

Abstract
Cryptochromes are flavoproteins that drive diverse developmental light-responses in plants and participate in the circadian clock in animals. Plant cryptochromes have found application as photoswitches in optogenetics. We have studied effects of pH and ATP on the functionally relevant photoreduction of the oxidized FAD cofactor to the semi-reduced FADH· radical in isolated Arabidopsis cryptochrome 1 by transient absorption spectroscopy on nanosecond to millisecond timescales. In the absence of ATP, the yield of light-induced radicals strongly decreased with increasing pH from 6.5 to 8.5. With ATP present, these yields were significantly higher and virtually pH-independent up to pH 9. Analysis of our data in light of the crystallographic structure suggests that ATP-binding shifts the pKa of aspartic acid D396, the putative proton donor to FAD·−, from ~7.4 to >9, and favours a reaction pathway yielding long-lived aspartate D396−. Its negative charge could trigger conformational changes necessary for signal transduction.
Cryptochromes (CRYs) exhibit diverse biological functions, yet share an essential, non-covalently bound flavin adenine dinucleotide (FAD) cofactor1,2. In mammalian CRYs, where the FAD-binding pocket can be open and dynamic3,4, replacement of FAD by a carbazole derivative affected the length of the circadian period5,6, suggesting therapeutic potential. The active redox states of FAD in CRYs are still a matter of debate and likely differ among organisms and/or for the given protein function7,8,9.
In Arabidopsis thaliana cryptochrome 1 (AtCRY1), the semi-reduced (semiquinone) neutral FAD radical (FADH·) appears to be the signalling state7. This signalling state is formed by photoreduction and subsequent protonation of the resting state, the fully-oxidized FAD (FADox), which strongly absorbs blue light (ε445 = 11 300 M−1cm−1)10. By analogy to the structurally related DNA repair enzyme photolyase11, in which fully reduced FAD is replenished by so-called photoactivation, the reducing electron in AtCRY1 is transferred to the excited FAD (*FADox) through a cascade of tryptophan residues12,13 (W400, W377 and W324, see Fig. 1; we denote them by Trp1H, Trp2H and Trp3H, respectively, when the protonation state is relevant) and possibly also a tyrosine (TyrOH)12 residue. The quantum yield of FADH· Trp· radical pair formation in isolated AtCRY1 was estimated to be as low as ~2%12. The facile reduction of Trp3· and TyrO· by extrinsic reductants stabilizes FADH· by preventing recombination of the radical pairs. The mechanism by which the FADox → FADH· transition might be sensed by signalling partners remains to be established.
Figure 1. Tertiary structure of AtCRY1.

(a) Overall structure of the photolyase homology region in Arabidopsis thaliana cryptochrome 1 with a bound non-hydrolyzable ATP analogue, adenylyl-imidodiphosphate (AMP-PNP), adapted from the crystal structure by Brautigam et al.20. The N-terminal α/β domain exhibits a Rossman fold; the FAD cofactor (yellow) and AMP-PNP (pink) are bound to the α-helical domain. The carboxy-terminal CCT domain comprising another ~220 amino acids in full-length AtCRY1 is absent in the crystallized protein20. (b) Detailed view of the surroundings of the FAD cofactor. The flavin-reducing tryptophan triad W400 (Trp1H), W377 (Trp2H) and W324 (Trp3H)13 is shown, together with the putative proton donor aspartic acid D396, and key residues interacting with the adenine moiety of AMP-PNP: leucine L296 and tyrosine Y402 (note that the α16-helix interconnects Y402 with D396 and W400). The H-bond between the aspartic acid D396 and the backbone carbonyl oxygen of methionine M381 interconnecting the helices α15 and α16 and the ultrafast proton transfer pathway25 between W400 and N5 of FAD through D396 are indicated by magenta dashed lines. The electron transfer (ET) steps are shown by red arrows.
Both plant and animal CRYs have been reported to bind ATP and exhibit autokinase activity in vitro14,15,16,17,18, though the implications of such enzymatic activity have yet to be elucidated in vivo. Furthermore, a prolonged lifetime of FADH·, the putative active redox form of FAD, and a higher resistance to denaturation and proteolysis have been reported for plant cryptochromes in the presence of ATP17,19. Isolated AtCRY1 binds ATP with a stoichiometry of 0.9–1.0 ATP/protein16,20 and a dissociation constant on the order of 10−5 M14,16,20. The photolyase homology region (PHR) of AtCRY1, lacking the poorly structured C-terminal region (CCT), was co-crystallized with a non-hydrolysable ATP analogue (adenylyl-imidodiphosphate, AMP-PNP). The crystal structure20 revealed that the ATP-binding site is located near the FAD cofactor. CRYs show a strikingly similar structural fold to photolyases21, consisting of N-terminal α/β and C-terminal α-barrel domains. FAD is buried in the α-barrel domain. Both CRYs and photolyases have a pocket leading to the FAD cofactor; ATP binds in the pocket of plant CRYs, whereas photolyases recognize pyrimidine-dimer lesions and restore normal bases at this site. When bound to AtCRY1, the adenine moiety of ATP is tightly sandwiched between leucine L296 and tyrosine Y402, which are highly conserved among plant CRYs. ATP phosphates are anchored by positively-charged residues on the protein surface.
Here we demonstrate by transient absorption spectroscopy that, in addition to the aforementioned effects, ATP binding to AtCRY1 leads also to an important increase in the yield of light-induced FAD·−–TrpH·+ and FADH·–Trp· radical pairs, and, accordingly, also in the yield of the metastable isolated FADH· radical. In the absence of ATP, we found that the efficiency of CRY photoreduction decreases substantially with increasing basicity between pH 6.5 and 8.5. ATP binding remediates this decrease, presumably by shifting the pH-sensitivity region towards the basic (to pH > 9.0).
Detailed analysis of the transient absorption kinetics observed in our study shows that both pH and ATP binding affect the protonation of FAD·−, formed by photo-induced electron transfer (ET) to *FADox from W400. Our data suggest that the observed pH-effect reflects the protonation state of D396(H), which is positioned opposite the N5 of the isoalloxazine ring of the FAD, and that ATP binding up-shifts the pKa of this aspartic acid. D396(H) is widely accepted to be the proton donor in the formation of FADH· from FAD·− in AtCRY122,23,24,25,26, however, there is currently a controversy on whether FAD·− protonation occurs in an ultrafast manner (<1 ns)25 or on a microsecond scale24 in plant cryptochromes. Our results indicate that both pathways exist and are affected by pH and by ATP.
Results
Transient absorption studies
Addressing the question of whether or not ATP directly enhances the photoreduction of FADox in CRYs (in addition to the reported stabilization of the FADH· form17,19), we have monitored the yields and kinetics of species involved in FADox photoreduction by transient absorption spectroscopy in the presence and absence of ATP.
Based on the absorption spectra of the expected transient species (Fig. 2), we have chosen three basic spectral regions to monitor the photoreactions subsequent to FADox excitation: 400–410 nm, ~450 nm and ~560 nm. At ~450 nm, FADox absorbs much stronger than both FAD·− and FADH· and the absorption of other expected species is negligible. Hence the major processes observed at this wavelength are FAD·−/FADH· formation (initial bleaching) and their recombination with tryptophan radicals back to FADox (recovery of bleaching). The region around 560 nm allows monitoring of TrpH·+ deprotonation to Trp· (decrease of the initially formed absorption) and the protonation of FAD·− to FADH· (absorption increase). Finally, at 400–410 nm, formation of FAD·− is accompanied by an absorption increase, and the subsequent protonation of FAD·− to FADH· is reflected by decay down to a slightly negative absorption difference (ΔA) signal (FADH· absorbs less than FADox in this region).
Figure 2. Nanosecond-microsecond kinetic traces for AtCRY1 PHR.

Transient absorption changes were recorded at 457, 562 and 403 nm upon excitation of FADox by a picosecond laser pulse (3.0 mJ at 355 nm, at time zero) in the absence (black traces) and in the presence of 1 mM ATP (red traces). The poorer signal to noise ratio at 403 nm is due to an inherently worse stability of the monitoring light source at this wavelength. The samples containing 47 μM protein, 0.05 M phosphate buffer of pH 7.4 and 0.5 M NaCl were kept at 1–2°C. Top-right segment: absorption spectra of FAD in three of its redox forms (FADox, FAD·− and FADH·) and other species potentially contributing to the absorption changes. The FADox spectrum is that of our AtCRY1 PHR, normalized to ε445 = 11 300 M−1cm−1 (from Ref. 10). The FADH· spectrum has been extracted from a spectrum of a mixture of FADox and FADH· of the same protein (see Supplementary Fig. 7 for more details). The FAD·− spectrum is that of FAD·− in a mosquito cryptochrome (AgCRY1) adopted from Liu et al.35. TrpH·+ and Trp· spectra are adopted from Solar et al.36, and TyrO· from Giese et al.37, normalized to ε (λmax) = 2 600 M−1cm−1 (from Dudley Bryant et al.38).
In order to obtain the best possible signal-to-noise ratios of sometimes weak transient absorption signals, we have used truncated AtCRY1 that lacks the flexible C-terminus and corresponds to the N-terminal PHR. This protein proved to be more stable and samples of higher concentration, higher purity and better batch-to-batch consistency could be produced in sufficient amounts. We have conducted several control experiments with full-length (wild-type) AtCRY1 to verify that the photochemistry of the two proteins is not substantially different (Supplementary Results, Supplementary Fig. 2). Indeed, we have not found any significant differences in their photoreactions and thus we conclude that the C-terminus has virtually no effect on the primary photophysical and photochemical events in AtCRY1.
The time resolution of the transient absorption set-up was limited to ~10 ns in order to obtain a good signal-to-noise ratio. Quantum yields of transient radical pairs were determined using the [Ru(bpy)3]2+ actinometer27.
All kinetic traces in Fig. 2 clearly show that ATP substantially increases the yield of the photogenerated transient species. In addition, ATP changes the evolution of their relative concentrations in time (best seen at 562 nm, but also at 457 nm), which is a clear evidence of a mechanistic change in the course of the photoreaction.
Two major kinetic phases are apparent in all signals (see fit in Supplementary Fig. 8a): The faster phase has a time constant τ ~ 200 ns and is assigned to deprotonation of TrpH·+ (contributing most significantly at 562 nm) parallel to recombination of FAD·− and TrpH·+ (contributing most significantly at 457 nm). The slower phase (τ ~ 1.5 μs) is assigned to the protonation of FAD·− to FADH· (absorption increase at 562 nm and decrease at 403 nm). Both phases are relatively more pronounced in the presence of ATP than in its absence. The absorption changes at the end of the time scale of Fig. 2 (10 μs) are assigned to mostly FADH· – Trp· radical pairs (~80%) with a small admixture of FAD·− – Trp· pairs (~20%; see Supplementary Information, Section 8).
Signals from ATP-free samples in Fig. 2 showing only a little evolution in the given time window seem to be consistent with the recently theoretically predicted25 ultrafast protonation of FAD·− by D396(H) and a subsequent protonation of D396− by Trp1H·+ (i.e., formation of an FADH· – Trp· pair already on the picosecond timescale). On the other hand, in samples containing ATP (and to a smaller but non-negligible extent also in samples lacking it) we clearly observed a much slower FAD·− protonation kinetics (τ ~ 1.5 μs; close to the 1.7 μs value observed in experiments on algal cryptochrome24) preceded by TrpH·+ deprotonation with an apparent τ ~ 200 ns (comparable to deprotonation of the terminal TrpH·+ in DNA photolyase from E. coli28 indicating a reaction pathway different from the one predicted by the theoretical study25).
Striving to better resolve and understand these protonation/deprotonation processes, we have recorded kinetic traces at 562 nm at different pH values (Fig. 3). The signals revealed another remarkable phenomenon: the yield of photogenerated species is very sensitive to pH change in the absence of ATP, diminishing dramatically with increasing basicity from pH 7.0 to 8.0. The signals from samples containing ATP, however, remain virtually unchanged within this pH range and decrease only at the highest pH tested (9.6). The pH-dependence of signal amplitudes from ATP-free samples after 10 μs (with major contribution to ΔA from the FADH· – Trp· radical pair) can be fitted by a titration curve for a monoprotic acid with pKa ~ 7.4 (Supplementary Fig. 3a).
Figure 3. Nanosecond-microsecond kinetic traces for AtCRY1 PHR at different pH values.

Transient absorption changes were recorded at 562 nm upon excitation of FADox by a picosecond laser pulse (3.5 mJ at 355 nm, at time zero) in the absence (black traces) and in the presence of ATP (red traces). While 1 mM ATP was largely sufficient to saturate the ATP effect on the signal amplitude at pH ≤ 8, more ATP had to be added to the sample at pH 9.6 in order to reach full saturation (possibly due to the fact that ATP hydrolyses rapidly at high pH). The samples contained 45 μM protein, 0.05 M phosphate buffer of pH 7.0 to 8.0 or 0.05 M Tris buffer of pH 9.6 and 0.5 M NaCl and were kept at 1–2°C.
The evolution of the transient species on time scales longer than 10 μs was monitored on a set-up with lower time resolution but higher amplitude sensitivity than the set-up used for Figs. 2 and 3. At all wavelengths studied (410, 450 and 560 nm), most of the detected absorption change decayed in a few milliseconds (Fig. 4). We attribute this decay essentially to recombination of the predominating pair of neutral radicals FADH· – Trp·. This recombination seems to be accelerated by ATP (from τ = 8.0 ms to 2.2 ms; see Supplementary Information, Section 4 for possible origins of this effect). The presence of even longer-lived absorption changes with spectral features reminiscent of FADH· indicates that part of FADH· was stabilized by reduction of Trp· by an intrinsic reductant different from FADH·, most likely a neighbouring tyrosine residue (see Fig. 4). FADH· – Trp· recombination can be virtually completely suppressed by addition of an extrinsic reductant (Supplementary Fig. 5). As a result of efficient reduction of Trp·, the overall quantum yield of the isolated metastable flavin radicals (FADH·/FAD·−) can attain ~14% in the presence of ATP (Supplementary Information, Sections 4 and 8).
Figure 4. Millisecond kinetic traces recorded for AtCRY1 PHR.
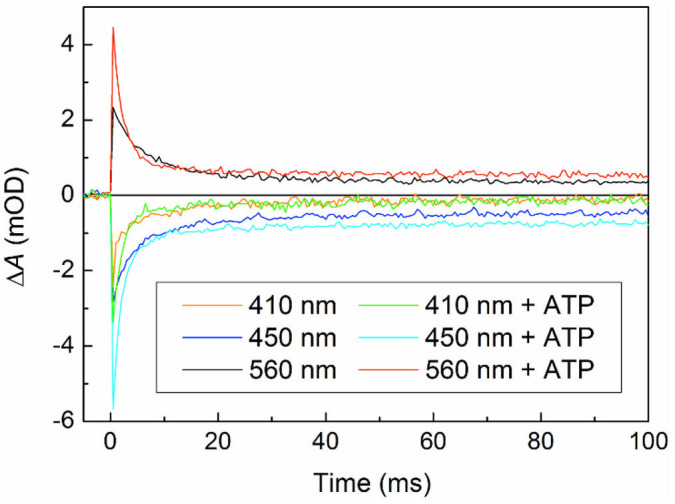
Transient absorption changes upon excitation of FADox by a picosecond laser pulse (2.2 mJ at 355 nm, at time zero) were recorded at 410, 450 and 560 nm in the absence/presence of 1 mM ATP. The samples containing 35 μM protein, 0.05 M phosphate buffer of pH 7.4 and 0.5 M NaCl were kept at 1–2°C. Note that despite ATP-accelerated recombination, the signal amplitudes remaining after 100 ms show still ~70% higher amounts of FADH· in samples containing ATP. At 410 nm, the signal bleaching due to the difference in ε of FADox and FADH· (see spectra in Fig. 2) is largely compensated by the absorption of the newly formed TyrO· radical.
Change in the UV/Vis spectrum associated with ATP binding and pH
While checking the quality and the state of our samples by UV-Vis spectroscopy, we have noticed that binding of ATP by AtCRY1 is accompanied by a reproducible change in the UV-Vis spectrum of FADox (Fig. 5a and Supplementary Fig. 2b). This effect is similar to the reported spectral change accompanying the substrate binding by a DNA photolyase containing an oxidized FAD cofactor29,30. While the molar absorption coefficients (ε) around the maximum in the blue spectral region (445 nm) slightly increase in the presence of ATP (by ~400 M−1cm−1 at pH 7.4) and the maxima exhibit a slight red shift (~1–2 nm), the two maxima in the near-UV (361 and 371 nm) shift towards shorter wavelengths (by ~4 and ~1 nm, respectively), and their amplitude ratio reverses. The ATP dependence of the spectral change can be described by a dissociation constant Kd = 1.4 ± 0.6 μM (see Supplementary Fig. 4).
Figure 5. Effects of ATP and pH on the absorption spectrum of FADox in AtCRY1 PHR.
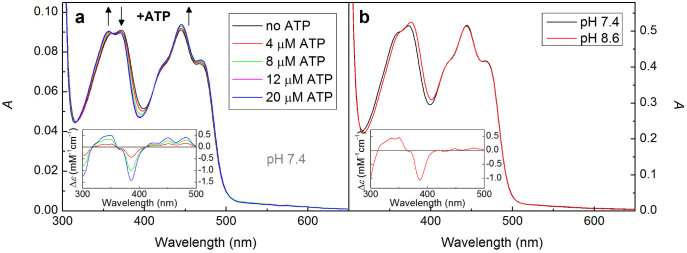
(a) Spectral change with increasing concentration of ATP (corrected for dilution due to the addition of ATP solution). Isosbestic points are situated at 317 nm, 362 nm and 411 nm. The inset shows ATP minus no ATP difference spectra scaled using ε445 = 11 300 M−1cm−1 for FADox. The sample contained 8 μM protein, 50 mM phosphate buffer of pH 7.4 and 0.5 M NaCl. (b) Comparison of spectra at pH 7.4 and pH 8.6 (50 μM protein, 50 mM phosphate buffer of pH 7.4/50 mM Tris buffer of pH 8.6; both containing 0.5 M NaCl). Isosbestic points are situated at 315 nm, 364 nm and 409 nm. The inset shows the pH 7.4 minus pH 8.6 difference spectrum scaled in the same way as in panel (a). See Supplementary Fig. 3b for more details on the pH-dependence of the AtCRY1 spectrum.
Interestingly, we have observed a similar spectral change when lowering the pH in the absence of ATP (Fig. 5b). Samples at lower pH showed blue shift of the UV bands with respect to samples at higher pH, but the red shift of the blue band was not so distinct. Again, like in the case of kinetic traces, also this pH-effect faded away upon ATP addition (in the studied pH range, i.e., between pH 6.5 and 9.6) and there was no significant difference in the spectra of samples saturated with ATP at different pH values. The spectral shift with pH observed for ATP-free samples is a strong indication of a change in the protonation state of an amino acid in the vicinity of the FADox cofactor. Plotting the absorption difference at 386 nm (minimum of the negative peak in the difference spectrum, inset Fig. 5b) against pH (Supplementary Fig. 3b) yields a titration curve of a monoprotic acid with the same pKa as the one obtained from the 562 nm-signal amplitudes after 10 μs, i.e., 7.4 (Supplementary Fig. 3a).
Discussion
Three major previously unreported phenomena were discovered in the present study of flavin photoreduction in isolated plant CRY on nanosecond to millisecond timescales:
In the absence of ATP, the yield of transient species formed upon photoexcitation of FADox dramatically decreases with increasing pH in the accessible pH range (6.5–9.6). The yield evolution can be fitted with a titration curve for a monoprotic acid of pKa ~ 7.4.
Addition of ATP strongly increases the yield of transient species and makes it pH-independent up to pH ~ 9. Moreover, ATP induces substantial qualitative changes in the species kinetics.
In the absence of ATP, the near-UV absorption spectrum of FADox in AtCRY1 changes with pH and this evolution also fits a pKa ~ 7.4. Addition of ATP and lowering the pH impacts the spectra in a similar way. When saturated with ATP, the spectrum of FADox in AtCRY1 no longer changes with pH (in the studied pH range, which was limited by fast aggregation of the protein at pH < 7 and by ATP hydrolysis at pH > 9).
In the following, we put our results into context with the current literature and propose a mechanistic model for FAD photoreduction combining photolyase-like ET through the tryptophan cascade11 with the theoretically predicted25 D396(H)-assisted ultrafast PTs from Trp1H·+ to FAD·− (Fig. 6).
Figure 6. Suggested global reaction scheme of flavin photoreduction in AtCRY1.

Upon photoexcitation of FADox, an ultrafast first ET forms the radical pair FAD·− – Trp1H·+. This pair can react through three parallel pathways: (i) direct back ET to the FADox ground state (left). (ii) ET through a cascade of three tryptophans, followed by deprotonation of the terminal tryptophan cation radical Trp3H·+ to the solvent and, in case D396 is present in its protonated form (D396 is in slow protonation equilibrium with the buffer), by (reversible) protonation of FAD·− by D396(H); the neutral terminal tryptophan radical Trp3· is readily reduced by one of the neighbouring tyrosine residues or by extrinsic reductants (centre). (iii) Sequential ultrafast PTs: from D396(H) to FAD·− (provided that D396 is present in its protonated form) and from Trp1H·+ to D396−, giving rise to a relatively stable pair of neutral radicals FADH· – Trp1· neighbouring neutral D396(H) (right). The partition among the three pathways is affected by pH and ATP. Trp3H·+ in pathway ii is in a dynamic equilibrium with Trp2H·+ and Trp1H·+ so that pathways i and iii remain accessible until Trp3H·+ is deprotonated. The indicated time constants for Trp3H·+ deprotonation (200 ns) and FAD·− protonation (1.5 μs) are observed values in the presence of competing or reverse reactions. The intrinsic time constants obtained from a model-based fit are somewhat slower (see text and Supplementary Information, Section 7). Trp1H = W400, Trp2H = W377, Trp3H = W324, B− = base (solvent), RH = tyrosine or extrinsic reductant.
Upon excitation of FADox, ultrafast (0.4 ps31) ET from the nearby Trp1H forms the first charge separated radical pair FAD·− – Trp1H·+. Subsequent ET through the tryptophan cascade (presumably in <1 ns) separates the charges farther. The resulting pair FAD·− – Trp3H·+ is stabilized by tryptophan deprotonation to the solvent within ~200 ns (observed in the present study), disfavouring further unproductive back ET in the FAD·− – TrpH·+ pairs. So far, the reaction scheme is analogous to photoreduction of FADH· to FADH− in DNA photolyase11. However, an additional PT to FAD·− occurs in plant CRY to yield the putative signalling state FADH·. Based on structural considerations, FTIR data and transient absorption data22,23,24,25,26 it appears established that FAD·− is protonated by a nearby aspartic acid (D396 in AtCRY1) in ~1.5 μs (observed in our study; 1.7 μs was reported for CRY from Chlamydomonas24).
In CRY/PL proteins that contain asparagine in the position corresponding to D396 in AtCRY1 (or even in the D396N mutant of AtCRY126), FAD·− can get eventually protonated via alternative pathways32. Nevertheless, the only time-resolved study of FAD·− protonation in such a protein32 (Methanosarcina mazei CPD II photolyase) reported a time constant of ~300 ms for this process at pH 8.0 (i.e., 200 000× slower than the ‘slow’ (~1.5 μs) direct protonation by D396(H) in AtCRY1). In insect CRYs, most of the FAD·− appear to remain unprotonated for at least a couple of minutes at pH 7.58. These CRYs contain cysteine in the position corresponding to D396 in AtCRY1. As even the replacement of this cysteine by an aspartic acid8 did not lead to formation of FADH·, it seems that the pKa of FADH· in insect CRYs is lower than 7.5.
In a recent computational study in AtCRY1, Solov'yov and coworkers25 predicted ultrafast protonation of FAD·− by a two-step PT from Trp1H·+ via D396, the first step being PT from D396(H) to FAD·−, and the second one PT from Trp1H·+ to D396−. These PTs would become possible by structural rearrangements triggered by formation of the pair FAD·− – Trp1H·+ that bring the groups involved in the PT closer to each other. It was argued25 that the observed 1.7 μs kinetics24 was wrongly assigned to protonation of FAD·− and may instead be attributed to deprotonation of D396(H) and protonation of Trp1· coupled to ET from Trp2H to Trp1· (the latter suggestion, however, is in contrast to the spectral features and amplitudes of the 1.5 μs kinetics observed in our study: such a process would yield virtually no absorption change at 403 nm where we observed a very prominent 1.5 μs phase). Nevertheless, analysis of our data (below) indicates that the ultrafast PT does indeed occur (contributing significantly in the absence of ATP), in competition with ET through the tryptophan cascade and subsequent microsecond protonation of FAD·− (strongly preferred in the presence of ATP). The products of the ultrafast PT process (FADH· – Trp1·) and those of the classical ET through the Trp triad followed by microsecond FAD·− protonation (FADH· – Trp3·) recombine with time constants of 8.0 ms and 2.2 ms, respectively (see Fig. 4), unless Trp· radicals are reduced by tyrosine residues or by extrinsic reducing agents (see Supplementary Information, Section 4 for more details).
The effect of pH on the stationary UV-Vis spectra of FADox and on the yield of photo-induced transients in the absence of ATP is a strong indication of a change in the dark state protonation of an amino acid (with pKa ~ 7.4) in the immediate vicinity of the chromophore. We cannot exclude that the protonation states of more remote groups indirectly influence the properties of FAD, e.g., by affecting the solvent access to the FAD cavity. However, it appears much more plausible that the observed pH effects are essentially due to the protonation state of a residue directly interacting with the isoalloxazine ring of the FAD chromophore. Looking at the known crystal structure of AtCRY1 PHR20, possible candidates include aspartic acid residues D359, D390, D392 and D396. Because of its hydrophobic environment, the best candidate appears to be D396, the putative proton donor to FAD·− 22,23,24,25,26. The observed pKa of ~7.4 is substantially higher than that of an aspartic acid in aqueous solution (pKa = 3.7). A hydrophobic environment destabilizes the negative charge of the deprotonated acid as compared to aqueous solution (i.e., increases the pKa of the given acid). D359, D390 and D392 are exposed20 to positively charged arginines (R360, R362 and R301, respectively) and are hence unlikely to have a pKa higher than that of the free acid. D396 seems to be protonated (H-bond to the backbone carbonyl oxygen of M381) in the crystal structure of AtCRY1 PHR20 determined at pH 5.5, suggesting a pKa > 5.5. An FTIR band attributed to light-induced deprotonation of D396 in AtCRY122 and of its equivalent in the PHR domain of Chlamydomonas CRY23 appears to be substantially more pronounced at pH 7.523 than at pH 8.022, which would be consistent with pKa ~7.4 obtained in our experiments.
We propose that binding of ATP induces a further increase of pKa of D396(H) from ~7.4 to >9 (likely ~10, as inferred from the decrease of signal amplitude at pH 9.6; Fig. 3), which assures that the protonated (uncharged) form of D396 prevails under physiological pH33. An increase of the pKa of D396 due to ATP binding could also explain the reported slower reoxidation of FADH· to FADox in the presence of ATP17,19, since FADH· needs to give off a proton in order to reoxidize back to FADox, which should be more difficult when D396 is protonated (see also Supplementary Information, Section 4).
ATP and D396(H) lie ~10 Å apart, separated by the isoalloxazine ring of FAD, thus the impact of ATP on the pKa of D396(H) appears to be indirect. Possible mechanisms include (i) a decrease in polarity of the D396 environment due to ATP restriction of solvent access to FAD, (ii) an electrostatic effect from negative charges of the ATP-phosphates, and/or (iii) strains resulting from ATP stacking. The adenine moiety of ATP is sandwiched between L296 and Y402 near the adenine of FAD (Fig. 1). The 6-membered ring of the ATP adenine is pi-stacked with the phenol ring of Y402. D396, W400 and Y402 are linked by helix α16. In the crystal structures of AtCRY1 PHR20 at pH 5.5, D396 appears to be protonated and H-bonded to the backbone carbonyl oxygen of M381 (α15), regardless of the presence of ATP. Since no crystal structures at higher pH are available, we can only speculate, consistent with the MD simulations34, that Y402-ATP interaction likely puts constraints on the entire α16 helix, resulting in a modified environment for D396. For example, placing the α16 helix in a position where the D396 carboxyl faces the electronegative backbone carbonyl oxygen and the sulphur atoms of M381 (α15) could increase the preference for protonation of D396 (i.e., its pKa) and the formation of an H-bond interconnecting the two helices α15 and α16.
We suggest two mutually non-exclusive mechanisms by which unprotonated (negatively charged) D396 might decrease the yield of long-lived radical pairs:
The ultrafast PT forming FADH· – Trp1· directly from FAD·− – Trp1H·+ requires protonated D396. With unprotonated D396, this pathway is lost and ET through the tryptophan cascade is the only productive pathway competing with back ET in the pair FAD·− – Trp1H·+.
By electrostatic interaction and/or by conformational changes, negatively charged (deprotonated) D396 might disfavour the first ET step (from Trp1H to *FADox) relative to the unproductive decay of *FADox, and/or disfavour the second ET step (from Trp2H to Trp1H·+) relative to back ET in the pair FAD·− – Trp1H·+, e.g., by repulsion between D396− of helix α16 (carrying Trp1H) and the electronegative oxygen of the M381 backbone carbonyl residing on helix α15 (carrying Trp2H), increasing the distance between Trp1H and Trp2H (weakening their electronic coupling).
In addition to the prominent effects of pH and ATP on the amplitudes of the transient absorption signals discussed so far, there are also effects on the time course of the signals which are less pronounced, but significant. Thus, at 562 nm (see Fig. 2), addition of ATP at pH 7.4 increased the contributions of both the 200 ns phase (deprotonation of Trp3H·+ competing with unproductive back ET) and the 1.5 μs phase (establishment of the equilibrium FAD·− + D396(H) [rlarr2 ] FADH· + D396−) relative to the end amplitude (attributed essentially to long-lived FADH· – Trp· pairs). In the framework of the suggested reaction scheme (Fig. 6), this implies that ATP favours the pathway involving ET through the tryptophan cascade over the ultrafast (<1 ns) PT pathway. Our observation that, in the absence of ATP, long-lived FADH· – Trp· can be formed with rather little Trp3H·+ deprotonation (200 ns) and ‘slow’ (1.5 μs) FAD·− protonation supports the (co-)existence of the theoretically predicted ultrafast PT pathway25.
The effect of ATP on the branching between the two productive pathways may be qualitatively explained by the proposed increase of the pKa of D396 upon binding of ATP, which would decrease the driving force of PT from D396(H) to FAD·− and hence disfavour the ultrafast PT pathway relative to ET through the tryptophan cascade. Of course, ATP may have other, more subtle effects on the branching ratio, such as conformational changes modifying ET and/or PT distances and changes of driving forces and/or reorganization energies of ET. It is conceivable that the ATP-induced constraint to the α16-helix (see above) hinders the rearrangement of D396(H) required for the ultrafast PT. By hindering the solvent access to FAD, ATP might also affect other properties of FAD that are relevant to its ET and PT reactions. In this context, we considered the possibility that ATP binding might have a major effect on the reduction potential of FADox in AtCRY1 but that does not seem to be the case (see Supplementary Information, Section 5).
For a quantitative analysis and verification of consistency with the proposed reaction scheme (Fig. 6), we tried to globally fit all transient absorption data in Fig. 2 (i.e., signals at 403, 457 and 562 nm in the presence and absence of ATP at pH 7.4) by a fitting function based on the reaction scheme (see Methods and Supplementary Information, Section 7). It was assumed that an (ATP-dependent) fraction of the sample contained unprotonated D396 at the moment of excitation and could only undergo the reactions shown on white ground in Fig. 6, while another fraction contained protonated D396 and could undergo all the reactions shown. Reactions supposed to be faster than 1 ns were not explicitly included in the model function but the products of these reactions are contained in the early absorption changes of the model functions. Reverse ET through the tryptophan cascade was considered implicitly by introducing: (i) an effective back-ET rate constant for the pair FAD·− – Trp3H·+ that includes thermal repopulation of the two preceding radical pairs, and (ii) an effective rate constant for formation of FADH· - Trp1· from FAD·− – Trp3H·+ via repopulation of the first radical pair and PT assisted by D396(H). Absorption spectra of the relevant species (Fig. 2) were taken from the literature35,36,37,38,39 and from our own determinations (Supplementary Fig. 7).
It turned out that the experimental data could be reasonably well fitted by our model (Supplementary Fig. 8b; fit parameters are summarized in Supplementary Tables 2 and 3). Of note, the intrinsic time constant of Trp3H·+ deprotonation (~350 ns) is slower than the apparent time constant from the multi-exponential fit (~200 ns) because the latter includes competing reactions (back ET and formation of FADH· – Trp1· via repopulation of FAD·− – Trp1H·+). Similarly, the intrinsic time constant of slow protonation of FAD·− (~2.5 μs) is slower than the apparent time constant of ~1.5 μs that includes the reverse reaction. Further significant outcomes of the model-based fits are:
The fraction of the sample containing unprotonated D396 at pH 7.4 disappeared upon addition of ATP, in line with a strong increase of the pKa of this aspartic acid.
In the fraction containing protonated D396, the branching ratio between the “ET through tryptophans” pathway (centre part of the proposed reaction scheme, Fig. 6) and the “ultrafast PT” pathway (right arrow in Fig. 6) changed from 2:1 in the absence of ATP to 7:1 in the presence of ATP. The “ultrafast PT” pathway is hence virtually negligible in the presence of ATP but it competes with the “ET through tryptophans” pathway in the absence of ATP.
The fit results further indicate that the PT from D396(H) to FAD·− in the “ET through tryptophans” pathway is reversible with a forward-to-backward rate ratio in the order of 2, i.e., it has only a weak driving force.
At first glance, it might seem puzzling that the same PT (from D396(H) to FAD·−) can occur on such different time scales as <1 ns (before ET through the tryptophan cascade) and ~2.5 μs (after this ET). There is, however, an important difference between the two situations: the nearby tryptophan W400 is in the radical cation state (Trp1H·+) in the former case, while it is neutral (Trp1H) in the latter case. By electrostatic interaction, the positive charge on W400 may strongly increase the otherwise weak driving force of PT from D396(H) to FAD·−. Alternatively, the positive charge on W400 may be specifically required for the ultrafast structural rearrangement that makes the PT possible. Once ET through the tryptophan cascade has occurred and W400 is neutral, the same structural rearrangement may be much slower.
The modifications of the UV absorption spectrum of FADox in AtCRY1 upon decrease of the pH and/or upon addition of ATP resemble those observed upon binding of cyclobutane pyrimidine dimer (CPD) lesion to a DNA photolyase29,30. The latter effect has been explained by the strong electric dipole field of the CPD present in the substrate binding pocket30. As adenine has a much weaker (~3.5×)40 electric dipole than the two nearly parallel thymines in the used CPD29,30, this explanation can hardly account for our observations in AtCRY1. We rather suggest that the electric field of point charges (most plausibly the negative charge on D396 at pH > pKa and possibly also the negative charges of ATP-phosphates) and the change of polarity of the flavin environment upon replacement of solvent in the binding pocket by adenine induce the spectral changes of FADox in AtCRY1. Such effects may also contribute to the spectral shifts accompanying substrate binding in DNA photolyase (see Supplementary Information, Section 2).
Given the high affinity of AtCRY1 for ATP (Kd ≤ 2 μM at pH 7.4, Supplementary Fig. 4) and the typical physiological concentration of ATP in plant cytoplasm (1.3 ± 0.3 mM)41, the ATP effects described here are unlikely to be used by the cells for regulation of cryptochrome photoreceptor activity. ATP may rather be considered as a coenzyme of AtCRY1, the roles of which include increasing the yield of FADH· (regardless of possible pH fluctuations) and prolonging the lifetime17,19 of this putative signalling redox state of flavin (see also Supplementary Information, Section 4). A more plausible way to regulate cryptochrome light response in vivo is by modulation of the redox environment within the cell affecting the yield of FADH· (scavenging of Trp·/TyrO· radicals prevents their recombination with FADH·).
We have shown that, in the presence of high concentrations of an extrinsic reductant, the in vitro quantum yield of long-lived FADH·/FAD·− can reach up to ~14% (in the presence of ATP; the previous estimate12 of only ~2% had been obtained in the absence of ATP and at higher pH; see Supplementary Information, Section 8). This new value, combined with the prolonged lifetime of FADH· in the presence of ATP17,19, seems much more congruent with the role of AtCRY1 as photoreceptor and with formation of FADH· as the process responsible for cryptochrome signalling.
Based on our analysis of the photoreduction pathways (Fig. 6) we would like to propose an alternative trigger of the signalling conformational change42,43 in AtCRY1. Unlike neutral FADH·, the long-lived negative charge of D396− should induce a strong perturbation of the hydrophobic protein environment and is hence much more likely to trigger a major conformational change (leading to transduction of the initial light signal), e.g., by electrostatic repulsion between D396− (helix α16) and the electronegative backbone carbonyl oxygen of M381 (helix α15), to which protonated D396 is hydrogen-bonded prior to photoexcitation20. Long-lived D396− is formed only by the ATP-enhanced “ET through tryptophans” pathway (centre of the reaction scheme, Fig. 6) but not by the “PT first” pathway, in which D396− is promptly protonated by Trp1H·+. Another effect of ATP, namely the up-shift of the pKa of D396 from 7.4 to >9, is crucial to make this photoswitch efficient as it assures that virtually all cryptochromes are turned “off” (D396(H) state) in the dark under physiological conditions. Future experiments and computer simulations should test these hypotheses.
Methods
Reagents and materials
Water was de-ionized (≥18.2 MΩcm specific resistance) by Elga ultrapure water purification system. Adenosine 5′-triphosphate disodium salt (≥99%), Imidazole (>99%) and [Ru(bpy)3]Cl2 (99.95%) were purchased from Sigma Aldrich Chemicals. Tris buffer was prepared by titration of tris(hydroxymethyl)aminomethane (EUROMEDEX, molecular biology grade) by HCl (Carlo Erba Reagenti, p.a. grade). Phosphate buffer was prepared from Na2HPO4 (p.a.) from Merck and NaH2PO4 2H2O (>99%) from Prolabo. Triton X-100 was purchased from Prolabo. Quartz cells for spectroscopic experiments with self-masking solid black walls and sample chambers of 2 × 8 × 10 mm (width × height × length) with 4 clear windows were obtained from Starna.
Protein preparation
The plasmid coding full-length AtCRY1 with a His Tag was inserted into the BacPAK™ 6 baculovirus expression system (Clontech Laboratories, Inc.). Sf21 insect cells were inoculated with the baculovirus and incubated at 28°C. After 48 h, the cells were spun down (2 600 × g) and lysed in a lysis buffer (50 mM Tris buffer at pH 7.6, 0.5 M NaCl, 1% Triton X-100) for 60 min on ice (lysis buffer volume ~10× the volume of the cell sediment). The cell lysate was spun down (19 500 × g) and the supernatant was incubated with NTA resin from Qiagen (0.5 to 1 mL per 50 mL) at 4°C for 30 minutes. The resin was washed with 50 mL of the wash buffer A (50 mM Tris buffer at pH 7.6, 0.5 M NaCl, 10 mM imidazole), with 10 mL of the wash buffer B (50 mM Tris buffer at pH 7.6, 0.5 M NaCl, 20 mM imidazole) and eluted with elution buffer (50 mM Tris buffer at pH 7.6, 0.5 M NaCl, 350 mM imidazole). Imidazole was removed on Micro Bio-Spin (Bio-Gel P-6) size-exclusion chromatography columns from Bio-Rad.
Gene coding the AtCRY1 lacking the CCT domain was inserted into pGEX-6P-1. The protein was expressed in E. coli and purified with glutathione Sepharose followed by Mono Q and Sephacryl S-300 after tag cleavage. The protein was dissolved in a buffer solution containing 50 mM Tris–HCl (pH 8.0), 50 mM NaCl, and 5% (vol/vol) glycerol and deep-frozen (−80°C). Before use, the buffer was exchanged to 50 mM phosphate (or Tris) buffer of desired pH (and 0.5 M NaCl) using Micro Bio-Spin (Bio-Gel P-6) size-exclusion chromatography columns.
UV-Vis and transient absorption spectroscopy
UV-Vis spectra of protein solutions and the reference compound [Ru(bpy)3]Cl2 were recorded using the Uvikon XS spectrophotometer (Secomam). Protein spectra were recorded prior to, several times during and after each experiment in order to verify there was no significant accumulation of FADH· or deterioration of the samples by multiple laser flashes.
In all transient absorption experiments, the samples were excited at 355 nm by a Nd:YAG laser (Continuum Leopard SS-10, pulse duration of 100 ps, repetition rate 0.25 Hz). The vertically polarized laser beam was passed through attenuation filters and shaped by lenses so that the cross section of the beam covered the whole 8 × 10 mm cell window. Laser energies (as a control for the actinometric quantum yield calculation) were measured behind a cell filled with H2O using an energy meter (Gentec QE25SP-H-MB-D0).
Experiments on timescales <20 μs were performed using a modification of the setup described previously44. Monitoring light was provided by the following light sources:
403 nm - 105 mW Laser diode SHARP GH04P21A2GE,
457 nm - 50 mW DPSS cw-Laser Cobolt Twist,
562 nm - 25 mW DPSS cw-Laser Oxxius 561-25-COL-002.
The monitoring light beam was perpendicular to the excitation beam and shaped such that the central part of the beam passed through the 2 × 8 mm cell window along the 10 mm path. In order to minimize sample excitation by monitoring light, attenuation filters and a rotating blade with a small opening was introduced between the light source and the sample, chopping the monitoring light to rectangular pulses of 140 μs duration (separated by ~30 ms dark intervals). The energy of one such 140 μs pulse of measuring light received by the sample was in the order of 1 μJ (beam cross-sectional area of ~5 mm2), which is negligible compared to ~4 mJ/cm2 of the excitation flash.
Nevertheless, the repeated monitoring light pulses could lead to some loss of dark state (FADox), particularly for monitoring light at 457 nm, where FADox absorbs most strongly (ε = 9.5 mM−1cm−1). Taking into account an AtCRY1 concentration of ~45 μM, a maximal quantum yield of ~15% (in the presence of ATP) of radical pairs at 10 μs after excitation (Supplementary Information, Table 4) and recombination of ~85% of these radical pairs during the dark time of 30 ms between two monitoring light pulses (Fig. 4), we estimate that at most 6.5% of the dark state could be lost due to the monitoring light during accumulation of one package of 16 transient absorption signals (total acquisition time of ~1 min). In fact, the observed accumulated loss of dark state (as estimated from the transmission increase at 457 nm) due to the combined effects of monitoring light and excitation flashes never exceeded 2% indicating that further radical pair recombination occurs in the time window from 30 ms to 1 min.
The system was synchronized so that the excitation pulse came 30 μs after the rise of the monitoring light. A λ/2 plate (Melles Griot 99E02 WRA 0000) and a polarizer (COLPA 10.0-425-675 set to 54.7° with respect to the vertical direction) were inserted between the light source and the sample to turn the polarization of the monitoring light source so that the excited molecules were probed at magic angle to the excitation light polarization (elimination of anisotropic artefacts). The position of the cell with respect to the excitation beam was unchanged for all experiments in order to guarantee comparable excitation conditions.
The detection system for the monitoring light consisted of a Si photodiode (Alphalas UPD-500-UP, rise time 500 ps, spectral range 170–1100 nm, sensitive area 1 mm2), connected via an electronic signal amplifier (Femto HCA; DC-325 MHz, 28 dB) to a digital oscilloscope (Tektronix DSA602A, with 11A52 plugin set to DC-20 MHz). Interference filters with transmittance maxima at the wavelengths of the monitoring light were placed in front of the photodiode to block stray light and possible sample fluorescence induced by the excitation pulse. The oscilloscope was triggered by a small fraction of the excitation pulse through a fast photodiode. The kinetic curves are averages of 64 to 128 signals recorded in packages of 16 signals with several minutes dark interval between the packages.
For experiments on time scales >20 μs, a 100 W-tungsten-halogen lamp was used as a light source, combined with interference filters with transmission maxima at 410, 450 or 560 nm and spectral bandwidths of ~5 nm placed between the lamp and the sample. The probe light intensity and its changes associated with the kinetics of the light-induced processes were monitored behind the sample by a Si photodiode (FND 100Q from EG&G, protected by another interference filter transmitting at the respective wavelength) connected to an amplifier (Tektronics AM502, upper bandwidth limit set to 1 or 10 kHz) and the amplified signal was fed to a digital oscilloscope (Tektronix DSA602A, with 11A33 plugin). The kinetic curves are averages of 4 signals (with 4 s intervals between two excitation pulses allowing for sample recovery between the individual pulses).
The samples were air-saturated and constantly kept on ice or in cooled sample holders (keeping the temperature between 0 and 2°C) to minimize the risk of protein aggregation and degradation. The pH values of the buffers refer to this temperature range.
Quantum yield determination
Quantum yields of the transient radical pairs were determined using the [Ru(bpy)3]2+ actinometer27,45,46. 44.7 μM [Ru(bpy)3]Cl2 (in a cell identical to that containing AtCRY1) was excited under the same conditions as the protein in the experiment, the results of which are shown in Fig. 2. The average signal amplitude of this actinometer allowed estimating the incident photon fluence per excitation pulse and the concentration of excited FADox in the AtCRY1 samples. Quantum yields of the different radical pairs in AtCRY1 were calculated by referring their respective concentrations (obtained from the analysis of the transient absorption data) to the concentration of excited FADox (see Supplementary Information, Section 8).
Spectroelectrochemistry
Spectroelectrochemical titrations of AtCRY1 PHR were performed essentially as described in Ref. 47. See Supplementary Information, Section 5.
Fluorescence spectroscopy
Fluorescence measurements (Supplementary Information, Section 1) were conducted using the FluoroMax®-4 spectrofluorometer from Horiba Scientific. Excitation was at 445 nm (bandpass 10 nm), detection was at 540 nm (bandpass 5 nm).
Global analysis of the transient absorption kinetics
For a phenomenological description, the six experimental curves in Fig. 2 were globally fitted by multi-exponential decays. A satisfactory fit (see Supplementary Fig. 8a) was obtained with the tri-exponential function:
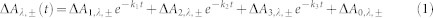 |
where ΔAλ,± denotes absorption change at wavelength λ in the presence (+) or absence (-) of ATP, t is time, ΔA1,λ,± to ΔA3,λ,± are the amplitudes of the individual kinetic phases at the given wavelengths and ATP condition, and k1 to k3 the exponential decay constants common to all six signals. The constants ΔA0,λ,± account for absorption changes that are much longer-lived than the 10 μs time window of the fit. The best fit yielded k1 = 4.1 × 107 s−1, k2 = 5.3 × 106 s−1 and k3 = 6.6 × 105 s−1 with amplitudes listed in Supplementary Table 1. Allowing the decay constants ki to depend on the ATP condition did not significantly improve the quality of the fit. The k2 and the k3 components were in first approximation assigned to deprotonation of a tryptophan cation radical TrpH·+ and protonation of FAD·−, respectively (see Results). The k1 component is close to the time resolution limit of the set-up and reflects the instrument response to processes that are too fast to be correctly monitored in the present experiments. The amplitudes ΔA1,λ,± were not used for further assignments.
The six experimental curves in Fig. 2 were also fitted by a function (equation (2)) that is based on the reaction model shown in Fig. 6. For the sake of simplicity, we introduce abbreviations for the states that could be observed at the present time resolution:
A = FAD·− – Trp3H·+; B = FAD·− – Trp3·; C = FADH· – Trp3·; C′ = FADH· – Trp1· and D = FADox – TrpH (ground state). Rate constants kAB, kAC', kAD, kBC and kCB refer to reactions A → B, A → C′, A → D, B → C and C → B, respectively. The species kinetics observable on the time scale of our experiments (~10 ns–10 μs) can be described as a superposition of three contributions corresponding to three different partial pathways (named α, β and γ in Supplementary Chart 1):
Proteins that contained unprotonated D396 at the moment of excitation and escape direct back ET in the first radical pair FAD·− – Trp1H·+ by ET through the tryptophan cascade. Protonation of FAD·− cannot occur. The initial (on our time scale) concentration of the pair FAD·− – Trp3H·+ (A) in the proteins following this pathway is denoted cα.
Proteins that contained protonated D396 at the moment of excitation and undergo ET through the tryptophan cascade (rather than immediate back ET or D396(H)-assisted ultra-fast PT in the first radical pair FAD·− – Trp1H·+). The initial (on our time scale) concentration of the pair FAD·− – Trp3H·+ (A) in the proteins following this pathway is denoted cβ.
Proteins that contained protonated D396 at the moment of excitation and undergo D396(H)-assisted ultrafast PT in the first radical pair FAD·− – Trp1H·+ (rather than direct back ET or ET through the tryptophan cascade). The initial (on our time scale) concentration of the pair FADH· – Trp1· (C′) in the proteins following this pathway is denoted cγ.
The differential equations governing the species kinetics, their analytical solutions and the conversion to absorption change kinetics are given in the Supplementary Chart 1. A term  was added to account for absorption changes that are too fast to be correctly resolved in the present experiments (see explanation to equation (1) above). The following equation was obtained:
was added to account for absorption changes that are too fast to be correctly resolved in the present experiments (see explanation to equation (1) above). The following equation was obtained:
 |
The indices + and − refer to the presence and absence of ATP, respectively. 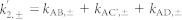 and
and 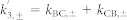 .
. 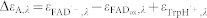 ,
, 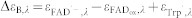 and
and 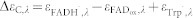 . εTrpH was omitted because neutral tryptophan in the ground state does not absorb at the wavelengths studied. C′ has the same absorption as C.
. εTrpH was omitted because neutral tryptophan in the ground state does not absorb at the wavelengths studied. C′ has the same absorption as C.
In globally fitting the six experimental curves in Fig. 2 with equation (2), k1' was fixed at the value obtained in the fit with equation (1). All other rate constants were free and allowed to depend on the ATP condition. Absorption coefficients were either fixed at the values from the spectra shown in Fig. 2 (for FADox, TrpH·+ and Trp· at all wavelengths and for FADH· at 457 and 562 nm) or allowed to vary in a limited range. FAD·− spectra in Ref. 35, as well as in other studies on insect cryptochromes (where FAD·− is much longer lived than in plant cryptochromes), have apparently been obtained by adding up-scaled photoreduction difference spectra to the spectrum of FADox. Because of an uncertainty with respect to the exact shape and scaling of these spectra, we have decided to use directly the photoreduction difference spectra of Drosophila cryptochrome from Ref. 39 to determine the ratio of 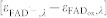 at the three wavelengths (403 nm/457 nm/562 nm): 15/–12/1 (note that because the blue absorption band of FADox in AtCRY1 (λmax = 445 nm) is blue-shifted by 5 nm with respect to insect CRY (λmax = 450 nm), ΔA462 instead of ΔA457 was read out from the figure). Maintaining this ratio was a condition in our fit and the absolute scaling at 403 nm was a free parameter within the range given by published spectra of protein-bound FADox and FAD·−
35,39,48,49: 5000 M−1cm−1 ≤
at the three wavelengths (403 nm/457 nm/562 nm): 15/–12/1 (note that because the blue absorption band of FADox in AtCRY1 (λmax = 445 nm) is blue-shifted by 5 nm with respect to insect CRY (λmax = 450 nm), ΔA462 instead of ΔA457 was read out from the figure). Maintaining this ratio was a condition in our fit and the absolute scaling at 403 nm was a free parameter within the range given by published spectra of protein-bound FADox and FAD·−
35,39,48,49: 5000 M−1cm−1 ≤ 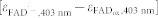 ≤ 7000 M−1 cm−1. The dip close to 400 nm in our FADH· spectrum (Fig. 2 and Supplementary Fig. 7) is less pronounced than in FADH· spectra from other flavoproteins50. We cannot exclude that this is due to some contamination of our spectrum by FADH− or by a species that had been oxidized by Trp· during photoreduction of FADox. Therefore,
≤ 7000 M−1 cm−1. The dip close to 400 nm in our FADH· spectrum (Fig. 2 and Supplementary Fig. 7) is less pronounced than in FADH· spectra from other flavoproteins50. We cannot exclude that this is due to some contamination of our spectrum by FADH− or by a species that had been oxidized by Trp· during photoreduction of FADox. Therefore, 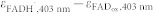 was allowed to vary between -1500 M−1cm−1 (value in Fig. 2) and -3000 M−1cm−1. All fits were done with user-defined functions in the Nonlinear Curve Fit algorithm of Origin (version 8.6).
was allowed to vary between -1500 M−1cm−1 (value in Fig. 2) and -3000 M−1cm−1. All fits were done with user-defined functions in the Nonlinear Curve Fit algorithm of Origin (version 8.6).
Author Contributions
P.M., K.B., J.P.B. and V.B. designed and performed experiments and analysed data; K.H. and P.M. prepared the protein samples, P.M. and K.B. wrote the manuscript; J.P.B., K.H., V.B., E.D.G. and T.R. co-edited the manuscript; T.R. and K.B. supervised the project.
Supplementary Material
Supplementary Information
Acknowledgments
We mourn the loss of our friend and scientific partner and collaborator Dr. Kenichi Hitomi who passed away on December 30, 2013. We thank Dr. Margaret Ahmad for her role in initiating the project, for preparing the full-length AtCRY1 for the initial studies, and for transferring this know-how to P.M. We acknowledge Dr. Aurélien de la Lande and Dr. Fabien Cailliez for stimulating discussions and the knowledgeable analysis of the published MD simulations. We also appreciate helpful discussions with Prof. Lars-Oliver Essen on X-ray crystallographic structures and we thank Dr. Pascal Plaza for precious advice on how to perform complex global analysis using Origin. This work was supported by Agence Nationale de la Recherche (grants #ANR-09-BLAN-0248 and #ANR-12-BSV8-0001-01), by Human Frontier (grant #RGP0045/2008-C) and by the French Infrastructure for Integrated Structural Biology (FRISBI): #ANR-10-INSB-05-01. We further acknowledge support by the National Science Foundation (grant #MCB-1330856).
References
- Chaves I. et al. The Cryptochromes: blue light photoreceptors in plants and animals. Annu. Rev. Plant. Biol. 62, 335–64 (2011). [DOI] [PubMed] [Google Scholar]
- Liu H., Liu B., Zhao C., Pepper M. & Lin C. The action mechanisms of plant cryptochromes. Trends in Plant Science 16, 684–91 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xing W. et al. SCFFBXL3 ubiquitin ligase targets cryptochromes at their cofactor pocket. Nature 496, 64–68 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Czarna A. et al. Structures of Drosophila Cryptochrome and Mouse Cryptochrome1 Provide Insight into Circadian Function. Cell 153, 1394–05 (2013). [DOI] [PubMed] [Google Scholar]
- Hirota T. et al. Identification of small molecule activators of cryptochrome. Science 337, 1094–97 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nangle S., Xing W. & Zheng N. Crystal structure of mammalian cryptochrome in complex with a small molecule competitor of its ubiquitin ligase. Cell Research 23, 1417–19 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bouly J. P. et al. Cryptochrome blue light photoreceptors are activated through interconversion of flavin redox states. J. Biol. Chem. 282, 9383–91 (2007). [DOI] [PubMed] [Google Scholar]
- Öztürk N., Song S. H., Selby C. P. & Sancar A. Animal type 1 cryptochromes. Analysis of the redox state of the flavin cofactor by site-directed mutagenesis. J. Biol. Chem. 283, 3256–63 (2008). [DOI] [PubMed] [Google Scholar]
- Liu Z. et al. Dynamic determination of the functional state in photolyase and the implication for cryptochrome. Proc. Natl. Acad. Sci. U.S.A. 110, 12972–77 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Macheroux P. UV-visible spectroscopy as a tool to study flavoproteins. In: Chapman, S. K. & Reid, G. A. eds. Flavoprotein Protocols. 131, 1–7 (Humana Press, Totowa, N.J., 1999). [DOI] [PubMed] [Google Scholar]
- Brettel K. & Byrdin M. Reaction mechanisms of DNA photolyase. Curr. Opin. Struc. Biol. 20, 693–701 (2010). [DOI] [PubMed] [Google Scholar]
- Giovani B., Byrdin M., Ahmad M. & Brettel K. Light-induced electron transfer in a cryptochrome blue-light photoreceptor. Nature Struct. Biol. 10, 489–90 (2003). [DOI] [PubMed] [Google Scholar]
- Zeugner A. et al. Light-induced electron transfer in Arabidopsis cryptochrome-1 correlates with in vivo function. J. Biol. Chem. 280, 19437–40 (2005). [DOI] [PubMed] [Google Scholar]
- Bouly J. P. et al. Novel ATP-binding and autophosphorylation activity associated with Arabidopsis and human cryptochrome-1. Eur. J. Biochem. 270, 2921–28 (2003). [DOI] [PubMed] [Google Scholar]
- Shalitin D., Yu X., Maymon M., Mockler T. & Lin C. Blue light dependent in vivo and in vitro phosphorylation of Arabidopsis cryptochrome 1. Plant Cell 15, 2421–29 (2003). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ozgür S. & Sancar A. Analysis of autophosphorylating kinase activities of Arabidopsis and human cryptochromes. Biochemistry 45, 13369–74 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Immeln D., Schlesinger R., Heberle J. & Kottke T. Blue light induces radical formation and autophosphorylation in the light-sensitive domain of Chlamydomonas cryptochrome. J. Biol. Chem. 282, 21720–28 (2007). [DOI] [PubMed] [Google Scholar]
- Ozturk N. et al. Comparative photochemistry of animal type 1 and type 4 cryptochromes. Biochemistry 48, 8585–93 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burney S. et al. Conformational change induced by ATP binding correlates with enhanced biological function of Arabidopsis cryptochrome. FEBS Letters 583, 1427–33 (2009). [DOI] [PubMed] [Google Scholar]
- Brautigam C. A. et al. Structure of the photolyase-like domain of cryptochrome 1 from Arabidopsis thaliana. Proc. Natl. Acad. Sci. U.S.A. 101, 12142–47 (2004). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hitomi K. et al. Functional motifs in the (6-4) photolyase crystal structure make a comparative framework for DNA repair photolyases and clock cryptochromes. Proc. Natl. Acad. Sci. U.S.A. 106, 6962–67 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kottke T., Batschauer A., Ahmad M. & Heberle J. Blue-light-induced changes in Arabidopsis cryptochrome 1 probed by FTIR difference spectroscopy. Biochemistry 45, 2472–79 (2006). [DOI] [PubMed] [Google Scholar]
- Immeln D. et al. Photoreaction of plant and DASH cryptochromes probed by infrared spectroscopy: the neutral radical state of flavoproteins. J. Phys. Chem. B 114, 17155–61 (2010). [DOI] [PubMed] [Google Scholar]
- Langenbacher T., Immeln D., Dick B. & Kottke T. Microsecond light-induced proton transfer to flavin in the blue light sensor plant cryptochrome. J. Am. Chem. Soc. 131, 14274–80 (2009). [DOI] [PubMed] [Google Scholar]
- Solov'yov I. A., Domratcheva T., Shahi A. R. M. & Schulten K. Decrypting cryptochrome: revealing the molecular identity of the photoactivation reaction. J. Am. Chem. Soc. 134, 18046–52 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burney S. et al. Single amino acid substitution reveals latent photolyase activity in Arabidopsis cry1. Angew. Chem. Int. Ed. 51, 9356–60 (2012). [DOI] [PubMed] [Google Scholar]
- Müller P. & Brettel K. [Ru(bpy)3]2+ as a reference in transient absorption spectroscopy: differential absorption coefficients for formation of the long-lived 3MLCT excited state. Photochem. Photobiol. Sci. 11, 632–36 (2012). [DOI] [PubMed] [Google Scholar]
- Aubert C., Vos M. H., Mathis P., Eker A. P. M. & Brettel K. Intraprotein radical transfer during photoactivation of DNA photolyase. Nature 405, 586–90 (2000). [DOI] [PubMed] [Google Scholar]
- Jorns M. S., Wang B., Jordan S. P. & Chanderkar L. P. Chromophore function and interaction in Escherichia coli DNA photolyase: reconstitution of the apoenzyme with pterin and/or flavin derivatives. Biochemistry 29, 552–61 (1990). [DOI] [PubMed] [Google Scholar]
- MacFarlane A. W. & Stanley R. J. Evidence of powerful substrate electric fields in DNA photolyase: implications for thymidine dimer repair. Biochemistry 40, 15203–14 (2001). [DOI] [PubMed] [Google Scholar]
- Immeln D., Weigel A., Kottke T. & Lustres J. L. P. Primary events in the blue light sensor plant cryptochrome: intraprotein electron and proton transfer revealed by femtosecond spectroscopy. J. Am. Chem. Soc. 134, 12536–46 (2012). [DOI] [PubMed] [Google Scholar]
- Kiontke S. Strukturbasierte Charakterisierung von Klasse II CPD Photolyasen. PhD Thesis, Philipps University of Marburg, Germany (2012). <http://archiv.ub.uni-marburg.de/diss/z2013/0246/pdf/dsk.pdf> Date of access: 10/04/2014. [Google Scholar]
- Shen J. et al. Organelle pH in the Arabidopsis endomembrane system. Mol. Plant 6, 1419–37 (2013). [DOI] [PubMed] [Google Scholar]
- Exner V. et al. A gain-of-function mutation of Arabidopsis cryptochrome1 promotes flowering. Plant Physiol. 154, 1633–45 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu B., Liu H., Zhong D. & Lin C. Searching for a photocycle of the cryptochrome photoreceptors. Curr. Opin. Struct. Biol. 13, 578–86 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Solar S., Getoff N., Surdhar P. S., Armstrong D. A. & Singh A. Oxidation of tryptophan an N-methylindole by N3, Br2−, and (SCN)2− radicals in light- and heavy-water Solutions: a pulse radiolysis study. J. Phys. Chem. 95, 3639–43 (1991). [Google Scholar]
- Giese B. et al. Electron relay race in peptides. J. Org. Chem. 74, 3621–25 (2009). [DOI] [PubMed] [Google Scholar]
- Dudley Bryant F., Santus R. & Grossweiner L. I. Laser flash photolysis of aqueous tryptophan. J. Phys. Chem. 79, 2711–16 (1975). [Google Scholar]
- Berndt A. et al. A novel photoreaction mechanism for the circadian blue light photoreceptor Drosophila cryptochrome. J. Biol. Chem. 282, 13011–21 (2007). [DOI] [PubMed] [Google Scholar]
- Berezhnoy A. Y. & Duplij S. A. Dependence of nucleotide physical properties on their placement in codons and determinative degree. J. Zhejiang. Univ. SCI. 6B, 948–60 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blatt M. R. Electrical characteristics of stomatal guard cells: the contribution of ATP-dependent, “electrogenic” transport revealed by current-voltage and difference-current-voltage analysis. J. Membrane Biol. 98, 257–74 (1987). [Google Scholar]
- Yang H. Q. et al. The C termini of Arabidopsis cryptochromes mediate a constitutive light response. Cell 103, 815–27 (2000). [DOI] [PubMed] [Google Scholar]
- Kondoh M. et al. Light-induced conformational changes in full-length Arabidopsis thaliana cryptochrome. J. Mol. Biol. 413, 128–37 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Byrdin M., Thiagarajan V., Villette S., Espagne A. & Brettel K. Use of ruthenium dyes for subnanosecond detector fidelity testing in real time transient absorption. Rev Sci. Intrum. 80, 043102 (2009). [DOI] [PubMed] [Google Scholar]
- Byrdin M. et al. Quantum yield measurements of short-lived photoactivation intermediates in DNA photolyase: Towards a detailed understanding of the triple tryptophan electron transfer chain. J. Phys. Chem. A 114, 3207–14 (2010). [DOI] [PubMed] [Google Scholar]
- Thiagarajan V., Byrdin M., Eker A. P. M., Müller P. & Brettel K. Kinetics of cyclobutane pyrimidine dimer splitting by DNA photolyase directly monitored in the UV. Proc. Natl. Acad. Sci. U.S.A. 108, 9402–07 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Balland V., Byrdin M., Eker A. P. M., Ahmad M. & Brettel K. What makes the difference between a cryptochrome and DNA photolyase? A spectroelectrochemical comparison of the flavin redox transitions. J. Am. Chem. Soc. 131, 426–27 (2009). [DOI] [PubMed] [Google Scholar]
- Kao Y. T. et al. Ultrafast dynamics and anionic active states of the flavin cofactor in cryptochrome and photolyase. J. Am. Chem. Soc. 130, 7695–701 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Damiani M. J., Nostedt J. J. & O'Neill M. A. Impact of the N5-proximal Asn on the thermodynamic and kinetic stability of the semiquinone radical in photolyase. J. Biol. Chem. 286, 4382–91 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Müller F., Brüstlein M., Hemmerich P., Massey V. & Walker W. H. Light-absorption studies on neutral flavin radicals. Eur. J. Biochem. 25, 573–80 (1972). [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Information