamidino-2-phenylindole (DAPI) was used.
Cells and myofibers were fixed with 4% paraformaldehyde/PBS and permeabilized with 0.5% (v/v) Triton X-100/PBS. After blocking with 20% normal goat serum, cells and myofibers were incubated with the following primary antibodies (overnight, 4°C): rat anti-BrdU (1:300, clone BU1/75; Abcam), mouse anti-myosin (1:100, clone MF20; DSHB), anti-Pax7 (1:100; DSHB), and rabbit anti-MyoD (1:50; Santa Cruz). Primary antibodies were detected and visualized with fluorochrome-conjugated secondary antibodies (1:200; Amersham) before mounting in DakoCytomation Faramount fluorescent mounting medium containing DAPI.
Image capture and quantification
Immunostained cells, myofibers, and tissue sections were viewed on a Zeiss Axiophot epifluorescence microscope. Digital images were acquired with an AxioCamMR3 camera (Zeiss). In addition, AxioVision software and ImageJ were used to analyze tissue sections or cells (ie, whole muscle transversal sections or entire areas occupied by cells). Positive staining for eMyHC was calculated, in each of the treated and control muscles, as the percentage of stained area divided by the total area of muscle. The percentage of positive area for eMyHC in treated TA was related to that found in their respective controls and expressed as a fold change in the % of eMyHC expression. The number of proliferating and nonproliferating satellite cells was calculated by counting the total number of Pax7+/Ki67+ and Pax7+/Ki67− satellite cells, which were then related to the total muscle area (mm2). Fusion index was calculated by counting the number of nuclei within myosin+ myotubes and expressed as a percentage. In addition, the mean number of nuclei composing each myotube was calculated by counting the DAPI+ nuclei in at least 50 myotubes in each experimental condition. The amount of proliferating satellite cells was determined by counting the number of BrdU+cells in relation to the number of DAPI+ cells. To establish the percentage of satellite cells with each phenotype, we analyzed at least 15–20 individual myofibers (along with their associated satellite cells). In at least four independent experiments, we counted Pax7−/MyoD+, Pax7+/MyoD+, and Pax7+/MyoD− satellite cells, which were subsequently related to the total number of stained cells within the same fiber.
Immunoblotting
TA muscles and cells were homogenized in ice-cold lysis buffer (1% Triton X-100), which was supplemented with a mixture of phosphatase inhibitors (50 mM sodium pyrophosphate, 0.1 M NaF, and 10 mM sodium orthovanadate). Protein concentrations were determined using the BCA Protein Assay Kit (Pierce).
Proteins in muscle homogenates (40 μg), cell lysates (15 μg), and cell supernatants (20 μg) were separated via SDS-PAGE (10% gels) electrophoresis and subsequently transferred to nitrocellulose membranes (Bio-Rad Laboratories). The membranes were then blocked for 1 h at room temperature in Tris-buffered saline (10 mM Tris-HCl, 150 mM NaCl, and pH 8) containing 0.05% Tween 20 (TBS-T) and 10% nonfat dry milk. The blocked membranes were then probed with the following primary antibodies in blocking buffer (overnight, 4°C): mouse anti-Pax7 (DSHB), anti-MyoD (DSHB), anti-myosin (DSHB), anti-PCNA, (Santa Cruz Biotechnology), rabbit polyclonal anti-SDF1 (Cell Signaling), and anti-CXCR4 (Abcam). Mouse antibodies against β-actin (Sigma) or β-tubulin (Promega) were used as loading controls. The antigen–antibody complexes were detected using peroxidase-labeled goat anti-rabbit or anti-mouse antibodies (Amersham) in TBS-T containing 2.5% nonfat dry milk for 1 h at room temperature. The results were visualized using the enhanced chemiluminescence ECL Plus detection system (Amersham), and the bands were analyzed via densitometric analysis with the Gel Doc-Quantity One 4.5.0 (Bio-Rad).
Quantitative real-time PCR
Total RNA was isolated from cells and purified using UltraSpec reagent (Biotecx Laboratories). The total RNA was then reverse transcribed using Superscript II RNase H reverse transcriptase (Invitrogen), and cDNA was amplified using the TaqManH Universal PCR Master Mix (Applied Biosystems). Transcript levels were quantified by real-time PCR (7300 Real-Time PCR System; Applied Biosystems) under the following conditions: 95°C for 3 min followed by 40 cycles consisting of 60 s at 90°C, 60 s at 60°C, and 60 s at 72°C. Calibration curves (1, 2, 5, and 10 ng of target cDNA) were included to specifically analyze CXCR4 and SDF1 expression. All measurements were related to the curves and normalized to GAPDH (Applied Biosystems). Specific primers and probes were designed using the Primer3 program (version 0.4.0) for CXCR4 (forward, 5′-ACGGCTGTAGAGCGAGTGTT-3′; reverse, 5′-AGGGTTCCTTGTTGGAGTCA-3′; probe, 5′-CATGGAACCGATCAGTGTGA-3′) and SDF1 (forward, 5′-CCAAACTGTGCCCTTCAGAT-3′; reverse, 5′-AAGTCCTTTGGGCTGTTGTG-3′; probe, 5′-TGTGCATTGACCCGAAATTA-3′).
Statistical analysis
All statistical analyses were performed using SPSS 15.0 (SPSS). The Kolmogorov–Smirnov test was used to verify the normality of the samples. If the variables passed the test, then we applied the parametric Student's t-test and one-way ANOVA followed by least significant difference (LSD) and honestly significant difference tests. However, when variables did not follow the normality criteria according to the Kolmogorov–Smirnov test, we applied the nonparametric Kruskal–Wallis and Mann–Whitney U tests. Data were expressed as means±standard error of the mean. Statistical significance was defined as P<0.05.
Results
The CXCR4/SDF1 axis is required for muscle regeneration
First, we confirmed that our experimental model of injury induction led to increases in CXCR4 and SDF1. We intramuscularly injected notexin into TA muscles of WT mice and assessed protein levels throughout the course of the degeneration–regeneration process via immunoblot (Supplementary Fig. S1A–C; Supplementary Data are available online at www.liebertpub.com/scd). In addition, we explored the functional relevance of the CXCR4/SDF1 axis in muscle regeneration. To this end, AMD3001 (AMD), a CXCR4 antagonist that blocks the SDF1–CXCR4 interaction [41], was injected into the right damaged TA muscle of WT mice, while the left muscle received PBS (control). Seven days later, muscle regeneration was analyzed via immunostaining for eMyHC, which is transitionally expressed in regenerating myofibers. Antibodies against Pax7 and Ki67 were used to detect proliferating (Pax7+/Ki67+) and nonproliferating (Pax7+/Ki67−) satellite cells. An examination of injured muscles revealed that the percentage of eMyHC+ regenerating fibers was significantly lower in AMD-treated muscles in comparison to controls (Fig. 1A, B). In contrast, the number of Pax7+ satellite cells was doubled by addition of AMD (Fig. 1C, D; arrows). We also observed an increase in the percentage of proliferating Pax7+ satellite cells (Ki67+) after AMD treatment of injured TA (Fig. 1E). Taken together, these findings indicated that blocking the interaction of CXCR4 with SDF1 delayed muscle regeneration.
FIG. 1.
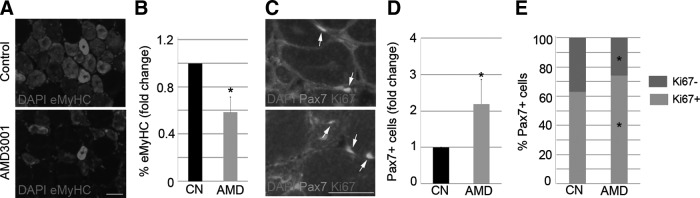
Inhibition of the CXCR4/SDF1 axis delays muscle regeneration. TA muscles of WT mice were injected with notexin. One day later, the right TA was treated with AMD3001 (AMD), and the left TA received PBS (Control, CN). The animals were sacrificed 7 days later, and the muscles were removed. Representative images of eMyHC (A) and Pax7/Ki67 (C) immunostaining are displayed. We quantitated the percentage of eMyHC+ regenerating fibers (B), the total number of Pax7+ satellite cells (C, D; arrows), and the percent distribution of Ki67 in Pax7+ satellite cells (E). Scale bar: 50 μm. Measurements in treated cells were related to those in controls and expressed as fold change. Values are presented as means±SEM from at least five independent experiments. An asterisk denotes a statistically significant difference compared with control (Mann–Whitney U test; P<0.05). eMyHC, embryonic myosin heavy chain; PBS, phosphate-buffered saline; SDF1, stromal cell-derived factor-1; SEM, standard error of the mean; TA, tibialis anterior; WT, wild type.
To assess whether delayed muscular repair is permanent, we analyzed injured TA 14 days after treatment with AMD. Despite finding a very few eMyHC+ regenerating fibers in injured muscles treated with AMD but not in the TA from controls (Supplementary Fig. S2A), the architecture of muscles was roughly similar, which, in turn, demonstrates that AMD temporally delays muscular regeneration.
Inhibition of CXCR4/SDF1 components impairs muscle regeneration
In order to verify the importance of the CXCR4/SDF1 axis in muscle regeneration and to better understand the individual roles of CXCR4 and SDF1 in the repair process, we depleted each of these components in damaged muscles. For this, both TA muscles of WT mice were treated with notexin. After 24 h, pooled siRNA duplexes (10 μM) against either CXCR4 (siCXCR4) or SDF1 (siSDF1) were intramuscularly delivered to the right damaged TA, while the left TA received control siRNA (siControl). Animals were sacrificed 7 days later, and the loss of CXCR4 and SDF1 proteins was confirmed by western blotting (Fig. 2A, E). Muscle regeneration was also analyzed.
FIG. 2.

Local modulation of CXCR4 or SDF1 impairs muscle regeneration. Right injured TA muscles of WT mice were treated with specific siRNA duplexes against SDF1 (siSDF1) or CXCR4 (siCXCR4), while contra-lateral damaged muscles received control siRNA (siCN). Seven days later, the animals were sacrificed. Western blot analysis revealed down-regulation of target genes (A, E). β-Tubulin was used as a loading control. Muscle differentiation was analyzed via eMyHC immunostaining (B, F) and counting of Pax7+ satellite cells (C, G). The percent distribution of Ki67 in Pax7+ satellite cells was also quantified (D, H). All measurements obtained from treated TA (siSDF1 or siCXCR4) were related to the control (siControl) and are expressed as fold change. Values are presented as mean±SEM from at least five independent experiments. An asterisk denotes a statistically significant difference compared with control (Mann–Whitney U test; P<0.05).
Similar to what we found in injured muscles treated with AMD, the administration of siCXCR4 into notexin-injured muscles led to a significant reduction of the percentage of new regenerating myofibers, as determined by the presence of eMyHC expression (Fig. 2B). In addition, the total number of Pax7+ satellite cells in siCXCR4-treated muscles was higher than that found in controls (Fig. 2C). An increased percentage of proliferating Pax7+ satellite cells was observed (Fig. 2D), suggesting a delay in muscle repair.
Interestingly, despite the fact that siSDF1-treated injured muscles displayed a lower percentage of eMyHC+ regenerating fibers compared with controls (Fig. 2F), and that the majority of Pax7+ satellite cells expressed Ki67 on SDF1 depletion (Fig. 2H), the number of total Pax7+ satellite cells was significantly decreased compared with the controls (Fig. 2G). These results suggest that SDF1 silencing impairs the differentiation of satellite cell progeny and is required to properly maintain the pool of satellite cells after muscle injury.
To assess whether modulation of CXCR4/SDF1 axis alters muscle repair kinetics, we analyzed TA 14 days after injury. Injured muscles treated with siCXCR4 or siSDF1 had a few eMyHC+ fibers as well as injured muscles treated with siControl (Supplementary Fig. S2B, C). These results suggest that transient alteration of CXCR4 and SDF1 expression in injured muscle alter muscular regeneration kinetics but not permanently.
Expression of CXCR4 and SDF1 is required for in vitro myogenesis
To characterize the role of the CXCR4/SDF1 axis in satellite cells, we cultured satellite cell-derived myoblasts and differentiated them through mitogen withdrawal. Throughout the course of myogenic differentiation, from myoblasts to myotubes with the ability to spontaneously contract, we observed increased expression and secretion of SDF1 by quantitative real-time PCR and western blot (Supplementary Fig. S3A–C). In contrast, we found that CXCR4 expression was down-regulated as myoblasts fused into myotubes (Supplementary Fig. S3D, E). These primary satellite cells were subsequently transfected with siCXCR4, siSDF1, or siControl. After 6 days in culture, the depletion of CXCR4 and SDF1 was confirmed by western blot (Fig. 3A, D). Notably, compared with controls, loss of CXCR4 or SDF1 led to a decreased number of BrdU+ satellite cells (Fig. 3B, E; arrows) and reduced ability of cells to fuse into myotubes, as determined by the fusion index (Fig. 3C, F). In addition, the mean number of nuclei in each myotube showed that CXCR4 and SDF1 silenced myoblasts had a defect in cell fusion (Fig. 3C, F), suggesting that these proteins are required for properly growth of myotubes. These findings were verified by immunoblotting for PCNA, MyoD, Pax7, and MyHC (Supplementary Fig. S4A–H). Collectively, these data confirm that both CXCR4 and SDF1 are required for efficient in vitro myogenesis.
FIG. 3.

Reduction of CXCR4 and SDF1 expression in myoblasts affects myogenesis. Satellite cell-derived myoblasts were transfected with siCXCR4 or siControl and cultivated for 6 days. Loss of CXCR4 (A) and SDF1 (D) expression was analyzed by western blot. Protein expression levels were normalized using β-actin. Myoblast proliferation was measured through quantification of BrdU+ cells (B, E; arrows), whereas cell differentiation was assessed via the fusion index and counting the mean number of nuclei within each myotube (C, F). DAPI was used to visualize nuclei. Scale bar: 20 μm. Measurements of protein levels and BrdU+ cells in siCXCR4- and siSDF1-treated cells were related to siControl and expressed as fold change. Values are presented as means±SEM from at least five independent experiments. An asterisk denotes a statistically significant difference compared with control (Mann–Whitney U test; P<0.05). DAPI, 4,6-diamidino-2-phenylindole.
Administration of SDF1 improves muscle regeneration
Based on our findings, we hypothesized that the CXCR4/SDF1 axis directly participates in the regenerative processes occurring in the muscle. In support of this theory, we injected notexin into both TA muscles of WT mice. One day later, we administered SDF1 (100 ng/mL) into the left muscle, while the right muscle served as the control. Strikingly, SDF1 treatment tripled the expression of eMyHC by day 7 (Fig. 4A), whereas the number of total Pax7+ satellite cells (Fig. 4B, C; arrows) was significantly decreased in comparison to controls. Accordingly, the administration of SDF1 led to a lower percentage of proliferating Pax7+satellite cells in damaged muscle (Fig. 4D). Overall, these results suggest that SDF1 directly accelerates muscle repair after injury.
FIG. 4.
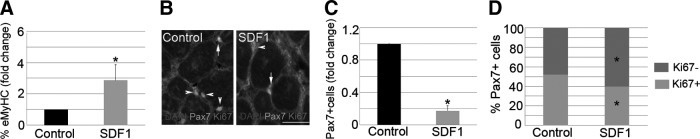
SDF1 accelerates muscle repair after injury. Right notexin-injured TA muscles of WT mice were treated with SDF1, while contra-lateral damaged muscles received PBS (control). Animals were sacrificed 7 days later. We quantitated the percentage of eMyHC+ cells (A), the total Pax7+ satellite cells (B, C), and the percent distribution of Ki67 in Pax7+ satellite cells (D). Arrows identify Pax7+/Ki67+ and arrowheads detect Pax7+/Ki67−. DAPI was used to visualize nuclei. Scale bar: 50 μm. All data from SDF1-treated muscles were related to the controls and expressed as fold change. Values are presented as mean±SEM from at least five independent experiments. An asterisk denotes a statistically significant difference from control (Mann–Whitney U test; P<0.05).
Exogenous SDF1 accelerates in vitro myogenic kinetics
To understand the effects of the SDF1 gain of function during muscle repair, we examined the role of SDF1 during the earlier steps of myogenic progression. To this end, single myofibers, along with their attached satellite cells, were cocultured with SDF1-secreting COS-1 cells (SDF1: 10 ng/mL). In addition, other fibers were cocultured with control COS-1 cells. After 72 h in suspension, SDF1 increased the percentage of Pax7+/MyoD+ satellite cells (Fig. 5A, C; arrows), while those with the Pax7−/MyoD+ phenotype were reduced (Fig. 5A, C; arrowheads). These findings support the notion that SDF1 participates in satellite cell proliferation rather than differentiation. In addition, similar data were obtained when exogenous SDF1 (10 ng/mL) was added to single myofiber-associated satellite cells (Fig. 5B). Notably, we found that the effect of SDF1 on myogenic progression was completely abolished on pretreatment with AMD (Fig. 5D). These findings demonstrate that the earlier steps of myogenic progression are dependent on the CXCR4–SDF1 interaction.
FIG. 5.
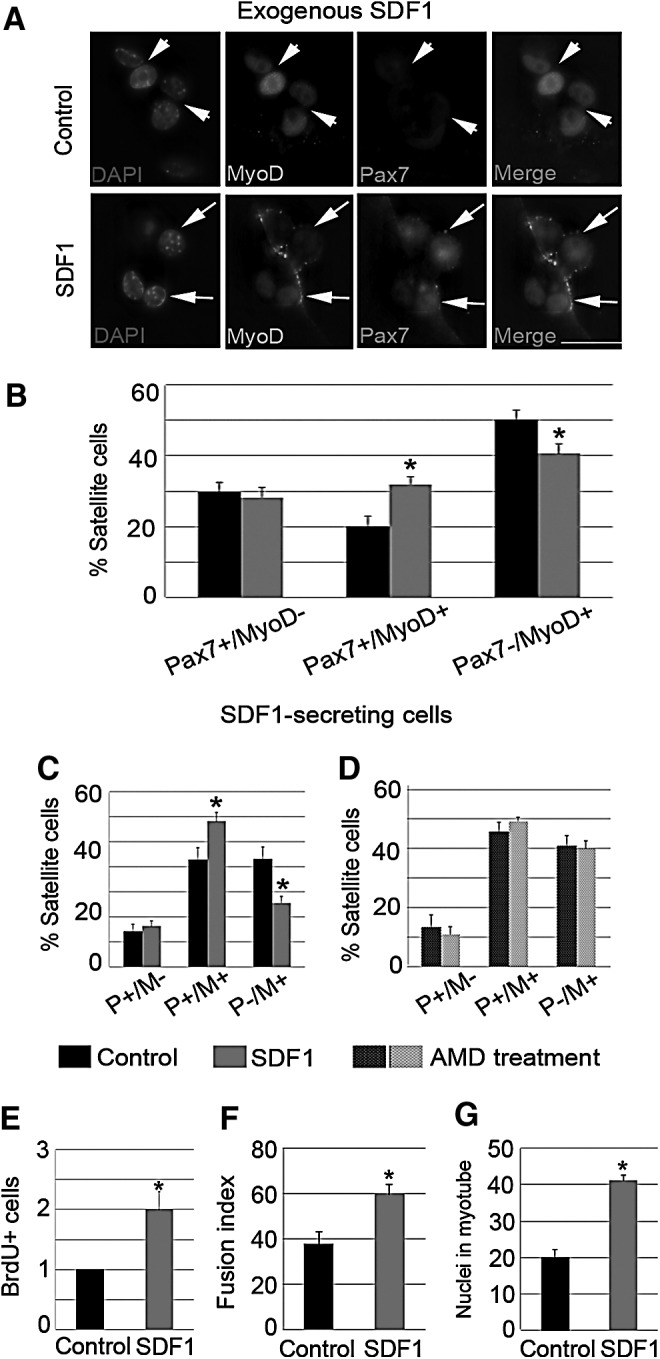
Exogenous SDF1 accelerates the myogenic kinetics of myoblasts. (A–D) SDF1 was added into suspension myofibers for 72 h (A, B), whereas other fibers were cocultured with SDF1-secreting COS-1 cells (C). In addition, some myofibers were pretreated with AMD3001 (AMD) before coculturing with SDF1-expressing cells (D). Pax7+/MyoD− (P+/M−), Pax7+/MyoD+ (P+/M+; arrows), and Pax7−/MyoD+ (P−/M+; arrowheads) phenotypes where calculated by coimmunostaining (Pax7 and MyoD). DAPI was used to visualize nuclei. Scale bar: 10 μm. (C, D) Satellite cell-derived myoblasts were cultured for 6 days with exogenous SDF1 or PBS (control). Proliferation (E) and myogenic differentiation, including the fusion index (F) and the mean number of nuclei in myotubes (G), were quantified. Scale bar: 20 μm. Values are presented as means±SEM from at least five independent experiments. An asterisk denotes a statistically significant difference from the control (Mann–Whitney U test; P<0.05).
In order to investigate the involvement of SDF1 in the later steps of myogenic progression, such as cell fusion, we added exogenous SDF1 (10 ng/mL) to primary satellite cell-derived myoblasts. Compared with control cells, treatment with SDF1 for 6 days led to a doubling in the number of proliferating myoblasts (Fig. 5E) and augmented fusion into myotubes (Fig. 5F). In addition, the increased mean number of nuclei in myotubes in SDF1- treated cell cultures showed that SDF1 improved growth of myotubes (Fig. 5G). These results correlated with higher protein levels of PCNA, MyoD, Pax7, and MyHC in SDF1-treated myoblasts compared with controls (Supplementary Fig. S5A–D). However, it remains possible that the effects of SDF1 on the later steps of myogenic differentiation are simply a consequence of increased proliferation during early myogenesis.
The CXCR4/SDF1 axis requires MMP-10 activity to induce muscle regeneration
It was previously reported that various MMPs, such as MMP-2 or MMP-9, could interact with the CXCR4/SDF1 axis during the regulation of specific cellular processes [30,42–45]. We recently described that the modulation of MMP-10 protein levels in injured muscle affects muscular regeneration [40]. In fact, the delivery of siMMP-10 into injured muscles delayed muscle regeneration; whereas rhMMP-10 accelerated the repair process. In addition, MMP-10 KO mice have delayed muscular regeneration compared with WT animals [40].
Since we had previously described a role for MMP-10 in muscle regeneration, we next sought to analyze whether MMP-10 modulation could affect the ability of the CXCR4/SDF1 axis to regulate repair. First, we assessed CXCR4 and SDF1 expression in injured muscles, which were treated with siMMP-10 or rhMMP-10 to delay or accelerate repair, respectively [40]. Western blot analysis revealed that siMMP-10 led to a significant reduction in CXCR4 and SDF1 levels (Supplementary Fig. S6A–C), while these proteins were significantly higher in rhMMP-10-treated muscles (Supplementary Fig. S6D–F). In addition, we assessed the expression of CXCR4 and SDF1 in muscles from MMP-10 KO mice. Indeed, the protein levels of both the receptor and ligand were significantly lower in MMP-10 KO TA compared with WT muscles (Supplementary Fig. S6G–I). These findings suggest that MMP-10 could mediate the effect of the CXCR4/SDF1 axis during muscle repair.
To corroborate our hypothesis, we induced injury in both TA muscles of MMP-10 KO mice and intramuscularly injected the right muscles with AMD or SDF1 (after 1 day of damage). The left TA muscles received PBS and served as controls. Seven days later, eMyHC staining revealed that SDF1 administration did not increase the percentage of newly regenerating myofibers in injured KO muscles (Fig. 6A), which was contrary to results observed with WT mice. Similarly, AMD treatment did not alter the expression of new eMyHC+ regenerating myofibers in damaged muscles from MMP-10 KO animals (Fig. 6B). Taken together, these findings suggest that the CXCR4/SDF1 axis requires MMP-10 activity to induce muscle repair.
FIG. 6.
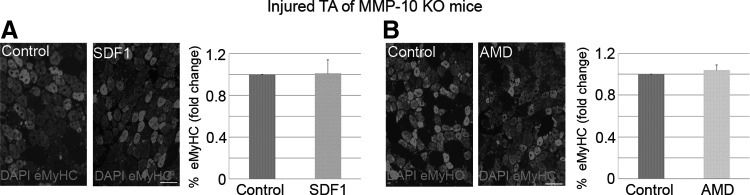
The CXCR4/SDF1 axis requires MMP-10 activity to induce muscle regeneration. (A, B) Right notexin-injured TA muscles of MMP-10 KO mice were treated with SDF1 or AMD3100 (AMD), while contra-lateral damaged muscles received PBS (control). Animals were sacrificed 7 days later, and the percentage of eMyHC+ cells was quantified. DAPI was used to visualize nuclei. Scale bar: 50 μm. All data from treated muscles were related to their respective controls and expressed as fold change. Values are presented as mean±SEM from at least five independent experiments. An asterisk denotes a statistically significant difference from control (Mann–Whitney U test; P<0.05). KO, knockout; MMP, matrix metalloproteinase.
Discussion
In the present study, we have examined the role of the CXCR4/SDF1 axis in muscle regeneration after injury. As previously reported [31,46], our findings have confirmed that skeletal muscle responds to damage by enhancing the expression of CXCR4 and SDF1, suggesting their participation in the muscle repair process. We found that AMD3001, an antagonist of CXCR4 [41], delayed muscle regeneration, reducing the number of eMyHC+ myofibers and increasing the percentage of proliferating Pax7+ satellite cells. In addition, transient depletion of CXCR4 or SDF1 significantly delayed muscle regeneration, as shown by reduced expression of eMyHC. In contrast, exogenous SDF1 accelerated the repair process. Furthermore, it appears that SDF1 is required to properly maintain the satellite cell pool after injury, as the total number of Pax7+ satellite cells decreased in SDF1-depleted injured muscles. In line with this, it was reported that local elevation of SDF1 in injured organs promotes cell recruitment that supports tissue repair and regeneration [18,47,48], an effect that is abrogated by AMD3100 [49,50]. Thus, in our injury model, we speculate that increased SDF1 is required for the recruitment of Pax7+ satellite cells, which subsequently differentiate to restore/replace damaged fibers.
It has been demonstrated that SDF1 expression at sites of injury can also recruit immune cells or endothelial progenitor cells, which can participate in muscle repair [51]. In addition, SDF1 participates in the release of ECM components, which are involved in muscle regeneration [52]. These factors might explain why our in vivo data were not efficiently reproduced in our in vitro models, as it is sometimes difficult to specifically attribute specific processes to particular cell types. In this regard, we have not determined which cells secrete SDF1 or which CXCR4+ cells are recruited to injured sites. However, our in vitro data confirmed that both CXCR4 and SDF1 had a direct effect on myogenic cells in the absence of other confounding cell types, and these myogenic cells are known to be directly responsible for muscle repair [4]. In addition, our data have verified that CXCR4 expression decreased during myogenesis, while expression and secretion of SDF1 increased [31,32]. These findings suggest that contractile myotubes produce SDF1 in order to attract satellite cells and facilitate fusion, which could mirror the process in vivo. Furthermore, we found that CXCR4 or SDF1 depletion diminished the ability of satellite cell-derived myoblasts to incorporate BrdU, subsequently reducing myotube differentiation. Accordingly, exogenous SDF1 increased satellite cell proliferation and fusion into myotubes. Therefore, since cell fusion constitutes a later event in the repair process, we propose that the CXCR4/SDF1 axis is required to satellite cells account the repair response of muscle after an episode of injury.
Although SDF1 mainly functions through CXCR4, it can also interact with CXCR7 [46,53]. Both CXCR4–SDF1 and CXCR7–SDF1 interactions regulate myogenesis, but at different stages of differentiation. SDF1 participates in the early steps of myogenic progression through CXCR4, whereas the CXCR7/SDF1 axis regulates later steps [46]. In suspension myofibers (ie, earlier steps of myogenic progression), we found that SDF1 increased the percentage of Pax7+/MyoD+ satellite cells, while the percentage of those with the Pax7−/MyoD+ phenotype decreased. This suggests that SDF1 influences satellite cell proliferation rather than differentiation. In addition, we found that this effect was abrogated on pretreatment of myofiber-associated satellite cells with AMD3100, confirming that SDF1 acts through CXCR4. These findings support a model in which increased satellite cell fusion into myotubes during the later steps of myogenic differentiation is secondary to enhanced proliferation.
Migration from distant myofibers to sites of injury requires satellite cells to cross the basal lamina of their resident myofibers [3], followed by directed movement to damaged areas [5]. This idea is in agreement with the notion that myogenic precursors not only respond to chemokines, but also should express and secrete MMPs [54]. In this regard, increased expression of MMPs and cytokines in damaged skeletal muscle seems to be indicative of their roles in muscle repair or disease progression [55–57]. However, the concomitant up-regulation of these factors could also suggest a functional interaction between them. In fact, several pieces of data support this idea: (1) MMPs and cytokines participate in the same cellular processes (eg, migration) [58–60]; (2) MMPs can regulate chemokine activity through direct proteolysis of chemokines; (3) and MMPs contribute to chemokine release from ECM [61–63]. In addition, some chemokines can affect MMP function by directly or indirectly [64,65] altering MMP activity. Thus, MMPs have the ability to modulate the locomotion machinery of cells through interaction with chemokines.
We recently demonstrated that MMP-10 has a pivotal role in skeletal muscle maintenance and repair [40]. In the present study, we have extended these previous findings by identifying that alterations in MMP-10 levels had parallel affects on the expression of CXCR4 and SDF1. Furthermore, we found that AMD3100 and SDF1 lost their effects on muscle regeneration (delayed or accelerated repair, respectively) in muscles from MMP-10 KO mice. This suggests that the CXCR4/SDF1 axis requires MMP-10 activity to induce efficient repair of skeletal muscle. Although the biochemical pathway triggered by MMP-10 during CXCR4/SDF1-induced muscle regeneration requires further investigation, it is clear that cross-talk exists between these two systems.
Taken together, our data suggest that the CXCR4/SDF1 axis is pivotal for efficient muscle regeneration after an episode of injury. In addition, it is possible that this system functions through MMP-10 activity where MMP-10 might regulate CXCR4/SDF1-mediated muscle repair via chemokine modulation. These findings suggest an important molecular mechanism that could be exploited to develop novel therapies for dystrophic diseases.
Supplementary Material
Acknowledgments
This work was supported by grants from the Spanish Ministry of Science and Technology (SAF2009-12039); Caja de Ahorros de Navarra (Programa Tú Eliges: Tú Decides); the Spanish Ministry of Health (PI08/1919, PS09/00143 and RD12/0019/0031); the Ministerio de Economia y Competitividades (PLE2009-0116); European Union's Seventh Framework Program for Research, Program (INELPY); and the Unión Temporal de Empresas-Centro de Investigación Médica Aplicada. The authors thank Virginia Izuriaga and María Jiménez for their technical assistance. They also thank Parks for providing MMP-10 KO mice. They are grateful to the Developmental Studies Hybridoma Bank for the antibodies used in this study.
Author Disclosure Statement
No competing financial interests exist.
References
- 1.Mauro A. (1961). Satellite cell of skeletal muscle fibers. J Biophys Biochem Cytol 9:493–495 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Moss FP. and Leblond CP. (1971). Satellite cells as the source of nuclei in muscles of growing rats. Anat Rec 170:421–435 [DOI] [PubMed] [Google Scholar]
- 3.Collins CA, Olsen I, Zammit PS, Heslop L, Petrie A, Partridge TA. and Morgan JE. (2005). Stem cell function, self-renewal, and behavioral heterogeneity of cells from the adult muscle satellite cell niche. Cell 122:289–301 [DOI] [PubMed] [Google Scholar]
- 4.Charge SB. and Rudnicki MA. (2004). Cellular and molecular regulation of muscle regeneration. Physiol Rev 84:209–238 [DOI] [PubMed] [Google Scholar]
- 5.Hughes SM. and Blau HM. (1990). Migration of myoblasts across basal lamina during skeletal muscle development. Nature 345:350–353 [DOI] [PubMed] [Google Scholar]
- 6.Broxmeyer HE. (2001). Regulation of hematopoiesis by chemokine family members. Int J Hematol 74:9–17 [DOI] [PubMed] [Google Scholar]
- 7.Nagasawa T, Nakajima T, Tachibana K, Iizasa H, Bleul CC, Yoshie O, Matsushima K, Yoshida N, Springer TA. and Kishimoto T. (1996). Molecular cloning and characterization of a murine pre-B-cell growth-stimulating factor/stromal cell-derived factor 1 receptor, a murine homolog of the human immunodeficiency virus 1 entry coreceptor fusin. Proc Natl Acad Sci U S A 93:14726–14729 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Lapidot T, Dar A. and Kollet O. (2005). How do stem cells find their way home?. Blood 106:1901–1910 [DOI] [PubMed] [Google Scholar]
- 9.Wang K, Zhao X, Kuang C, Qian D, Wang H, Jiang H, Deng M. and Huang L. (2012). Overexpression of SDF-1alpha enhanced migration and engraftment of cardiac stem cells and reduced infarcted size via CXCR4/PI3K pathway. PLoS One 7:e43922. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Percherancier Y, Berchiche YA, Slight I, Volkmer-Engert R, Tamamura H, Fujii N, Bouvier M. and Heveker N. (2005). Bioluminescence resonance energy transfer reveals ligand-induced conformational changes in CXCR4 homo- and heterodimers. J Biol Chem 280:9895–9903 [DOI] [PubMed] [Google Scholar]
- 11.Sharma M, Afrin F, Satija N, Tripathi RP. and Gangenahalli GU. (2011). Stromal-derived factor-1/CXCR4 signaling: indispensable role in homing and engraftment of hematopoietic stem cells in bone marrow. Stem Cells Dev 20:933–946 [DOI] [PubMed] [Google Scholar]
- 12.Kucia M, Jankowski K, Reca R, Wysoczynski M, Bandura L, Allendorf DJ, Zhang J, Ratajczak J. and Ratajczak MZ. (2004). CXCR4-SDF-1 signalling, locomotion, chemotaxis and adhesion. J Mol Histol 35:233–245 [DOI] [PubMed] [Google Scholar]
- 13.Wong D. and Korz W. (2008). Translating an antagonist of chemokine receptor CXCR4: from bench to bedside. Clin Cancer Res 14:7975–7980 [DOI] [PubMed] [Google Scholar]
- 14.Broxmeyer HE, Cooper S, Kohli L, Hangoc G, Lee Y, Mantel C, Clapp DW. and Kim CH. (2003). Transgenic expression of stromal cell-derived factor-1/CXC chemokine ligand 12 enhances myeloid progenitor cell survival/antiapoptosis in vitro in response to growth factor withdrawal and enhances myelopoiesis in vivo. J Immunol 170:421–429 [DOI] [PubMed] [Google Scholar]
- 15.Lataillade JJ, Clay D, Bourin P, Herodin F, Dupuy C, Jasmin C. and Le Bousse-Kerdiles MC. (2002). Stromal cell-derived factor 1 regulates primitive hematopoiesis by suppressing apoptosis and by promoting G(0)/G(1) transition in CD34(+) cells: evidence for an autocrine/paracrine mechanism. Blood 99:1117–1129 [DOI] [PubMed] [Google Scholar]
- 16.Konoplyannikov M, Haider KH, Lai VK, Ahmed RP, Jiang S. and Ashraf M. (2013). Activation of diverse signaling pathways by ex-vivo delivery of multiple cytokines for myocardial repair. Stem Cells Dev 22:204–215 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Askari AT, Unzek S, Popovic ZB, Goldman CK, Forudi F, Kiedrowski M, Rovner A, Ellis SG, Thomas JD, et al. (2003). Effect of stromal-cell-derived factor 1 on stem-cell homing and tissue regeneration in ischaemic cardiomyopathy. Lancet 362:697–703 [DOI] [PubMed] [Google Scholar]
- 18.Hu X, Dai S, Wu WJ, Tan W, Zhu X, Mu J, Guo Y, Bolli R. and Rokosh G. (2007). Stromal cell derived factor-1 alpha confers protection against myocardial ischemia/reperfusion injury: role of the cardiac stromal cell derived factor-1 alpha CXCR4 axis. Circulation 116:654–663 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Schober A. (2008). Chemokines in vascular dysfunction and remodeling. Arterioscler Thromb Vasc Biol 28:1950–1959 [DOI] [PubMed] [Google Scholar]
- 20.Ho TK, Tsui J, Xu S, Leoni P, Abraham DJ. and Baker DM. (2012). Angiogenic effects of stromal cell-derived factor-1 (SDF-1/CXCL12) variants in vitro and the in vivo expressions of CXCL12 variants and CXCR4 in human critical leg ischemia. J Vasc Surg 51:689–699 [DOI] [PubMed] [Google Scholar]
- 21.Toupadakis CA, Wong A, Genetos DC, Chung DJ, Murugesh D, Anderson MJ, Loots GG, Christiansen BA, Kapatkin AS. and Yellowley CE. (2012). Long-term administration of AMD3100, an antagonist of SDF-1/CXCR4 signaling, alters fracture repair. J Orthop Res 30:1853–1859 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Libura J, Drukala J, Majka M, Tomescu O, Navenot JM, Kucia M, Marquez L, Peiper SC, Barr FG, Janowska-Wieczorek A. and Ratajczak MZ. (2002). CXCR4-SDF-1 signaling is active in rhabdomyosarcoma cells and regulates locomotion, chemotaxis, and adhesion. Blood 100:2597–2606 [DOI] [PubMed] [Google Scholar]
- 23.Fernandis AZ, Prasad A, Band H, Klosel R. and Ganju RK. (2004). Regulation of CXCR4-mediated chemotaxis and chemoinvasion of breast cancer cells. Oncogene 23:157–167 [DOI] [PubMed] [Google Scholar]
- 24.Ratajczak MZ, Majka M, Kucia M, Drukala J, Pietrzkowski Z, Peiper S. and Janowska-Wieczorek A. (2003). Expression of functional CXCR4 by muscle satellite cells and secretion of SDF-1 by muscle-derived fibroblasts is associated with the presence of both muscle progenitors in bone marrow and hematopoietic stem/progenitor cells in muscles. Stem Cells 21:363–371 [DOI] [PubMed] [Google Scholar]
- 25.Vasyutina E, Stebler J, Brand-Saberi B, Schulz S, Raz E. and Birchmeier C. (2005). CXCR4 and Gab1 cooperate to control the development of migrating muscle progenitor cells. Genes Dev 19:2187–2198 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Odemis V, Lamp E, Pezeshki G, Moepps B, Schilling K, Gierschik P, Littman DR. and Engele J. (2005). Mice deficient in the chemokine receptor CXCR4 exhibit impaired limb innervation and myogenesis. Mol Cell Neurosci 30:494–505 [DOI] [PubMed] [Google Scholar]
- 27.Odemis V, Boosmann K, Dieterlen MT. and Engele J. (2007). The chemokine SDF1 controls multiple steps of myogenesis through atypical PKCzeta. J Cell Sci 120:4050–4059 [DOI] [PubMed] [Google Scholar]
- 28.Bae GU, Gaio U, Yang YJ, Lee HJ, Kang JS. and Krauss RS. (2008). Regulation of myoblast motility and fusion by the CXCR4-associated sialomucin, CD164. J Biol Chem 283:8301–8309 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Melchionna R, Di Carlo A, De Mori R, Cappuzzello C, Barberi L, Musaro A, Cencioni C, Fujii N, Tamamura H, et al. (2010). Induction of myogenic differentiation by SDF-1 via CXCR4 and CXCR7 receptors. Muscle Nerve 41:828–835 [DOI] [PubMed] [Google Scholar]
- 30.Brzoska E, Kowalewska M, Markowska-Zagrajek A, Kowalski K, Archacka K, Zimowska M, Grabowska I, Czerwinska AM, Czarnecka-Gora M, et al. (2012). Sdf-1 (CXCL12) improves skeletal muscle regeneration via the mobilisation of Cxcr4 and CD34 expressing cells. Biol Cell 104:722–737 [DOI] [PubMed] [Google Scholar]
- 31.Griffin CA, Apponi LH, Long KK. and Pavlath GK. (2010). Chemokine expression and control of muscle cell migration during myogenesis. J Cell Sci 123:3052–3060 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Vincenti MP. and Brinckerhoff CE. (2007). Signal transduction and cell-type specific regulation of matrix metalloproteinase gene expression: can MMPs be good for you?. J Cell Physiol 213:355–364 [DOI] [PubMed] [Google Scholar]
- 33.Schultz GS. and Wysocki A. (2009). Interactions between extracellular matrix and growth factors in wound healing. Wound Repair Regen 17:153–162 [DOI] [PubMed] [Google Scholar]
- 34.Mott JD. and Werb Z. (2004). Regulation of matrix biology by matrix metalloproteinases. Curr Opin Cell Biol 16:558–564 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Huang W, Wang T, Zhang D, Zhao T, Dai B, Ashraf A, Wang X, Xu M, Millard RW, et al. (2012). Mesenchymal stem cells overexpressing CXCR4 attenuate remodeling of postmyocardial infarction by releasing matrix metalloproteinase-9. Stem Cells Dev 21:778–789 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Christov C, Chretien F, Abou-Khalil R, Bassez G, Vallet G, Authier FJ, Bassaglia Y, Shinin V, Tajbakhsh S, Chazaud B. and Gherardi RK. (2007). Muscle satellite cells and endothelial cells: close neighbors and privileged partners. Mol Biol Cell 18:1397–1409 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Montero I, Orbe J, Varo N, Beloqui O, Monreal JI, Rodriguez JA, Diez J, Libby P. and Paramo JA. (2006). C-reactive protein induces matrix metalloproteinase-1 and -10 in human endothelial cells: implications for clinical and subclinical atherosclerosis. J Am Coll Cardiol 47:1369–1378 [DOI] [PubMed] [Google Scholar]
- 38.Rodriguez JA, Orbe J, Martinez de Lizarrondo S, Calvayrac O, Rodriguez C, Martinez-Gonzalez J. and Paramo JA. (2008). Metalloproteinases and atherothrombosis: MMP-10 mediates vascular remodeling promoted by inflammatory stimuli. Front Biosci 13:2916–2921 [DOI] [PubMed] [Google Scholar]
- 39.Gill JH, Kirwan IG, Seargent JM, Martin SW, Tijani S, Anikin VA, Mearns AJ, Bibby MC, Anthoney A. and Loadman PM. (2004). MMP-10 is overexpressed, proteolytically active, and a potential target for therapeutic intervention in human lung carcinomas. Neoplasia 6:777–785 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Bobadilla M, Sáinz N, Rodriguez JA, Abizanda G, Orbe J, De Martino A, García Verdugo JM, Páramo JA, Prósper F. and Pérez-Ruiz A. (2013). MMP-10 is required for efficient muscle regeneration in mouse models of injury and muscular dystrophy. Stem Cells 32:447–461 [DOI] [PubMed] [Google Scholar]
- 41.De Clercq E. (2005). Potential clinical applications of the CXCR4 antagonist bicyclam AMD3100. Mini Rev Med Chem 5:805–824 [DOI] [PubMed] [Google Scholar]
- 42.Zhang R, Pan X, Huang Z, Weber GF. and Zhang G. (2011). Osteopontin enhances the expression and activity of MMP-2 via the SDF-1/CXCR4 axis in hepatocellular carcinoma cell lines. PLoS One 6:e23831. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Gong Y, Fan Y. and Hoover-Plow J. (2011). Plasminogen regulates stromal cell-derived factor-1/CXCR4-mediated hematopoietic stem cell mobilization by activation of matrix metalloproteinase-9. Arterioscler Thromb Vasc Biol 31:2035–2043 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Bhoopathi P, Chetty C, Gogineni VR, Gujrati M, Dinh DH, Rao JS. and Lakka SS. (2011). MMP-2 mediates mesenchymal stem cell tropism towards medulloblastoma tumors. Gene Ther 18:692–701 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Zhang H, Trivedi A, Lee JU, Lohela M, Lee SM, Fandel TM, Werb Z. and Noble-Haeusslein LJ. (2011). Matrix metalloproteinase-9 and stromal cell-derived factor-1 act synergistically to support migration of blood-borne monocytes into the injured spinal cord. J Neurosci 31:15894–15903 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Hunger C, Odemis V. and Engele J. (2012). Expression and function of the SDF-1 chemokine receptors CXCR4 and CXCR7 during mouse limb muscle development and regeneration. Exp Cell Res 318:2178–2190 [DOI] [PubMed] [Google Scholar]
- 47.Misao Y, Takemura G, Arai M, Ohno T, Onogi H, Takahashi T, Minatoguchi S, Fujiwara T. and Fujiwara H. (2006). Importance of recruitment of bone marrow-derived CXCR4+ cells in post-infarct cardiac repair mediated by G-CSF. Cardiovasc Res 71:455–465 [DOI] [PubMed] [Google Scholar]
- 48.Saxena P, Kejriwal N. and Newman MA. (2007). Use of flexible silastic drains in thoracic surgery: a word of caution. Ann Thorac Surg 83:2258–2259; author reply 2259 [DOI] [PubMed] [Google Scholar]
- 49.Abbott JD, Huang Y, Liu D, Hickey R, Krause DS. and Giordano FJ. (2004). Stromal cell-derived factor-1alpha plays a critical role in stem cell recruitment to the heart after myocardial infarction but is not sufficient to induce homing in the absence of injury. Circulation 110:3300–3305 [DOI] [PubMed] [Google Scholar]
- 50.Dai S, Yuan F, Mu J, Li C, Chen N, Guo S, Kingery J, Prabhu SD, Bolli R. and Rokosh G. (2010). Chronic AMD3100 antagonism of SDF-1alpha-CXCR4 exacerbates cardiac dysfunction and remodeling after myocardial infarction. J Mol Cell Cardiol 49:587–597 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Ghadge SK, Muhlstedt S, Ozcelik C. and Bader M. (2010). SDF-1alpha as a therapeutic stem cell homing factor in myocardial infarction. Pharmacol Ther 129:97–108 [DOI] [PubMed] [Google Scholar]
- 52.Kim DS, Kang SI, Lee SY, Noh KT. and Kim EC. (2014). Involvement of SDF-1 and monocyte chemoattractant protein-1 in hydrogen peroxide-induced extracellular matrix degradation in human dental pulp cells. Int Endod J 47:298–308 [DOI] [PubMed] [Google Scholar]
- 53.Naumann U, Cameroni E, Pruenster M, Mahabaleshwar H, Raz E, Zerwes HG, Rot A. and Thelen M. (2010). CXCR7 functions as a scavenger for CXCL12 and CXCL11. PLoS One 5:e9175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Brinckerhoff CE. and Matrisian LM. (2002). Matrix metalloproteinases: a tail of a frog that became a prince. Nat Rev Mol Cell Biol 3:207–214 [DOI] [PubMed] [Google Scholar]
- 55.Sachidanandan C, Sambasivan R. and Dhawan J. (2002). Tristetraprolin and LPS-inducible CXC chemokine are rapidly induced in presumptive satellite cells in response to skeletal muscle injury. J Cell Sci 115:2701–2712 [DOI] [PubMed] [Google Scholar]
- 56.Porter JD, Guo W, Merriam AP, Khanna S, Cheng G, Zhou X, Andrade FH, Richmonds C. and Kaminski HJ. (2003). Persistent over-expression of specific CC class chemokines correlates with macrophage and T-cell recruitment in mdx skeletal muscle. Neuromuscul Disord 13:223–235 [DOI] [PubMed] [Google Scholar]
- 57.Hirata A, Masuda S, Tamura T, Kai K, Ojima K, Fukase A, Motoyoshi K, Kamakura K, Miyagoe-Suzuki Y. and Takeda S. (2003). Expression profiling of cytokines and related genes in regenerating skeletal muscle after cardiotoxin injection: a role for osteopontin. Am J Pathol 163:203–215 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Stebler J, Spieler D, Slanchev K, Molyneaux KA, Richter U, Cojocaru V, Tarabykin V, Wylie C, Kessel M. and Raz E. (2004). Primordial germ cell migration in the chick and mouse embryo: the role of the chemokine SDF-1/CXCL12. Dev Biol 272:351–361 [DOI] [PubMed] [Google Scholar]
- 59.Rucci N, Sanita P. and Angelucci A. (2011). Roles of metalloproteases in metastatic niche. Curr Mol Med 11:609–622 [DOI] [PubMed] [Google Scholar]
- 60.Raffo D, Pontiggia O. and Simian M. (2011). Role of MMPs in metastatic dissemination: implications for therapeutic advances. Curr Pharm Biotechnol 12:1937–1947 [DOI] [PubMed] [Google Scholar]
- 61.McQuibban GA, Gong JH, Wong JP, Wallace JL, Clark-Lewis I. and Overall CM. (2002). Matrix metalloproteinase processing of monocyte chemoattractant proteins generates CC chemokine receptor antagonists with anti-inflammatory properties in vivo. Blood 100:1160–1167 [PubMed] [Google Scholar]
- 62.Overall CM, Tam E, McQuibban GA, Morrison C, Wallon UM, Bigg HF, King AE. and Roberts. CR. (2000). Domain interactions in the gelatinase A.TIMP-2.MT1-MMP activation complex. The ectodomain of the 44-kDa form of membrane type-1 matrix metalloproteinase does not modulate gelatinase A activation. J Biol Chem 275:39497–39506 [DOI] [PubMed] [Google Scholar]
- 63.Van Lint P. and Libert C. (2007). Chemokine and cytokine processing by matrix metalloproteinases and its effect on leukocyte migration and inflammation. J Leukoc Biol 82:1375–1381 [DOI] [PubMed] [Google Scholar]
- 64.Hatfield KJ, Reikvam H. and Bruserud O. (2010). The crosstalk between the matrix metalloprotease system and the chemokine network in acute myeloid leukemia. Curr Med Chem 17:4448–4461 [DOI] [PubMed] [Google Scholar]
- 65.Chinni SR, Sivalogan S, Dong Z, Filho JC, Deng X, Bonfil RD. and Cher ML. (2006). CXCL12/CXCR4 signaling activates Akt-1 and MMP-9 expression in prostate cancer cells: the role of bone microenvironment-associated CXCL12. Prostate 66:32–48 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.