

Abstract
A DNA macroarray containing 465 intragenic amplicons was designed to identify Staphylococcus aureus at the species level and to type S. aureus isolates. The genes selected included those encoding (i) S. aureus-specific proteins, (ii) staphylococcal and enterococcal proteins mediating antibiotic resistance and factors involved in their expression, (iii) putative virulence proteins and factors controlling their expression, and (iv) proteins produced by mobile elements. The macroarray was hybridized with the cellular DNAs of 80 S. aureus clinical isolates that were previously typed by analyses of their antibiograms and SmaI patterns. The set selected contained unrelated, endemic, and outbreak-related isolates belonging to 45 SmaI genotypes. In a gene content dendrogram, the 80 isolates were distributed into 52 clusters. The outbreak-related isolates were linked in the same or a closely related cluster(s). Clustering based on gene content provided a better discrimination than SmaI pattern analysis for the tested mecA+ isolates that were endemic to Europe. All of the antibiotic resistance genes detected could be correlated with their corresponding phenotypes, except for one isolate which carried a mecA gene without being resistant. The 16 isolates responsible for bone infections were distinguishable from the 12 isolates from uninfected nasal carriers by a significantly higher prevalence of the sdrD gene coding for a putative SD (serine-aspartate) adhesin (in 15 and 7 isolates, respectively). In conclusion, the macroarray designed for this study offers an attractive and rapid typing method which has the advantage of providing additional information concerning the gene content of the isolate of interest.
The best known staphylococcal species is Staphylococcus aureus, by virtue of its frequent and highly versatile pathogenicity in humans and animals. Isolates belonging to this species are responsible for suppurative infections and syndromes provoked by toxins. Excluding pathologies caused by toxins such as enterotoxins and exfoliative or toxic shock syndrome toxins (20), the pathology of a staphylococcal infection is attributable not to a single factor but to the coordinated actions of several factors whose expression is controlled by several regulatory systems (3, 26, 29, 30). S. aureus is one of the most common causes of nosocomial infections. The emergence of such infections is of particular concern since most isolates, such as methicillin-resistant S. aureus (MRSA), are resistant to several antibiotics (4, 28) and because the spread of these strains in hospitals often increases the overall incidence of nosocomial S. aureus infections in the institution. MRSA clinical isolates with decreased susceptibilities to glycopeptides (1, 17) threaten to compromise our ability to treat hospital-acquired S. aureus infections.
S. aureus typing is a useful adjunct in several clinical settings, in addition to its use during dramatic acute outbreaks. Despite the use of several phenotypic and genotypic methods (antibiotyping, phage typing, multilocus enzyme electrophoresis, restriction analysis of cellular DNA, analysis of PCR products, and multilocus sequence typing) (10, 13, 22, 24, 31, 32, 35, 36), indistinguishable or closely related isolates have been detected not only among those responsible for outbreaks, but also among those isolated in different countries, at time intervals of several years, and without any obvious epidemiological links. Indeed, Oliveira et al. (27) identified five major pandemic MRSA clones that accounted for almost 70% of the 3,000 isolates analyzed.
The whole genome sequencing of seven S. aureus strains (N315 [19], Mu50 [19], COL [http://www.tigr.org/tdb/], MW2 [2], NCTC8325 [http://www.genome.ou.edu/staph.html], methicillin-susceptible S. aureus strain 476 [http://www.sanger.ac.uk/Projects/S_aureus/], and epidemic MRSA (EMRSA) 16 strain 252 [http://www.sanger.ac.uk/Projects/S_aureus/]) revealed the presence of large amounts of well-conserved DNA regions in the chromosomes. Fitzgerald et al. (11) demonstrated that 2,198 (78%) of the 2,817 COL chromosomal open reading frames (ORFs) represented on a DNA microarray were shared by the 36 analyzed S. aureus isolates from various sources, which belonged to 14 multilocus enzyme electrophoretic types. Ten of the 18 large regions of difference carry genes that encode putative virulence factors and proteins that mediate antibiotic resistance.
The aim of the present study was to design a DNA macroarray with several intragenic PCR amplicons to identify S. aureus at the species level and to type S. aureus isolates. To evaluate the DNA macroarray's usefulness for typing and for the investigation of a putative pathogenicity index correlated with bone infections (BIs), we probed it with cellular DNAs from 80 clinical isolates that were previously typed by the determination of their antibiograms and SmaI restriction patterns. These included unrelated isolates responsible for BIs and isolates from nasal samples of uninfected carriers to check whether these two categories of isolates could be distinguished.
MATERIALS AND METHODS
Bacterial isolates.
The relevant characteristics of the 80 S. aureus clinical isolates used to validate the DNA macroarray designed in this study are given in Table 1. The 44 staphylococcal, enterococcal, and Escherichia coli stains used as substrates for PCR amplification of the genes chosen for the construction of the macroarray are reported in Table S1 in the supplemental material at http://genopole.pasteur.fr/staph/.
TABLE 1.
Relevant characteristics of S. aureus clinical isolates
| Isolate designation or characteristics | Year of isolation | Source (city/country/hospital) | SmaI genotypeb | Reference | Antibiotic resistance marker(s)a,c | Antibiotic resistance gene(s) (on DNA macroarrays) |
|---|---|---|---|---|---|---|
| Outbreak-related isolates from the same ward | This study | |||||
| PEN + | blaZ + | |||||
| IPF735 | 101 | MLSi | ermC | |||
| IPF736 | 101 | MLSi | ermC | |||
| IPF738 | 2000 | Calais/France/A | 101 | No additional marker | No additional gene | |
| IPF741 | 101 | MLSi | ermC | |||
| IPF743 | 101 | MLSi | ermC | |||
| Outbreak-related isolates producing exfoliative toxin A and responsible for scalded skin syndrome in neonates | This study | |||||
| PEN + | blaZ | |||||
| IPF308 | 100 | No additional marker | No additional gene | |||
| IPF310 | 2001 | Villencuve St Georges/France/B | 100 | No additional marker | No additional gene | |
| IPF311 | 100 | No additional marker | No additional gene | |||
| IPF313 | 100 | No additional marker | No additional gene | |||
| Outbreak-related h-VISAb isolates, indistinguishable from isolates previously detected at low frequencies | Paris/France/C | 14 | ||||
| PEN, OXA, STR, TET, MIN, SPT, KAN, NEO, TOB, GEN, MLSc, PEF, RIF, FUC, h-VISA + | blaZ, mecA, tetM, spc, aadD, aac A-aph D, ermA | |||||
| IPF555 | 39aA | No additional marker | No additional gene | |||
| IPF557 | 1999 | 39aA | FOF | No additional gene | ||
| IPF562 | 39aA | FOF | No additional gene | |||
| MSRA isolates with decreased susceptibility to glycopeptides, endemically spread in several European cities | ||||||
| PEN, OXA, STR, TET, MIN, SPT, KAN, NEO, TOB, GEN, MLSc, PEF, RIF + | blaZ, mecA, tetM, spc, aadD, aacA-aphD, ermA | |||||
| BM12612(CIP106757) | 1998 | Villiers St Denis/France/D | 39aA | 7 | GISA, FOF | No additional gene |
| BM10828(CIP106759) | 1993 | Bordeaux/France/E | 39aA | 22 | h-VISA,b SUL, FUC, FOF | No additional gene |
| SPAIN E1 | 1989 | Seville/Spain/- | 39aA | 23 | h h-VISA,b SUL, FUC | No additional gene |
| FINLAND E7 | 1990 | Turku/Finland/- | 39bA | 23 | No additional marker | No additional gene |
| 97130(CIP106761) | 1997 | Toulouse/France/F | 39cA | 7 | h-VISAb, FUC, FOF | No additional gene |
| 96145(CIP106762) | 1996 | Blois/France/G | 39dA | 7 | h-VISAb, FOF | No additional gene |
| BM10829 | 1993 | Bordeaux/France/E | 39eA | 22 | h h-VISAb, SUL, FUC | No additional gene |
| Phage-type 77 MSRA isolates endemically spread in European cities | ||||||
| OXA, STR, TET, MIN, SPT, KAN, TOB, GEN, MLSc, PEF + | mecA, tetM, spc, aacA-aphD, ermA + | |||||
| BM9290 | 1987 | Paris/France/H | 45aA | PEN, RIF | blaZ | |
| BM9586 | 1987 | Paris/France/C | 45aA | PEN, RIF, FOF | blaZ, fos | |
| BM12184 | 1987 | Paris/France/C | 45aA | PEN, RIF, FOF | blaZ, fos | |
| BM12188 | 1987 | Paris/France/C | 45aA | RIF | No additional gene | |
| BM10761 | 1993 | Toulouse/France/F | 45bA | PEN, NEO, RIF, FUC, FOF, PRI, SGA, SXT | blaZ, aadD | |
| BM10759 | 1993 | Toulouse/France/F | 45cA | PEN, NEO, RIF, FUC, FOF, PRI, SGA, SXT | blaZ, aadD | |
| BM9343 | 1987 | Toulouse/France/F | 45dA | PEN, RIF, FUC, FOF | blaZ, fos | |
| BM10872 | 1992 | Aalst/Belgium/I | 45eA | PEN | blaZ | |
| BM10888 | 1993 | Aalst/Belgium/I | 45eA | PEN | blaZ | |
| BM10896 | 1994 | Ghent/Belgium/J | 45fA | PEN, NEO | blaZ, aadD | |
| BM10914 | 1991 | Paris/France/C | 45fA | PEN, NEO, RIF, FOF | blaZ, aadD | |
| BM10130 | 1989 | Barcelona/Spain/K | 45gA | PEN, NEO, RIF | blaZ, aadD | |
| BM10138 | 1989 | Barcelona/Spain/K | 45gA | PEN, NEO, RIF | blaZ, aadD | |
| BM12152 | 1989 | Barcelona/Spain/K | 45iA | PEN, NEO, RIF | blaZ, aadD | |
| SGAr isolates whose streptogramin-resistant genes were previously investigated by PCR | ||||||
| PEN, KAN, NEO, SGA + | ||||||
| BM3364 | 1981 | Paris/France/C | 13 | OXA, TET, MIN, SPT, TOB, GEN, MLSc | blaZ, mecA, tetM, spc, aadE, aacA-aphD, ermA, aphA-3, vgaAv, vgaB, vatB | |
| BM12828 | 1999 | Paris/France/C | 16aB | OXA, SPT, TOB, MLSc, PRI, SXT, PEF | mecA, spc, aadD, ermA, vatA, vgbA | |
| BM12830 | 1999 | Paris/France/C | 16aB | OXA, SPT, TOB, GEN, MLSc, PRI, PEF, RIF | mecA, spc, aadD, aacA-aphD, ermA, vatA, vgbA | |
| BM12714 | 1996 | Grenoble/France/L | 17bB | OXA, SPT, TOB, GEN, MLSc, PRI, SXT, PEF, FOF | blaZ, mecA, spc, aadD, aacA-aphD, ermA, vgaAv, vatB, vgaB, dfrA | |
| BM12942 | 1999 | Paris/France/C | 19 | STR, MLSc, PRI, SUL, SXT, FUC | blaZ, aadE, aph A-3, ermC, vatA, vgbA | |
| BM12286 | 1996 | Paris/France/M | 36a | STR, PRI | blaZ, aadE, aph A-3, vatA, vgbA | |
| BM12827 | 1996 | Paris/France/M | 36a | STR, PRI | blaZ, aadE, aphA-3, vatA, vgbA | |
| 97233 | 1997 | Paris/France/C | 24hF | OXA, TOB, MLSc, PRI, SXT, PEF | blaZ, mecA, aadD, vga Av | |
| IPF083 | 1998 | Toulouse/France/F | 24aF | OXA, TOB, LIN, PEF | blaZ, mecA, aadD, vga Av | |
| 93184 | 1993 | Paris/France/N | 26F | OXA, TOB, PRI, PEF, RIF | blaZ, mecA, aadD, vatA, vgbA | |
| Isolates from uninfected NCs | Tunis/Tunisia/O | This study | ||||
| IPF139 | 2000 | 133 | PEN, FOF | blaZ, fos | ||
| IPF140 | 2001 | 139 | PEN, | blaZ | ||
| IPF143 | 2001 | 149aC | PEN | blaZ | ||
| IPF145 | 2001 | 155a | PEN, MLSi, FUC, RIF | blaZ, ermC | ||
| IPF147 | 2001 | 121b | PEN, MLSi | blaZ, ermC | ||
| IPF150 | 2001 | 137a | PEN | blaZ | ||
| IPF153 | 2001 | 128 | PEN, | blaZ | ||
| IPF157 | 2001 | 127 | PEN, TET, MIN, STR, KAN, NEO | blaZ, tetK; tetM, aadE, aphA-3 | ||
| IPF159 | 2001 | 108D | PEN | blaZ | ||
| IPF511 | 2001 | 103 | PEN | blaZ | ||
| IPF520 | 2002 | 161 | No resistance marker | No resistance gene | ||
| IPF524 | 2002 | 104 | PEN, TET | blaZ, tetK | ||
| Isolates responsible for BIs | ||||||
| BM12623 | 1998 | Tunis/Tunisia/O | 163 | This study | PEN, TET, STR, KAN NEO, ERY | blaZ, tetK, aadE, aphA-3, msrA |
| BM12633 | 1998 | Tunis/Tunisia/O | 136a | This study | PEN, STR, KAN NEO, ERY | blaZ, aadE, aphA-3, msrA |
| BM12681 | 1997 | Tunis/Tunisia/O | 125a | This study | PEN, OXA, TET, MIN, STR, SPT, KAN, GEN, TOB, RIF | blaZ, mecA, tetM, spc, aacA-aphD, ermA |
| BM12685 | 1997 | Tunis/Tunisia/O | 151a | This study | PEN | blaZ |
| BM12718 | 1998 | Tunis/Tunisia/O | 120a | This study | PEN, TET, MIN | blaZ, tetM |
| BM12881 | 1999 | Tunis/Tunisia/O | 140i | This study | PEN, MLSi | blaZ, ermC |
| BM12889 | 1999 | Tunis/Tunisia/O | 117a | This study | PEN, TET, MIN | blaZ, tetM |
| BM12987 (O24) | 1989 | Sweden | 144eC | 33 | PEN | blaZ |
| IPF161 | 2000 | Tunis/Tunisia/O | 141b | This study | STR, KAN, NEO, FUC | mecA, aadE, aphA-3 |
| IPF166 | 2000 | Tunis/Tunisia/O | 116a | This study | PEN, TET, MIN, LIN | tetM, lnuA |
| IPF488 | 2001 | Tunis/Tunisia/O | 102 | This study | PEN | blaZ |
| IPF490 | 2001 | Tunis/Tunisia/O | 111D | This study | PEN | blaZ |
| IPF493 | 2001 | Tunis/Tunisia/O | 143 | This study | PEN | blaZ |
| IPF494 | 2001 | Tunis/Tunisia/O | 132 | This study | PEN | blaZ |
| IPF497 | 2001 | Tunis/Tunisia/O | 119 | This study | TET, MIN | tetM |
| IPF498 | 2001 | Tunis/Tunisia/O | 162a | This study | PEN | blaZ |
| Isolates responsible for cutaneous infections | Tunis/Tunisia/O | E | This study | |||
| BM12666 | 1998 | 105 | PEN | blaZ | ||
| BM12755 | 1998 | 123 | PEN, OXA, TET, MIN, SPT, KAN, GEN, TOB, MLSc, RIF | blaZ, mecA, tetK, tetM, spc, aacA-aphD, ermA, ermC | ||
| BM12764 | 1998 | 131 | TET, MIN | tetM | ||
| BM12766 | 1998 | 117a | TET, MIN | tetM | ||
| BM12771 | 1997 | 120a | PEN, TET, MIN | blaZ, tetM | ||
| BM12816 | 1998 | 118 | PEN, OXA, TET, MIN, KAN, TOB, MLSi, RIF | mecA, tetM, aadD, ermC | ||
| BM12863 | 1998 | 130 | PEN | blaZ | ||
| BM12947 | 1999 | 144aC | PEN, MLSi | blaZ, msrA | ||
| IPF505 | 2001 | 126 | PEN, OXA, TET, MIN, SPT, STR, KAN, GEN, TOB, MLSc, SXT, RIF | blaZ, mecA, tetK, tetM, spc, aacA-aphD, ermA, ermC |
Abbreviations ERY, erythromycin; FUC, fucidic acid; FOF, fosfomycin; h-VISA, heterogeneous vancomycin-intermediate S. aureus; GEN, gentamicin; GISA, glycopeptide intermediate S. aureus; KAN, kanamycin; LIN, lincomycin; MLSci, macrolides-lincosamides-streptogramin B-inducible resistance; MLSc, macrolides-lincosamides-streptogramin B constitutive resistance; MIN, minocycline; NEO, neomycin; OXA, oxacillin; PEF, pefloxacin; PEN, penicillinase; PRI, pristinamycin; RIF, rifampin; SGA, streptogramin A; SPT, spectinomycin; STR, streptomycin; SUL, sulfonamides; SXT, trimethoprim-sulfamethoxazole; TET, tetracycline; TOB, tobramycin.
Strains were clustered according to the following criteria proposed by Tenover et al. (32). (i) Strains were grouped in the same major genotype if their patterns differed by no more than three bands (these strains were considered to be closely related and monoclonal). (ii) If patterns differed by between four and six bands, the strains were scored as being possibly related but were nevertheless classified into distinct genotypes to discriminate them from the closely related strains. (iii) If patterns differed by seven or more bands, strains were considered to be different. Major genotypes are designated by arabic numerals. Strains with indistinguishable patterns were classified within the same subtype. Subtypes are designated by arabic numerals with letter suffixes. Genotypes which include strains that are possibly related (less than seven bands with differences) are marked with a superscript letter.
All h-VISA strains were either intermediate or resistant to teicoplanin.
DNA extraction.
Total cellular DNAs were extracted and purified by use of a QIAamp DNA mini kit (Qiagen, Hilden, Germany). The method described by the supplier was modified by the inclusion of lysostaphin (Applied Microbiology), at a final concentration of 100 mg/liter, in the lysis step. RNAs were removed after 30 min of incubation at 37°C by the addition of 5 mg of RNase (DNase-free) (Roche, Meylan, France)/liter.
Comparative genome analysis, primer design, and PCR amplification.
For the annotation and comparative analysis of the available genome sequences from the seven S. aureus isolates cited above, the program CAAT-Box (12) was used. Genes whose nucleotide sequences exhibited <80% similarity were considered distinct. CAAT-Box uses the BLAST program, which presents the area of least similarity with the rest of the genome. The Primer3 program (http://www.broad.mit.edu/cgi-bin/primer/primer3-www.cg) identifies primer pairs in this specific area which are unlikely to produce nonspecific amplifications with regard to the seven sequenced S. aureus genomes. The criteria used by CAAT-Box and Primer3 were as follows: match threshold, 21; maximum length of nonspecific PCR products, 3,000 bases; minimum PCR product length, 250 bases; optimum PCR product length, 400 to 500 bases; primer size, 18, 20, or 25 bases (minimum, optimum, and maximum sizes); primer melting temperature (Tm), 51, 55, or 60°C; % G+C, 25, 50, or 80%; maximum difference in Tm for a primer pair, 5°C.
Each of the 478 selected genes encoded at least 150 amino acids. Primers were designed to amplify a fragment of 400 to 500 bp specific for each gene. Each PCR was performed in a 100-μl reaction volume containing 10 to 20 ng of DNA and a 1 μM concentration of each primer (Eurogentec, Liege, Belgium). The conditions used were an initial cycle of 5 min at 94°C, followed by 35 cycles of 1 min at 94°C, 1 min at 50°C, and 1 min at 72°C, with a final extension step of 7 min at 72°C. The concentration and size of each PCR product were verified by electrophoresis using agarose gels.
Array construction.
For array preparation, high-density nylon Performa membranes (Genetix, New Milton, United Kingdom) were soaked in TE solution (10 mM Tris [pH 7.6], 1 mM EDTA). Double spot blots of each PCR product were printed (50 ng of DNA in PCR buffer per spot) by a Qpix robot (Genetix). After spot deposition, DNAs were denatured and fixed on the membranes by incubation for 15 min in 0.5 M NaOH-1.5 M NaCl. The membranes were then washed briefly in distilled water and stored wet at −20°C until use.
Hybridization.
The cellular DNAs of the S. aureus strains (50 ng) were labeled by use of a random priming DNA labeling kit (Roche Diagnostics GmbH, Penzberg, Germany) and 50 μCi of 5′-[α-33P]dCTP (Amersham, Piscataway, N.J.). Labeled probes were purified by use of a QIAquick nucleotide removal kit (Qiagen). The membranes were moistened in 2× SSC (0.3 M NaCl, 0.03 M sodium citrate) and prehybridized for 1 h in 10 ml of 5× SSPE (0.9 M NaCl, 6 mM NaH2PO4, 7.5 mM EDTA, pH 8), 4% sodium dodecyl sulfate, 1× Denhardt solution (0.02% Ficoll, 0.02% polyvinylpyrrolidone, 0.02% bovine serum albumin), and 1 mg of denatured salmon sperm DNA. Hybridization was performed overnight at 65°C. Membranes were washed twice at room temperature and twice at 65°C in 0.5× SSPE-0.2% sodium dodecyl sulfate. Arrays were then sealed in polypropylene bags and exposed to a PhosphorImager screen for 24 h.
Verification of specificity of DNA macroarray.
Of the 478 DNA fragments amplified, 106 were randomly chosen and sequenced. Sequencing of the PCR products was done with an ABI3700 capillary sequencer. For a test of correct spotting, the membranes loaded with the amplicons were hybridized with the cellular DNAs of the S. aureus strains used as substrates in PCR amplifications. For 465 amplicons, the results were as expected, i.e., specific. Thirteen of the 478 genes selected were eliminated, either because two nonspecific DNA bands were amplified (1 gene) or because hybridization experiments revealed false-positive or -negative results (10 and 2 genes, respectively). The characteristics of the amplicons and the strains used as substrates, as well as the sequences of the primers and their positions on the genome, are shown in Table S1 (http://genopole.pasteur.fr/staph/).
Data analysis.
For scanning, a Typhoon 9400 PhosphorImager (Molecular Dynamics) was used. Array Vision software (Imaging Research) was used for the quantification of the hybridization intensities and for normalization. For each spot, the hybridization intensity value was normalized by dividing it by the average of all significant intensity values on each membrane. For gene content analysis, a reference array was built by combining the average normalized data of two replicate hybridization experiments with the cellular DNAs of the strains used as substrates for PCR amplification. When a gene was known to be present either as a single copy or as multiple copies, the lowest significant intensity value corresponding to a single-copy gene was chosen. When a gene was known to be present in the tested strain used as a substrate, such as in the five strains whose genomes have been sequenced (N315 [19], Mu50 [19], COL [http://www.tigr.org/tdb/], MW2 [2], and NCTC8325 [http://www.genome.ou.edu/staph.html]), the ratio between the normalized signal intensity of the gene hybridized with the tested strain and that of the reference array was always higher than 0.3. Thus, the threshold for the presence of a gene or a variant related by at least 80% similarity was defined as 0.3. The data were then converted into a binary score as follows: at ≥0.3, a gene was scored as present (score = 1), and at <0.3, a gene was scored as absent (score = 0).
The binary data were used to cluster the isolates hierarchically, using the program J-Express (9). The threshold adopted to distribute the isolates into clusters was that which enabled each of the outbreak-related isolates belonging to SmaI genotypes 100 or 101 (Table 1) to be grouped and distinguished from any of the other isolates.
Comparative analysis of the gene contents for different categories of isolates.
When categories of n and m isolates are compared, the probability that a given gene is present by chance in n1 isolates of the first category of isolates and n2 isolates of the second category is given by the following binomial formula:
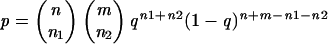 |
where q is estimated by maximum likelihood, using the equation q = (n1 + n2)/(n + m). A Bayesian approach based on the integration over q with a uniform prior gives results similar to those presented in the sequel. pg is the normalized probability, with g representing the total number of genes investigated. The gene distribution was considered significant if the normalized probability, or pg, was <0.10.
RESULTS
Choice of genes for construction of DNA macroarray.
Based on a comparative analysis of the seven S. aureus genomes sequenced (N315 [19], Mu50 [19], COL [http://www.tigr.org/tdb/], MW2 [2], NCTC8325 [http://www.genome.ou.edu/staph.html], methicillin-susceptible S. aureus strain 476 [http://www.sanger.ac.uk/Projects/S_aureus/], and EMRSA 16 strain 252 [http://www.sanger.ac.uk/Projects/S_aureus/]), we selected 397 genes for the macroarray. Among these strains, 305 of the genes were not shared by all of them and thus were candidate probes for typing. Although they were shared by the seven sequenced genomes, 92 additional genes were used. They included genes such as nuc (6) and sodM (34) for identification at the species level, genes encoding putative virulence proteins and factors involved in their regulation, and genes encoding proteins involved in antibiotic transport and resistance expression.
Furthermore, 67 genes that were not detected in these seven S. aureus genomes were also spotted on the array because they encoded specific groups of proteins. (i) Genes encoding staphylococcal and enterococcal proteins mediating drug resistance were included. Thirteen antibiotic resistance genes were identified in gram-positive species other than S. aureus, as follows: Staphylococcus hyicus, tetL; Staphylococcus cohnii, vatC and vgbB; Staphylococcus epidermidis, fos and lnuA; Enterococcus faecium, vatD, vatE, msrA, lnuB, and vanA; Enterococcus faecalis, vanB and lsa; and Enterococcus gallinarum, vanC. These genes were chosen because of their possible transfer to S. aureus. (ii) Genes encoding factors known to be involved in S. aureus pathogenicity and structurally related proteins (e.g., toxins, adhesins, and enzymes involved in the biosynthesis of capsule or slime) were also included. (iii) Finally, genes encoding proteins produced by mobile elements (transposons, insertion sequences, and plasmids) were spotted on the array. The negative control consisted of an amplicon corresponding to the Staphylococcus intermedius-specific nucI gene (6).
Thus, a total of 465 amplicons were spotted on the membranes. S. aureus strains N315, Mu50, COL, MW2, and NCTC8325 were used to amplify 385 intragenic fragments. The 80 other genes were previously amplified from 39 other strains (see Table S1 in the supplemental material [http://genopole.pasteur.fr/staph/]).
Distribution of the 465 genes among the 80 S. aureus clinical isolates analyzed.
The gene content of each of the 80 isolates is given in Table S2 in the supplemental material (http://genopole.pasteur.fr/staph/). Of the 92 genes shared by the seven sequenced genomes and used in the macroarray, 76, including S. aureus nuc and sodM, were detected in all isolates analyzed. Therefore, a total of 388 genes of this set were useful for typing.
Antibiotic resistance genes and phenotypes.
An analysis of the data reported in Table 1 enabled us to check whether the genes detected by hybridization were correlated with their phenotypic expression in the isolates. As shown in Table 2, for 79 of the 80 isolates, each antibiotic resistance gene detected was associated with the corresponding phenotype. A single mecA+ isolate was susceptible to β-lactams.
TABLE 2.
Antibiotic resistance genes and their corresponding phenotypes in each of the 80 isolates
| Antibiotic resistance gene(s) | No. of isolates | Antibiotic resistance phenotypea | No. of isolates |
|---|---|---|---|
| mecA | 4 | OXA + PEN | 4 |
| mecA, blaZ | 31 | OXA + PEN | 31 |
| mecA | 1 | No resistance | 1 |
| blaZ | 39 | PEN | 39 |
| aacA-aphD | 30 | GEN | 30 |
| aadD | 24 | NEO | 27 |
| aphA-3 | 3 | ||
| spc | 31 | SPT | 31 |
| tetK | 2 | TET | 2 |
| tetM | 34 | TET + MIN | 37 |
| tetM, tetK | 3 | ||
| ermA | 31 | MLS | 43 |
| ermC | 11 | ||
| ermA, ermC | 1 | ||
| msrA | 3 | ERY | 3 |
| lnuA | 1 | LIN | 1 |
| vgaAv, vgaB, vatB | 2 | SGA | 12 |
| vgaAv | 2 | ||
| vatA, vgbA | 6 | ||
| fos | 3 | FOF | 12 |
| aadE | 4 | STR | 27 |
| dfrA | 1 | TMP | 7 |
| far1 | 0 | FUC | 12 |
See Table 1 for explanation of abbreviations. The phenotypes which are conferred by acquired genes in S. aureus are reported.
The streptogramin resistance genes previously found by PCR with the isolates that were resistant to streptogramin A (15) (Table 1) were detectable by hybridization with the DNA macroarrays designed for this study. The intragenic amplicons from vgaA and vgaAv appear to be specific to each variant despite the 83.2% similarity relating them. This is due to the fact that the divergence is distributed along the entire sequence of the gene variants, without >29 consecutive matching nucleotides between the amplicon and the variant gene. Resistance to fosfomycin, streptomycin, trimethoprim, and fusidic acid, which can result from mutations in preexisting genes, are rarely associated with acquired genes (Table 2). In contrast, resistance to β-lactams, aminocyclitols (except streptomycin), tetracycline, minocycline, macrolides, lincosamides, and streptogramin B was correlated with the presence of at least one acquired gene (Table 2). Two of the 12 isolates that were resistant to streptogramin A (BM10761 and BM10759; Table 1) did not carry any of the investigated genes encoding resistance to this antibiotic.
The S. aureus fosB gene, included in the arrays because of its similarity to fos, was found in 69 of the isolates, independent of their phenotypes of resistance to fosfomycin.
The combinations of genes carried by the transposons Tn554 (spe, ermA, tnpA, and tnpB), Tn5406 (vgaAv, tnpA, and tnpB), and Tn4001 (aacA-aphD and IS256 tnp) were found in the isolates exhibiting the antibiotic resistance phenotypes mediated by these transposons. The genes blaZ and tnp480, which are cocarried by Tn552, were associated with only 28 of the 70 isolates containing blaZ. As was stated previously (8), the genes aadE, sat4, and aphA-3, initially found in Tn5405, were always combined, and they were found in seven isolates in this study. This last combination was occasionally associated with other Tn5405 genes, i.e., orfX (two isolates), orfX and IS1182 tnp (four isolates), or orfX, IS1182 tnp, and IS1181 tnp (one isolate).
Distribution of genes in mecA+ isolates and isolates lacking mecA.
As shown in Table 1, 36 of the 80 tested isolates were mecA+ and 44 lacked mecA. Several genes, including those coding for antibiotic resistance and putative virulence factors, had a distribution which was significantly different (pg < 0.1) for the two categories of isolates. The distribution of genes encoding putative toxins or adhesins is reported in Table 3. Interestingly, the enterotoxin-encoding genes seg, sei, sem, sen, and seo, codetected in the same pathogenicity island of the S. aureus N315 and Mu50 strains (19), were always associated with each other in our isolates and were significantly predominant in the mecA-negative isolates (30 of 44 isolates) compare to the mecA+ isolates (1 of 36 isolates).
TABLE 3.
Comparative analysis of the mecA+ and mecA-negative isolates included in this study
| Category | No. of isolates | No. of SmaI genotypes | No. of clusters (based on gene content) | No. of isolates harboring gene(s)a
|
||||
|---|---|---|---|---|---|---|---|---|
| seg, sei, sem, sen, seo | entA | cna | bbp | sask (SAV2595) | ||||
| mecA+ | 36 | 12 | 20 | 1 | 32 | 4 | 10 | 3 |
| mecA mutant | 44 | 33 | 32 | 30 | 9 | 20 | 38 | 21 |
The pg values for the sets of genes were 1.0 × 10−8, 1.4 × 10−8, 0.023, 2.9 × 10−6, and 0.003, respectively. Three hundred eighty-eight genes were used for typing.
Distribution of genes in 16 BI isolates and 12 NC isolates.
Unrelated isolates were selected for a comparative analysis of BIs and nasal carriers (NCs) (Table 1). No significant differences in the gene contents were observed between the BI and NC isolates when the 388 genes were taken into account for calculations of the probability that a given gene is present by chance. However, taking into account only 11 genes that were not shared by all isolates and that encode adhesins (sdrD, sdrC, fnbA, fnbB, efb, map, cna, bbp, vwb, bap, and ebpS), the two categories of isolates became significantly distinguishable (pg = 0.059) by the presence of the sdrD gene, which codes for a putative SD (serine-aspartate) adhesin (18) and was detected in 15 of the 16 BI isolates compared to 7 of the 12 NC isolates.
Clustering of the 80 S. aureus clinical isolates on the basis of their gene contents, as investigated with the DNA macroarray designed for this study.
The hierarchical clustering of the isolates by neighbor joining is represented in the dendrogram shown in Fig. 1. First we checked whether the outbreak-related isolates (shown in gray boxes in the figure) were more closely linked to each other than to any of the other isolates.
FIG. 1.

Hierarchical clustering of the 80 S. aureus isolates investigated according to their gene contents by the J-Express program (9). The threshold was chosen to distinguish each of the outbreak-related isolates belonging to SmaI genotype 100 or 101 (clusters 9 and 28, respectively) from any of the other isolates.
Within SmaI genotype 100 or 101 (Table 1), the isolates were more closely linked to each other. These isolates were included in this study because they were responsible for documented acute outbreaks in the hospitals of Villeneuve St. Georges and Calais, France, respectively. Such isolates were not detected in the hospitals before the outbreaks. An analysis of their gene contents revealed the absence of two or seven widespread genes, respectively, which were detected in at least 84% of the other isolates. The four SmaI type 100 isolates lacked fnbB and MW2409, while the five SmaI type 101 isolates lacked set14, lukM, splcC, splD, vwb, emp, and SA0276. The absence of widespread genes confirmed the hypothesis of a close relationship between the isolates belonging to each of the two SmaI genotypes. As was found previously by PCR, the four isolates from Villeneuve St. Georges, responsible for scalded skin syndrome in newborns (Table 1), carried the eta gene encoding the exfoliative toxin A. Moreover, the single SmaI 101 isolate that was distinguishable from the other four SmaI 101 isolates by its susceptibility to erythromycin lacked the ermC gene that was present in the latter isolates (Table 1).
The other outbreak-related isolates belonged to SmaI subtype 39aA (IPF 555, IPF 557, and IPF 562) (14) or 45aA (BM 9586, BM 12184, nad BM 12188) (22) and were isolated in hospital C (Paris) in 1999 and 1987, respectively (Table 1). Isolates belonging to SmaI genotype 39A were phenotypically recognizable because of their decreased susceptibility to glycopeptides. Those belonging to SmaI genotype 45A and phage type 77 were initially discovered in 1987, during the emergence of resistance to fluoroquinolones in French hospitals. Such isolates preexisted in European hospitals before these outbreaks, but at very low frequencies. In this study, we analyzed 24 mecA+ isolates belonging to SmaI genotypes 39A and 45A that were isolated in several European countries and at time intervals of several years. These endemic isolates, which are considered putatively related according to their SmaI genotypes, were more linked to each other than to any of the 56 other isolates (Fig. 1). Note that some of them are clearly divergent in the dendrogram and that the mode of their linkage is not correlated to their SmaI genotype, but those considered to be outbreak related are closely linked.
Clustering of the 80 clinical isolates after choice of threshold for hierarchical clustering dendrogram.
For the distribution of the isolates into clusters, it was necessary to choose a threshold for the dendrogram. For this purpose, the threshold adopted was that which enabled each of the outbreak-related isolates belonging to SmaI genotype 100 or 101 to be distinguished from any of the other isolates. These isolates were taken into consideration because they were not detected before the outbreaks, in contrast to the SmaI subtype 39aA or 45aA outbreak-related isolates. The choice of this threshold enabled the discrimination of 52 clusters belonging to 45 SmaI genotypes among the 80 isolates (Fig. 1). In Table S2 in the supplemental material (http://genopole.pasteur.fr/staph/), the genes are listed according to the clusters to which they belong.
With the selected threshold, a total of five clusters were found among the 10 SmaI type 39A isolates and eight clusters were found among the 14 SmaI type 45A isolates (Fig. 1). Among these isolates, which are endemic in European cities, those collected in the same hospital or city were not necessarily the most closely linked. The three outbreak-related SmaI subtype 39aA isolates collected in hospital C (Paris) in 1999 (IPF 555, IPF 557, and IPF 562) are linked in cluster 41, which includes another SmaI subtype 39aA isolate (BM 12612) collected at Villiers St. Denis in 1998. Moreover, four of five isolates belonging to two SmaI subtypes, 45aA and 45dA, and collected in three French hospitals in 1987 are within cluster 46 (BM 9290, BM 9343, BM 9586, and BM 12184). The fifth isolate, BM 12188, located in the separate but close cluster 47, was distinguishable by the lack of five drug resistance genes, namely blaZ, qacA, qacC, CZ040, and CZ041, encoding β-lactamase, resistance to antiseptics, organomercurial lyase, and mercuric reductase, respectively. Figure 2 shows the images resulting from scanning of the two DNA macroarrays hybridized with the total cellular DNAs from the BM9290 and BM12188 isolates (Table 1).
FIG. 2.

Images resulting from scanning of the DNA macroarrays hybridized with the total cellular DNAs from two isolates. (A) Isolate BM9290 (cluster 46). (B) Isolate BM12188 (cluster 47). Even though they belonged to the same SmaI subtype (45aA), the two isolates were found in two close but separate clusters (Fig. 1) due to the lack, in BM12188, of the following five drug resistance genes: blaZ, qacA, qacC, CZ040, and CZ041, encoding β-lactamase, resistance to antiseptics, organomercurial lyase, and mercuric reductase, respectively.
Each of the isolates linked in clusters 6, 13, 37, and 40 belonged to the same SmaI genotypes. In contrast, the isolates linked in clusters 11, 14, and 33 belonged to unrelated SmaI genotypes, and cluster 31 contained two distinct but related SmaI genotypes. In addition, isolates with the same SmaI genotype, if it was 117 or 144, were separated. Note that the two isolates belonging to SmaI genotype 144 had no epidemiological link since they were from distinct sources (Tunisia and Sweden) and were collected over a 10-year time interval. For this last case, the use of the DNA macroarray is more appropriate than the analysis of SmaI patterns for discrimination between the two isolates.
DISCUSSION
DNA macroarrays offer a rapid, robust, and easily standardizable method for the simultaneous detection of several hundred genes of interest and may be used for analyses of transcriptional expression in isolates grown under different in vitro and in vivo conditions. The 465 genes spotted on the DNA macroarray used in this study were chosen as probes in order to identify S. aureus at the species level and to type S. aureus isolates. They included, in particular, genes encoding antibiotic resistance and putative virulence factors.
The detection of antibiotic resistance genes is particularly interesting when these genes mediate low antibiotic resistance levels that are not reproducibly detectable by antibiograms. This level of detection also contributes to the selection of isolates that carry genes that have not yet been described. By hybridization with 400- to 500-bp amplicons, mutations in preexisting genes associated with antibiotic resistance cannot be visualized and would necessitate hybridization with oligonucleotides. For 79 of the 80 clinical isolates tested, the resistance phenotype conferred by each of the detected resistance genes was expressed, whereas one mecA+ isolate was susceptible to β-lactams. This high correlation demonstrated an extensive and satisfactory choice of antibiotic resistance genes spotted on the membranes. For the two related streptogramin A-resistant isolates, the lack of any known staphylococcal or enterococcal gene conferring resistance to this antibiotic is probably due to the presence of a gene(s) that has not yet been described.
The assessment of the presence of all known S. aureus genes encoding putative virulence factors may contribute to the determination of the pathogenic potential correlated with particular types of infection and to the identification of emerging pathotypes. In this study, we checked whether some genes were more prevalent in isolates responsible for BIs than in isolates from uninfected NCs. For this purpose, only unrelated isolates from our collection were included. This constraint explains why the numbers of isolates analyzed were 16 BI isolates and 12 NC isolates. Despite the fact that BIs were contracted by children outside the hospital, several patients were infected by S. aureus isolates that were considered monoclonal on the basis of their SmaI patterns. Although a few genes, including sdrD, encoding a putative SD adhesin, appeared predominant in one of the two categories of isolates, the differences were not significant when the 388 genes used for typing were taken into account for the calculation of the probability that a given gene is present by chance. Thus, a larger number of unrelated isolates from various sources merits further analysis. However, when only the 11 genes encoding putative adhesins were taken into account, the higher prevalence of sdrD in BI isolates than in NC isolates became significant. Some SD proteins were shown to bind fibrinogen (ClfA [21], ClfB [25], and SdrG [16]) or bone sialoprotein (Bbp) (33), but the ability of SdrD to bind a matrix protein(s) has not been investigated. The impact of sdrD inactivation merits evaluation in an animal model of BIs.
The significantly distinct distribution of some genes encoding enterotoxins or adhesins among the mecA+ and mecA- negative isolates in this study (Table 3) may not be the case among isolates from various sources. Indeed, most of the 80 isolates tested were collected in France and Tunisia, and the mecA+ isolates belonged to a limited number of SmaI genotypes. Nevertheless, the low frequency of cna detection in mecA+ isolates has been reported already by Booth et al. (5).
Due to the use of a large number of genes for typing (388), all 80 isolates tested were typeable. A method based on the analysis of a large number of genes was expected to yield more discrimination between the isolates than the typing methods based on sequencing of a limited number of genes or on the analysis of SmaI patterns, which depends on the number and locations of SmaI sites in the genome. This was confirmed by this study, for the mecA+ isolates were endemic to several European cities and were collected at large time intervals (SmaI genotypes 39A and 45A). Among the latter isolates, those considered to be outbreak related in the same hospital were found in the same or in a close cluster(s): cluster 41 or 46-47. In such a context, the typing method proposed in this study provides more discrimination of the isolates responsible for acute outbreaks than the determination of SmaI patterns. For the other isolates, if we excluded the three pairs which were linked in the same cluster despite belonging to unrelated SmaI types, our results revealed a correlation between the modes of isolate clustering based on the two typing methods, i.e., the analysis of gene contents and the SmaI patterns. Indeed, the isolates belonging to the same or related SmaI types appeared to be more linked to each other than to those belonging to unrelated SmaI types.
In conclusion, the typing method proposed here performed better than that based on the analysis of SmaI patterns, in particular for distinguishing outbreak-related isolates from those that are endemic to a particular area. It also has the advantages of being faster and providing additional information concerning the gene contents of interest. This macroarray should be updated when additional genes are described and also needs to be validated for the analysis of the transcription of genes in order to evaluate the levels of gene expression which may be correlated with particular types of infections. The method described here can also be performed with glass slides and fluorescent labeling in order to be more amenable to automation for routine analyses.
Acknowledgments
For part of this work, S. Trad received a grant from Fondation pour la Recherche Médical (FRM). The isolate MW2 was obtained through the Network on Antimicrobial Resistance in Staphylococcus aureus (NARSA) Program supported under NIAID, NIH, contract no. N01-AI-95359.
We thank the biologists and NARSA who provided several of the strains used in this study and Iain Old for reviewing the manuscript.
REFERENCES
- 1.Anonymous. 2002. First U.S. case of vancomycin-resistant Staphylococcus aureus infection reported; patient has chronic renal failure. Dialysis Transplant. 2002:602-603. [Google Scholar]
- 2.Baba, T., F. Takeuchi, M. Kuroda, H. Yuzawa, K. Aoki, A. Oguchi, Y. Nagai, N. Iwama, K. Asano, T. Naimi, H. Kuroda, L. Cui, K. Yamamoto, and K. Hiramatsu. 2002. Genome and virulence determinants of high virulence community-acquired MRSA. Lancet 359:1819-1827. [DOI] [PubMed] [Google Scholar]
- 3.Becker, K., A. W. Friedrich, G. Lubritz, M. Weilert, G. Peters, and C. Von Eiff. 2003. Prevalence of genes encoding pyrogenic toxin superantigens and exfoliative toxins among strains of Staphylococcus aureus isolated from blood and nasal specimens. J. Clin. Microbiol. 41:1434-1439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Berger-Bächi, B. 1997. Resistance not mediated by β-lactamase (methicillin-resistance), p. 158-174. In K. B. Crossley and G. L. Archer (ed.), The staphylococci in human disease. Churchill Livingstone, New York, N.Y.
- 5.Booth, M. C., L. M. Pence, P. Mahasreshti, M. C. Callegan, and M. S. Gilmore. 2001. Clonal associations among Staphylococcus aureus isolates from various sites of infection. Infect. Immun. 69:345-352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Chesneau, O., J. Allignet, and N. El Solh. 1994. Three thermonuclease gene probes designed for rapid identification of S. aureus, S. hyicus, and S. intermedius, p. 83-85. In R. Möllby, J.-I. Flock, C. E. Nord, and B. Christensen (ed.), Staphylococci and staphylococcal infections. Gustav Fischer Verlag, Stuttgart, Germany.
- 7.Chesneau, O., A. Morvan, and N. E. Solh. 2000. Retrospective screening for heterogeneous vancomycin resistance in diverse Staphylococcus aureus clones disseminated in French hospitals. J. Antimicrob. Chemother. 45:887-890. [DOI] [PubMed] [Google Scholar]
- 8.Derbise, A., S. Aubert, and N. El Solh. 1997. Mapping the regions carrying the three contiguous antibiotic resistance genes aadE, sat4, and aphA-3 in the genomes of staphylococci. Antimicrob. Agents Chemother. 41:1024-1032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Dysvik, B., and I. Jonassen. 2001. J-Express: exploring gene expression data using Java. Bioinformatics 17:369-370. [DOI] [PubMed] [Google Scholar]
- 10.Enright, M. C., N. P. Day, C. E. Davies, S. J. Peacock, and B. G. Spratt. 2000. Multilocus sequence typing for characterization of methicillin-resistant and methicillin-susceptible clones of Staphylococcus aureus. J. Clin. Microbiol. 38:977-986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Fitzgerald, J. R., D. E. Sturdevant, S. M. Mackie, S. R. Gill, and J. M. Musser. 2001. Evolutionary genomics of Staphylococcus aureus: insights into the origin of methicillin-resistant strains and the toxic shock syndrome epidemic. Proc. Natl. Acad. Sci. USA 98:8821-8826. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Frangeul, L., P. Glaser, C. Rusniok, C. Buchrieser, E. Duchaud, P. Dehoux, and F. Kunst. 2004. CAAT-box, contigs-assembly and annotation tool-box for genome sequencing projects. Bioinformatics 20:790-797. [DOI] [PubMed] [Google Scholar]
- 13.Grundmann, H., S. Hori, M. C. Enright, C. Webster, A. Tami, E. J. Feil, and T. Pitt. 2002. Determining the genetic structure of the natural population of Staphylococcus aureus: a comparison of multilocus sequence typing with pulsed-field gel electrophoresis, randomly amplified polymorphic DNA analysis, and phage typing. J. Clin. Microbiol. 40:4544-4546. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Guerin, F., A. Buu-Hoi, J. Mainardi, G. Kac, N. Colardelle, S. Vaupre, L. Gutmann, and I. Podglajen. 2000. Outbreak of methicillin-resistant Staphylococcus aureus with reduced susceptibility to glycopeptides in a Parisian hospital. J. Clin. Microbiol. 38:2985-2988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Haroche, J., A. Morvan, M. Davi, J. Allignet, F. Bimet, and N. El Solh. 2003. Clonal diversity among streptogramin A-resistant Staphylococcus aureus isolates collected in French hospitals. J. Clin. Microbiol. 41:586-591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Hartford, O., L. O'Brien, K. Schofield, J. Wells, and T. Foster. 2001. The Fbe (SdrG) protein of Staphylococcus epidermidis HB promotes bacterial adherence to fibrinogen. Microbiology 147:2545-2552. [DOI] [PubMed] [Google Scholar]
- 17.Hiramatsu, K. 2001. Vancomycin-resistant Staphylococcus aureus: a new model of antibiotic resistance. Lancet Infect. Dis. 1:147-155. [DOI] [PubMed] [Google Scholar]
- 18.Josefsson, E., D. O'Connell, T. Foster, I. Durussel, and J. A. Cox. 1998. The binding of calcium to the B-repeat segment of SdrD, a cell surface protein of Staphylococcus aureus. J. Biol. Chem. 273:31145-31152. [DOI] [PubMed] [Google Scholar]
- 19.Kuroda, M., T. Ohta, I. Uchiyama, T. Baba, H. Yuzawa, I. Kobayashi, L. Cui, A. Oguchi, K. Aoki, Y. Nagai, J. Lian, T. Ito, M. Kanamori, H. Matsumaru, A. Maruyama, H. Murakami, A. Hosoyama, Y. Mizutani-Ui, N. K. Takahashi, T. Sawano, R. Inoue, C. Kaito, K. Sekimizu, H. Hirakawa, S. Kuhara, S. Goto, J. Yabuzaki, M. Kanehisa, A. Yamashita, K. Oshima, K. Furuya, C. Yoshino, T. Shiba, M. Hattori, N. Ogasawara, H. Hayashi, and K. Hiramatsu. 2001. Whole genome sequencing of methicillin-resistant Staphylococcus aureus. Lancet 357:1225-1240. [DOI] [PubMed] [Google Scholar]
- 20.McCormick, J. K., J. M. Yarwood, and P. M. Schlievert. 2001. Toxic shock syndrome and bacterial superantigens: an update. Annu. Rev. Microbiol. 55:77-104. [DOI] [PubMed] [Google Scholar]
- 21.McDevitt, D., P. Francois, P. Vaudaux, and T. Foster. 1994. Molecular characterization of the clumping factor (fibrinogen receptor) of Staphylococcus aureus. Mol. Microbiol. 11:237-248. [DOI] [PubMed] [Google Scholar]
- 22.Morvan, A., S. Aubert, C. Godard, and N. El Solh. 1997. Contribution of a typing method based on IS256-probing of SmaI-digested cellular DNA to discrimination of European phage-type 77 methicillin-resistant Staphylococcus aureus strains. J. Clin. Microbiol. 35:1415-1423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Murchan, S., M. Kaufmann, A. Deplano, R. de Ryck, M. Struelens, C. E. Zinn, V. Fussing, S. Salmenlinna, J. Vuopio-Varkila, N. El Solh, C. Cuny, W. Witte, P. Tassios, N. Legakis, W. van Leeuwen, A. van Belkum, A. Vindel, I. Laconcha, J. Garaizar, S. Haeggman, B. Olsson-Liljequist, U. Ransjo, G. Coombes, and B. Cookson. 2003. Harmonization of pulsed-field gel electrophoresis protocols for epidemiological typing of strains of methicillin-resistant Staphylococcus aureus: a single approach developed by consensus in 10 European laboratories and its application for tracing the spread of related strains. J. Clin. Microbiol. 41:1574-1585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Musser, J. M., and R. K. Selander. 1990. Genetic analysis of natural populations of Staphylococcus aureus, p. 59-67. In R. P. Novick (ed.), Molecular biology of the staphylococci. VCH Publishers, New York, N.Y.
- 25.Ni Eidhin, D., S. Perkins, P. Francois, P. Vaudaux, M. Hook, and T. Foster. 1998. Clumping factor B (ClfB), a new surface-located fibrinogen-binding adhesin of Staphylococcus aureus. Mol. Microbiol. 30:245-257. [DOI] [PubMed] [Google Scholar]
- 26.Novick, R. P. 2003. Autoinduction and signal transduction in the regulation of staphylococcal virulence. Mol. Microbiol. 48:1429-1449. [DOI] [PubMed] [Google Scholar]
- 27.Oliveira, D. C., A. Tomasz, and H. de Lencastre. 2001. The evolution of pandemic clones of methicillin-resistant Staphylococcus aureus: identification of two ancestral genetic backgrounds and the associated mec elements. Microb. Drug Resist. 7:349-361. [DOI] [PubMed] [Google Scholar]
- 28.Paulsen, I. T., N. Firth, and R. A. Skurray. 1997. Resistance to antimicrobial agents other than β-lactams, p. 175-212. In K. B. Crossley and G. L. Archer (ed.), The staphylococci in human disease. Churchill Livingstone, New York, N.Y.
- 29.Peacock, S. J., C. E. Moore, A. Justice, M. Kantzanou, L. Story, K. Mackie, G. O'Neill, and N. P. Day. 2002. Virulent combinations of adhesin and toxin genes in natural populations of Staphylococcus aureus. Infect. Immun. 70:4987-4996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Projan, S. J., and R. P. Novick. 1997. The molecular basis of pathogenicity, p. 55-81. In K. B. Crossley and G. L. Archer (ed.), The staphylococci in human disease. Churchill Livingstone, New York, N.Y.
- 31.Sabat, A., J. Krzyszton-Russjan, W. Strzalka, R. Filipek, K. Kosowska, W. Hryniewicz, J. Travis, and J. Potempa. 2003. New method for typing Staphylococcus aureus strains: multiple-locus variable-number tandem repeat analysis of polymorphism and genetic relationships of clinical isolates. J. Clin. Microbiol. 41:1801-1804. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Tenover, F. C., R. D. Arbeit, R. V. Goering, P. A. Mickelsen, B. E. Murray, D. H. Persing, and B. Swaminathan. 1995. Interpreting chromosomal DNA restriction patterns produced by pulsed-field gel electrophoresis: criteria for bacterial strain typing. J. Clin. Microbiol. 33:2233-2239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Tung, H., B. Guss, U. Hellman, L. Persson, K. Rubin, and C. Ryden. 2000. A bone sialoprotein-binding protein from Staphylococcus aureus: a member of the staphylococcal Sdr family. Biochem. J. 345:611-619. [PMC free article] [PubMed] [Google Scholar]
- 34.Valderas, M. W., J. W. Gatson, N. Wreyford, and M. E. Hart. 2002. The superoxide dismutase gene sodM is unique to Staphylococcus aureus: absence of sodM in coagulase-negative staphylococci. J. Bacteriol. 184:2465-2472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.van Leeuwen, W., C. Jay, S. Snijders, N. Durin, B. Lacroix, H. A. Verbrugh, M. C. Enright, A. Troesch, and A. van Belkum. 2003. Multilocus sequence typing of Staphylococcus aureus with DNA array technology. J. Clin. Microbiol. 41:3323-3326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.van Leeuwen, W., H. Verbrugh, J. van der Velden, N. van Leeuwen, M. Heck, and A. van Belkum. 1999. Validation of binary typing for Staphylococcus aureus strains. J. Clin. Microbiol. 37:664-674. [DOI] [PMC free article] [PubMed] [Google Scholar]