

Abstract
Background
Understanding the complex, multistep process of metastasis remains a major challenge in cancer research. Metastasis models can reveal insights in tumor development and progression and provide tools to test new intervention strategies.
Methods
To develop a new cancer metastasis model, we used DU145 human prostate cancer cells and performed repeated rounds of orthotopic prostate injection and selection of subsequent lymph node metastases. Tumor growth, metastasis, cell migration and invasion were analyzed. Microarray analysis was used to identify cell migration- and cancer-related genes correlating with metastasis. Selected genes were silenced using siRNA, and their roles in cell migration and invasion were determined in transwell migration and Matrigel invasion assays.
Results
Our in vivo cycling strategy created cell lines with dramatically increased tumorigenesis and increased ability to colonize lymph nodes (DU145LN1-LN4). Prostate tumor xenografts displayed increased vascularization, enlarged podoplanin-positive lymphatic vessels and invasive margins. Microarray analysis revealed gene expression profiles that correlated with metastatic potential. Using gene network analysis we selected 3 significantly upregulated cell movement and cancer related genes for further analysis: EPCAM (epithelial cell adhesion molecule), ITGB4 (integrin β4) and PLAU (urokinase-type plasminogen activator (uPA)). These genes all showed increased protein expression in the more metastatic DU145-LN4 cells compared to the parental DU145. SiRNA knockdown of EpCAM, integrin-β4 or uPA all significantly reduced cell migration in DU145-LN4 cells. In contrast, only uPA siRNA inhibited cell invasion into Matrigel. This role of uPA in cell invasion was confirmed using the uPA inhibitors, amiloride and UK122.
Conclusions
Our approach has identified genes required for the migration and invasion of metastatic tumor cells, and we propose that our new in vivo model system will be a powerful tool to interrogate the metastatic cascade in prostate cancer.
Keywords: Prostate cancer, Invasion, Migration, Metastasis, Angiogenesis, Lymphangiogenesis, Lymph node, EpCAM, Integrin, Beta4, uPA, New model
Background
Prostate cancer affects 1 in 6 males in their lifetime, and is the second leading cause of cancer death in men in the U.S. [1]. Almost 2.8 million men are currently living with a diagnosis of prostate cancer [2], yet the ability to discern whose cancer will progress to metastatic disease remains a challenge. A better understanding of the metastatic process could lead to enhanced prognostic ability and subsequent improvements in patient care and outcome. Cancer cells can escape the primary tumor via blood vessels or lymphatic vessels and travel to distant organs. The presence of tumor cell-positive lymph nodes from biopsy indicates the tumor has already spread from the primary site. Lymph node metastasis is an important prognostic indicator in many cancers, such as breast, melanoma and prostate [3-6]. Lymph node metastasis correlates with poor prognosis in prostate cancer, as compared to those without lymph node involvement [7]. Even before evidence of lymph node metastasis, lymphovascular invasion (LVI), defined as the unequivocal presence of tumor cells within an endothelium-lined space, can act as an independent risk factor in prostate cancer [5]. Since all lymphatic drainage eventually empties into the venous system, tumor extravasation into lymphatic vessels may lead to more widespread metastasis via the vascular circulatory system to distant organs like bone [8,9].
As many patients now opt for an active surveillance or ‘watchful waiting’ period during the management of organ-confined disease [10,11], the development of new biomarkers and therapeutic options is greatly needed. The identification of genes important in the metastatic cascade may facilitate our development of such therapies.
Animal models of metastasis are important tools that allow us to interrogate steps in this process. Spontaneous and experimental models of metastasis in mice have allowed us to discover and analyze new genes and biomarkers and to test anti-cancer drugs within complex microenvironments. Studies have shown that when human cancer cell xenografts are implanted into the orthotopic site, as compared to an ectopic (usually subcutaneous) site, enhanced tumorigenicity and metastasis followed [12-14]. The microenvironment is well documented to influence tumor cell behavior and is capable of stimulating or repressing cell plasticity, proliferation, migration and invasion [15-17]. Orthotopically implanted tumor cells and their spontaneously metastasizing counterparts are exposed to many of the same environmental influences and selective pressures that human prostate cancer cells undergo in the prostate and lymph nodes. In addition, human xenografts allow one to interrogate the efficacy of human-specific drugs such as proteins (eg, interferons) or antibodies (eg, bevacizumab). Xenograft models provide a complement to genetically engineered mouse models which develop over a longer time and reside in an immunocompetent host but do not always capture all aspects of human cancer.
In vivo cycling of cancer cells has been demonstrated to be a useful method to select for highly aggressive cell lines. The human prostate cancer cell lines, PC-3 and LNCaP, were previously cycled in vivo to select for highly metastatic variants from sentinel lymph node metastasis [12,18]. These human cancer models have proven highly beneficial to the prostate cancer research community [19]. Herein, we describe a similar method to create a novel prostate cancer model developed in our laboratory using the DU145 human prostate cancer cell line. Originally isolated by Stone, et. al., from a human brain metastasis, DU145 is a “classical” and widely-used prostate cancer cell line [20]. DU145 cells do not express detectable levels of prostate specific antigen and are not hormone sensitive.
This report describes the development and characterization of this model and our studies investigating molecular changes that correlate with metastatic potential.
Methods
Cell culture and transfection
DU145 human prostate cancer cells were obtained from ATCC (HTB-81) and maintained in high glucose DMEM with 10% fetal bovine serum (FBS), 1% glutamine, penicillin and streptomycin (GPS), and 1% sodium pyruvate (Invitrogen, Carlsbad, CA). Phase contrast microscopy was performed using a TE2000 microscope (Nikon) and RT SPOT camera with SPOT Advanced v4.0.9. software (Diagnostic Instruments, Inc., Sterling Heights, MI). Cells were transfected with siRNA using SilentFect (Biorad) in Opti-MEM I Reduced Serum Medium (Invitrogen), incubated for 4 hours, media changed, and cells used for assays at 48-72 hr. siRNAs were obtained from Thermo Scientific: ON-TARGETplus non-targeting control siRNA pool (D-001818-10-05), ON-TARGETplus human EPCAM siRNA pool (L-004568-01-0005), ON-TARGETplus human PLAU siRNA (L-006000-00-0005), ON-TARGETplus human ITGB4 siRNA pool (L-008011-00-0005). EPCAM and ITGB4 siRNAs were used at 30nM and PLAU siRNA used at 90nM for effective knockdown without toxicity.
Cell migration, invasion and proliferation assays
Cell migration was measured using Corning transwell inserts (BD Biosciences) with 8.0 μm pore polycarbonate membrane. Membranes were coated with Collagen I (BD Biosciences) at 100 μg/ml. 1% FBS in DMEM was used in the lower wells as chemoattractant. Cells were trypsinized, trypsin inactivated with soybean trypsin inhibitor and washed in DMEM. 6×104 cells were added to the top transwell chamber and allowed to migrate for 4 hours. Cells were fixed and stained with Diff-Quik (Fisher Scientific) and a cotton swab used to remove non-migrated cells from the upper chamber. Migrated cells were counted in 3–5 fields/well with 2–3 wells/condition. Cells were used for experiments 48 hours after transfection. For invasion assays, BD BioCoat Matrigel Invasion Chambers, with 8.0 μm pore PET membrane in 24-well cell culture inserts (BD Biosciences) were used with 5% FBS as the chemoattractant. Cells were allowed to invade for 12 hours and were fixed, stained and counted as described above. For uPA inhibitor experiments, cells were treated with 0.1% DMSO vehicle, 10 μM amiloride or UK122 (EMD Millipore, Billerica, MA). In vitro cell number was measured using CyQUANT Cell Proliferation Assay kit (Life Technologies). Cells were plated in a 96 well plate at 2.5×103 cells per well and incubated for 1–4 days. Plates were frozen and processed together at the end of the experiment. Fluorescent signal correlated with cell number and was measured with 450 nm excitation and 520 nm emission filters.
Western blot analysis
Whole cell lysates were collected in modified RIPA buffer with EGTA and EDTA (Boston Bioproducts, Ashland, MA) with protease inhibitor cocktail (P8340, Sigma-Aldrich). Conditioned media was collected from serum-free cell cultures, cells removed by centrifugation at 200 × g and protein concentrated using Amicon Ultra-15 3 kDa Centrifugal Filter Units (Millipore) at 3000 × g. Protein concentration was measured using a BCA (bicinchoninic acid) assay kit (Pierce/Thermo Scientific). Reduced protein in Laemmli sample buffer was resolved using SDS-PAGE and transferred to Immobilon-P 0.45 μm PVDF membrane (EMD Millipore, Billerica, MA). Membranes were blocked with 5% non-fat dry milk in PBS, incubated with primary antibody, followed by the appropriate secondary IgG antibody; sheep anti-mouse IgG HRP or donkey anti-rabbit IgG HRP linked (GE Healthcare). Membranes were washed thoroughly between steps using PBS containing 0.05% Tween-20, and developed using ECL Plus western blotting detection kit (GE Healthcare). Primary antibodies used for western blot analysis were as follows: EpCAM (C10, sc-25308), Integrin β4 (H-101, sc-9090), uPA (H-140, sc-14019) from Santa Cruz Biotechnology; AKT (#9272), p-AKT (#9271), S6K (#9202), p-S6K (#9205) from Cell Signaling. GAPDH (6C5) antibody was obtained from Abcam. Membranes were stripped using ReBlot Plus Strong Antibody stripping solution (EMD Millipore) before reprobing.
Immunohistochemistry
Paraffin-embedded tumor tissue and lymph nodes were dewaxed, rehydrated, and stained with hematoxylin and eosin (H&E) or immunostained to detect human cytokeratin-18 (K18, Epitomics), EpCAM (Santa Cruz), E-Cadherin (BD Bioscience), mouse blood vessels (CD31, Pharmingen), or mouse lymphatic vessels (podoplanin, Reliatech). Antigen retrieval was performed with boiling citrate buffer (pH 6) for K18, EpCAM and E-cadherin or with proteinase K for podoplanin and CD31. Endogenous peroxidases were blocked with 3% peroxide in methanol. Tissues were blocked using normal serum and incubated with primary antibodies overnight at 4°C, biotinylated secondary antibodies (Vector Laboratories, Burlingame, CA) for one hour, and Vectastain Elite (avidin-HRP; Vector) for 30 min, and finally developed with diaminobenzidine chromogen (DAB, Vector). To detect human epithelial cell metastases, sentinel lymph node sections were stained with K18, counterstained with hematoxylin, examined by microscopy and K18-positive cells in small foci were scored as metastases. Single K18-positive cells in the lymph node were not scored as metastases. Three different tissue levels from each of two lymph nodes (when available) were examined per mouse.
In vivo tumor experiments
Eight week old male Balb/c Nu/Nu mice were purchased from Massachusetts General Hospital and housed in the Animal Resource at Children’s Hospital (ARCH) facility accredited by the American Association for Accreditation of Laboratory Animal Care (AAALAC). All experiments were conducted in accordance with the principles and procedures outlined in the NIH Guide for the Care and Use of Laboratory Animals and approved by an Institutional Animal Care and Use Committee (IACUC) at Boston Children’s Hospital. For orthotopic prostate injections, mice were anesthetized and an abdominal incision was made to expose the prostate. 2×106 cells ( suspended in 40 μl HBSS) were injected into the prostate using a Hamilton mini-injector, and the incision was closed with 9 mm wound clips. Tumor growth was monitored by palpation. After 4–12 weeks (5 weeks for direct comparison experiment), mice were sacrificed and necropsied. Tumors (and lymph nodes in 5 wk experiment) were removed, weighed and measured with calipers, fixed in formalin and processed for paraffin blocks. Orthotopic tumor volumes were calculated as widthSuperscript> × /Superscript> × length × 0.5. Sentinel paraaortic lymph nodes were washed with PBS, filtered through a 100 μm cell strainer (BD Biosciences), and plated in complete media on tissue culture dishes. The following day, cells were washed thoroughly with PBS, replaced with fresh complete media and re-named DU145-LN1 (from lymph node). After expansion in culture, in vivo orthotopic prostate injection was repeated for additional rounds of selection with subsequent cells named DU145-LN2, then DU145-LN3, and finally DU145-LN4.
For skin tumors, 5×106 cells were injected subcutaneously into the right dorsal flank of 8 week old male Balb/c Nu/Nu mice. Tumor size was measured externally with calipers, and tumor volume was calculated as V = widthSuperscript> × /Superscript> × length × 0.5.
Gene expression analysis
RNA for cDNA microarray analysis was purified using RNeasy mini kits (Qiagen). Purity and integrity was confirmed by spectrophotometer and agarose gel. Total RNA was labeled and amplified according to manufacturer’s instructions by the Microarray Core Facility of the Molecular Genetics Core Facility at Boston Children’s Hospital supported by NIH-P50-NS40828 and NIH-P30-HD18655. DU145, DU145-LN1, DU145-LN2 and DU145-LN4 RNA samples were run on Illumina HumanRef-8 BeadChips (Illumina, San Diego, CA). Raw data were analyzed in BRB-ArrayTools (Biometric Research Branch, National Cancer Institute, Bethesda, MD, USA, http://linus.nci.nih.gov/BRB-ArrayTools.html).
Signal intensity data was subject to rank invariant normalization. Duplicated probes on the array were treated independently during normalization and statistical analyses. Negative or low intensity signals <10 were corrected to 10 to prevent extreme fold change artifacts.
Samples were subject to Hierarchical cluster analysis using Euclidian distance. Differentially expressed genes were identified using a time course analysis (DU145 as time = 0, and DU145LN1, LN2 and LN4 as time = 1, 2 and 3 respectively), with a cut-off minimum of 1.5-fold change in DU145-LN4 relative to DU145. For functional gene analysis, the entire dataset was imported into Ingenuity IPA Network Analysis software (Ingenuity Systems, Redwood City, CA), and we selected Cancer and Cellular Movement categories for further analysis. Cluster analysis of the relationship between cell types within these categories or of the entire gene probe population using one minus Pearson correlation, produced essentially indistinguishable dendrograms. We cross-referenced back to probe intensity values and genes were removed if all data points had low intensities of <100 Arbitrary Intensity Units. Selected genes were represented by heat map using GENE-E software (http://www.broadinstitute.org/cancer/software/GENE-E). For analysis of Cell Signaling, data were excluded if Illumina probe values were negative, <10, or less than the probe signal in the control group (DU145).
Statistical analyses
Data from cell proliferation, migration and invasion assays were analyzed using unpaired two-sample student’s t-test. Statistical significance was considered at p ≤ 0.05. Specific p-values for each experiment are indicated in Figure Legends.
Results
Development and characterization of a new prostate cancer metastasis model in vivo
In order to select for prostate cancer cells with increased metastatic potential we used an in vivo cycling approach [12,18]. The DU145 human prostate cancer cell line [20] was used to establish a series of metastatic variants. DU145 cells were injected orthotopically into the prostate of immunodeficient Nu/Nu (nude) male mice. After the tumor was palpable (4–8 weeks), mice were euthanized and the sentinel paraaortic lymph nodes were removed and minced sterilely, and the cells placed into culture as described in Methods (Figure 1 left side). If no tumor cell outgrowth occurred, tumors in the remaining mice were allowed to grow for additional 2 week intervals before lymph nodes were removed and a cell line was established. Cells, now called DU145-LN1, were expanded in culture for several passages to eliminate fibroblast contamination, and re-injected orthotopically into the ventral lobes of the prostate of subsequent nude male mice. Repeated rounds of in vivo cycling were performed to establish the DU145-LN2, DU145-LN3 and DU145-LN4 cell lines. All cell lines were analyzed by RT-PCR for mouse and human GAPDH expression to ensure that only human cells and no mouse stromal cells were injected [21].
Figure 1.
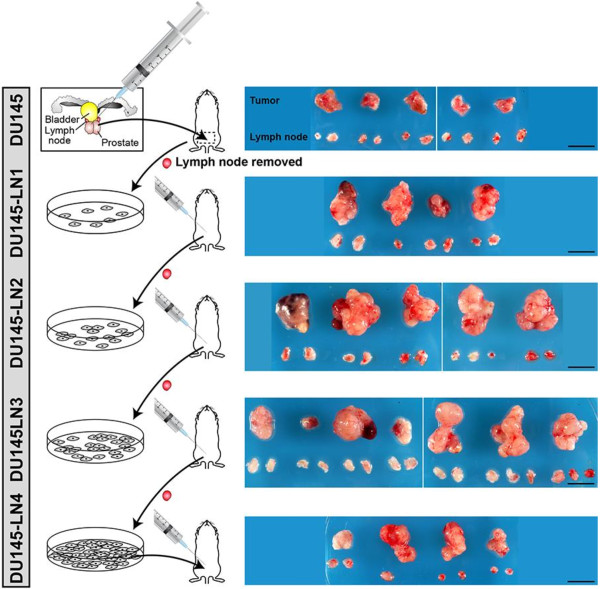
Development of a model of metastatic prostate cancer through repeated selection of spontaneous lymph node metastases from orthotopic DU145 human prostate tumors. Schematic (left panels) of the experimental approach shows orthotopic prostate inoculation of DU145 cells. Lymph nodes were removed and cultured, and selected tumor cells subject to repeated rounds of orthotopic injection. Right panels show gross anatomy of tumors and lymph nodes 5 weeks after all DU145 sublines were reinjected (this figure is modified with permission from [21]). Scale bar = 1 cm.
Once all cell lines were established, our in vivo metastatic model was tested and characterized by injecting each cell line orthotopically into the prostate of mice simultaneously (n = 4-7 mice per group). Tumors and lymph nodes were removed after 5 weeks, as shown in Figure 1 (right side). Tumor incidence was 100% in all groups, but local metastatic incidence varied (Table 1). To quantify metastases, paraffin-embedded lymph node sections (3 levels per lymph node, 4–5 mice per group) were analyzed by H&E and human K18 immunostaining. Lymph node metastasis was recorded as incidence of K18-positive foci per mouse. Repeated rounds of metastatic selection increased the incidence of K18-positive metastatic foci from 0% in parental DU145 lymph nodes, to ≥75% in DU145-LN2, DU145-LN3 and DU145-LN4 lymph nodes (Figure 2A, Table 1). In addition to enhancing the metastatic potential of the DU145-LN sublines, our in vivo cycling approach also increased the growth of these tumors. Orthotopic prostate tumor size was significantly increased from DU145 to DU145-LN1 and further to DU145-LN2 (Figure 1 right side, Table 1). Interestingly, we found that DU145-LN2 was consistently the largest tumor when injected into the prostate, with rapid tumor growth compared to that of the DU145 parental cell line. While increased tumorigenic and metastatic ability appeared to have been established by the LN2 generation, we also employed DU145-LN4 cells in many of our studies as we surmised it was likely to represent the most stable and homogenous cell line.
Table 1.
In vivo orthotopic growth and metastasis of DU145 sublines
| Cell line injected | % Tumor incidence | Mean tumor weight (g) (±SD) | Mean tumor diameter (mm) (range) | % Metastatic incidence* |
|---|---|---|---|---|
| DU145 |
100 |
0.66 (0.09) |
7.3 (6.3-9.3) |
0 |
| DU145-LN1 |
100 |
0.99 (0.25) |
11.9 (8.3-14.1) |
50 |
| DU145-LN2 |
100 |
1.80 (0.37) |
15.8 (12.9-18.2) |
75 |
| DU145-LN3 |
100 |
1.16 (0.45) |
12.1 (5.5-16.1) |
83 |
| DU145-LN4 | 100 | n.d. | 10.2 (7.3-12.8) | 75 |
Cells were injected into the prostate. After 5 weeks, tumors and lymph nodes were removed. *K18-positive metastatic foci in lymph nodes. All available lymph nodes were evaluated.
Figure 2.
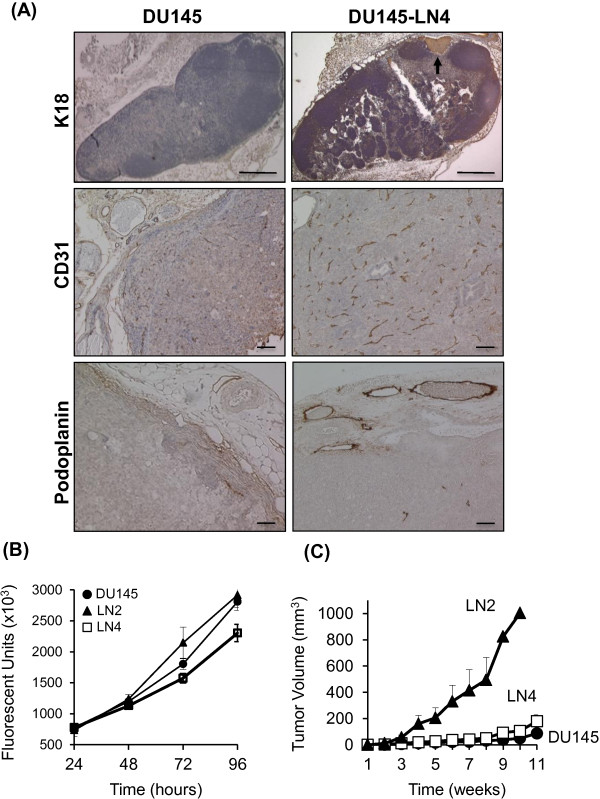
DU145 LN tumors show increased growth, angiogenesis, lymphangiogenesis, and metastasis. (A) Metastasis was measured by K18 staining of lymph nodes from mice bearing DU145 tumor (top left panel) and DU145-LN4 tumor (top right panel). K18-positive (brown color) tumor foci (arrow) were counted as positive incidence of metastasis. Scale bars = 0.5 mm. Tumor vascularization was assessed by CD31 IHC (brown color) of DU145 (middle left panel) and DU145-LN4 (middle right panel) prostate tumors. Increased vascularization was observed in DU145-LN4 tumors relative to DU145 tumors. Scale bars = 100 μm. Lymphangiogenesis was measured by podoplanin staining. Enlarged podoplanin-positive vessels (brown color) were observed in DU145-LN4 orthotopic tumors (lower right panel), compared to DU145 tumors (lower left panel). All sections were counterstained with hematoxylin (blue color). Scale bars = 100 μm. (B) In vitro proliferation assays of the DU145LN sublines indicate similar proliferation rates with slight reduced proliferation of DU145-LN4. 2.5x103 cells plated/well, absorbance measured using Cyquant dye (Ex = 485 nm). Data in arbitrary fluorescence units x1000. Filled circles: parental DU145, filled triangles: DU145-LN2, empty squares: DU145-LN4. Error bars indicate S.D. of triplicate wells. (C) DU145-LN2 shows increased tumor growth compared to parental DU145 when injected subcutaneously into nude mice. 5x106 cells were injected into the flank. Symbols as above.
The increase in tumor size in the DU145-LN model was not explained by changes in the cell proliferation rate in vitro. The proliferation rate of DU145-LN2 cells was not significantly different than parental DU145 cells, while DU145-LN4 consistently showed a slightly reduced proliferation rate in vitro (Figure 2B). We further investigated tumor growth potential by assessing subcutaneous tumor growth over time. Subcutaneous tumor size is more accurate and straightforward to measure compared to intraprostatic tumor size. Tumor cells were injected subcutaneously into the dorsal right flank of nude male mice, and tumor size was measured externally with calipers. DU145 cells were less tumorigenic when injected into the skin of nude mice. In independent experiments, we found that an average of 7/10 mice that received DU145 cells showed tumor-take. To our surprise, DU145-LN2 cells were highly proliferative when injected subcutaneously, relative to parental DU145 control cells (Figure 2C). DU145-LN2 cells showed tumor take in 9/10 and 10/10 mice, more rapid tumor take and more rapid tumor growth. DU145-LN4 cells injected into the skin showed tumor take in 10/10 mice and similar growth rate relative to parental DU145 cells (Figure 2C). Metastatic potential was not evaluated in the ectopic experiments.The increase in lymph node metastasis and tumor size in our DU145-LN model was accompanied by greater vessel density in the DU145-LN4 as compared to DU145 prostate tumors, as observed by CD31 immunostaining (Figure 2A, middle panels). Despite remaining relatively small, DU145 tumors were often observed to be necrotic in their center (Figure 2A middle left). This observation is likely related to their low recruitment of supporting CD31-positive blood vessels. Since we “selected” for metastasis to regional lymph nodes in our model, we anticipated that these cells would use lymphatic vessels as a conduit. In fact, LVI and lymphangiogenesis can predict metastatic potential. The more aggressive tumors DU145-LN2 (not shown) and DU145-LN4 showed more numerous, enlarged peri-tumoral lymphatic vessels (as detected by podoplanin staining) compared to DU145 tumors, indicating increased lymphangiogenesis (Figure 2A, lower panels).
Metastatic selection changes prostate tumor cell phenotype
The selection of DU145 metastatic variants resulted in a progressive change in cell phenotype. DU145 human prostate tumor cells are a heterogeneous epithelial cell population in in vitro culture [22]. We recently showed that the more metastatic DU145-LN cells undergo mesenchymal to epithelial transition (MET)-like changes in gene expression [21]. As seen in phase contrast microscopy, metastatic DU145-LN4 cells form more clusters in vitro, with more cell-cell interactions than DU145 cells (Figure 3A). The intermediate DU145-LN cell lines displayed intermediate phenotypes. These expression changes were maintained in in vivo tumors. Immunohistochemical staining of paraffin embedded subcutaneous tumors showed higher expression of E-cadherin and EpCAM by DU145-LN4 compared to DU145 (Figure 3B).
Figure 3.
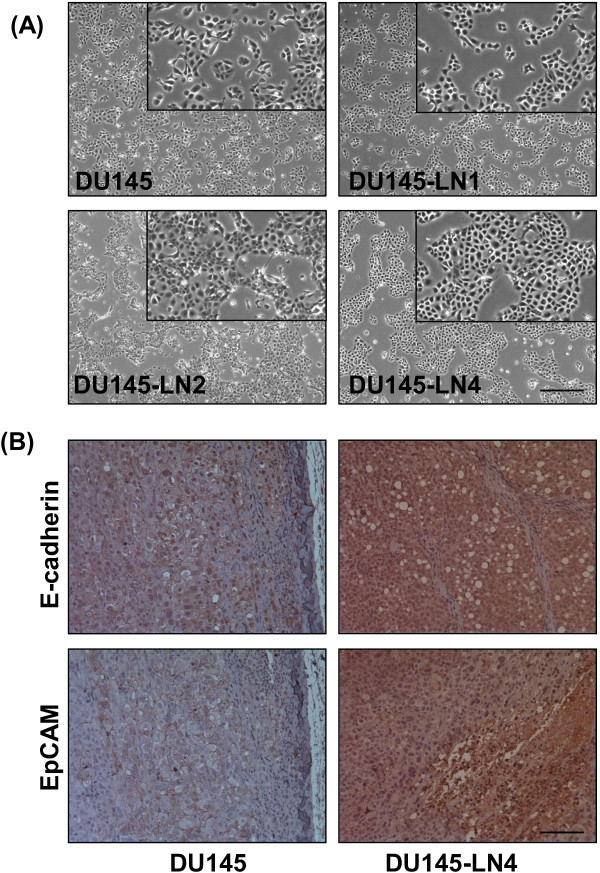
Phenotype of DU145-LN cells with increased metastatic ability. (A) Phase contrast microscopy images of parental DU145 cells, DU145-LN1, DU145-LN2, and DU145-LN4 cells. Cells exhibit progressive phenotypic changes after selection, with increased clustering and cell-cell adhesions from parental DU145 (top left panel) to DU145-LN4 (bottom right panel). Insets are larger images of lower panels. Scale bar = 0.5 mm (B) DU145-LN4 tumor cells maintain their mesenchymal-epithelial transition (MET) phenotype in vivo. IHC of DU145 and DU145-LN4 subcutaneous tumor tissue with the epithelial markers, E-cadherin and EpCAM. High expression of E-cadherin and EpCAM was maintained in the tumor tissue. Scale bar = 100 μm.
Increased migration and invasion in metastatic DU145-LN cells
To further characterize our new prostate cancer model, we examined the effect of this metastatic selection on cell behavior in vitro. Cell migration and invasion are important steps in the process of metastasis [23]. The effect of metastatic selection on DU145 cell migration was determined in the transwell migration assay. 1% fetal bovine serum (FBS) was used in the lower wells as a chemoattractant, and cells were allowed to migrate for 4 hours. We found that there was a progressive increase in cell migration from the DU145 parental cell line to the metastatic DU145-LN4 cells. Parental DU145 exhibited a low level of migration toward 1% FBS on collagen-coated membranes, while metastatic DU145-LN4 cells displayed over 2.5-fold higher migration (Figure 4A). The migration of DU145-LN2, LN-3 and LN-4 was significantly greater than DU145.We next looked at the invasive behavior of the DU145-LN cells using the transwell Matrigel invasion assay. In this assay, cells are required to invade through an extracellular matrix barrier. 5% FBS was used as a chemoattractant and cell invasion was assessed after 12 hours. The metastatic DU145-LN sublines also showed significantly increased invasive abilities compared to parental DU145 cells. Figure 4B demonstrates that DU145-LN1 to LN4 all showed over 2.5 fold higher invasion, compared to parental DU145 cells.
Figure 4.
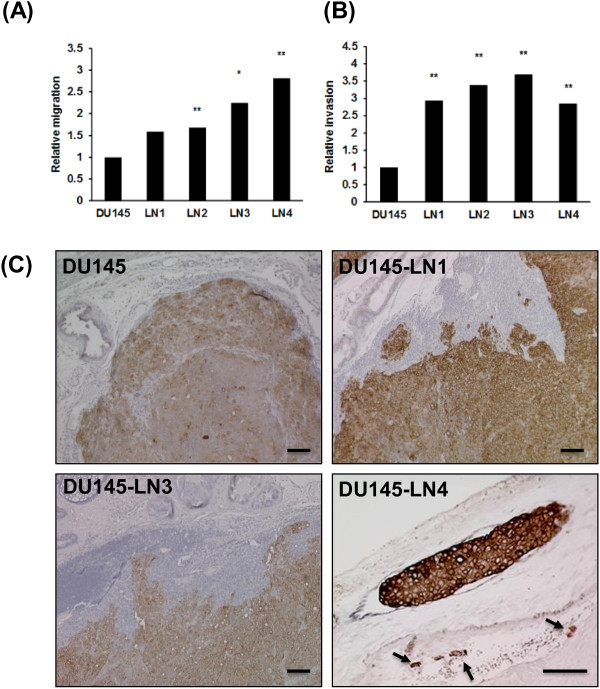
DU145-LN sublines exhibit increased migration and invasion with metastatic ability, relative to DU145 cells. DU145-LN metastatic sublines show increased (A) cell migration in the transwell migration assay and (B) increased invasion in the Matrigel invasion assay. For both assays: Mean of triplicate assays ± S.D. Student t-test, *p < 0.05, **p < 0.01, (C) IHC of orthotopic prostate tumors with K18 (brown) and counterstained with hematoxylin (blue). Margins were smooth and well-defined in the DU145 tumors (top left panel), while invasive margins were observed in DU145-LN1 (top right panel) and DU145-LN3 (lower left panel) tumors. Right lower panel shows magnified image of DU145-LN4 tumor seen in Figure 2A. Double staining of K18 (brown) and podoplanin (black) shows tumor foci present in an enlarged lymphatic vessel and in a tumor-associated blood vessel (arrows) (lower right panel). Scale bars = 100 μm.
The increased invasion of DU145-LN1-4 cells was also observed in vivo. Orthotopic prostate tumor tissue was stained with human K18 to visualize tumor margins. DU145 tumor margins were largely defined and well circumscribed (Figure 4C top left). DU145-LN subline tumors showed highly invasive edges with human K18-positive tumor cells protruding into the mouse prostate gland (Figure 4C top right, bottom left). Interestingly, in DU145-LN4 tumors K18-positive tumor emboli were also clearly visible inside lymphatic vessels in the peritumoral stroma, as visualized by double staining with podoplanin (black color) and K18 (brown color) (Figure 4C). Tumor cells were also seen inside tumor-associated blood vessels (Figure 4C bottom right, arrows).
Identification and analysis of genes involved in cell invasion and migration
To investigate the molecular changes underlying the gain of metastatic potential in our new model, we analyzed the gene expression profile of these cells using an Illumina cDNA Ref6 bead expression array. RNA was isolated from these cells between passage 6–8. We confirmed through RT-PCR that cell cultures were not contaminated with cells of mouse origin that might share identity in gene probe sequence [21]. Gene expression data was normalized as described in Methods. Hierarchical Cluster analysis confirmed that DU145-LN1 was most closely related to the parental DU145 cells in gene expression profile. The more metastatic cells, DU145-LN2 and DU145-LN4, clustered together and were progressively more divergent from DU145 parental cells, as visualized by dendrogram (Figure 5A).
Figure 5.
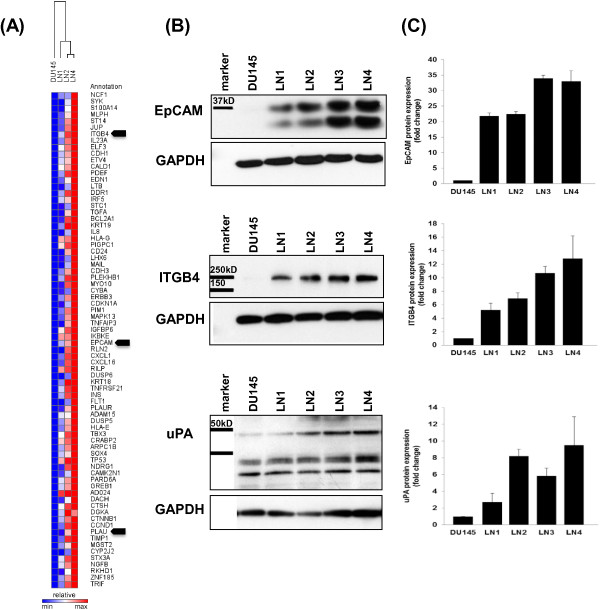
Gene expression analysis in the DU145-LN metastatic sublines and validation of increased EpCAM, integrin β4 and uPA protein expression. (A) Heat map of cell-movement and cancer genes increased in the more metastatic DU145-LN sublines, relative to DU145 cells. Relative expression color scale from high (red) to low (blue). Arrowheads indicate genes selected for further analysis. (B) Western blot analysis of EpCAM, integrin β4 and uPA expression in whole cell lysate showed progressive increase in expression in the DU145-LN metastatic sublines. 20 μg protein was loaded to detect EpCAM, 70 μg protein was used for integrin β4 and uPA western blots. GAPDH shown as protein loading controls. (C) Band intensity for each protein was evaluated using ImageJ software and averaged from 2–4 western blots each.
To identify genes related to metastasis we applied a continuous scale time course analysis using BRB Array Tools. Our analysis revealed a pattern of gene expression changes that showed progressively increased or decreased expression across the cell lines, from parental DU145 cells to DU145-LN2 or DU145-LN4 cells. These gene expression changes correlated with the increased migration, invasion and metastatic potential of the cell lines. We used Ingenuity software analysis to select for genes upregulated in cancer and cellular movement, as described in Methods. Figure 5A shows a heat map generated using cancer and cell movement genes significantly increased (red color) in DU145-LN4 cells. The genes included ITGB4 (integrin β4), ST14 (Matriptase), EPCAM (Epithelial Cell Adhesion Molecule, (EpCAM)), CDH1 (E-cadherin), JUP (junction plakoglobin/desmoplakin 3/γ-catenin) and PLAU (urokinase plasminogen activator (uPA)). Three of these genes were further analyzed in this study: Epithelial Cell Adhesion Molecule (EpCAM), urokinase plasminogen activator, (uPA, gene name PLAU), and integrin β4 (ITGB4). Relative cDNA expression levels are shown in Additional file 1: Table S1. In addition, Ingenuity software was used to select for cellular signaling genes differentially regulated among the cell lines (Additional file 1: Figure S1).We investigated whether protein expression levels correlated to the RNA expression profiles using immunoblotting of whole cell lysates. EpCAM, integrin β4 and uPA all showed a progressive increase in protein expression from parental DU145 to DU145-LN4 cells (Figure 5B). GAPDH immunoblotting was used to confirm equal protein loading. ImageJ software was used to measure protein band intensity and averaged from 2–4 western blots for each protein and graphed in Figure 5C. The protein expression levels showed good correlation with the microarray data for these selected genes.We investigated whether these proteins were involved in the increased migration observed in the metastatic DU145-LN cells. DU145-LN4 cells were transfected with siRNA against EpCAM, uPA or integrin β4, and the effect on cell migration was measured in the transwell assay after 48 hours. Our data show that siRNA knockdown of either EpCAM, integrin β4 or uPA using siRNA significantly inhibited cell migration (Figure 6A-C). We also examined whether silencing these genes would affect cell invasion. uPA silencing significantly inhibited tumor cell invasion, while both EpCAM or integrin β4 knockdown had no significant effect on DU145-LN4 cell invasion (Figure 6D-F). Whole cell lysates were collected in parallel and effective protein knockdown by siRNA treatment was confirmed by immunoblotting (Figure 6G-H). In addition, two chemical inhibitors of uPA were able to inhibit cell migration and invasion of DU145-LN4 cells (Figure 6J-K, respectively). Downstream cell signaling pathways were evaluated following uPA knockdown in DU145-LN4 cells. Specifically, phosphorylation of AKT and phosphorylation of p70 S6 kinase (S6K) were upregulated in control cells following serum stimulation, but these proteins were not activated in uPA-lacking cells (Figure 6I).
Figure 6.
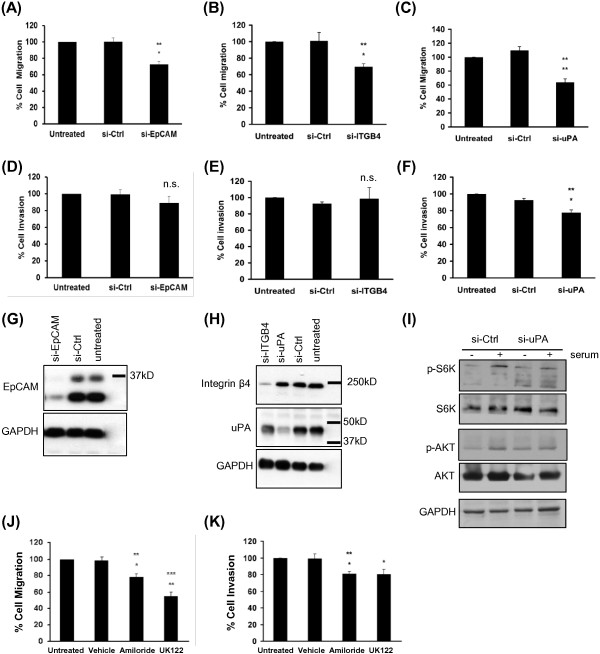
The role of EpCAM, integrin β4 and uPA in migration and invasion of DU145-LN4 cells. Cell migration of DU145-LN4 cells in a transwell migration assay (A-C) and a Matrigel invasion assay (D-F). siRNA knockdown of (A) EpCAM, (B) integrin β4, or (C) uPA significantly inhibited cell migration relative to control siRNA and untreated cells. siRNA knockdown of (D) EpCAM or (E) ITGB4 did not significantly affect cell invasion in a Matrigel invasion assay. (F) uPA siRNA significantly inhibited cell invasion relative to control siRNA and untreated cells. Western blot analysis of whole cell lysates from cells treated with siRNA in parallel with migration/invasion assays: (G) EpCAM, (H) integrin β4, and uPA western blots. Blots were probed with GAPDH as loading controls. (I) Western blot analysis of whole cell lysates from cells treated with control siRNA or si-uPA and serum starved (−) or serum stimulated (+). p-AKT and p-S6K were induced after serum addition in control cells but not in cells lacking uPA. Total AKT, S6K, and GAPDH served as controls. DU145-LN4 cell migration (J), and cell invasion (K) were significantly inhibited by treatment with 10 μM amiloride or 10 μM UK122. Students t-test, *p ≤ 0.05, **p ≤ 0.01, n.s.-not significant. Top asterisked p value represents analysis between untreated cells and specific siRNA, lower value indicates analysis between control siRNA treated cells and specific siRNA.
In summary, we have established a new model of human prostate cancer metastasis using DU145 cells, a widely used androgen-independent human prostate cancer cell line. This model represents an advance upon other widely available prostate metastasis cell models, because the intermediate cell lines are available for analysis. Our selection approach has produced more highly metastatic, migratory and invasive sublines. Our initial analysis has revealed a subset of genes that correlate with metastatic potential. Cell migration and invasion are important steps in metastasis; we have shown that three molecules upregulated in our model: EpCAM, integrin-β4 and uPA play roles in these processes.
Discussion
Our goal was to create a new reliable human prostate cancer model system that would use human prostate tumor cells and result in rapidly growing (non-necrotic) tumors in 100% of the mice injected and consistently recapitulate the invasive and metastatic properties seen in patients. Many prostate cancer research studies use one of three human cell lines: PC3, LNCaP or DU145. While each cell line has its benefits and drawbacks, we focused on the DU145 cell line since it did not have well-used metastatic sublines reported in the literature. In our search for metastasis-related pathways, we had also wanted to select an androgen independent cell line. These cells grow robustly in vitro and express many prostate and epithelial markers, yet they grow poorly in mice even when injected into the mouse prostate gland. Therefore, many labs resort to injecting high numbers (>2×106) of cells and co-injecting ECM components or fibroblasts to enhance tumor-take and angiogenic potential. We chose to select for highly metastatic variants of DU145 using an in vivo cycling strategy that was previously successful for PC-3 M and LNCaP [12,18].
Herein, we have presented the establishment and characterization of our new model of human prostate cancer, the DU145-LN metastatic series. The DU145-LN cells show enhanced in vivo growth as well as migratory, invasive and metastatic abilities. These cell lines represent new tools to explore the process of metastasis. Our approach to developing the DU145-LN metastatic series via spontaneous metastasis from orthotopic organ sites provided the tumor with appropriate micro-environmental signals [14]. Cells with the ability to spontaneously metastasize from the prostate tumor and survive in the lymph node were repeatedly selected. The role of lymphatic versus hematologic metastasis has been debated [24,25]. However, the presence of tumor-positive lymph nodes continues to be an important predictor of distant metastases and patient survival in many cancers [9,26]. Studies have shown that lymphovascular invasion is significantly associated with PSA biochemical recurrence and patient survival in prostate cancer [5,27], although LVI may not significantly improve predictive accuracy above standard clinicopathological features in prostate cancer [11]. We clearly observed tumor foci in the enlarged lymphatics of the DU145-LN4 orthotopic tumors, indicating that our model recapitulates steps common in human prostate cancer progression.
After completing four rounds of cycling the DU145 cells in mice (prostate to lymph node), we compared all five cell lines in a head-to-head comparison for tumorigenicity and metastatic potential in a 5 week period (Figure 1 shows gross images of resulting tumors). DU145 was poorly angiogenic and had a low vascular density, therefore resulting in small tumors. Cycled tumors had higher microvessel densities, and vessel density has been shown to correlate with metastatic potential in human prostate cancer [28]. In addition, the DU145-LN4 tumors had increased lymphangiogenesis surrounding the tumors and invasive leading edges. Lymphangiogenesis has been shown to be an important mechanism of prostate cancer metastasis [26,29], and has been our focus in this study.
Most human prostate cancer cells do not grow well subcutaneously; however, our new DU145-LN2 cell line represents a useful and rapid non-surgical xenograft model for tumor growth studies in the skin, e.g. drug screening. Metastatic cycling of DU145 prostate cancer cells also resulted in cells that were more motile and invasive. By examining the gene expression profiles of these cells we revealed many genes correlating with their metastatic ability. In this report we have demonstrated the involvement of EpCAM, integrin β4 and uPA in tumor cell migration and/or invasion--key steps in the metastatic cascade. Although each of these genes may not be individually competent to induce metastasis in parental cells, we propose that our model represents a valuable and relevant system, as the genes we have identified have been shown to be clinically important in prostate cancer.
EpCAM (also known as CD326) is well established as a tumor marker in many carcinomas, and is widely used to purify circulating tumor cells from blood [30]. EpCAM is a transmembrane glycoprotein and has diverse functions in cell-cell adhesion, migration, proliferation and differentiation [31]. In human prostate cancer, several tissue studies have shown upregulated EpCAM in the tumor epithelium and in metastatic lesions [32-35]. EpCAM expression in prostate tumor tissue is also a significant predictor of shorter biochemical recurrence free-survival [35]. The mechanism of EpCAM activity has not yet been well defined. EpCAM can be cleaved in its ectodomain to release an extracellular fragment, (EpEX) and this may affect E-cadherin mediated cell-cell adhesion [36]. It is possible that this fragment may be involved in the increased migration and invasion observed in the DU145-LN4 cells. High expression of the epithelial marker E-cadherin has been associated with stronger cell-cell interaction and subsequent reduced cell motility [36]. However, the presence of EpEX may modulate this role. Our Western blot analysis (Figure 5B) indicates that both full length EpCAM and EpEX is present at high levels in the DU145-LN4 (and DU145-LN2) cells. EpCAM also associates with the tight junction protein, claudin 7, to promote tumor cell migration rather than cell-cell adhesion that leads to lymphatic spread [37]. Claudin 7 expression was also dramatically upregulated in the DU145-LN cell series in microarray data (relative to DU145, DU145-LN1 had 9.6X, DU145-LN2 had 21X and DU145-LN4 had 28X fold higher claudin-7 expression). Antisense knockdown of either EpCAM or claudin-7 reduces tumor growth and metastasis in mice, and knockdown of both is more effective [38].
The cell surface EpCAM complexes can also involve an additional partner identified in our gene expression analysis, β4 integrin. In normal epithelial cells α6β4 resides in hemidesmosomes. In tumor cells, integrin β4 can relocate from hemiodesmosomes to the leading edge of migrating cells where it is involved in the signaling of many receptor tyrosine kinases, including ErbB2, ErbB3, EGFR and Met [39-41]. β4 integrin therefore impacts cell signaling, migration and invasion through multiple pathways. β4 expression also influences multiple miRNAs impacting cell motility [42]. High levels of β4-integrin have been found across many prostate cancer tissue expression studies, and in metastatic and castrate-resistant prostate cancer metastases [41]. Transgenic mice with a β4-integrin signaling domain mutation showed reduced prostate tumor formation and progression, thus supporting our data that ITGB4 is involved in tumor cell migration and metastasis [41].
We also showed that uPA expression positively correlated with metastatic potential in the DU145-LN cell series. uPA silencing significantly inhibited both tumor cell migration and invasion. Serine proteases, such as uPA play an important role in tumor progression. By degrading the extracellular matrix and basement membrane they can promote cell invasion, angiogenesis and metastasis [43]. Circulating levels of uPA, and its receptor uPAR (urokinase-type plasminogen activator receptor), are significantly elevated in prostate cancer patients, and are higher in patients with lymph node and bone metastases, compared to those with non-metastatic disease [44,45]. uPA and uPAR levels correlate with Gleason score, extracapsular extension, LVI, seminal vesicle and lymph node invasion and are also associated with biochemical progression and poor prognosis [45,46]. Both uPA and uPAR are involved in Matrigel invasion in PC-3 cells [47,48], and RNAi or shRNA knockdown of uPA and uPAR reduced orthotopic prostate tumor size via apoptosis. In DU145 cells, uPAR over-expression increased Matrigel invasion in vitro which was inhibited by uPA antibody or inhibitor. In addition, stable overexpression of uPAR was accompanied by uPA upregulation [49], providing additional evidence for the interdependence of the protease and receptor activities.
The uPA protease axis appears to play an important role in the invasive and metastatic behavior of our metastatic model. The uPA receptor, uPAR (gene name PLAUR) also showed increased gene expression as DU145 cells become more metastatic; with 1.3X fold increased expression in LN1, 2.0X fold in LN2, and 2.7X fold in LN4, relative to parental DU145 cells. Furthermore, one of the key activators of uPA activity is the protease Matriptase (gene name ST14). Matriptase was also highly upregulated in our model of prostate cancer metastasis; 4.5X fold in DU145-LN1, 11X fold in DU145-LN2 and 16X fold increased in DU145-LN4, relative to DU145 (Figure 5A). Antagonists of the uPA/uPAR axis have been suggested for use as anti-tumor agents with targeted monoclonal antibodies and nanoparticles currently under development [50].
Our model has identified a network of gene and pathway changes spontaneously arising as cells became more metastatic. These include EpCAM, β4-integrin and uPA. Clearly many of these pathways may interact and feedback upon each other. There may be master regulators in this system, such as transcription factors and/or microRNAs that influence expression of these and many other genes. Indeed, ZEB1 has been reported to regulate EpCAM, β4-integrin and uPA [51-53]. In turn, the miR-200 family regulates the epithelial phenotype and ZEB1 [54-56]. In addition, there are other transcription factors related to cancer and cell movement that are significantly upregulated in this model, including ELF3 (8.7X higher in DU145-LN4) and ETV4 (7.5X fold higher level in DU145-LN4 compared to DU145 cells). These and other genes may present new targets for intervention in metastatic cell behavior.
Conclusions
Using one of the “classical” human prostate cancer cell lines, DU145, we have developed a series of new metastatic variants, DU145-LN1 to DU145-LN4, through in vivo cycling of spontaneous lymph node metastases. The metastatic cells are more migratory and invasive. Gene expression analysis revealed many genes correlating with metastatic ability. We show that EpCAM and integrin β4 are involved in migration, while uPA is involved in migration and invasion of metastatic prostate cancer cells.
Our analysis of the role of these genes has demonstrated the relevance of our new system. We expect that further study of downregulated genes and as yet uncharacterized cDNAs with strong correlation to metastatic ability will bring new discoveries. We propose that our new model system will be a powerful and additional tool to interrogate the metastatic cascade in prostate cancer.
Abbreviations
EpCAM: Epithelial cell adhesion molecule; ITGB4: Integrin β4; uPA: Urokinase plasminogen activator; uPAR: Urokinase plasminogen activator receptor; siRNA: Small interfering RNA; IHC: Immunohistochemistry; FBS: Fetal bovine serum; K18: Cytokeratin 18; MET: Mesenchymal to epithelial transition.
Competing interests
The authors declare that they have no competing interests.
Authors’ contributions
JB conceived of the study, developed the animal model and cell lines, performed microarray analyses and wrote the manuscript; IC participated in in vivo experiments and edited the manuscript; MM performed immunohistochemistry; DTP carried out the migration, invasion and western blotting; AMW performed the proliferation assay, immunohistochemistry and participated in subcutaneous tumor experiments. BRZ participated in study design and edited the manuscript. DRB conceived of the study, developed the animal model and cell lines, performed immunohistochemistry and western blotting, and edited the manuscript. All authors read and approved the final manuscript.
Pre-publication history
The pre-publication history for this paper can be accessed here:
Supplementary Material
Heat map using Ingenuity Analysis software and the “Cell Signaling” category. The range of difference between DU145 and DU145LN4 groups are 1.8-16.4 fold. Table S1. Relative expression levels of PLAU, EPCAM, ITGB4 and housekeeping genes in the metastatic DU145-LN sublines.
Contributor Information
Jacqueline Banyard, Email: jacqueline.banyard@childrens.harvard.edu.
Ivy Chung, Email: ivychung@ummc.edu.my.
Matthew Migliozzi, Email: matthew.migliozzi@childrens.harvard.edu.
Derek T Phan, Email: dungpt87@gmail.com.
Arianne M Wilson, Email: wilson.arianne@gmail.com.
Bruce R Zetter, Email: bruce.zetter@childrens.harvard.edu.
Diane R Bielenberg, Email: diane.bielenberg@childrens.harvard.edu.
Acknowledgements
We thank Melissa Anderson for editing and administrative assistance and Kristin Johnston for preparation of graphics and photography. We acknowledge Ricardo Sanchez, HTL (Ricasan Histology Consultants) for tissue sections. The content of this article is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health. Research reported in this publication was supported by the National Cancer Institute of the National Institutes of Health under award numbers P50CA09381 (JB), R21CA155728; K01CA118732 (DRB), and RO1CA37393 (BRZ). This study was also supported by The Prostate Cancer Foundation (BRZ).
References
- American Cancer Society. Cancer Facts & Figures 2012. Atlanta: American Cancer Society; 2012. [Google Scholar]
- Siegel R, DeSantis C, Virgo K, Stein K, Mariotto A, Smith T, Cooper D, Gansler T, Lerro C, Fedewa S, Lin C, Leach C, Cannady RS, Cho H, Scoppa S, Hachey M, Kirch R, Jemal A, Ward E. Cancer treatment and survivorship statistics, 2012. CA Cancer J Clin. 2012;62(4):220–241. doi: 10.3322/caac.21149. [DOI] [PubMed] [Google Scholar]
- Smith JA Jr, Seaman JP, Gleidman JB, Middleton RG. Pelvic lymph node metastasis from prostatic cancer: influence of tumor grade and stage in 452 consecutive patients. J Urol. 1983;130(2):290–292. doi: 10.1016/s0022-5347(17)51112-x. [DOI] [PubMed] [Google Scholar]
- Dadras SS, Paul T, Bertoncini J, Brown LF, Muzikansky A, Jackson DG, Ellwanger U, Garbe C, Mihm MC, Detmar M. Tumor lymphangiogenesis: a novel prognostic indicator for cutaneous melanoma metastasis and survival. Am J Pathol. 2003;162(6):1951–1960. doi: 10.1016/S0002-9440(10)64328-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheng L, Jones TD, Lin H, Eble JN, Zeng G, Carr MD, Koch MO. Lymphovascular invasion is an independent prognostic factor in prostatic adenocarcinoma. J Urol. 2005;174(6):2181–2185. doi: 10.1097/01.ju.0000181215.41607.c3. [DOI] [PubMed] [Google Scholar]
- Rinderknecht M, Detmar M. Tumor lymphangiogenesis and melanoma metastasis. J Cell Physiol. 2008;216(2):347–354. doi: 10.1002/jcp.21494. [DOI] [PubMed] [Google Scholar]
- Karakiewicz PI, Hutterer GC. Predictive models and prostate cancer. Nat Clin Pract Urol. 2008;5(2):82–92. doi: 10.1038/ncpuro0972. [DOI] [PubMed] [Google Scholar]
- Sleeman JP, Thiele W. Tumor metastasis and the lymphatic vasculature. Int J Cancer. 2009;125(12):2747–2756. doi: 10.1002/ijc.24702. [DOI] [PubMed] [Google Scholar]
- Zwaans BM, Bielenberg DR. Potential therapeutic strategies for lymphatic metastasis. Microvasc Res. 2007;74(2–3):145–158. doi: 10.1016/j.mvr.2007.08.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilt TJ, Brawer MK, Jones KM, Barry MJ, Aronson WJ, Fox S, Gingrich JR, Wei JT, Gilhooly P, Grob BM, Nsouli I, Iyer P, Cartagena R, Snider G, Roehrborn C, Sharifi R, Blank W, Pandya P, Andriole GL, Culkin D, Wheeler T. Prostate Cancer Intervention versus Observation Trial (PIVOT) Study Group. Radical prostatectomy versus observation for localized prostate cancer. N Engl J Med. 2012;367(3):203–213. doi: 10.1056/NEJMoa1113162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aizer AA, Paly JJ, Zietman AL, Nguyen PL, Beard CJ, Rao SK, Kaplan ID, Niemierko A, Hirsch MS, Wu CL, Olumi AF, Michaelson MD, D'Amico AV, Efstathiou JA. Multidisciplinary care and pursuit of active surveillance in low-risk prostate cancer. J Clin Oncol. 2012;30(25):3071–3076. doi: 10.1200/JCO.2012.42.8466. [DOI] [PubMed] [Google Scholar]
- Stephenson RA, Dinney CP, Gohji K, Ordonez NG, Killion JJ, Fidler IJ. Metastatic model for human prostate cancer using orthotopic implantation in nude mice. J Natl Cancer Inst. 1992;84(12):951–957. doi: 10.1093/jnci/84.12.951. [DOI] [PubMed] [Google Scholar]
- Perrotte PJR, Bielenberg DR, Eve BY, Dinney CPN. Organ-specific angiogenesis and metastasis of human bladder carcinoma growing in athymic mice. Mol Urol. 1997;1(4):299–307. [Google Scholar]
- Bielenberg DR, Fidler IJ. In: Antiangiogenic Agents in Cancer Therapy. 6. Teicher BA, editor. Totowa: Humana Press; 1999. Regulation of Angiogenesis by the Organ Microenvironment; pp. 77–91. [Google Scholar]
- Fidler IJ. The organ microenvironment and cancer metastasis. Differentiation. 2002;70(9–10):498–505. doi: 10.1046/j.1432-0436.2002.700904.x. [DOI] [PubMed] [Google Scholar]
- Hanahan D, Weinberg RA. Hallmarks of cancer: the next generation. Cell. 2011;144(5):646–674. doi: 10.1016/j.cell.2011.02.013. [DOI] [PubMed] [Google Scholar]
- Sottnik JL, Zhang J, Macoska JA, Keller ET. The PCa tumor microenvironment. Cancer Microenviron. 2011;4(3):283–297. doi: 10.1007/s12307-011-0073-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pettaway CA, Pathak S, Greene G, Ramirez E, Wilson MR, Killion JJ, Fidler IJ. Selection of highly metastatic variants of different human prostatic carcinomas using orthotopic implantation in nude mice. Clin Cancer Res. 1996;2(9):1627–1636. [PubMed] [Google Scholar]
- Sobel RE, Sadar MD. Cell lines used in prostate cancer research: a compendium of old and new lines–part 1. J Urol. 2005;173(2):342–359. doi: 10.1097/01.ju.0000141580.30910.57. [DOI] [PubMed] [Google Scholar]
- Stone KR, Mickey DD, Wunderli H, Mickey GH, Paulson DF. Isolation of a human prostate carcinoma cell line (DU 145) Int J Cancer. 1978;21(3):274–281. doi: 10.1002/ijc.2910210305. [DOI] [PubMed] [Google Scholar]
- Banyard J, Chung I, Wilson AM, Vetter G, Le Béchec A, Bielenberg DR, Zetter BR. Regulation of epithelial plasticity by miR-424 and miR-200 in a new prostate cancer metastasis model. Sci Rep. 2013;3:3151. doi: 10.1038/srep03151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chunthapong J, Seftor EA, Khalkhali-Ellis Z, Seftor RE, Amir S, Lubaroff DM, Heidger PM Jr, Hendrix MJ. Dual roles of E-cadherin in prostate cancer invasion. J Cell Biochem. 2004;91(4):649–661. doi: 10.1002/jcb.20032. [DOI] [PubMed] [Google Scholar]
- Yilmaz M, Christofori G. Mechanisms of motility in metastasizing cells. Mol Cancer Res. 2010;8(5):629–642. doi: 10.1158/1541-7786.MCR-10-0139. [DOI] [PubMed] [Google Scholar]
- Wong SY, Hynes RO. Lymphatic or hematogenous dissemination: how does a metastatic tumor cell decide? Cell Cycle. 2006;5(8):812–817. doi: 10.4161/cc.5.8.2646. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Christiansen A, Detmar M. Lymphangiogenesis and cancer. Genes Cancer. 2011;2(12):1146–1158. doi: 10.1177/1947601911423028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Datta K, Muders M, Zhang H, Tindall DJ. Mechanism of lymph node metastasis in prostate cancer. Future Oncol. 2010;6(5):823–836. doi: 10.2217/fon.10.33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yee DS, Shariat SF, Lowrance WT, Maschino AC, Savage CJ, Cronin AM, Scardino PT, Eastham JA. Prognostic significance of lymphovascular invasion in radical prostatectomy specimens. BJU Int. 2010;108(4):502–507. doi: 10.1111/j.1464-410X.2010.09848.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weidner N, Carroll PR, Flax J, Blumenfeld W, Folkman J. Tumor angiogenesis correlates with metastasis in invasive prostate carcinoma. Am J Pathol. 1993;143(2):401–409. [PMC free article] [PubMed] [Google Scholar]
- Mumprecht V, Detmar M. Lymphangiogenesis and cancer metastasis. J Cell Mol Med. 2009;13(8A):1405–1416. doi: 10.1111/j.1582-4934.2009.00834.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Diamond E, Lee GY, Akhtar NH, Kirby BJ, Giannakakou P, Tagawa ST, Nanus DM. Isolation and characterization of circulating tumor cells in prostate cancer. Front Oncol. 2012;2:131. doi: 10.3389/fonc.2012.00131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ni J, Cozzi PJ, Duan W, Shigdar S, Graham PH, John KH, Li Y. Role of the EpCAM (CD326) in prostate cancer metastasis and progression. Cancer Metastasis Rev. 2012;31(3–4):779–791. doi: 10.1007/s10555-012-9389-1. [DOI] [PubMed] [Google Scholar]
- Went P, Vasei M, Bubendorf L, Terracciano L, Tornillo L, Riede U, Kononen J, Simon R, Sauter G, Baeuerle PA. Frequent high-level expression of the immunotherapeutic target Ep-CAM in colon, stomach, prostate and lung cancers. Br J Cancer. 2006;94(1):128–135. doi: 10.1038/sj.bjc.6602924. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Poczatek RB, Myers RB, Manne U, Oelschlager DK, Weiss HL, Bostwick DG, Grizzle WE. Ep-Cam levels in prostatic adenocarcinoma and prostatic intraepithelial neoplasia. J Urol. 1999;162(4):1462–1466. doi: 10.1016/S0022-5347(05)68341-3. [DOI] [PubMed] [Google Scholar]
- Zellweger T, Ninck C, Bloch M, Mirlacher M, Koivisto PA, Helin HJ, Mihatsch MJ, Gasser TC, Bubendorf L. Expression patterns of potential therapeutic targets in prostate cancer. Int J Cancer. 2005;113(4):619–628. doi: 10.1002/ijc.20615. [DOI] [PubMed] [Google Scholar]
- Benko G, Spajic B, Kruslin B, Tomas D. Impact of the EpCAM expression on biochemical recurrence-free survival in clinically localized prostate cancer. Urol Oncol. 2013;31(4):468–474. doi: 10.1016/j.urolonc.2011.03.007. [DOI] [PubMed] [Google Scholar]
- Denzel S, Maetzel D, Mack B, Eggert C, Barr G, Gires O. Initial activation of EpCAM cleavage via cell-to-cell contact. BMC Cancer. 2009;9:402. doi: 10.1186/1471-2407-9-402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thuma F, Zoller M. EpCAM-associated claudin-7 supports lymphatic spread and drug resistance in rat pancreatic cancer. Int J Cancer. 2013;133(4):855–866. doi: 10.1002/ijc.28085. [DOI] [PubMed] [Google Scholar]
- Nubel T, Preobraschenski J, Tuncay H, Weiss T, Kuhn S, Ladwein M, Langbein L, Zoller M. Claudin-7 regulates EpCAM-mediated functions in tumor progression. Mol Cancer Res. 2009;7(3):285–299. doi: 10.1158/1541-7786.MCR-08-0200. [DOI] [PubMed] [Google Scholar]
- Mercurio AM, Rabinovitz I, Shaw LM. The alpha 6 beta 4 integrin and epithelial cell migration. Curr Opin Cell Biol. 2001;13(5):541–545. doi: 10.1016/S0955-0674(00)00249-0. [DOI] [PubMed] [Google Scholar]
- Giancotti FG. Targeting integrin beta4 for cancer and anti-angiogenic therapy. Trends Pharmacol Sci. 2007;28(10):506–511. doi: 10.1016/j.tips.2007.08.004. [DOI] [PubMed] [Google Scholar]
- Yoshioka T, Otero J, Chen Y, Kim YM, Koutcher JA, Satagopan J, Reuter V, Carver B, de Stanchina E, Enomoto K, Greenberg NM, Scardino PT, Scher HI, Sawyers CL, Giancotti FG. Beta4 Integrin signaling induces expansion of prostate tumor progenitors. J Clin Invest. 2013;123(2):682–699. doi: 10.1172/JCI60720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gerson KD, Maddula VS, Seligmann BE, Shearstone JR, Khan A, Mercurio AM. Effects of beta4 integrin expression on microRNA patterns in breast cancer. Biol Open. 2012;1(7):658–666. doi: 10.1242/bio.20121628. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Andreasen PA, Kjoller L, Christensen L, Duffy MJ. The urokinase-type plasminogen activator system in cancer metastasis: a review. Int J Cancer. 1997;72(1):1–22. doi: 10.1002/(SICI)1097-0215(19970703)72:1<1::AID-IJC1>3.0.CO;2-Z. [DOI] [PubMed] [Google Scholar]
- Hienert G, Kirchheimer JC, Pfluger H, Binder BR. Urokinase-type plasminogen activator as a marker for the formation of distant metastases in prostatic carcinomas. J Urol. 1988;140(6):1466–1469. doi: 10.1016/s0022-5347(17)42074-x. [DOI] [PubMed] [Google Scholar]
- Shariat SF, Roehrborn CG, McConnell JD, Park S, Alam N, Wheeler TM, Slawin KM. Association of the circulating levels of the urokinase system of plasminogen activation with the presence of prostate cancer and invasion, progression, and metastasis. J Clin Oncol. 2007;25(4):349–355. doi: 10.1200/JCO.2006.05.6853. [DOI] [PubMed] [Google Scholar]
- Miyake H, Hara I, Yamanaka K, Gohji K, Arakawa S, Kamidono S. Elevation of serum levels of urokinase-type plasminogen activator and its receptor is associated with disease progression and prognosis in patients with prostate cancer. Prostate. 1999;39(2):123–129. doi: 10.1002/(SICI)1097-0045(19990501)39:2<123::AID-PROS7>3.0.CO;2-2. [DOI] [PubMed] [Google Scholar]
- Pulukuri SM, Gondi CS, Lakka SS, Jutla A, Estes N, Gujrati M, Rao JS. RNA interference-directed knockdown of urokinase plasminogen activator and urokinase plasminogen activator receptor inhibits prostate cancer cell invasion, survival, and tumorigenicity in vivo. J Biol Chem. 2005;280(43):36529–36540. doi: 10.1074/jbc.M503111200. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- Conn EM, Botkjaer KA, Kupriyanova TA, Andreasen PA, Deryugina EI, Quigley JP. Comparative analysis of metastasis variants derived from human prostate carcinoma cells: roles in intravasation of VEGF-mediated angiogenesis and uPA-mediated invasion. Am J Pathol. 2009;175(4):1638–1652. doi: 10.2353/ajpath.2009.090384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mamoune A, Kassis J, Kharait S, Kloeker S, Manos E, Jones DA, Wells A. DU145 human prostate carcinoma invasiveness is modulated by urokinase receptor (uPAR) downstream of epidermal growth factor receptor (EGFR) signaling. Exp Cell Res. 2004;299(1):91–100. doi: 10.1016/j.yexcr.2004.05.008. [DOI] [PubMed] [Google Scholar]
- O’Halloran TV, Ahn R, Hankins P, Swindell E, Mazar AP. The many spaces of uPAR: delivery of theranostic agents and nanobins to multiple tumor compartments through a single target. Theranostics. 2013;3(7):496–506. doi: 10.7150/thno.4953. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sanchez-Tillo E, de Barrios O, Siles L, Amendola PG, Darling DS, Cuatrecasas M, Castells A, Postigo A. ZEB1 Promotes invasiveness of colorectal carcinoma cells through the opposing regulation of uPA and PAI-1. Clin Cancer Res. 2013;19(5):1071–1082. doi: 10.1158/1078-0432.CCR-12-2675. [DOI] [PubMed] [Google Scholar]
- Drake JM, Barnes JM, Madsen JM, Domann FE, Stipp CS, Henry MD. ZEB1 coordinately regulates laminin-332 and {beta}4 integrin expression altering the invasive phenotype of prostate cancer cells. J Biol Chem. 2010;285(44):33940–33948. doi: 10.1074/jbc.M110.136044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gemmill RM, Roche J, Potiron VA, Nasarre P, Mitas M, Coldren CD, Helfrich BA, Garrett-Mayer E, Bunn PA, Drabkin HA. ZEB1-responsive genes in non-small cell lung cancer. Cancer Lett. 2010;300(1):66–78. doi: 10.1016/j.canlet.2010.09.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park SM, Gaur AB, Lengyel E, Peter ME. The miR-200 family determines the epithelial phenotype of cancer cells by targeting the E-cadherin repressors ZEB1 and ZEB2. Genes Dev. 2008;22(7):894–907. doi: 10.1101/gad.1640608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Korpal M, Lee ES, Hu G, Kang Y. The miR-200 family inhibits epithelial-mesenchymal transition and cancer cell migration by direct targeting of E-cadherin transcriptional repressors ZEB1 and ZEB2. J Biol Chem. 2008;283(22):14910–14914. doi: 10.1074/jbc.C800074200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gregory PA, Bert AG, Paterson EL, Barry SC, Tsykin A, Farshid G, Vadas MA, Khew-Goodall Y, Goodall GJ. The miR-200 family and miR-205 regulate epithelial to mesenchymal transition by targeting ZEB1 and SIP1. Nat Cell Biol. 2008;10(5):593–601. doi: 10.1038/ncb1722. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Heat map using Ingenuity Analysis software and the “Cell Signaling” category. The range of difference between DU145 and DU145LN4 groups are 1.8-16.4 fold. Table S1. Relative expression levels of PLAU, EPCAM, ITGB4 and housekeeping genes in the metastatic DU145-LN sublines.