

Abstract
Addictive drugs have in common that they target the mesocoticolimbic dopamine (DA) system. This system originates in the ventral tegmental area (VTA) and projects mainly to the nucleus accumbens (NAc) and prefrontal cortex (PFC). Here we review the effects that such drugs leave on glutamatergic and GABAergic synaptic transmission in these three brain areas. We refer to these changes as drug-evoked synaptic plasticity, which outlasts the presence of the drug in the brain and contributes to the reorganization of neural circuits. While in most cases these early changes are not sufficient to induce the disease, with repetitive drug exposure, they may add up and cause addictive behavior.
From acute drug effects to persistent synaptic adaptations
Addictive drugs mediate their reinforcing properties by targeting the mesocorticolimbic dopamine (DA) system, which we define as including the ventral tegmental area (VTA) and its major targets, the nucleus accumbens (NAc) and prefrontal cortex (PFC). Despite their chemical diversity and individual molecular targets, all addictive drugs have in common that they increase DA concentrations in projection areas of the VTA as well as the VTA itself (Di Chiara and Imperato, 1988; Nestler, 2005). In brief (for a more extensive review on the pharmacology of addictive drugs see review by D. Sulzer in the present issue of Neuron and (Lüscher and Ungless, 2006), nicotine can directly increase firing of DA neurons through α4β2 containing nicotinic receptors that are expressed on DA neurons (Maskos et al., 2005). Opioids (Johnson and North, 1992), cannabinoids (Szabo et al., 2002), the club-drugγ-hydoxybutyrate (GHB) (Cruz et al., 2004) and benzodiazepines (Tan et al., 2010) primarily target GABAergic interneurons in the VTA and decrease their activity, which leads to an indirect increase of DA neuron activity. Such disinhibition can occur because of the cell-type specific expression of the respective receptors (e.g. μ-opioid receptors are expressed on GABA- but not on DA neurons) or because GABA neurons are more sensitive to the drug than DA neurons. Benzodiazepines for example primarily silence interneurons because unitary GABA-A receptor-mediated currents in these cells are larger than in DA neurons. This observation correlates with the interneuron-specific expression of the α1 receptor subunit isoform (Tan et al., 2010). Finally, the psychostimulants cocaine, amphetamines and ecstasy, target the DA transporter (DAT), which is normally responsible for the reuptake of DA (Sulzer et al., 2005). Since midbrain DA neurons also release DA from their dendrites (Cheramy et al., 1981; Beckstead et al., 2004), DAT inhibition causes an increase of DA in the VTA as well as in the NAc and PFC. Important mechanistic differences exist between the individual members of this class. Cocaine directly inhibits the DAT, while amphetamines are transporter substrates that are taken up into the cell to enhance non-vesicular release of DA. Psychostimulants have in common that they decrease the firing rate of DA neurons through D2 receptor mediated autoinhibition (Groves et al., 1975) (Chen and Reith, 1994). The Gi/o coupled D2 receptors hyperpolarize DA neurons by activating GIRK/Kir3 channels. Because the block of DA reuptake exceeds the consequences of reducing DA cell firing frequency there is nevertheless a net increase of ambient DA.
These initial actions of addictive drugs have been extensively studied and remain important targets for possible therapeutic interventions in the treatment of addiction1 (Fig. 1). For example, the drug varenicline, which is used in the treatment of nicotine addiction, targets nicotinic receptors on VTA DA neurons (Coe et al., 2005). However, the acute actions of drugs of abuse dissipate as the drug leaves the brain and therefore, alone, cannot explain the development of addictive behaviors. To understand addiction we must elucidate the specific traces the drug experience leaves in the brain and which of these are causally related to the development of addiction. Here, we will focus on some of the key synaptic adaptations that occur after single or repetitive exposures to an addictive drug. This focus is based on the assumption that like virtually all forms of adaptive experience-dependent plasticity, the neural circuit adaptations that underlie drug-induced behavioral changes will involve drug-induced synaptic changes. Indeed, converging evidence from many studies suggests that addictive drugs modify synaptic transmission in the mesocorticolimbic DA system by hijacking mechanisms normally used for adaptive forms of experience-dependent synaptic plasticity; hence the term “drug-evoked synaptic plasticity”. However, the term should not imply that drug exposure alone is necessarily sufficient to elicit synaptic plasticity. On the contrary, many forms of drug-evoked synaptic plasticity appear to depend on the context in which the drug has been experienced, presumably because the final synaptic adaptation will depend both on the molecular action of the drug and the pattern of neural activity in the brain at the time the drug is experienced. It is also important to note that a single drug experience is certainly not sufficient to induce addiction. However, the synaptic and neural circuit adaptations caused by a drug experience often persist and lay the foundation upon which further drug-induced adaptations occur.
Figure 1. The mesocorticolimbic dopamine system as target of addictive drugs.
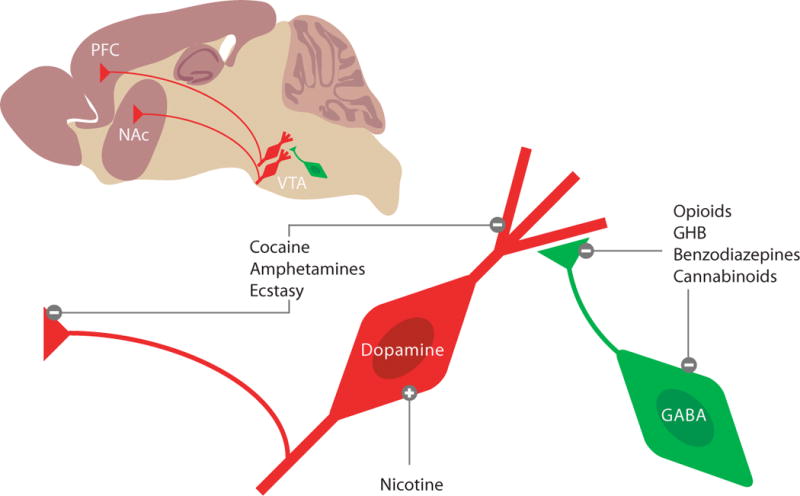
On a saggital slice, the vental tegmental area (VTA), the nucleus accumbens (NAc) and the prefrontal cortex (PFC) can be visualized. The projection neurons are mostly dopaminergic, and under inhibitory control of local GABA neurons (i.e. interneurons). Enlaged schematics to illustrate three cellular mechanims, by which addictive drugs increase mesolimbic DA levels. Nicotine can directly depolarize DA neurons, while opioids, GHB, benzodiazepines and cannabinoids act indirectly via pre- and postsynaptic inhibition of interneurons (i.e. disinhibition). Cocaine, amphetamines and ecstasy target the dopamine transporter (DAT) on axon terminals as well as on dendrites of DA neurons. While cocaine act as an inhibitor of the DAT, promote amphetamines and ecstasy non-vesicular release. In both cases DA levels in the VTA, NAc and PFC increase.
Focusing on the mesocorticolimbic DA system makes sense not only because it is well established to be a major site of action of addictive drugs but also because it has long been considered a structure that is essential for translating motivations into goal-directed actions (Phillips et al., 2003; Zweifel et al., 2009). The synaptic adaptations within this system that occur in response to addictive drugs and how these may contribute to addiction-related behaviors in rodent models have been the subject of a number of recent reviews (Kauer and Malenka, 2007; Thomas et al., 2008; Russo et al., 2010; Kalivas et al., 2009; Schmidt and Pierce, 2010; Wolf and Ferrario, 2010; Bowers et al., 2010). Rather than comprehensively reviewing this same material, we will instead highlight some of the most salient findings with an emphasis on the most recent results that point to important avenues of future research. We will also exhibit the strong bias that changes in synaptic function can best be assayed using electrophysiological techniques. Biochemical and imaging based measurements certainly provide important information that is critical for understanding the mechanisms underlying the synaptic and circuit adaptations caused by drugs of abuse. However, by definition, changes in synaptic function can only be unequivocally provided by directly measuring function and this requires recording the postsynaptic responses to afferent stimulation.
Molecular mechanisms of drug-evoked plasticity
VTA
Excitatory transmission
In the 90’s, based primarily on pharmacological manipulations and biochemical assays as well as the excitement generated by the elucidation of some of the mechanisms and putative function of hippocampal LTP and LTD, it was proposed that modifications of excitatory synaptic transmission in the mesolimbic DA system are important for the neural circuit adaptations underlying some of the long-lasting behavioral consequences of administration of drugs of abuse, in particular psychostimulants (Overton et al., 1999; Wolf, 1998; Kalivas, 1995). A direct test of this hypothesis was subsequently performed and resulted in the first characterization of a form of drug-evoked synaptic plasticity (Ungless et al., 2001). The experimental approach, now standard, involved preparing acute midbrain slices from an animal that 24 hours earlier had received a single non-contingent injection of cocaine (or saline) and recording from VTA DA neurons. As a surrogate measure of synaptic strength, the authors measured the ratio of the AMPA receptor-mediated ESPC (AMPAR EPSC) to the NMDA receptor-mediated EPSC (NMDAR/EPSC); the so-called AMPAR/NMDAR ratio. This ratio was significantly increased for approximately a week following the injection of cocaine and several electrophysiological measures suggested that it was caused, at least in part, by an increase in the AMPAR EPSCs. A recent study reexamined this question by measuring unitary synaptic responses evoked by a highly localized glutamate source (2 photon photolysis of caged glutamate, Mameli et al. 2011 in press). The study concludes that both AMPAR transmission and NMDAR transmission are altered. A population of synapses shows strong rectification of the AMPAR-EPSC along with a decrease of the amplitude of the NMDAR-EPSC. Consistent with this conclusion, the magnitude of LTP at these synapses was reduced and that of LTD increased (Ungless et al., 2001; Dong et al., 2004). This transient enhancement of excitatory synaptic strength in VTA DA cells also occurs following administration of other addictive drugs, including morphine, nicotine, ethanol, and benzodiazepines, but not with non-addictive psychoactive substances such as fluoxetine or carbamazepine (Saal et al., 2003)2. Furthermore, prolonged self-administration of cocaine, unlike passive injections of cocaine or self-administration of food or sucrose, elicits an increase in the AMPAR/NMDAR ratio lasting 3 months (Chen et al., 2008). Cues predicting reward also caused this increase suggesting that this modification of excitatory synaptic function on VTA DA neurons has important roles in shaping behavioral adaptations (Stuber et al., 2008).
Several results suggest that activation of VTA DA neurons alone is sufficient to elicit this form of drug-evoked synaptic plasticity when paired with spontaneous activity. Applying cocaine to isolated midbrain slices caused an increase in the AMPAR/NMDAR ratio in DA cells when measured several hours later (Argilli et al., 2008). In vivo, driving burst firing in DA cells using light activation of channelrhodopsin (ChR2) also caused a synaptic adaptations in DA cells when measured 24 hours later (Brown et al., 2010). The synaptic plasticity induced by both of these manipulations was prevented by pharmacological blockade of D1/D5 receptors in the VTA suggesting that an increase in DA within the VTA is a critical trigger (Schilstrom et al., 2006). Consistent with this conclusion is the finding that this plasticity is not elicited in mice expressing a mutated DAT that is insensitive to cocaine (Brown et al., 2010). An additional induction requirement for this drug-evoked synaptic plasticity is activation of NMDARs on the DA neurons. This conclusion is based on the observations that systemic administration of an NMDAR antagonist blocks the plasticity (Ungless et al., 2001) as does ablation of the critical NMDAR subunit NR1 selectively in DA neurons (Engblom et al., 2008; Zweifel et al., 2008). A simple model that can explain all of these results is that DA receptor activation on VTA DA neurons leads to an increase in NMDAR EPSCs and that this in turn facilitates the generation of NMDAR-dependent LTP (Schilstrom et al., 2006).
The expression mechanisms of the drug-evoked plasticity at excitatory synapses on VTA DA neurons have also been explored. Because the original report of a cocaine-induced increase in the AMPAR/NMDAR ratio suggested that this was due to an upregulation of AMPARs, it was surprising to find that in mice treated with cocaine the AMPAR EPSCs exhibited a partial inward rectification (Bellone and Lüscher, 2006) which is a hallmark of GluA2-lacking AMPARs (Isaac et al., 2007; Liu and Zukin, 2007). The presence of such AMPARs was confirmed by the sensitivity of the EPSCs to external polyamine toxines such as Joro spider toxin as well as an increased single channel conductance estimated using non-stationary fluctuation analysis (Mameli et al., 2007). While immunohistochemical staining with light-microscopic resolution failed to reveal changes in AMPAR subunit expression (Lu et al., 2002), immunogold labeling at the electron microscopy level did show an increase of GluA1 after morphine treatment (Lane et al., 2008) and that cocaine treatment causes a decrease of GluA2 content at synapses (Mameli et al., 2007; Brown et al., 2010). These results are consistent with a scenario whereby GluA2 containing receptors are exchanged for GluA2 lacking ones and no significant increase in the number of synaptic AMPARs occurs. The insertion of GluA2 lacking AMPARs which are highly conductive at negative potentials but carry minimal current at positive potentials can explain an increase of the AMPAR/NMDAR ratio when calculated at -70 mV / +40 mV but not when both components are measured at +40 mV. This raises the possibility that cocaine administration may also decrease the number and/or function of synaptic NMDARs, which is supported by the recent observation of a decrease in unitary NMDAR-EPSCs following cocaine treatment (Mameli et al., 2011). This study also examined the functional consequences for further activity-dependent synaptic plasticity. While in slices an induction protocol that depolarizes DA neurons led to LTP, thus obeying a Hebbien induction rule, this protocol was inefficient after cocaine treatment. Conversely a slight hyperpolarization of the DA neurons during afferent stimulation (i.e. “anti-Hebbian” coincidence) induced a strengthening of AMPAR-transmission only in slices from mice that had received cocaine. Thus, cocaine administration not only causes a lasting change in the basal properties of excitatory synapses but may also inverses of the rules of activity-dependent synaptic plasticity.
As mentioned above drug-evoked synaptic plasticity at this synapse persists for about a week after a single injection. Beyond this time, the receptor redistribution is reversed and the initial state of the synapse restored (Fig. 2). Several lines of evidence implicate metabotropic mGluR1 receptors in this reversal. In fact, pharmacological or synaptic activation of mGluR1 in slices from cocaine treated mice quickly removes GluA2-lacking AMPARs and replaces them by GluA2 containing ones, leading to an overall depression of synaptic transmission (Bellone and Lüscher, 2006; Bellone and Lüscher, 2005). Such mGluR-LTD relies on mammalian target of rapamycin (mToR) signaling and rapid synthesis of GluA2 subunits, via local translation from prefabricated mRNA present in dendrites of DA neurons (Mameli et al., 2007). Moreover interfering with mGluR1 function in vivo by introducing a TAT conjugated dominant negative peptide that disrupts mGluR1-Homer interaction selectively in the VTA significantly prolongs the persistence of cocaine-evoked plasticity (Mameli et al., 2009). Taken together, these results suggest that mGluR1 triggers an endogenous defense mechanism that ensures the removal of calcium permeable AMPARs, which were inserted in response to drug-exposure. It will be important to determine why such a mechanism does not appear to function following prolonged self-administration of cocaine (Chen et al., 2008).
Figure 2. Drug-evoked synaptic plasticity in excitatory synapses onto VTA DA neurons.
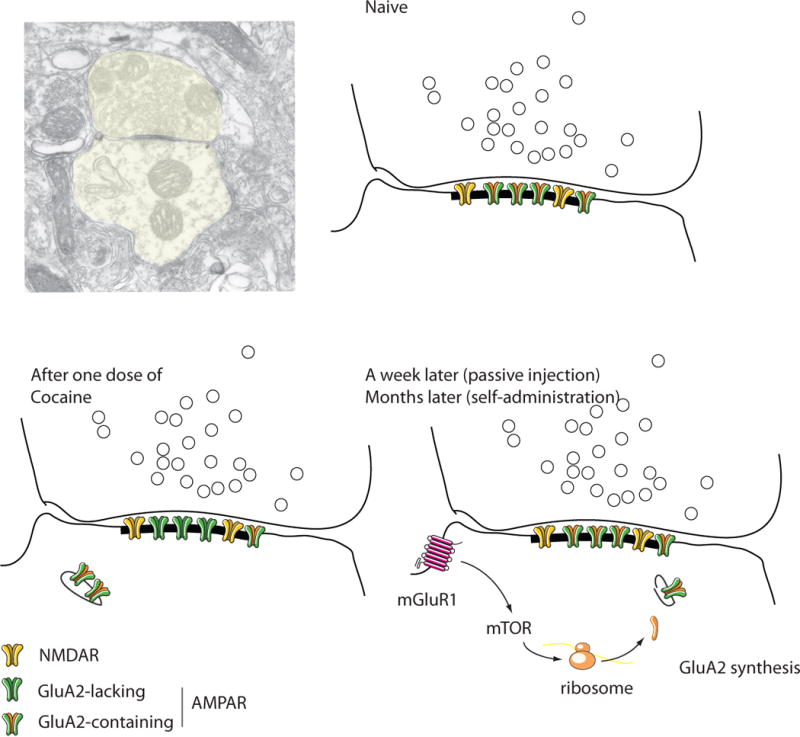
The schematics are drawn from a postembedding EM micrograph of a drug-naive mouse (courtesy Rafael Lujan, Albacete). Note that these asymetrical synapses are made directly onto the shaft of the dendrite (aspiny shaft synapse). In naïve animals NMDARs and AMPARs are present, the latter all containing GluA2. After one dose of cocaine, some GluA2 containing AMPARs are exchanged for GluA2 lacking ones through mechanisms involving endo- and exocytosis. After a week of a passive injection (months after self-administration) the baseline composition is restored through mGluR1 activation, mTOR signaling and de novo synthesis of GluA2 from prefabricated mRNA. Throughout the whole process the total number of receptors remains constant.
Although the drug-evoked synaptic plasticity in VTA DA neurons is robust, recent studies have demonstrated that these cells are not homogeneous but instead exhibit different electrophysiological and molecular properties depending on the specific brain area to which they project (Lammel et al., 2008; Margolis et al., 2006). This distinction between different subtypes of VTA DA neurons may be important because the presence of a large Ih current has commonly been used as a feature to identify DA neurons yet, for example, VTA DA neurons projecting to the medial PFC and the amygdala lack Ih currents and also express low levels of DAT and D2 receptors. Indeed, recent work has found that these mesocortical DA neurons do not exhibit an increase in the AMPAR/NMDAR ratio following single or chronic administration of cocaine but do express an increase following a salient, but aversive stimulus (Lammel and Malenka, 2010). The exclusively DAergic nature of VTA projection neurons has also been questioned. Using optogenetic tools unambiguous functional evidence for co-release of DA and glutamate from VTA projection neurons has been provided (Tecuapetla et al., 2010; Stuber et al., 2010). In addition, pure glutamatergic and GABAergic projection neurons also exist in the VTA (Yamaguchi et al., 2007; Margolis et al., 2008). Whether the activity of the latter is affected by addictive drugs and how co-release of glutamate affects drug action is largely unknown.
An additional important question is whether the drug-evoked synaptic plasticity in VTA DA neurons is induced uniformly at all excitatory inputs onto a given DA neuron. These arise from many origins including the PFC, the amygdala, the lateral hypothalamus and brain stem nuclei such as the pedunculopontine tegmentum and the basal nucleus of the stria terminalis. Optogenetic approaches involving expression of ChR2 in specific brain regions should help provide an answer to this question in the near future. Identifying both the specific synapses that are subject to drug-evoked plasticity and how the site to which DA cells project influence their synaptic adaptations will be a necessary prerequisite to fully understanding the complex neural circuit adaptations caused by drug experiences.
Orexin and CRF modulation of drug-evoked synaptic plasticity
Both corticotropin releasing factor (CRF) and orexin A/hypocretin-1 (oxA/hcrt-1) can modulate drug-evoked synaptic plasticity, presumably through their effect on firing rates of VTA DA neurons and potentiation of NMDAR-mediated synaptic transmission (for a recent review see Borgland et al., 2010). While either neuropeptide, when administered in the VTA, can lead to dopamine release in target regions of the VTA, distinct subpopulations of DA neurons seem to be the target for these substances. Cells that synthesize orexin are located in lateral hypothalamus and project to VTA DA neurons, where they release OxA/hcrt-1 and OxB/hcrt-2. DA neurons in turn express both orexin 1 and orexin 2 receptors and are generally excited by their activation. It has been suggested that orexin preferentially activates caudomedial VTA neurons that primarily project to the PFC and NAc shell (Vittoz et al., 2008) and that this effect contributes to the behaviors elicited by drugs of abuse as well as natural rewards. Indeed, the increase in the AMPAR/NMDAR ratio following in vivo cocaine administration was blocked when an OXR1 antagonist was administered systemically before the cocaine injection (Borgland et al., 2006).
CRF neurons projecting to the VTA are found in the limbic forebrain and the paraventricular nucleus of the hypothalamus (Rodaros et al., 2007). About one quarter of VTA DA neurons, mostly located in the parabrachial pigmented (PBP) region of the VTA express both CRF-R2 and the CRF binding protein (CRF-BP). These are both required for a slowly developing, transient potentiation of NMDAR-mediated synaptic transmission elicited by application of CRF (Ungless et al., 2003). Whether, like oxA/hcrt-1, CRF is important for drug-evoked synaptic plasticity in VTA DA neurons has not been investigated. Furthermore, genetically modified mice have not been used thus far to study the role of these peptide systems in drug-evoked synaptic plasticity.
Inhibitory transmission
Under physiological conditions, these synapses are capable of expressing a form of potentiation called LTP(GABA), which is modulated by addictive drugs (Nugent et al., 2007). NMDAR activation by a train of high frequency stimulation causes a rise in intracellular calcium, which triggers the release of nitric oxide (NO). NO then acts as a retrograde messenger to activate guanylate cyclase, eventually increasing release from GABAergic terminals. In vivo exposure to morphine prevents LTP(GABA) via interruption of NO to guanylate cyclase signaling (Nugent et al., 2007; Nugent et al., 2009). This inhibition of LTP(GABA) can also be observed following cocaine and nicotine administration, albeit with a distinct time course for each of the drugs tested (Niehaus et al., 2010). Inhibition by morphine can be observed within a couple of hours and lasts approximately 5 days. The effects of nicotine wear off quicker, within 24 hours, while the cocaine-mediated inhibition needs more time to be fully established. Ethanol also blocks LTP(GABA), an effect that is reversed by naloxone, a mu-opioid receptor (MOR) antagonist (Guan and Ye, 2010), suggesting that ethanol may exert its block via the opioid system. To understand the consequences of LTP(GABA) inhibition on mesolimbic DA circuitry it will be important to determine the precise connectivity of GABAergic neurons and which specific inputs are being modulated. VTA DA neurons receive inhibitory input from medium spiny neurons of the NAc, the rostromedial tegmental nucleus (RMTg) as well as from interneurons within the VTA, which themselves receive projections form the NAc.
Chronic drug exposure leads to adaptive changes of inhibitory transmission onto VTA DA neurons (Liu et al., 2005). After several daily injections of cocaine baseline inhibitory transmission becomes depressed. The induction requirements of this plasticity remain largely unknown, but appear to involve priming by BDNF (Pu et al., 2006). Its expression relies on a redistribution of GABA-ARs in the postsynaptic membrane. The major consequence of this downregulation of GABAergic transmission has been proposed to be a lower threshold for the induction of subsequent spike-timing dependent LTP at glutamatergic synapses (Liu et al., 2005). However, other groups have found that this form of LTP is reduced following cocaine administration (Luu and Malenka, 2008; Argilli et al., 2008). Once again, to understand the circuit repercussions of these drug-evoked synaptic adaptations it will be crucial to determine which specific projections are modified, the one directly arising from the NAc, the RMTg or local inputs from interneurons.
Nucleus accumbens (NAc)
Over 95% of the cells within the NAc are medium-sized spiny neurons (MSNs), which receive excitatory inputs from four major brain regions, the prefrontal cortex, the ventral subiculum of the hippocampus, the basolateral amygdala and the thalamus (Sesack and Grace, 2010). Surprisingly, little is known about potential differences in the synaptic properties of these different inputs and whether these different synapses are modified in an identical fashion by drugs of abuse during the transition to addiction. This is an important topic given that these different inputs must serve different behavioral functions. Another critical feature of NAc MSNs is that as in the dorsal striatum, they can be divided into two major classes, direct pathway and indirect pathway MSNs with the former primarily expressing D1 DA receptors and the latter D2 DA receptors (Kreitzer and Malenka, 2008). Finally, the NAc contains two subregions known as the core and the shell, which differ in their anatomical connectivity and their presumptive functions. The complexity of NAc circuitry generated by its multiple different inputs, different cell types and different subregions provides a major challenge for understanding the neural circuit modifications that underlie the transition to addiction. Nevertheless, as will be described later in this review, recent advances should allow a much more sophisticated and meaningful approach to this topic.
Excitatory transmission
Analogous to synapses in other brain regions, excitatory synapses in the NAc can express several different forms of synaptic plasticity including NMDAR-dependent LTD and LTP, endocannabinoid-dependent LTD (eCB LTD) as well as a presynaptic form of mGluR-triggered LTD (for reviews see Thomas and Malenka, 2003; Surmeier et al., 2007; Lüscher and Huber, 2010). Similar to the VTA, evidence is accumulating that, in vivo, drugs of abuse can activate or “hijack” some of these synaptic plasticity mechanisms as well as influence the subsequent generation of synaptic plasticity in slices prepared from animals with a history of drug intake. Assuming that plasticity at NAc excitatory synapses has evolved to serve adaptive behavioral functions, drug-induced synaptic adaptations will have important influences on subsequent behaviors.
As was the case for modifications within the VTA, pharmacological and biochemical studies first suggested that alterations in excitatory, glutamatergic synaptic function within the NAc may be important for the behavioral adaptations elicited by administration of drugs of abuse (Vanderschuren and Kalivas, 2000; Wolf, 1998). Direct support for this hypothesis came from electrophysiological studies that used the same strategy as the ex vivo experiments performed in the VTA. Non-contingent administration of cocaine for 5 days caused a decrease in the AMPAR/NMDAR ratio at synapses on NAc MSNs when measured 24h later or after a single challenge dose of cocaine following a 10–14 day withdrawal period (Kourrich 2007; Thomas et al., 2001). This decrease was accompanied by a reduction in the magnitude of NMDAR-dependent LTD. Prolonged cocaine self-administration also reduced excitatory synaptic responses recorded extracellularly in the NAc shell (Schramm-Sapyta et al., 2006) and was associated with an inability to elicit LTD in both the core and the shell (Martin et al., 2006). LTD in the NAc core could still not be elicited even following 21 days of abstinence yet was present in NAc slices prepared from animals that self-administered food or received cocaine passively in a yoked-design (Martin et al., 2006). These results suggest that some specific aspects of the cocaine self-administration protocol results in a long-lasting adaptation in NAc MSNs that impairs LTD.
A confusing aspect of the results summarized thus far is that an extensive body of evidence suggests that activation, not inhibition, of NAc MSNs by prefrontal cortical afferents is important for the reinstatement of drug seeking (Knackstedt and Kalivas, 2009). Indeed, surface levels of AMPARs in the NAc are increased for several weeks during withdrawal following repeated non-contingent cocaine administration (Boudreau et al., 2007; Boudreau and Wolf, 2005) and the AMPAR/NMDAR ratio recorded from NAc shell MSNs was increased 10–14 days following 5 days of repetitive cocaine administration (Kourrich et al., 2007). Importantly, in all these studies, a challenge dose of cocaine was not administered 24 hours before the electrophysiological recordings. A day after a challenge dose of cocaine terminating a protracted withdrawal period both the surface levels of AMPARs and the AMPAR/NMDAR ratio in NAc MSNs are decreased (Boudreau et al., 2007; Kourrich et al., 2007; Wolf and Ferrario, 2010; Bachtell and Self, 2008). Within a week the surface levels of AMPARs recovered and eventually stabilized at an enhanced level.
An additional drug-induced synaptic adaptation in the NAc occurs during protracted (6–7 weeks) withdrawal from cocaine self-administration, a period that is associated with an increase in cue-induced cocaine seeking (a model for craving) (Grimm et al., 2001). While normally synapses on NAc MSNs contain the AMPAR subunit GluA2, rendering AMPARs Ca-impermeable, 6 weeks following cessation of cocaine self-administration, a proportion of synaptic AMPARs now lack GluA2 making them Ca-permeable (Conrad et al., 2008). As discussed below, this change in the stoichiometry of AMPARs has important behavioral ramifications.
Another intriguing finding with potential functional importance is that 1–2 days following repetitive non-contingent cocaine administration, NAc shell MSNs express a relatively large proportion of so-called silent synapses due to the insertion of NR2B-containing NMDARs (Huang et al., 2009). Because silent synapses contain NMDARs but no or very few AMPARs (Malenka and Nicoll, 1997), they are ideal substrates for LTP (Marie et al., 2005) and thus may importantly contribute to the increase in AMPAR surface expression and AMAPR/NMDAR ratios that occur during prolonged withdrawal periods. All of the findings reviewed thus far can be incorporated into a relatively simple model of the changes that occur in excitatory synaptic strength during different time periods of withdrawal/challenge injections from cocaine administration (Fig. 3). As recently pointed out (Russo et al., 2010), these correlate well with the cocaine-elicited structural changes in dendritic spines on NAc MSNs, and will be further discussed below.
Figure 3. Drug-evoked synaptic plasticity in excitatory synapses onto medium spiny neurons in the NAc.

The schematics are drawn from postembedding EM micrograph of a drug-naive mouse (courtesy Rafael Lujan, Albacete). Note that these neurons have prominent spines. In naive animals NMDARs and GluA2-containing AMPARs are present. After several dose of cocaine (or a challenge dose that terminates withdrawal), some AMPARs are endoytosed and the synapse depressed. After weeks of withdrawal, GluA-lacking AMPARs appear, which lead to a potentation of this synapse.
PFC
Only a handful of studies report drug-evoked synaptic plasticity in neurons of the PFC. Applying biochemical methods to PFC tissue, and isolating a synaptic membrane preparation, it was proposed that GluA2 was endocytosed when cues previously associated with heroin self-administration were presented to a rat (Van den Oever et al., 2008). These findings were paralleled by a decrease in the AMPAR/NMDAR ratio measured in acute PFC slices, while self-administration of heroin alone did not affect synaptic strength. Other studies focusing on withdrawal from cocaine performed immunoblotting assays for several synaptic proteins in the dorsal medial PFC and reported an increased expression of NR2B at 14 days, and NR2A at 60 days of withdrawal (Ben-Shahar et al., 2009). Subcellular fractionation techniques revealed a significant increase in the trafficking of these receptors into the synaptosomal compartment (Ghasemzadeh et al., 2009).
More recently, a study observed a gradual increase in the expression of brain-derived neurotrophic factor (BDNF) in the mPFC during acute withdrawal from repetitive cocaine exposure in rats (Lu et al., 2010). This led to a downregulation of GABA-AR-mediated transmission and a subsequent facilitation of activity-induced long-term potentiation (LTP) of excitatory synapses on layer V pyramidal neurons. Taken together, these studies suggest that exposure to addictive drugs also causes glutamate receptor redistribution in PFC neurons, which, given the extensive literature implicating the PFC in drug-related behavior, is not surprising. In the future, direct characterizations of drug-evoked synaptic plasticity in identified synapse will allow for a better understanding of the circuit adaptations in the PFC in response to addictive drugs.
Drug-evoked synaptic plasticity and neural circuit adaptations
To understand the repercussion of drug-evoked synaptic plasticity on the behavior of mesocorticolimbic DA system neural circuitry, synaptic transmission must be studied in a cell-type specific manner on identified inputs. In other words, future studies will have to resolve the origin of the synapses being monitored (i.e. which afferents are being stimulated), the detailed identity of the recorded neurons and the targets to which these neurons project. Approaches such as cell-type specific expression of fluorescent markers, retrograde labeling of neurons using in vivo injection of retrobeads or viruses, and expression of ChR2 or the inhibitory halorhodopsin in restricted sets of afferents by vector delivery into upstream brain areas will have to be applied in combination so that control of identified sets of synapses can be achieved (Witten et al., 2010; Stuber, 2010; Wall et al., 2010; Haubensak et al., 2010; Ciocchi et al., 2010).
An example of the types of studies that will advance our understanding of the neural circuit adaptations that underlie addiction comes from experiments using BAC transgenic mice in which specific subpopulations of cells can be labeled with fluorescent markers (Gong et al., 2003). As mentioned above, NAc MSNs are not homogeneous but, like MSNs in the dorsal striatum, can be divided into two major subpopulations (Kreitzer and Malenka, 2008; Sesack and Grace, 2010; Gertler et al., 2008). Direct pathway MSNs express D1 DA receptors and project directly to midbrain DA areas whereas indirect pathway MSNs expresses D2 DA receptors and project to the ventral pallidum. In the dorsal striatum, there is growing evidence that these two subpopulations of MSNs exhibit different physiological and synaptic properties including different forms of synaptic plasticity (Kreitzer and Malenka, 2008; Shen et al., 2008). Indeed, independent manipulation of the activity of direct and indirect pathway MSNs in the dorsal striatum directly demonstrated that they participate in distinct circuits that mediate dramatically different behaviors (Hikida et al., 2010; Kravitz et al., 2010).
Very little is known about differences in the properties of NAc indirect and direct pathway MSNs. Using targeted recordings from BAC transgenic mice that express fluorescent proteins in one or the other MSN populations (Gong et al., 2003; Shuen et al., 2008), it was recently reported that the basal properties of indirect and direct pathway MSNs in the NAc core differ in a manner quite similar to the differences observed in dorsal striatal MSNs (Kreitzer and Malenka, 2008; Grueter et al., 2010). D2 DA receptor-expressing indirect pathway MSNs are more excitable and the excitatory synapses on these cells exhibit a higher probability of release. Furthermore, while NMDAR-dependent LTD can be generated to the same degree in both subtypes of MSNs, as in the dorsal striatum eCB LTD is generated much more robustly in indirect pathway NAc MSNs (Kreitzer and Malenka, 2007; Grueter et al., 2010). This was surprising giving that eCB LTD in the NAc had been reported previously in slices from wildtype mice in which it was not possible to make targeted recordings (Robbe et al., 2002). However, the techniques used in this previous report, extracellular field recordings, do not allow the source of the postsynaptic responses to be determined. An additional surprising finding was that the eCB LTD in NAc indirect pathway MSNs was not completely blocked by a CB1 receptor antagonist. This observation led to an extensive set of experiments demonstrating that, in addition to standard eCB LTD, NAc indirect pathway MSNs express a novel form of LTD triggered by activation of postsynaptic TRPV1 channels (Grueter et al., 2010). A very similar form of postsynaptic TRPV1-triggered LTD was found in medial perforant path synapses on dentate gyrus granule cells (Chavez et al., 2010) suggesting that this may be a ubiquitous, albeit highly input specific, form of synaptic plasticity.
The potential importance of the restriction of these forms of LTD to one MSN subtype is that in contrast to NMDAR-dependent LTD, the eCB LTD observed in the NAc is dramatically impaired 24 hours following only a single dose of either cocaine or 9-THC (Fourgeaud et al., 2004; Mato et al., 2004). Similarly, the eCB- and TRPV1-dependent LTD in indirect pathway NAc MSNs was absent 24 hours after administration of a single dose of cocaine (Grueter et al., 2010). The inhibition of these forms of LTD by cocaine is consistent with the suggestion that the cocaine treatment impairs postsynaptic mGluR signaling, which is required for the generation of eCB’s in NAc MSNs (Fourgeaud et al., 2004). The behavioral function of these forms of LTD and their block by cocaine is unknown. Interestingly, although basal locomotor activity was normal in knockout mice lacking TRPV1, these mice exhibited an enhanced locomotor response to cocaine that was maintained as behavioral sensitization developed with repeated daily administration of cocaine (Grueter et al., 2010). Assuming that the absence of TRPV1 specifically in NAc indirect pathway MSNs was responsible for the enhanced cocaine-induced locomotor activity, the LTD triggered by TRPV1 may function as a brake on cocaine-elicited locomotor activity. This role is opposite to that proposed for NMDAR-dependent LTD. However, since the TRPV1-dependent LTD is restricted to indirect pathway MSNs while NMDAR-dependent LTD occurs in both subtypes of MSNs, it is not unexpected that these forms of synaptic plasticity may have different behavioral roles.
The results summarized thus far emphasize the importance of defining the cell type in which drug-induced adaptations have occurred. As mentioned above, of equal importance will be defining the source of the synapses being studied. Currently, using ex vivo approaches that involve preparing acute brain slices from animals that have had drug experience; it is very difficult to know definitively which sets of inputs are being activated. For the NAc, these arise primarily from the prefrontal cortex, subiculum or amygdala. Furthermore, with standard techniques, it is impossible to prevent the activation of GABAergic, dopaminergic or other modulatory inputs within the slice. The potential importance of distinguishing the different NAc afferents is exemplified by in vivo experiments during which it is possible to activate specific sets of inputs independently. For example, at hippocampal-NAc synapses, LTP recorded using extracellular field potentials was reported to be impaired 2–3 weeks following repetitive non-contingent cocaine administration whereas LTP at prefrontal-NAc synapses was unaffected (Goto and Grace, 2005). On the other hand, in acute slices prepared two weeks after repetitive cocaine administration, LTP, also assayed using field potentials and generated by presumptive prefrontal-NAc synapses, was modestly enhanced (Yao et al., 2004). Finally, during withdrawal from cocaine self-administration, both LTP and LTD at prefrontal-NAc synapses in vivo were impaired (Moussawi et al., 2009). The difference in these results may reflect that different sets of inputs were in fact stimulated as well as the differences in the drug administration protocols. A significant limitation of using in vivo preparations is the technical difficulty of recording synaptic responses from individual cells. Instead, extracellular field potentials are normally recorded, changes in which are difficult to interpret since the potentials are generated both by synaptic responses and voltage-dependent conductances. A further limitation of in vivo extracellular recordings is the difficulty in performing assays of basal synaptic properties in a manner that permits cross preparation comparisons. Ideally, it would be valuable to combine the advantages of both in vivo and ex vivo approaches while minimizing their limitations. An obvious approach to accomplish this in the service of elucidating the modifications caused by drugs of abuse at known sets of synapses will be to express channelrhodopsin (ChR2) in the specific brain regions that project to NAc in BAC transgenic mice and then prepare acute slices from these animals following in vivo drug exposure. Many labs worldwide, including the authors’ labs, are taking this approach with good success.
It will also be important to understand how the different forms of drug-evoked plasticity in the mesocorticolimbic system relate to each other. Anatomical analysis in rodents and non-human primates suggest that the midbrain and striatal components of the mesocorticolimbic DA system are connected in a spiraling manner (Haber et al., 2000; Ikemoto, 2007). DA neurons of the medial VTA project to the medial shell, from where MSNs back-project to more lateral parts of the VTA. This backprojection most likely involves a relay via an interneuron in the VTA (Maeda and Mogenson, 1981; Xia et al., 2010). These DA neurons then project to the NAc core, from where MSNs backproject to even more lateral VTA, and, after several additional loops, the circuit reaches the substantia nigra (SNc) which projects to the dorsal striatum. The ascending projections are in general dopaminergic along with a glutamatergic component for a subset of neurons that co-release both transmitters (Dobi et al., 2010). NAc MSNs that send GABAergic projections to the VTA are likely to be direct pathway or D1 expressing neurons, but most MSNs reach DA neurons only via inhibitory interneurons located in the VTA. Enhanced activity of MSN neurons may therefore actually excite DA neurons. An appealing hypothesis is that drug-evoked synaptic plasticity reinforces the connectivity within the spiral, which would lead to the recruitment of more and more dorsal projections as drug use becomes chronic. In line with this idea are the observations that a single injection of cocaine triggers drug-evoked synaptic plasticity in VTA DA neurons that project to the medial NAc shell while several injections are require to downregulate inhibitory transmission onto DA neurons.
One study has attempted to directly link early changes induced by cocaine in the VTA (i.e. increased AMPAR/NMDAR ratios and appearance of inward rectification in AMPAR EPSCs) to later cocaine-evoked plasticity in the NAc (i.e. decreased AMPAR/NMDAR ratios followed by inward rectification after prolonged withdrawal) (Mameli et al., 2009). Consistent with a causal link, abolishing the drug-evoked synaptic plasticity only in the VTA by inducible deletion of the obligatory NMDAR subunit NR1 only in DA cells (see above) prevented drug-evoked synaptic changes in the NAc. Furthermore, manipulating the persistence of the cocaine-evoked plasticity in the VTA controlled synaptic plasticity in the NAc. These results are reminiscent of earlier findings suggesting that the induction of psychostimulant-induced behavioral sensitization requires the VTA but that its expression is independent of the VTA and requires modifications within the NAc (Wolf, 1998). They also support the idea that there is a hierarchical relationship between these forms of drug-evoked synaptic plasticity in the early phases of the spiral.
Future studies will have to identify the “master signals” that control this hierarchical organization between VTA and NAc and test whether similar mechanisms control the circuit remodeling in later stages of the “spiral”. Will chronic drug exposure eventually affect transmission in the substantia nigra compacta (SNc) and the dorsal striatum? One study implicated the recruitment of these dorsal parts of the dopamine system in cocaine-seeking habits by demonstrating that the surgical disconnection of the spiral via a lesion of the NAc core, blocked the development of cocaine seeking in rats (Belin and Everitt, 2008). Thus, the spiral appears to be engaged sequentially during the development of addiction. Drug-evoked synaptic plasticity in various parts of the mesoorticolimbic system may constitute the underlying cellular correlate confirming the hypothesis that “staged neuroplasticity” underlies the disease (Kalivas and O’Brien, 2008). A model of the mesocorticolimbic circuitry that is subject to drug-evoked synaptic plasticity is emerging (Fig. 4). While this model is based on a decade’s work, it is apparent that many questions regarding the anatomical connections and precise cellular mechanisms remain unanswered.
Figure 4. Emerging model of mesolimbic circuit known to be affected by drug-evoked synaptic plasticity.
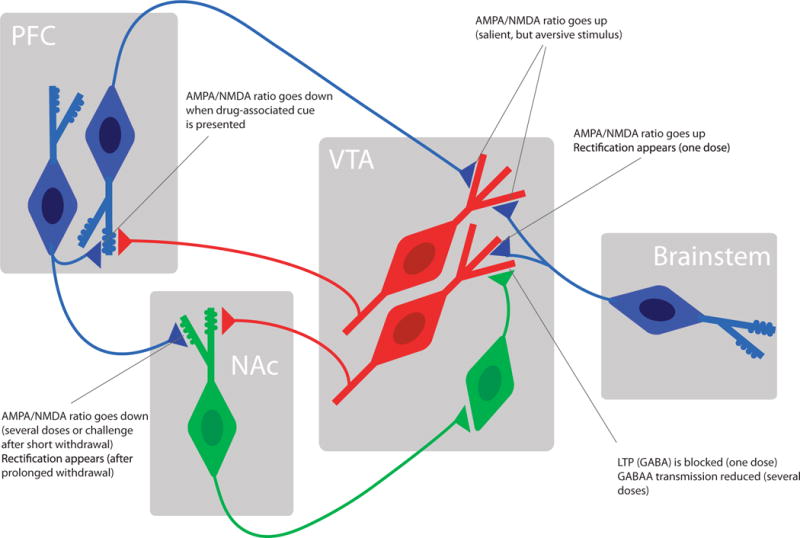
For simplicity several projections have been omitted to highlight the synapses that undergo drug-evoked adaptations. For further explanation see text. Dopamine neurons (red) GABA neurons (green) and glutamate neurons (blue).
Beyond synaptic adptations
Of course, synaptic adaptations are not the only mechanisms by which drugs of abuse can modify mesocorticolimbic DA system circuitry to cause long-lasting behavioral changes. In vivo exposure to addictive substances activate complex intracellular signaling cascades including transcription factors, cause changes in intrinsic membrane excitability, affect dendrite and spine structure, and influence the levels of extracellular glutamate via impairments in cysteine-glutamate exchange. These additional drug-induced adaptations have been the subject of several recent reviews, which attempt to integrate all of these findings into a coherent view of the neural circuit adaptations that mediate addiction (Russo et al., 2010; Kalivas et al., 2009; Wolf, 2010).
Linking synaptic plasticity and behavior
The toughest challenge for all forms of synaptic plasticity is the demonstration of a causal link between a specific synaptic modification and a specific behavior. Indeed, the specific behavioral role of the original form of long-lasting synaptic plasticity, NMDAR-dependent LTP in the hippocampus, is still not unequivocally demonstrated. Space constraints prevent us from reviewing all the behavioral literature implicating drug-evoked synaptic plasticity in addiction-related behaviors. Instead, we will focus on studies that have used ex vivo approaches to correlate drug-elicited behavioral changes with mechanistic investigations of the underlying synaptic plasticity. The behavioral models mimicking core components of drug addiction that have been studied most extensively in this context are self administration, locomotor sensitization, conditioned place preference (CPP), drug-evoked relapse, cue-induced cocaine seeking and incubation of craving.
Given that the VTA has been implicated in both behavioral sensitization and CPP, several studies have attempted to link these drug-induced behavioral changes with drug-evoked plasticity of excitatory transmission in the VTA. Initial findings indeed seemed to confirm the link; pharmacological inhibition of NMDARs in the VTA blocks both the drug-evoked synaptic plasticity and behavioral sensitization (Dunn et al., 2005; Kalivas and Alesdatter, 1993; Vezina and Queen, 2000) as well as CPP (Harris and Aston-Jones, 2003; Harris et al., 2004). Furthermore, the expression via viral vectors of the AMPAR subunit GluA1 in the VTA led to behavioral sensitization in animals that had never been exposed to cocaine (Carlezon and Nestler, 2002). Surprisingly, however, behavioral sensitization to cocaine could still be elicited in GluA1−/− mice even though the drug evoked increase in the AMPAR/NMDAR ratio was abolished (Dong et al., 2004). The GluA1−/− mice did exhibit impaired CPP, which suggested that the drug-evoked plasticity of excitatory synaptic transmission in VTA DA neurons, although not required for behavioral sensitization per se, may contribute to the attribution of incentive value to drug-associated cues.
More selective genetic manipulations, however, call this interpretation into question. Specifically, conditionally deleting the NMDAR subunit NR1 selectively in DA neurons of adult mice caused a loss of NMDAR EPSCs as well as the cocaine-evoked synaptic plasticity yet behavioral sensitization and CPP were still normal (Engblom et al., 2008). A behavioral repercussion of this genetic manipulation became apparent only during the withdrawal period following drug self-administration, a time period during which the normal reinstatement of self-administration triggered by a priming dose of cocaine was abolished or when cue-induced cocaine seeking after more than a month of withdrawal was significantly reduced (Mameli et al., 2009). The postnatal, rather than constitutive, deletion of NR1 from DA neurons may explain an apparent difference with another study in which mice with a constitutive removal of functional NMDARs from DA neurons exhibited impaired CPP (Zweifel et al., 2008). In these mice the absence of NMDARs during development led to an increase/decrease in AMPAR EPSCs, presumably due to a form of synaptic scaling (Adesnik et al., 2008). Interestingly in the same DATCre-NR1 mouse behavioral sensitization was abolished when the NMDAR-antagonist AP5 was directly applied into the VTA, implicating NMDARs on non-DA neurons in this behavior (Luo et al., 2010).
The behavioral relevance of drug-evoked synaptic plasticity in the NAc has been examined in several studies. To test whether LTD in the NAc was required for amphetamine-induced behavioral sensitization, a membrane permeable peptide that prevents endocytosis of AMPARs and thus NMDAR-dependent LTD was infused into the NAc of sensitized rats immediately prior to a challenge dose of amphetamine (Brebner et al., 2005). This manipulation prevented the increase in locomotor activity normally elicited by amphetamine suggesting that drug-elicited LTD in the NAc is required for the expression of behavioral sensitization. More recently, the behavioral importance of the impairment of NMDAR-dependent LTD in the NAc caused by cocaine has been addressed by taking advantage of the observation that following prolonged cocaine self-administration, only a modest proportion of rats develop behaviors analogous to human addicts (Deroche-Gamonet et al., 2004; Kasanetz et al., 2010). Although approximately two weeks following the cessation of cocaine self-administration, LTD was impaired in all animals, the ability to generate LTD slowly recovered in “non-addicted” animals. In contrast, “addicted” animals expressed persistently impaired LTD (Kasanetz et al., 2010). If LTD in the NAc is indeed important for adaptive forms of behavioral plasticity, its long lasting impairment may contribute to the inflexible, compulsive behaviors that are a hallmark of addiction.
Another study presented strong evidence that synaptic adaptations within the NAc are important for cue-induced reinstatement of drug self-administration. Withdrawal after prolonged cocaine-self administration is associated with a so-called incubation of craving (Grimm et al., 2001). Specifically, as time passes during the withdrawal period, the presentation of a cue previously associated with the availability of drug leads to increasing efforts (i.e. number of lever presses) to obtain the drug. Over approximately this same time period, the initial depression of excitatory transmission in the NAc reverses into a potentiation and the appearance of calcium permeable AMPARs (see above). The behavioral importance of this change in the stoichiometry of AMPAs was directly tested by infusing into the NAc a compound that blocks GluA2-lacking AMPARs (Conrad et al., 2008). This manipulation prevented the enhanced cue-elicited drug seeking that normally occurs 6–7 weeks after withdrawal from cocaine self-administration. Importantly, infusing this compound into the NAc had no effect on cue-elicited drug seeking the first day of withdrawal nor on sucrose self-administration. Because GluA2-lacking AMPARs have a higher conductance than GluA2-containing AMPARs and this withdrawal period is associated with an increase in the AMPAR/NMDAR ratio, these findings are consistent with a general increase in excitatory synaptic strength in NAc MSNs during prolonged withdrawal from either self-administered or non-contingent administered cocaine (Russo et al., 2010; Kalivas et al., 2009; Wolf, 2010).
Although synaptic adaptations were not examined, a study worth mentioning directly tested the behavioral roles of NAc indirect and direct pathway MSNs (Lobo et al., 2010). Optogenetic activation of D1 receptor-expressing direct pathway NAc MSNs dramatically enhanced the rewarding effects of a threshold dose of cocaine in a conditioned place preference protocol whereas activation of D2 receptor-expressing indirect pathway NAc MSNs dramatically attenuated cocaine reward. Other molecular manipulations of direct versus indirect pathway MSNs in the striatum also support the hypothesis that the specific circuits in which these two subpopulations of MSNs participate have profoundly different behavioral roles in addiction related behaviors (Hikida et al., 2010; Bateup et al., 2010).
Conclusions
We have reviewed a growing body of evidence indicating that drug-evoked synaptic plasticity in the mesocorticolimbic DA system is common to all addictive drugs and is rooted in their common pharmacological effect of increasing DA in specific target structures. An appealing hypothesis therefore posits that drug-evoked changes in the VTA may constitute an initial permissive step for changes in the NAc and PFC, which then mediate many of the core behavioral changes that define addiction. The well-documented of the different classes of addictive drugs on other brain regions may in turn mediate specific symptoms that make the addiction to a specific drug unique. This model proposes a hierarchical system of several forms of nested drug-evoked plasticity, which in part underlie the circuit reorganization that eventually leads to a transition from thoughtful, balanced decisions to the automatic, compulsive decisions, which are a hallmark of addiction. Future studies will have to work out the functional connectivity and identify the signals orchestrating the various forms of drug-evoked plasticity. Of equal importance will be determining the basis for the individual vulnerability to develop addiction. Only with a more sophisticated understanding of these topics will mechanistically solid and clinically efficient treatments become available.
Table I.
Key forms of drug-evoked synaptic plasticity
| Brain area | Postsynaptic cell | Transmissi on | Drug treatment required | Induction | Expression | Examples In the literature3 |
|---|---|---|---|---|---|---|
| VTA | DA neurons | excitatory | 1 injection of cocaine, and other addictive drugs, as well as acute stress | Activation of NMDAR on DA neurons and D1-like receptors, presumably on excitatory afferents | Increase of AMPAR/NMDAR and rectification: insertion of GluA2-lacking AMPARs and decrease of NMDAR function | (Ungless et al., 2001) (Saal et al., 2003) (Bellone and Lüscher, 2006) (Mameli et al., 2011) |
| DA neurons | inhibitory | 1 injection of morphine, cocaine, nicotine and stress | Nitric oxyde | Decreased GABA release. | (Nugent et al., 2007) (Nugent et al., 2009) | |
| DA neurons | excitatory | Only present in slices of mice that have received cocaine injection | mGluR1 | Removel of GluA2-lacking AMPARs | (Bellone & Lüscher, 2006) (Mameli et al., 2007) | |
| DA neurons | inhibitory | 5 daily injections of cocaine | BDNF | Decrease of the number of GABAA receptors | (Liu et al., 2005) | |
| NAc | MSN | excitatory | Weeks after several days of cocaine self-administration (incubation of craving) | GluA2 lacking AMPARs | (Conrad et al., 2008) | |
| MSN | excitatory | 7 injections of cocaine, followed by one week of withdrawal & one challenge injection | Decrease of AMPAR/NM DAR ratio | NMDAR dependent LTD, silent synapses | (Thomas et al., 2001) | |
| MSN | excitatory | 5 daily injections, followed by 10–14 days of withdrawal | Increase of AMPAR/NMDAR ratio | (Kourrich et al., 2007) | ||
| MSN | excitatory | Several injections of cocaine inhibit mGluR5 LTD Also 24h after a single exposure to cocaine |
Post mGluR5 | Presynaptic via endocannabinoids | (Moussawi et al., 2009) | |
| MSN (D2 expressing) | excitatory | Blocked 24h after oneinjection of cocaine | TrpV1 channel | Postsynaptic | (Grueter 2010) | |
| Medial PFC | Pyramidal neurons | excitatory | 3 week withdrawal and then cue presentations | Cue induced decrease AMPAR/NMDAR | (Van den Oever et al., 2008) | |
| Layer V pyramidal neurons | Inhibitory/excit atory | 7 daily injections, followed by 5–17 days of withdrawal | BDNF-dependent decrease of GABAA transmission | facilitated LTP | (Lu et al., 2010) |
Acknowledgments
We thank members of both our labs for critically reading earlier versions of the manuscript. We are indebted to Rafael Lujan, Albacete for providing the EM images. This work has been made possible, in part, by the University of Geneva’s program for sabbatical leaves and CL’s appointment as a visiting scholar at Stanford University.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Addiction is normally defined as the compulsive use of a drug despite the negative consequences. An addictive drug has the potential to induce the disease, but does so only in a fraction of consumers. Here we focus on the initial adaptive changes required, but not sufficient, to cause addition (Redish et al., 2008).
The case however seems more complicated for ethanol. When comparing two mouse strains, one injection of ethanol did not affect the AMPAR/NMDAR ratio in C57BL/6, and led to a decrease in DAB mice (Wanat et al., 2009). The authors propose that in DAB a decreased NMDAR-signaling is at the origin of this adaptation.
The cited literature here is not exhaustive. We apologize for not being able to cite all the work because of space constraints.
References
- Adesnik H, Li G, During MJ, Pleasure SJ, Nicoll RA. NMDA receptors inhibit synapse unsilencing during brain development. Proc Natl Acad Sci U S A. 2008;105:5597–5602. doi: 10.1073/pnas.0800946105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Argilli E, Sibley DR, Malenka RC, England PM, Bonci A. Mechanism and time course of cocaine-induced long-term potentiation in the ventral tegmental area. J Neurosci. 2008;28:9092–9100. doi: 10.1523/JNEUROSCI.1001-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bachtell RK, Self DW. Renewed cocaine exposure produces transient alterations in nucleus accumbens AMPA receptor-mediated behavior. J Neurosci. 2008;28:12808–12814. doi: 10.1523/JNEUROSCI.2060-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bateup HS, Santini E, Shen W, Birnbaum S, Valjent E, Surmeier DJ, Fisone G, Nestler EJ, Greengard P. Distinct subclasses of medium spiny neurons differentially regulate striatal motor behaviors. Proc Natl Acad Sci U S A. 2010;107:14845–14850. doi: 10.1073/pnas.1009874107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beckstead MJ, Grandy DK, Wickman K, Williams JT. Vesicular dopamine release elicits an inhibitory postsynaptic current in midbrain dopamine neurons. Neuron. 2004;42:939–946. doi: 10.1016/j.neuron.2004.05.019. [DOI] [PubMed] [Google Scholar]
- Belin D, Everitt BJ. Cocaine seeking habits depend upon dopamine-dependent serial connectivity linking the ventral with the dorsal striatum. Neuron. 2008;57:432–441. doi: 10.1016/j.neuron.2007.12.019. [DOI] [PubMed] [Google Scholar]
- Bellone C, Lüscher C. mGluRs induce a long-term depression in the ventral tegmental area that involves a switch of the subunit composition of AMPA receptors. Eur J Neurosci. 2005;21:1280–1288. doi: 10.1111/j.1460-9568.2005.03979.x. [DOI] [PubMed] [Google Scholar]
- Bellone C, Lüscher C. Cocaine triggered AMPA receptor redistribution is reversed in vivo by mGluR-dependent long-term depression. Nat Neurosci. 2006;9:636–641. doi: 10.1038/nn1682. [DOI] [PubMed] [Google Scholar]
- Ben-Shahar O, Obara I, Ary AW, Ma N, Mangiardi MA, Medina RL, Szumlinski KK. Extended daily access to cocaine results in distinct alterations in Homer 1b/c and NMDA receptor subunit expression within the medial prefrontal cortex. Synapse. 2009;63:598–609. doi: 10.1002/syn.20640. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Borgland SL, Taha SA, Sarti F, Fields HL, Bonci A. Orexin A in the VTA is critical for the induction of synaptic plasticity and behavioral sensitization to cocaine. Neuron. 2006;49:589–601. doi: 10.1016/j.neuron.2006.01.016. [DOI] [PubMed] [Google Scholar]
- Borgland SL, Ungless MA, Bonci A. Convergent actions of orexin/hypocretin and CRF on dopamine neurons: Emerging players in addiction. Brain Res. 2010;1314:139–144. doi: 10.1016/j.brainres.2009.10.068. [DOI] [PubMed] [Google Scholar]
- Boudreau AC, Wolf ME. Behavioral sensitization to cocaine is associated with increased AMPA receptor surface expression in the nucleus accumbens. J Neurosci. 2005;25:9144–9151. doi: 10.1523/JNEUROSCI.2252-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boudreau AC, Reimers JM, Milovanovic M, Wolf ME. Cell surface AMPA receptors in the rat nucleus accumbens increase during cocaine withdrawal but internalize after cocaine challenge in association with altered activation of mitogen-activated protein kinases. J Neurosci. 2007;27:10621–10635. doi: 10.1523/JNEUROSCI.2163-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bowers MS, Chen BT, Bonci A. AMPA receptor synaptic plasticity induced by psychostimulants: the past, present, and therapeutic future. Neuron. 2010;67:11–24. doi: 10.1016/j.neuron.2010.06.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brebner K, Wong TP, Liu L, Liu Y, Campsall P, Gray S, Phelps L, Phillips AG, Wang YT. Nucleus accumbens long-term depression and the expression of behavioral sensitization. Science. 2005;310:1340–1343. doi: 10.1126/science.1116894. [DOI] [PubMed] [Google Scholar]
- Brown MT, Bellone C, Mameli M, Labouebe G, Bocklisch C, Balland B, Dahan L, Lujan R, Deisseroth K, Lüscher C. Drug-driven AMPA receptor redistribution mimicked by selective dopamine neuron stimulation. PLoS ONE. 2010;5:e15870. doi: 10.1371/journal.pone.0015870. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carlezon WA, Jr, Nestler EJ. Elevated levels of GluR1 in the midbrain: a trigger for sensitization to drugs of abuse? Trends Neurosci. 2002;25:610–615. doi: 10.1016/s0166-2236(02)02289-0. [DOI] [PubMed] [Google Scholar]
- Chavez AE, Chiu CQ, Castillo PE. TRPV1 activation by endogenous anandamide triggers postsynaptic long-term depression in dentate gyrus. Nat Neurosci. 2010;13:1511–1518. doi: 10.1038/nn.2684. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen BT, Bowers MS, Martin M, Hopf FW, Guillory AM, Carelli RM, Chou JK, Bonci A. Cocaine but not natural reward self-administration nor passive cocaine infusion produces persistent LTP in the VTA. Neuron. 2008;59:288–297. doi: 10.1016/j.neuron.2008.05.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen NH, Reith ME. Autoregulation and monoamine interactions in the ventral tegmental area in the absence and presence of cocaine: a microdialysis study in freely moving rats. J Pharmacol Exp Ther. 1994;271:1597–1610. [PubMed] [Google Scholar]
- Cheramy A, Leviel V, Glowinski J. Dendritic release of dopamine in the substantia nigra. Nature. 1981;289:537–542. doi: 10.1038/289537a0. [DOI] [PubMed] [Google Scholar]
- Ciocchi S, Herry C, Grenier F, Wolff SB, Letzkus JJ, Vlachos I, Ehrlich I, Sprengel R, Deisseroth K, Stadler MB, Muller C, Luthi A. Encoding of conditioned fear in central amygdala inhibitory circuits. Nature. 2010;468:277–282. doi: 10.1038/nature09559. [DOI] [PubMed] [Google Scholar]
- Coe JW, Brooks PR, Vetelino MG, Wirtz MC, Arnold EP, Huang J, Sands SB, Davis TI, Lebel LA, Fox CB, Shrikhande A, Heym JH, Schaeffer E, Rollema H, Lu Y, Mansbach RS, Chambers LK, Rovetti CC, Schulz DW, Tingley FDr, O’Neill BT. Varenicline: an alpha4beta2 nicotinic receptor partial agonist for smoking cessation. J Med Chem. 2005;48:3474–3477. doi: 10.1021/jm050069n. [DOI] [PubMed] [Google Scholar]
- Conrad KL, Tseng KY, Uejima JL, Reimers JM, Heng LJ, Shaham Y, Marinelli M, Wolf ME. Formation of accumbens GluR2-lacking AMPA receptors mediates incubation of cocaine craving. Nature. 2008;454:118–121. doi: 10.1038/nature06995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cruz HG, Ivanova T, Lunn ML, Stoffel M, Slesinger PA, Lüscher C. Bi-directional effects of GABA(B) receptor agonists on the mesolimbic dopamine system. Nat Neurosci. 2004;7:153–159. doi: 10.1038/nn1181. [DOI] [PubMed] [Google Scholar]
- Deroche-Gamonet V, Belin D, Piazza PV. Evidence for addiction-like behavior in the rat. Science. 2004;305:1014–1017. doi: 10.1126/science.1099020. [DOI] [PubMed] [Google Scholar]
- Di Chiara G, Imperato A. Drugs abused by humans preferentially increase synaptic dopamine concentrations in the mesolimbic system of freely moving rats. Proc Natl Acad Sci U S A. 1988;85:5274–5278. doi: 10.1073/pnas.85.14.5274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dobi A, Margolis EB, Wang HL, Harvey BK, Morales M. Glutamatergic and nonglutamatergic neurons of the ventral tegmental area establish local synaptic contacts with dopaminergic and nondopaminergic neurons. J Neurosci. 2010;30:218–229. doi: 10.1523/JNEUROSCI.3884-09.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dong Y, Saal D, Thomas M, Faust R, Bonci A, Robinson T, Malenka RC. Cocaine-induced potentiation of synaptic strength in dopamine neurons: behavioral correlates in GluRA(−/−) mice. Proc Natl Acad Sci U S A. 2004;101:14282–14287. doi: 10.1073/pnas.0401553101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dunn JM, Inderwies BR, Licata SC, Pierce RC. Repeated administration of AMPA or a metabotropic glutamate receptor agonist into the rat ventral tegmental area augments the subsequent behavioral hyperactivity induced by cocaine. Psychopharmacology (Berl) 2005;179:172–180. doi: 10.1007/s00213-004-2054-9. [DOI] [PubMed] [Google Scholar]
- Engblom D, Bilbao A, Sanchis-Segura C, Dahan L, Perreau-Lenz S, Balland B, Rodriguez Parkitna J, Lujan R, Halbout B, Mameli M, Parlato R, Sprengel R, Lüscher C, S G, Spanagel R. Glutamate receptors on dopamine neuons control the persistence of cocaine seeking. Neuron. 2008;59:497–508. doi: 10.1016/j.neuron.2008.07.010. [DOI] [PubMed] [Google Scholar]
- Fourgeaud L, Mato S, Bouchet D, Hemar A, Worley PF, Manzoni OJ. A single in vivo exposure to cocaine abolishes endocannabinoid-mediated long-term depression in the nucleus accumbens. J Neurosci. 2004;24:6939–6945. doi: 10.1523/JNEUROSCI.0671-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gertler TS, Chan CS, Surmeier DJ. Dichotomous anatomical properties of adult striatal medium spiny neurons. J Neurosci. 2008;28:10814–10824. doi: 10.1523/JNEUROSCI.2660-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ghasemzadeh MB, Windham LK, Lake RW, Acker CJ, Kalivas PW. Cocaine activates Homer1 immediate early gene transcription in the mesocorticolimbic circuit: differential regulation by dopamine and glutamate signaling. Synapse. 2009;63:42–53. doi: 10.1002/syn.20577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gong S, Zheng C, Doughty ML, Losos K, Didkovsky N, Schambra UB, Nowak NJ, Joyner A, Leblanc G, Hatten ME, Heintz N. A gene expression atlas of the central nervous system based on bacterial artificial chromosomes. Nature. 2003;425:917–925. doi: 10.1038/nature02033. [DOI] [PubMed] [Google Scholar]
- Goto Y, Grace AA. Dopamine-dependent interactions between limbic and prefrontal cortical plasticity in the nucleus accumbens: disruption by cocaine sensitization. Neuron. 2005;47:255–266. doi: 10.1016/j.neuron.2005.06.017. [DOI] [PubMed] [Google Scholar]
- Grimm JW, Hope BT, Wise RA, Shaham Y. Neuroadaptation. Incubation of cocaine craving after withdrawal Nature. 2001;412:141–142. doi: 10.1038/35084134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Groves PM, Wilson CJ, Young SJ, Rebec GV. Self-inhibition by dopaminergic neurons. Science. 1975;190:522–528. doi: 10.1126/science.242074. [DOI] [PubMed] [Google Scholar]
- Grueter BA, Brasnjo G, Malenka RC. Postsynaptic TRPV1 triggers cell type-specific long-term depression in the nucleus accumbens. Nat Neurosci. 2010;13:1519–1525. doi: 10.1038/nn.2685. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guan YZ, Ye JH. Ethanol blocks long-term potentiation of GABAergic synapses in the ventral tegmental area involving mu-opioid receptors. Neuropsychopharmacology. 2010;35:1841–1849. doi: 10.1038/npp.2010.51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haber SN, Fudge JL, McFarland NR. Striatonigrostriatal pathways in primates form an ascending spiral from the shell to the dorsolateral striatum. J Neurosci. 2000;20:2369–2382. doi: 10.1523/JNEUROSCI.20-06-02369.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harris GC, Aston-Jones G. Critical role for ventral tegmental glutamate in preference for a cocaine-conditioned environment. Neuropsychopharmacology. 2003;28:73–76. doi: 10.1038/sj.npp.1300011. [DOI] [PubMed] [Google Scholar]
- Harris GC, Wimmer M, Byrne R, Aston-Jones G. Glutamate-associated plasticity in the ventral tegmental area is necessary for conditioning environmental stimuli with morphine. Neuroscience. 2004;129:841–847. doi: 10.1016/j.neuroscience.2004.09.018. [DOI] [PubMed] [Google Scholar]
- Haubensak W, Kunwar PS, Cai H, Ciocchi S, Wall NR, Ponnusamy R, Biag J, Dong HW, Deisseroth K, Callaway EM, Fanselow MS, Luthi A, Anderson DJ. Genetic dissection of an amygdala microcircuit that gates conditioned fear. Nature. 2010;468:270–276. doi: 10.1038/nature09553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hikida T, Kimura K, Wada N, Funabiki K, Nakanishi S. Distinct roles of synaptic transmission in direct and indirect striatal pathways to reward and aversive behavior. Neuron. 2010;66:896–907. doi: 10.1016/j.neuron.2010.05.011. [DOI] [PubMed] [Google Scholar]
- Huang YH, Lin Y, Mu P, Lee BR, Brown TE, Wayman G, Marie H, Liu W, Yan Z, Sorg BA, Schluter OM, Zukin RS, Dong Y. In vivo cocaine experience generates silent synapses. Neuron. 2009;63:40–47. doi: 10.1016/j.neuron.2009.06.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ikemoto S. Dopamine reward circuitry: two projection systems from the ventral midbrain to the nucleus accumbens-olfactory tubercle complex. Brain Res Rev. 2007;56:27–78. doi: 10.1016/j.brainresrev.2007.05.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Isaac JT, Ashby M, McBain CJ. The role of the GluR2 subunit in AMPA receptor function and synaptic plasticity. Neuron. 2007;54:859–871. doi: 10.1016/j.neuron.2007.06.001. [DOI] [PubMed] [Google Scholar]
- Johnson SW, North RA. Opioids excite dopamine neurons by hyperpolarization of local interneurons. J Neurosci. 1992;12:483–488. doi: 10.1523/JNEUROSCI.12-02-00483.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kalivas PW. Interactions between dopamine and excitatory amino acids in behavioral sensitization to psychostimulants. Drug Alcohol Depend. 1995;37:95–100. doi: 10.1016/0376-8716(94)01063-q. [DOI] [PubMed] [Google Scholar]
- Kalivas PW, Alesdatter JE. Involvement of N-methyl-D-aspartate receptor stimulation in the ventral tegmental area and amygdala in behavioral sensitization to cocaine. J Pharmacol Exp Ther. 1993;267:486–495. [PubMed] [Google Scholar]
- Kalivas PW, Lalumiere RT, Knackstedt L, Shen H. Glutamate transmission in addiction. Neuropharmacology. 2009;56(Suppl 1):169–173. doi: 10.1016/j.neuropharm.2008.07.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kalivas PW, O’Brien C. Drug addiction as a pathology of staged neuroplasticity. Neuropsychopharmacology. 2008;33:166–180. doi: 10.1038/sj.npp.1301564. [DOI] [PubMed] [Google Scholar]
- Kasanetz F, Deroche-Gamonet V, Berson N, Balado E, Lafourcade M, Manzoni O, Piazza PV. Transition to addiction is associated with a persistent impairment in synaptic plasticity. Science. 2010;328:1709–1712. doi: 10.1126/science.1187801. [DOI] [PubMed] [Google Scholar]
- Kauer JA, Malenka RC. Synaptic plasticity and addiction. Nat Rev Neurosci. 2007;8:844–858. doi: 10.1038/nrn2234. [DOI] [PubMed] [Google Scholar]
- Knackstedt LA, Kalivas PW. Glutamate and reinstatement. Curr Opin Pharmacol. 2009;9:59–64. doi: 10.1016/j.coph.2008.12.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kourrich S, Rothwell PE, Klug JR, Thomas MJ. Cocaine experience controls bidirectional synaptic plasticity in the nucleus accumbens. J Neurosci. 2007;27:7921–7928. doi: 10.1523/JNEUROSCI.1859-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kravitz AV, Freeze BS, Parker PR, Kay K, Thwin MT, Deisseroth K, Kreitzer AC. Regulation of parkinsonian motor behaviours by optogenetic control of basal ganglia circuitry. Nature. 2010;466:622–626. doi: 10.1038/nature09159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kreitzer AC, Malenka RC. Endocannabinoid-mediated rescue of striatal LTD and motor deficits in Parkinson’s disease models. Nature. 2007;445:643–647. doi: 10.1038/nature05506. [DOI] [PubMed] [Google Scholar]
- Kreitzer AC, Malenka RC. Striatal plasticity and basal ganglia circuit function. Neuron. 2008;60:543–554. doi: 10.1016/j.neuron.2008.11.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lammel S, Hetzel A, Hackel O, Jones I, Liss B, Roeper J. Unique properties of mesoprefrontal neurons within a dual mesocorticolimbic dopamine system. Neuron. 2008;57:760–773. doi: 10.1016/j.neuron.2008.01.022. [DOI] [PubMed] [Google Scholar]
- Lammel S, Malenka RC. Aversive but not rewarding stimuli modulate synaptic properties of mesoprefrontal dopamine neurons. Society for Neuroscience Annual Meeting 2010 [Google Scholar]
- Lane DA, Lessard AA, Chan J, Colago EE, Zhou Y, Schlussman SD, Kreek MJ, Pickel VM. Region-specific changes in the subcellular distribution of AMPA receptor GluR1 subunit in the rat ventral tegmental area after acute or chronic morphine administration. J Neurosci. 2008;28:9670–9681. doi: 10.1523/JNEUROSCI.2151-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu QS, Pu L, Poo MM. Repeated cocaine exposure in vivo facilitates LTP induction in midbrain dopamine neurons. Nature. 2005;437:1027–1031. doi: 10.1038/nature04050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu SJ, Zukin RS. Ca(2+)-permeable AMPA receptors in synaptic plasticity and neuronal death. Trends Neurosci. 2007 doi: 10.1016/j.tins.2007.01.006. [DOI] [PubMed] [Google Scholar]
- Lobo MK, Covington HEr, Chaudhury D, Friedman AK, Sun H, Damez-Werno D, Dietz DM, Zaman S, Koo JW, Kennedy PJ, Mouzon E, Mogri M, Neve RL, Deisseroth K, Han MH, Nestler EJ. Cell type-specific loss of BDNF signaling mimics optogenetic control of cocaine reward. Science. 2010;330:385–390. doi: 10.1126/science.1188472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu H, Cheng PL, Lim BK, Khoshnevisrad N, Poo MM. Elevated BDNF after cocaine withdrawal facilitates LTP in medial prefrontal cortex by suppressing GABA inhibition. Neuron. 2010;67:821–833. doi: 10.1016/j.neuron.2010.08.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu W, Monteggia LM, Wolf ME. Repeated administration of amphetamine or cocaine does not alter AMPA receptor subunit expression in the rat midbrain. Neuropsychopharmacology. 2002;26:1–13. doi: 10.1016/S0893-133X(01)00272-X. [DOI] [PubMed] [Google Scholar]
- Luo Y, Good CH, Diaz-Ruiz O, Zhang Y, Hoffman AF, Shan L, Kuang SY, Malik N, Chefer VI, Tomac AC, Lupica CR, Backman CM. NMDA receptors on non-dopaminergic neurons in the VTA support cocaine sensitization. PLoS ONE. 2010;5:e12141. doi: 10.1371/journal.pone.0012141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lüscher C, Huber KM. Group 1 mGluR-dependent synaptic long-term depression: mechanisms and implications for circuitry and disease. Neuron. 2010;65:445–459. doi: 10.1016/j.neuron.2010.01.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lüscher C, Ungless MA. The Mechanistic Classification of Addictive Drugs. PLoS Med. 2006;3:e437. doi: 10.1371/journal.pmed.0030437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luu P, Malenka RC. Spike timing-dependent long-term potentiation in ventral tegmental area dopamine cells requires PKC. J Neurophysiol. 2008;100:533–538. doi: 10.1152/jn.01384.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maeda H, Mogenson GJ. Electrophysiological responses of neurons of the ventral tegmental area to electrical stimulation of amygdala and lateral septum. Neuroscience. 1981;6:367–376. doi: 10.1016/0306-4522(81)90130-5. [DOI] [PubMed] [Google Scholar]
- Malenka RC, Nicoll RA. Silent synapses speak up. Neuron. 1997;19:473–476. doi: 10.1016/s0896-6273(00)80362-1. [DOI] [PubMed] [Google Scholar]
- Mameli M, Balland B, Lujan R, Lüscher C. Rapid synthesis and synaptic insertion of GluR2 for mGluR-LTD in the ventral tegmental area. Science. 2007;317:530–533. doi: 10.1126/science.1142365. [DOI] [PubMed] [Google Scholar]
- Mameli M, Bellone C, Brown MT, Lüscher C. Cocaine inverst rules for synaptic plasticity of glutamate transmission in the VTA. Nat Neurosci. 2011 doi: 10.1038/nn.2763. in press. [DOI] [PubMed] [Google Scholar]
- Mameli M, Halbout B, Creton C, Engblom D, Parkitna JR, Spanagel R, Lüscher C. Cocaine-evoked synaptic plasticity: persistence in the VTA triggers adaptations in the NAc. Nat Neurosci. 2009;12:1036–1041. doi: 10.1038/nn.2367. [DOI] [PubMed] [Google Scholar]
- Margolis EB, Lock H, Hjelmstad GO, Fields HL. The ventral tegmental area revisited: is there an electrophysiological marker for dopaminergic neurons? J Physiol. 2006;577:907–924. doi: 10.1113/jphysiol.2006.117069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Margolis EB, Mitchell JM, Ishikawa J, Hjelmstad GO, Fields HL. Midbrain dopamine neurons: projection target determines action potential duration and dopamine D(2) receptor inhibition. J Neurosci. 2008;28:8908–8913. doi: 10.1523/JNEUROSCI.1526-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marie H, Morishita W, Yu X, Calakos N, Malenka RC. Generation of silent synapses by acute in vivo expression of CaMKIV and CREB. Neuron. 2005;45:741–752. doi: 10.1016/j.neuron.2005.01.039. [DOI] [PubMed] [Google Scholar]
- Martin M, Chen BT, Hopf FW, Bowers MS, Bonci A. Cocaine self-administration selectively abolishes LTD in the core of the nucleus accumbens. Nat Neurosci. 2006;9:868–869. doi: 10.1038/nn1713. [DOI] [PubMed] [Google Scholar]
- Maskos U, Molles BE, Pons S, Besson M, Guiard BP, Guilloux JP, Evrard A, Cazala P, Cormier A, Mameli-Engvall M, Dufour N, Cloez-Tayarani I, Bemelmans AP, Mallet J, Gardier AM, David V, Faure P, Granon S, Changeux JP. Nicotine reinforcement and cognition restored by targeted expression of nicotinic receptors. Nature. 2005;436:103–107. doi: 10.1038/nature03694. [DOI] [PubMed] [Google Scholar]
- Mato S, Chevaleyre V, Robbe D, Pazos A, Castillo PE, Manzoni OJ. A single in-vivo exposure to delta 9THC blocks endocannabinoid-mediated synaptic plasticity. Nat Neurosci. 2004;7:585–586. doi: 10.1038/nn1251. [DOI] [PubMed] [Google Scholar]
- Moussawi K, Pacchioni A, Moran M, Olive MF, Gass JT, Lavin A, Kalivas PW. N-Acetylcysteine reverses cocaine-induced metaplasticity. Nat Neurosci. 2009;12:182–189. doi: 10.1038/nn.2250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nestler EJ. Is there a common molecular pathway for addiction? Nat Neurosci. 2005;8:1445–1449. doi: 10.1038/nn1578. [DOI] [PubMed] [Google Scholar]
- Niehaus JL, Murali M, Kauer JA. Drugs of abuse and stress impair LTP at inhibitory synapses in the ventral tegmental area. Eur J Neurosci. 2010;32:108–117. doi: 10.1111/j.1460-9568.2010.07256.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nugent FS, Niehaus JL, Kauer JA. PKG and PKA signaling in LTP at GABAergic synapses. Neuropsychopharmacology. 2009;34:1829–1842. doi: 10.1038/npp.2009.5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nugent FS, Penick EC, Kauer JA. Opioids block long-term potentiation of inhibitory synapses. Nature. 2007;446:1086–1090. doi: 10.1038/nature05726. [DOI] [PubMed] [Google Scholar]
- Overton PG, Richards CD, Berry MS, Clark D. Long-term potentiation at excitatory amino acid synapses on midbrain dopamine neurons. Neuroreport. 1999;10:221–226. doi: 10.1097/00001756-199902050-00004. [DOI] [PubMed] [Google Scholar]
- Phillips PE, Stuber GD, Heien ML, Wightman RM, Carelli RM. Subsecond dopamine release promotes cocaine seeking. Nature. 2003;422:614–618. doi: 10.1038/nature01476. [DOI] [PubMed] [Google Scholar]
- Pu L, Liu QS, Poo MM. BDNF-dependent synaptic sensitization in midbrain dopamine neurons after cocaine withdrawal. Nat Neurosci. 2006;9:605–607. doi: 10.1038/nn1687. [DOI] [PubMed] [Google Scholar]
- Redish AD, Jensen S, Johnson A. A unified framework for addiction: vulnerabilities in the decision process. Behav Brain Sci. 2008;31:415–37. doi: 10.1017/S0140525X0800472X. discussion 437–87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robbe D, Kopf M, Remaury A, Bockaert J, Manzoni OJ. Endogenous cannabinoids mediate long-term synaptic depression in the nucleus accumbens. Proc Natl Acad Sci U S A. 2002;99:8384–8388. doi: 10.1073/pnas.122149199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rodaros D, Caruana DA, Amir S, Stewart J. Corticotropin-releasing factor projections from limbic forebrain and paraventricular nucleus of the hypothalamus to the region of the ventral tegmental area. Neuroscience. 2007;150:8–13. doi: 10.1016/j.neuroscience.2007.09.043. [DOI] [PubMed] [Google Scholar]
- Russo SJ, Dietz DM, Dumitriu D, Morrison JH, Malenka RC, Nestler EJ. The addicted synapse: mechanisms of synaptic and structural plasticity in nucleus accumbens. Trends Neurosci. 2010;33:267–276. doi: 10.1016/j.tins.2010.02.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saal D, Dong Y, Bonci A, Malenka RC. Drugs of abuse and stress trigger a common synaptic adaptation in dopamine neurons. Neuron. 2003;37:577–582. doi: 10.1016/s0896-6273(03)00021-7. [DOI] [PubMed] [Google Scholar]
- Schilstrom B, Yaka R, Argilli E, Suvarna N, Schumann J, Chen BT, Carman M, Singh V, Mailliard WS, Ron D, Bonci A. Cocaine enhances NMDA receptor-mediated currents in ventral tegmental area cells via dopamine D5 receptor-dependent redistribution of NMDA receptors. J Neurosci. 2006;26:8549–8558. doi: 10.1523/JNEUROSCI.5179-05.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmidt HD, Pierce RC. Cocaine-induced neuroadaptations in glutamate transmission: potential therapeutic targets for craving and addiction. Ann N Y Acad Sci. 2010;1187:35–75. doi: 10.1111/j.1749-6632.2009.05144.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schramm-Sapyta NL, Olsen CM, Winder DG. Cocaine self-administration reduces excitatory responses in the mouse nucleus accumbens shell. Neuropsychopharmacology. 2006;31:1444–1451. doi: 10.1038/sj.npp.1300918. [DOI] [PubMed] [Google Scholar]
- Sesack SR, Grace AA. Cortico-Basal Ganglia reward network: microcircuitry. Neuropsychopharmacology. 2010;35:27–47. doi: 10.1038/npp.2009.93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shen W, Flajolet M, Greengard P, Surmeier DJ. Dichotomous dopaminergic control of striatal synaptic plasticity. Science. 2008;321:848–851. doi: 10.1126/science.1160575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shuen JA, Chen M, Gloss B, Calakos N. Drd1a-tdTomato BAC transgenic mice for simultaneous visualization of medium spiny neurons in the direct and indirect pathways of the basal ganglia. J Neurosci. 2008;28:2681–2685. doi: 10.1523/JNEUROSCI.5492-07.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stuber GD. Dissecting the neural circuitry of addiction and psychiatric disease with optogenetics. Neuropsychopharmacology. 2010;35:341–342. doi: 10.1038/npp.2009.102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stuber GD, Hnasko TS, Britt JP, Edwards RH, Bonci A. Dopaminergic terminals in the nucleus accumbens but not the dorsal striatum corelease glutamate. J Neurosci. 2010;30:8229–8233. doi: 10.1523/JNEUROSCI.1754-10.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stuber GD, Klanker M, de Ridder B, Bowers MS, Joosten RN, Feenstra MG, Bonci A. Reward-predictive cues enhance excitatory synaptic strength onto midbrain dopamine neurons. Science. 2008;321:1690–1692. doi: 10.1126/science.1160873. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sulzer D, Sonders MS, Poulsen NW, Galli A. Mechanisms of neurotransmitter release by amphetamines: a review. Prog Neurobiol. 2005;75:406–433. doi: 10.1016/j.pneurobio.2005.04.003. [DOI] [PubMed] [Google Scholar]
- Surmeier DJ, Ding J, Day M, Wang Z, Shen W. D1 and D2 dopamine-receptor modulation of striatal glutamatergic signaling in striatal medium spiny neurons. Trends Neurosci. 2007;30:228–235. doi: 10.1016/j.tins.2007.03.008. [DOI] [PubMed] [Google Scholar]
- Szabo B, Siemes S, Wallmichrath I. Inhibition of GABAergic neurotransmission in the ventral tegmental area by cannabinoids. Eur J Neurosci. 2002;15:2057–2061. doi: 10.1046/j.1460-9568.2002.02041.x. [DOI] [PubMed] [Google Scholar]
- Tan KR, Brown M, Labouebe G, Yvon C, Creton C, Fritschy JM, Rudolph U, Lüscher C. Neural bases for addictive properties of benzodiazepines. Nature. 2010;463:769–774. doi: 10.1038/nature08758. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tecuapetla F, Patel JC, Xenias H, English D, Tadros I, Shah F, Berlin J, Deisseroth K, Rice ME, Tepper JM, Koos T. Glutamatergic signaling by mesolimbic dopamine neurons in the nucleus accumbens. J Neurosci. 2010;30:7105–7110. doi: 10.1523/JNEUROSCI.0265-10.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thomas MJ, Beurrier C, Bonci A, Malenka RC. Long-term depression in the nucleus accumbens: a neural correlate of behavioral sensitization to cocaine. Nat Neurosci. 2001;4:1217–1223. doi: 10.1038/nn757. [DOI] [PubMed] [Google Scholar]
- Thomas MJ, Malenka RC. Synaptic plasticity in the mesolimbic dopamine system. Philos Trans R Soc Lond B Biol Sci. 2003;358:815–819. doi: 10.1098/rstb.2002.1236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thomas MJ, Kalivas PW, Shaham Y. Neuroplasticity in the mesolimbic dopamine system and cocaine addiction. Br J Pharmacol. 2008;154:327–342. doi: 10.1038/bjp.2008.77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ungless MA, Whistler JL, Malenka RC, Bonci A. Single cocaine exposure in vivo induces long-term potentiation in dopamine neurons. Nature. 2001;411:583–587. doi: 10.1038/35079077. [DOI] [PubMed] [Google Scholar]
- Ungless MA, Singh V, Crowder TL, Yaka R, Ron D, Bonci A. Corticotropin-releasing factor requires CRF binding protein to potentiate NMDA receptors via CRF receptor 2 in dopamine neurons. Neuron. 2003;39:401–407. doi: 10.1016/s0896-6273(03)00461-6. [DOI] [PubMed] [Google Scholar]
- Van den Oever MC, Goriounova NA, Li KW, Van der Schors RC, Binnekade R, Schoffelmeer AN, Mansvelder HD, Smit AB, Spijker S, De Vries TJ. Prefrontal cortex AMPA receptor plasticity is crucial for cue-induced relapse to heroin-seeking. Nat Neurosci. 2008;11:1053–1058. doi: 10.1038/nn.2165. [DOI] [PubMed] [Google Scholar]
- Vanderschuren LJ, Kalivas PW. Alterations in dopaminergic and glutamatergic transmission in the induction and expression of behavioral sensitization: a critical review of preclinical studies. Psychopharmacology (Berl) 2000;151:99–120. doi: 10.1007/s002130000493. [DOI] [PubMed] [Google Scholar]
- Vezina P, Queen AL. Induction of locomotor sensitization by amphetamine requires the activation of NMDA receptors in the rat ventral tegmental area. Psychopharmacology (Berl) 2000;151:184–191. doi: 10.1007/s002130000463. [DOI] [PubMed] [Google Scholar]
- Vittoz NM, Schmeichel B, Berridge CW. Hypocretin /orexin preferentially activates caudomedial ventral tegmental area dopamine neurons. Eur J Neurosci. 2008;28:1629–1640. doi: 10.1111/j.1460-9568.2008.06453.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wall NR, Wickersham IR, Cetin A, De La Parra M, Callaway EM. Monosynaptic circuit tracing in vivo through Cre-dependent targeting and complementation of modified rabies virus. Proc Natl Acad Sci U S A. 2010 doi: 10.1073/pnas.1011756107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wanat MJ, Sparta DR, Hopf FW, Bowers MS, Melis M, Bonci A. Strain specific synaptic modifications on ventral tegmental area dopamine neurons after ethanol exposure. Biol Psychiatry. 2009;65:646–653. doi: 10.1016/j.biopsych.2008.10.042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Witten IB, Lin SC, Brodsky M, Prakash R, Diester I, Anikeeva P, Gradinaru V, Ramakrishnan C, Deisseroth K. Cholinergic interneurons control local circuit activity and cocaine conditioning. Science. 2010;330:1677–1681. doi: 10.1126/science.1193771. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wolf ME. The role of excitatory amino acids in behavioral sensitization to psychomotor stimulants. Prog Neurobiol. 1998;54:679–720. doi: 10.1016/s0301-0082(97)00090-7. [DOI] [PubMed] [Google Scholar]
- Wolf ME. The Bermuda Triangle of cocaine-induced neuroadaptations. Trends Neurosci. 2010;33:391–398. doi: 10.1016/j.tins.2010.06.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wolf ME, Ferrario CR. AMPA receptor plasticity in the nucleus accumbens after repeated exposure to cocaine. Neurosci Biobehav Rev. 2010 doi: 10.1016/j.neubiorev.2010.01.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xia YF, Driscoll JR, Margolis EB, Wilbrecht L, Fields HL, Hjelmstad GO. GABAergic afferents from the nucleus accumbens target non-dopaminergic neurons in the ventral tegmental area. Society for Neuroscience Annual Meeting. 2010 doi: 10.1523/JNEUROSCI.1504-11.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamaguchi T, Sheen W, Morales M. Glutamatergic neurons are present in the rat ventral tegmental area. Eur J Neurosci. 2007;25:106–118. doi: 10.1111/j.1460-9568.2006.05263.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yao WD, Gainetdinov RR, Arbuckle MI, Sotnikova TD, Cyr M, Beaulieu JM, Torres GE, Grant SG, Caron MG. Identification of PSD-95 as a regulator of dopamine-mediated synaptic and behavioral plasticity. Neuron. 2004;41:625–638. doi: 10.1016/s0896-6273(04)00048-0. [DOI] [PubMed] [Google Scholar]
- Zweifel LS, Argilli E, Bonci A, Plamiter RD. Role of NMDA receptors in dopamine neurons for plasticity and addictive behaviors. Neuron. 2008;59:486–96. doi: 10.1016/j.neuron.2008.05.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zweifel LS, Parker JG, Lobb CJ, Rainwater A, Wall VZ, Fadok JP, Darvas M, Kim MJ, Mizumori SJ, Paladini CA, Phillips PE, Palmiter RD. Disruption of NMDAR-dependent burst firing by dopamine neurons provides selective assessment of phasic dopamine-dependent behavior. Proc Natl Acad Sci U S A. 2009;106:7281–7288. doi: 10.1073/pnas.0813415106. [DOI] [PMC free article] [PubMed] [Google Scholar]