

Abstract
Preeclampsia occurs more frequently in women of African ancestry. The cause of this hypertensive complication is unclear, but placental oxidative stress may play a role. Because mitochondria are the major sites of oxidative phosphorylation, we hypothesized that placentas of preeclamptic pregnancies harbor mitochondrial DNA (mtDNA) mutations. Next-generation sequencing of placental mtDNA in African American preeclamptics (N = 30) and controls (N = 38) from Chicago revealed significant excesses in preeclamptics of nonsynonymous substitutions in protein-coding genes and mitochondrially encoded nicotinamide adenine dinucleotide dehydrogenase 5 gene and an increase in the substitution rate (P = .0001). Moreover, 88% of preeclamptics and 53% of controls carried at least one nonsynonymous substitution (P = .005; odds ratio [OR] = 6.36, 95% confidence interval [CI]: 1.5-39.1). These results were not replicated in a sample of African American preeclamptics (N = 162) and controls (N = 171) from Detroit. Differences in study design and heterogeneity may account for this lack of replication. Nonsynonymous substitutions in mtDNA may be risk factors for preeclampsia in some African American women, but additional studies are required to establish this relationship.
Keywords: preeclampsia, mtDNA, African American women, oxidative phosphorylation
Introduction
Preeclampsia, a leading cause of maternal and fetal morbidity and mortality worldwide,1 complicates ∼5% of pregnancies in the United States,2 where African ancestry is a significant risk factor for the disease.3–5 Preeclampsia is a hypertensive proteinuric disorder with multisystem manifestations.6 Currently, the only effective treatment is delivery of the placenta, one reason why the pathogenesis of preeclampsia has been attributed to defects in placentation.7
The etiology of preeclampsia remains elusive, but 3 lines of evidence suggest a role for aberrations in oxidative phosphorylation due to mutations in mitochondrial genes. First, both increased generation of reactive oxygen species (ROS) and decreased levels of the antioxidants that normally scavenge ROS have been observed both in placental tissue and in the maternal circulation during preeclampsia.8–11 Second, mitochondria are the major sites of ROS production and removal, and mutations in mitochondrial genes result in increased ROS production.12 Lastly, an increased prevalence of preeclampsia in families and individuals with myopathies due to mitochondrial mutations has been reported.13–15 Collectively, these observations suggest that mitochondrial dysfunction may contribute directly to the pathobiology of preeclampsia.13,16–18
The mitochondrial genome, comprising a circular, double-stranded, maternally inherited chromosome of 16 569 bp, encodes only 37 genes, all involved in mitochondrial oxidative phosphorylation pathways. Thirteen encode enzymatic subunits of the complexes participating in the respiratory chain; the remaining encode transfer (t)RNA and ribosomal (r)RNA that are involved in protein synthesis in the mitochondria. Because the products of all mitochondrial genes participate in critical and overlapping pathways, mutations in any of these can result in disease and mutations in different mitochondrial genes can cause the same disease.19
We hypothesized that the placentas of preeclamptic pregnancies harbor mtDNA mutations that disrupt these critical pathways. Previous studies of mtDNA in preeclampsia interrogated only a few known common mtDNA polymorphisms and failed to detect associations between these common sequence variants and preeclampsia.8 , 20 However, because mutations in any of the mitochondrial genes could contribute to pathology, an interrogation of variation in all the mitochondrial genes is necessary to reveal an enrichment of the mitochondrial mutations in pregnancies with preeclampsia. Importantly, deep sequencing of the entire mitochondrial chromosome will allow the detection of rare, and previously unknown, variants, as well as heteroplasmy, a unique property of mtDNA reflecting the presence of nonidentical mtDNA genomes in a single individual, analogous to mosaicism in the nuclear genome. Therefore, to comprehensively survey mitochondrial mutations and to specifically identify novel variants and heteroplasmic nucleotides, we used next-generation high-throughput sequencing21 of the entire mitochondrial chromosome in placental DNA from African American pregnancies with preeclampsia and normotensive term deliveries. The results of those studies are reported here.
Materials and Methods
Participants in the Discovery Sample
African American women were identified through retrospective surveys of the Chicago Lying-In Pregnancy Program (CLIPP) Biobank. These women, all self-identified as African American, had pregnancies that were either diagnosed with preeclampsia or normotensive term deliveries. Preeclampsia was defined as blood pressure of 140/90 mm Hg with readings at least 6 hours apart and proteinuria (300 mg/24 h or ≥30 mg/dL on random urine analysis).22 , 23 Controls, selected from normotensive women with term deliveries (weeks 37-41), were matched to the patients with preeclampsia for age, parity, and type of insurance (public or private). Women with chronic hypertension, gestational hypertension, preconceptual or gestational diabetes mellitus, multiple gestation, or fetal anomalies were excluded. Placental DNA from 30 preeclamptic and 38 normotensive (control) pregnancies had sufficient amounts of high-quality DNA and was included in the mitochondrial sequencing study.
This study was approved by the University of Chicago Institutional Review Board; all participants gave written informed consent.
Isolation of Trophoblast Tissue and DNA
Placental villous tissue (∼100 mg), dissected manually from the placenta and rinsed in sterile Hank’s balanced salt solution (HBSS; Life Technologies, Carlsbad, CA 92008), was dissected under a microscope at 40× power and decidual or other maternally derived tissues were removed. The cleaned tissue was rinsed a final time in HBSS to remove any traces of blood. Whole genome DNA, including mtDNA, was extracted using the Pure-Gene kit (Gentra Systems, Minneapolis, Minnesota). DNA concentration and purity were measured by UV spectrophotometry and automated fluorimetric quantification (PicoGreen dsDNA Quantitation Kit; Molecular Probes, Eugene, Oregon).
Mitochondrial Genome Sequencing
Mitochondrial genome libraries were prepared by amplification of the complete mtDNA genome using 2 sets of overlapping primers. These fragments were pooled in molar equivalent amounts, sonicated to randomly fragment the DNA, and then prepared for sequencing by Illumina's recommended protocol (Illumina, San Diego, California), with minor modifications to the adaptors to introduce base-balanced indexes that allowed 12 samples to be simultaneously sequenced in the same sequencing lane at the HudsonAlpha Institute for Biotechnology (Huntsville, Alabama).
The completed libraries were sequenced to produce millions of short 27 to 37 bp sequence reads using the Illumina Genome Analyzer IIx (Illumina). Two quality filters were used on the reads. First, exact duplicate reads were reduced to 10 to reduce polymerase chain reaction-based amplification error. Second, only reads with Phred scores for each base greater than 23 were used to reduce base-calling error to less than 5%. Reads that passed both quality filters were then used to determine alignment and read depth. To determine the consensus sequence for each sample, we aligned the short reads to the revised mitochondrial Cambridge Reference Sequence (rCRS, GenBank accession NC_012920) using Bowtie software.24 Samples with less than 95% of reads aligning to the reference sequence were further excluded.
Analysis of Sequence Data
Human mitochondrial chromosomes evolved from a common ancestral chromosome that accumulated mutations along descendant branches.25 The result of this hierarchical tree-like structure is the clustering of mtDNA into groups, called haplogroups, based on shared genetic variants due to common ancestry.26 As a result, haplogroups are geographically based such that certain haplogroups occur at higher frequencies in specific geographical regions.27 The most common haplogroup in Africa, and the deepest branch of the mitochondrial tree, is the “L” haplogroup.26–29 Because we only studied African Americans, and to avoid spurious associations arising from mitochondrial population structure, our analyses were restricted to individuals carrying variants that “tag” the L haplogroup (T10873C and C10400C).26 Haplogroups were assigned using the program mtPhyl v.2.9030 using the full mitochondrial sequence. All subsequent analyses focused only on the coding region (ie, excluding sequences in the hypervariable regions) because we were interested in variants that might directly affect mitochondrial function.
Sequence variants (point mutations and insertion/deletions) were identified and annotated with respect to the rCRS. Nonsynonymous and synonymous substitutions were annotated with respect to an L haplotype from 1 Yoruban individual (GenBank accession AF347015.1).31
To test our hypothesis, we first examined all sequence variants, and then all nonsynonymous and synonymous substitutions. Insertions or deletions causing frameshifts were counted as single substitutions and included in the analyses of nonsynonymous substitutions. We conducted each of these analyses (all variants, nonsynonymous substitutions, synonymous substitutions) on (i) all genes grouped together, (ii) genes grouped by membership in mitochondrial complexes, and (iii) individual genes. Complex I includes the mitochondria-encoded nicotinamide adenine dinucleotide (NADH) dehydrogenase genes, MT-ND1, MT-ND2, MT-ND3, MT-ND4, MT-ND4L, MT-ND5, MT-ND6, which comprise part of the intermembrane enzyme complex I. Complex III includes the cytochrome B gene, MT-CYB, which contributes to the ubiquinol–cytochrome c reductase complex. Complex IV includes the cytochrome c oxidase I to III genes, MT-COI, MT-COII, and MT-COIII, which contribute to the cytochrome oxidase complex. The adenosine triphosphate (ATP) synthase 6 and 8 genes, MT-ATP6 and MT-ATP8, make up part of complex V and are referred to here as ATP synthase. The 22 tRNA genes and the 3 rRNA were each grouped for analysis.
Heteroplasmic sites were identified from the reads of each mtDNA sample aligned to their fully assembled sequence, and were called as such when both reference and mutant alleles were present. To distinguish true heteroplasmy from sequencing error, we used a Bayesian method based on the frequency and quality of the reads, base location within the read, and a prior based on the rCRS base call (provided by the HudsonAlpha Institute for Biotechnology). We considered sites to be heteroplasmic only if their Bayesian posterior probability was greater than 0.1 and the credible interval was greater than or included 0.1.
Association with the phenotype was tested using the number of non-reference alleles in a gene or complex and significance was empirically assessed using 10 000 permutations of the sample under the null hypothesis. A Kolmogorov-Smirnov test was used to test for differences in the distribution of missing data in cases compared to controls in the Detroit sample. The Fisher exact test and Wilcoxon rank sum test were used where indicated. To account for tables with low counts, Haldane’s continuity correction was applied to calculate the odds ratio and 95% confidence interval (CI); the one instance of this is noted in the text.
Sanger Sequencing of the NADH Dehydrogenase 5 (MT-ND5) Gene in a Replication Cohort
To replicate our findings in the Chicago samples, we performed Sanger sequencing of the MT-ND5 gene in umbilical cord blood DNA from 162 African American pregnancies with preeclampsia and 171 African American uncomplicated pregnancies who delivered in Detroit, Michigan. These pregnancies were identified by searching the clinical database and the bank of biological specimens of the Perinatology Research Branch and Wayne State University, Detroit, Michigan. All patients provided written informed consent for the collection and use of samples for research purposes (including DNA) under the protocols approved by the institutional review boards of Wayne State University and the Eunice Kennedy Shriver National Institute of Child Health and Human Development, National Institutes of Health, Department of Health and Human Services (NICHD/NIH/DHHS).
Patients with preeclampsia and uncomplicated pregnancies who had cord blood available were included. Ethnicity was based on self-reporting by the mother. Pregnancies were excluded if they were associated with chronic hypertension in the mother, known major fetal or chromosomal anomaly in the current pregnancy, or multiple gestations. All women delivered at Hutzel Women’s Hospital, Detroit, Michigan. Preeclampsia was diagnosed using the similar criteria to that used in the Chicago sample. The control pregnancies were considered uncomplicated if there were no major medical, obstetrical, or surgical complications, and delivery occurred at term (≥37 weeks) with an infant whose birth weight was appropriate for gestational age (10th-90th percentile), according to the reference range of Alexander et al32 for the United States.
Genomic DNA was isolated from 150 μL of buffy coat with EZ1 DNA Blood 350 μL kit (Qiagen) using EZ1 instrument (Qiagen). The whole genome amplification from genomic DNA was performed using the REPLI-g Midi kit (Qiagen), according to manufacturer’s instruction. The amplified DNA was stored at −20°C.
The MT-ND5 gene was amplified in 8 overlapping segments, which were sequenced by dideoxy Sanger sequencing.33 Sequencing protocols are available upon request. Individuals and sites with more than 5% missing data were excluded from all analyses. L haplogroup (African) membership of the infants was confirmed using a custom TaqMan assay (Applied Biosystems, Foster City, California) for the T10873C variant that tags this haplogroup, and participants with a non-L haplogroup mitochondrial chromosome were removed from further analyses.
Results
The clinical characteristics of the participants in the discovery sample after exclusion of women with non-L haplogroup mitochondria are shown in Table 1. There were no significant differences between the preeclamptic and normotensive term (control) pregnancies in this sample for the variables examined, except for an increase in the maximum systolic and diastolic blood pressures in labor in the preeclamptics (P < .02), as expected.
Table 1.
Clinical Characteristics of the Sequencing Study Sample After Excluding Participants With Non-L Haplogroup Mitochondriaa
| Preeclampsia | Control | |
|---|---|---|
| Sample size | 25 | 36 |
| Mean age, SD (years) | 23.1 (5.4) | 23.9 (4.3) |
| Mean weight at first visit, SD (lbs)b | 204.9 (57.9) | 188.5 (41.2) |
| Nulliparous, % | 60.0 | 58.3 |
| Parity (range) | 0-5 | 0-3 |
| Nulligravidous, % | 48.0 | 41.6 |
| Gravidity (range) | 1-6 | 1-4 |
| Mean gestational age at delivery, SD (weeks) | 36.5 (4.5) | 39.0 (1.1) |
| Mean maximum systolic (SD) blood pressure in labor (mm Hg)b | 150.2 (23.4) | 136.0 (16.7) |
| Average maximum diastolic (SD) blood pressure in labor (mm Hg)b | 92.5 (19.1) | 84.7 (13.8) |
a None of the differences between women with preeclampsia and normotensive and term (control) pregnancies are significant (P > .05), except for an increase in the maximum systolic and diastolic blood pressures in labor in the preeclamptics (P < .02).
bData only available for 13 preeclamptic women (see Supplement Table 1).
Whole mtDNA sequencing in 30 preeclamptic and 38 control placentas resulted in an average of 1.9 million reads per sample and an average alignment to rCRS of 95.47%. The mean read depth per sample did not significantly differ between preeclamptic and control placentas (P = .86, Wilcoxon rank sum test). Five preeclamptics (16.7%) and 2 controls (5.3%) were excluded from analyses because they carried a non-L haplogroup mitochondrial chromosome. However, including these participants did not alter any of the significant findings (data not shown). The distribution of mitochondrial L haplogroups was similar in cases and controls (Fisher exact test, P = 0.14; using Haldane’s continuity correction), although the L0 haplogroup was present only in the preeclamptics ( Supplement Figure 1).
The overall mean number of variants per individual was greater in the preeclamptic compared with control pregnancies, with significant excesses in complex I genes (P = .02), ATP synthase genes (P = .004), all protein-coding genes combined (P = .04), and all rRNA genes (0.03; Table 2). The mean number of nonsynonymous substitutions was increased in the preeclamptics for complex I genes (P = .02), ATP synthase genes (P = .02), and all protein-coding genes (P = .02; Table 2); and the mean number of synonymous substitutions was increased in the preeclamptics for the ATP synthase genes (P = .02) and all protein-coding genes (P = .02; Table 2).
Table 2.
Mean Number (Standard Deviation) of Variants Per Individual by Gene Groups for Protein-Coding and Noncoding (RNA) Genesa
| Complex I | Complex III | Complex IV | ATP Synthase | All Protein Coding | All rRNA | All tRNA | ||
|---|---|---|---|---|---|---|---|---|
| All variants | Controls | 16.06 (6.36) | 4.06 (1.17) | 7.31 (3.26) | 2 (1.14) | 29.42 (10.11) | 5 (3.43) | 1.94 (1.31) |
| Preeclampsia | 19.72 (8.14) | 4.04 (1.21) | 8.16 (3.65) | 3.24 (1.98) | 35.16 (12.85) | 6.44 (3.48) | 2.52 (1.64) | |
| P value | .04b | .39 | .17 | .004b | .04b | 0.03 | 0.11 | |
| Nonsynonymous substitutions | Controls | 3.91 (2.66) | 0.77 (0.79) | 0.67 (0.98) | 0.44 (0.77) | 5.81 (3.71) | ||
| Preeclampsia | 5.08 (1.85) | 1 (0.58) | 1.2 (1.41) | 0.72 (0.61) | 8 (3.30) | |||
| P value | .02b | .05 | .08 | .02 | .01b | |||
| Synonymous substitutions | Controls | 12.92 (5.29) | 2.75 (2.64) | 5.78 (2.87) | 0.92 (1.27) | 22.36 (9.64) | ||
| Preeclampsia | 15.12 (5.82) | 3.44 (3.04) | 6.84 (3.88) | 2.40 (2.78) | 27.80 (12.19) | |||
| P value | .08 | .15 | .20 | .02 | .02 |
Abbreviations: ATP, adenosine triphosphate; rRNA, ribosomal RNA; tRNA, transfer RNA.
a Protein-coding genes are shown on a shaded background. P values based on 10 000 permutations.
b P value remains significant (P < .05) when carriers of the L0 haplogroup are removed (see text for details).
Among the 13 protein-coding genes, the overall mean number of variants was significantly increased in the preeclamptics for MT-ND2 (P = .01), MT-ND4 L (P = .03), MT-ATP6 (P = .01), and MT-ATP8 (P = .01; Table 3); nonsynonymous substitutions were increased for MT-ND2 (P = .02), MT-ND5 (P = .0001), MT-CYB (P = .04), and MT-ATP6 (P = .04; Table 3); and synonymous substitutions were increased for MT-ND2 (P = .02), MT-ND3 (P = .01), MT-ND4L (P = .02), MT-ATP8 (P = .03; Table 3).
Table 3.
Mean Number (Standard Deviation) of Variants Per Individual for mtDNA Protein-Coding Genesa
| MT-ND1 | MT-ND2 | MT-ND3 | MT-ND4L | MT-ND4 | MT-ND5 | MT-ND6 | MT-CO1 | MT-CO2 | MT-CO3 | MT-CYB | MT-ATP6 | MT-ATP8 | ||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| All variants | Controls | 2.47 (1.82) | 1.17 (1.08) | 1.86 (0.64) | 0.36 (0.59) | 4 (1.21) | 4.91 (2.79) | 1.27 (0.97) | 4.14 (2.37) | 1.36 (1.07) | 1.81 (0.67) | 4.06 (1.17) | 1.64 (0.68) | 0.36 (0.59) |
| Preeclampsia | 2.84 (2.03) | 2.2 (1.87) | 1.68 (0.90) | 1.04 (1.27) | 4.76 (2.28) | 5.88 (2.54) | 1.32 (1.11) | 4.64 (2.94) | 1.08 (1.04) | 2.44 (1.39) | 4.04 (1.21) | 2.32 (1.25) | 0.92 (0.99) | |
| P value | .21 | .01 | .09 | .03 | .16 | .06 | .52 | .32 | .14 | .09 | .39 | .01 | .01 | |
| Nonsynonymous substitutions | Controls | 0.39 (0.69) | 0.36 (0.59) | 0.36 (0.90) | 0.03 (0.17) | 0.24 (0.43) | 2.25 (1.5) | 0.25 (0.44) | 0.53 (0.84) | 0.08 (0.28) | 0.05 (0.23) | 0.74 (0.79) | 0.33 (0.68) | 0.11 (0.32) |
| Preeclampsia | 0.24 (0.52) | 0.76 (0.78) | 0.44 (0.77) | 0 | 0.25 (0.44) | 3.12 (0.88) | 0.28 (0.46) | 0.88 (1.67) | 0.16 (0.37) | 0.16 (0.37) | 1 (0.58) | 0.52 (0.51) | 0.20 (0.41) | |
| P value | .14 | .02 | .38 | 1 | .33 | .0001b | .53 | .10 | .28 | .20 | .04 | .04 | .26 | |
| Synonymous substitutions | Controls | 2.36 (1.71) | 1.67 (2.17) | 1.25 (2.12) | 0.34 (0.58) | 2.91 (1.49) | 2.07 (1.89) | 2.69 (0.98) | 2.59 (1.65) | 1.28 (1.06) | 1.91 (1.20) | 2.75 (2.64) | 0.55 (1.03) | 0.36 (0.59) |
| Preeclampsia | 2.64 (1.85) | 2.56 (3.75) | 0.6 (1.60) | 1.04 (1.27) | 2.92 (1.61) | 2.76 (1.89) | 2.8 (0.98) | 2.8 (2.14) | 1.2 (1.73) | 2.84 (2.36) | 3.44 (3.04) | 1.48 (2.24) | 0.92 (1.18) | |
| P value | .22 | 0.02 | 0.01 | 0.02 | 0.56 | 0.09 | 0.30 | 0.45 | 0.17 | 0.09 | 0.15 | 0.02 | 0.06 |
a P values based on10 000 permutations.
b P value remains significant (P < .05) when carriers of the L0 haplogroup are removed (see text for details).
The overall mean number of heteroplasmic sites per individual was 6.39 (standard deviation [SD] = 8.24) in the combined sample, 6.64 (SD = 9.22) in the preeclamptics, and 6.22 (SD = 7.61) in the control placentas. The mean number of heteroplasmic sites per individual did not differ significantly between the 2 groups overall or for any complexes or individual genes (data not shown).
The only association that remained significant after adjusting for the number of tests using a Bonferroni correction for 13 gene tests was the excess of nonsynonymous mutations in the MT-ND5 gene (0.05/13 = .0038; observed P = .0001, Table 3). This excess included an enrichment of a single nonsynonymous substitution (rs2853502, Met314Val) in cases compared with controls (Fisher exact test, P = .007; odds ratio [OR] = 15.74, 95% CI 1.42-166.36; using Haldane’s continuity correction). The Val allele at this variant site tags the L0 mitochondrial haplogroup, which was present only in 5 preeclamptics (Figure 1). Of note, however, is that there are proportionally more nonsynonymous substitutions in MT-ND5 in the preeclamptics on nearly all haplogroup backgrounds (Figure 1). As a result, more cases (22 of 25; 88%) than controls (19 of 36; 53%) carried at least 1 nonsynonymous substitution in MT-ND5 (excluding the common Ile172Gly and Val257Ile variants; Fisher exact test, P = .005; OR = 6.36, 95% CI: 1.5-39.1). The clinical characteristics, MT-ND5 mutations, and haplogroups for the Chicago pregnancies with preeclampsia are shown in Supplement Table 1.
Figure 1.
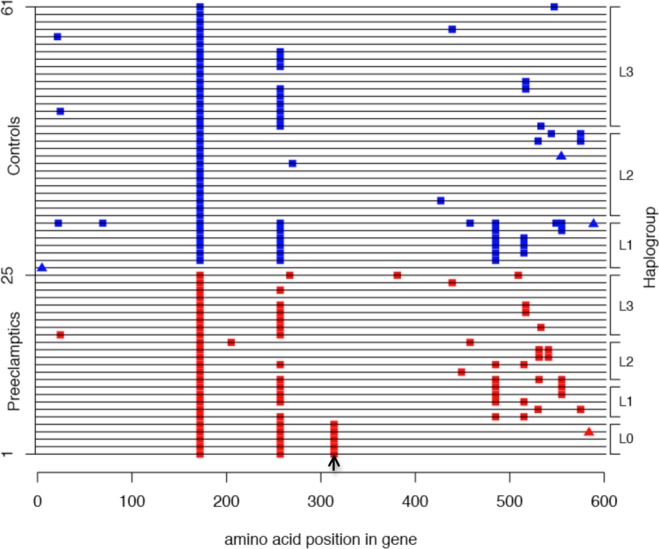
Haplotypes of the MT-ND5 gene in the Chicago sample; only nonsynonymous substitutions are shown. The haplotype for each of the individuals is displayed as a single horizontal line. Nonsynonymous substitutions that differ from the Yoruban L haplotype are shown as filled colored squares. Filled triangles show insertions causing frameshift mutations. Preeclamptics are individuals 1 to 25 (red squares) and controls are individuals 26 to 61 (blue squares). The site of the Met314Val variant is shown by an arrow. Haplogroups are shown on the right y-axis. See Supplement Table 1 for additional details.
Because the Val allele occurs only on the L0 haplogroup background, and the L0 haplogroup was present only in preeclamptics, it was possible that the excess of nonsynonymous variants for other genes or complexes also occurred on this same background and did not represent independent associations with preeclampsia. To explore this possibility, we excluded the 5 cases with the Val allele (L0 haplogroup) and repeated analyses of all results in Tables 2 and 3 with P < .05. Excesses of nonsynonymous substitutions remained for all protein-coding genes, for complex I genes, and for the MT-ND5 gene (P < .02) indicating an overall enrichment of nonsynonymous substitutions in the preeclamptics that are independent of the L0 haplotype and the Met314Val polymorphism. Next, all nonsynonymous substitutions in the MT-ND5 gene were excluded. An excess of nonsynonymous substitutions in protein-coding genes overall remained (P = .01), primarily due to an enrichment of mutations in the MT-ND2, MT-ATP6, and MT-CYB genes (data not shown). Taken together, these analyses reveal independent enrichments of nonsynonymous substitutions in MT-ND5 (in addition to the Met314Val variant), MT-ND2, MT-CYB, and MT-ATP6 in African American placentas from preeclamptic pregnancies.
To replicate our findings of the increased number of nonsynonymous sites in the MT-ND5 gene, we sequenced this gene in African American preeclamptic infant cases and infant controls from Detroit. Prior to sequencing, we genotyped the 607 samples for the T1087C variant that tags the L haplogroup and excluded 9 patients (3.5%) and 26 controls (7.6%) because their mitochondrial chromosome did not belong to the L haplogroup. Among the remaining 361 participants (179 patients with preeclampsia and 182 controls), 28 (17 patients, 11 controls) were excluded after sequencing because they had >5% missing data (Supplement Figure 3). The overall distribution of missing sites was not significantly different between cases and controls (Kolmogorov-Smirnov test, P = .38, Supplement Figure 3). The final sample included 162 infants from preeclamptic pregnancies and 171 infants from normal, term pregnancies. In this sample, the mean number of MT-ND5 nonsynonymous substitutions were 2.26 (SD = 1.54) and 2.31 (SD = 1.24 P = 0.67) and synonymous substitutions were 2.35 (SD = 1.84) and 2.37 (SD = 1.88, P = .84) in preeclamptic and control pregnancies, respectively. The Val allele at amino acid 314 in the MT-ND5 gene occurred in 3.4% of cases and 6.6% of controls (Fisher exact test, P = .13, OR = 0.41 [95% CI: 0.11-1.28]).
Thus, the excess of nonsynonymous substitutions in the MT-ND5 gene observed in the Chicago patients with preeclampsia compared with controls was not replicated in the Detroit sample, in which 51.2% of cases and 53.8% of controls carried at least 1 nonsynonymous substitution in the MT-ND5 gene compared to 88% and 53% of Chicago cases and controls, respectively. The mean numbers of nonsynonymous substitutions in MT-ND5 in the Detroit preeclamptic infants (2.26) and control infants (2.31) were also similar to the Chicago control placentas (2.25), all of which were significantly lower than the mean number of nonsynonymous variants in the Chicago patients with preeclampsia (3.12). Moreover, the frequency of the Val allele at the Met314Val polymorphism occurred in 3.4% and 6.6% of Detroit patients and controls, respectively, compared with 25.0% and 0% in the Chicago patients and controls, respectively.
Discussion
The underlying cause of the component phenotypes of preeclampsia has been ascribed to an antiangiogenic state characterized by excessive concentrations of antiangiogenic factors, including soluble vascular endothelial growth factor receptor-1 and soluble endoglin, and low concentrations of the angiogenic placental growth factors,34–38 but why the placenta overproduces these proteins remains obscure. One plausible hypothesis is that this results from excess ROS in the placentas of preeclamptics due to mtDNA mutations that disrupt critical pathways in oxidative phosphorylation. We tested this hypothesis by sequencing the entire mitochondrial chromosome in a discovery sample of African American pregnancies with preeclampsia and normotensive term pregnancies from Chicago, and then sequencing the MT-ND5 gene in African American pregnancies with preeclampsia and normal term pregnancies from Detroit.
Our studies in the Chicago sample revealed a significant excess of nonsynonymous sites in the preeclamptic compared to control placentas, especially in the MT-ND5 gene. Mutations in MT-ND5 are frequently involved in mitochondrial diseases with defects in oxidative phosphorylation, such as Leber's hereditary optic neuropathy,39 , 40 mitochondrial encephalomyopathy, lactic acidosis, stroke-like episode syndrome,41 , 42 and Leigh syndrome.42 This has led to the suggestion that MT-ND5 is a mutational hot spot for oxidative phosphorylation defects.41 , 42 Although MT-ND5 participates as only one of the enzymatic subunits of complex I in the respiratory chain, in vivo studies have shown that MT-ND5 is not only necessary for complex I assembly and function, but that its synthesis is a rate-limiting step in respiration.43 However, the increase in nonsynonymous substitutions was not replicated in the larger Detroit sample, in which both cases and controls had rates of variation similar to the Chicago controls.
There are a number of potential explanations for these discrepant results. In particular, our discovery sample was quite small, consisting of only 25 patients with preeclampsia cases and 36 normotensive controls. It is possible, therefore, that we selected by chance a subset of the cases with a skewed distribution of nonsynonymous sites in their mitochondrial genome. The preeclamptic pregnancies included in the discovery sample were selected from the larger CLIPP Biobank samples because they met the definition of preeclampsia used in this study and had a sufficient amount of high-quality placenta DNA available for sequencing. Subsequent to sequencing, participants with mitochondrial chromosomes that did not belong to the L-haplogroup (ie, non–African mitochondrial chromosomes) were excluded. Thus, there are no obvious biases in the selection of the Chicago preeclampsia pregnancy samples compared to those from Detroit, yet it remains possible that the significant results in the discovery sample were type 1 errors in a small sample.
On the other hand, differences in the study designs in the Chicago and Detroit samples could have influenced these results. First, sequencing studies in the Chicago samples were performed in placental-derived DNA, whereas sequencing studies in the Detroit samples were performed in DNA derived from cord blood. While we expect the placental genome to reflect the fetal (cord blood) genome, it is possible that mutation rates are higher in rapidly dividing placental cells compared to cord blood cells, particularly in mitochondrial DNA (mtDNA), which has higher mutation rates than nuclear DNA.44–46 However, the fact that we did not see increased rates of heteroplasmy or of synonymous substitutions in the placental DNA in the Chicago samples, suggests that increased de novo mutations rates in preeclamptic placentas are not likely to underlie the observed differences. Moreover, the occurrence of the same variants in multiple individuals further argues against this theory.
A second difference between the studies in the Chicago and Detroit samples was that we used next-generation sequencing of the entire mitochondrial chromosome in the Chicago samples and dideoxy Sanger sequencing of just the MT-ND5 gene in the Detroit samples. In addition, the studies in the Detroit samples were performed in whole genome amplified DNA, resulting in an overall higher frequency of missing data in the Detroit compared to Chicago samples (Supplement Figures 2 and 3). Although these differences could result in overall different rates of detected variants in the MT-ND5 gene between the 2 samples, it is unlikely that sequencing technology or DNA quality would result in differences between cases and controls in the Chicago samples only. However, we cannot exclude the possibility that the Detroit preeclamptic pregnancies harbor excesses nonsynonymous variants in mitochondrial genes other than MT-ND5. Due to practical considerations, we could not sequence the entire mitochondrial chromosome in the Detroit samples, so we selected the MT-ND5 gene for targeted sequencing because it harbored the greatest excesses of variation in the Chicago cases. However, the Chicago patients with preeclampsia also showed significant excesses of nonsynonymous variation in the MT-ND2, MT-ATP6, and MT-CYB genes that were independent of the MT-ND5 findings. Therefore, it is possible that the patients with preeclampsia from Detroit harbor increased rates of nonsynonymous substitutions in mitochondrial genes other than MT-ND5.
Lastly, cryptic heterogeneity may exist between the Chicago and Detroit samples, such that mutations in mitochondrial genes contribute more to the onset of preeclampsia in the Chicago cases than in the Detroit cases. Preeclampsia is a heterogeneous condition with many different potential etiologies, all of which result in the onset of hypertension and proteinuria during pregnancy. We focused these studies in African American pregnancies both because African ancestry is a risk factor for preeclampsia3 - 5 and because mitochondrial chromosomes from Africa harbor more variations due their deep ancestral roots.47 , 48 However, African Americans are genetically very heterogeneous, with respect both to proportions of European admixture, ranging from 6.8% to 22.5%,49 and to specific ancestry within Africa.50 Although our study included African American participants from 2 Midwestern US cites, the proportions of mitochondrial chromosomes that were not members of the L (African) haplogroup were 10.3% of the Chicago sample (16.7% of cases, 5.3% of controls) and 5.8% of the Detroit sample (3.5% of cases, 7.6% of controls), suggesting more European admixture in the Chicago preeclamptic pregnancies. Because mtDNA reveals ancestry just through maternal lineages it provides an incomplete picture of overall admixture rates in the 2 samples, but nonetheless suggests differences in European admixture that could have influenced our results. For example, if the risk for preeclampsia associated with mitochondrial nonsynonymous substitutions requires interaction with an autosomal European genetic background, increased rates of European admixture could enhance the effects of mitochondrial mutations. Such interactions have been demonstrated for other mitochondrial-associated diseases, for which mutations in both autosomal and mitochondrial genes result in more severe phenotypes51 and mutations in autosomal and mitochondrial genes result in the same phenotype.52–54 It is possible therefore that mutations in autosomal genes as well as in all mitochondrial genes involved in oxidative phosphorylation pathways need to be considered in future studies.
Whether a higher background rate of mitochondrial mutations among women of African descent contributes to the increased risk for preeclampsia and related health disparities among African American women3 - 5 remains to be determined. Future studies of mitochondrial sequence variants from larger samples of African American placentas from preeclamptic pregnancies and normotensive pregnancies are required to address this important outstanding question.
Acknowledgements
We acknowledge the Chicago Lying-In Women’s Board and the Departments of OB/GYN and Pediatrics for supporting the CLIPP Biobank, and the University of Chicago Institute of Medicine CTSA for core services. This research was supported, in part, by the Perinatology Research Branch, Division of Intramural Research, Eunice Kennedy Shriver National Institute of Child Health and Human Development, NIH, DHHS.
Footnotes
Authors’ Note: David Ding, Nicole M. Scott, and Emma E. Thompson contributed equally to this work. The online data supplements are available at http:/rsx.sagepub.com/supplemental.
Declaration of Conflicting Interests: The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding: The author(s) disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: Preeclampsia Foundation Vision Grant and Stanley W. Marion Family’s Maternal-Fetal Medicine Fellow Research Grant to DD, National Institutes of Health grants R01 HD21244 and P01 HD49480 to CO, and UL1 RR024999 to the Institute Translational Medicine at the University of Chicago.
References
- 1. Ness R, Roberts J. Epidemiology of pregnancy-related hypertension. In: Lindheimer M, Roberts J, Cunningham F, eds. Chesley’s Hypertensive Disorders in Pregnancy. 3 rd ed San Diego, CA: Academic Press; 2009:37–50 [Google Scholar]
- 2. Villar J, Say L, Gulmezohlu A, et al. Eclampsia and preeclampsia: a worldwide health problem for 2000 years. In: Critchley H, Maclean A, Poston L, Walker J, eds. Preeclampsia. RCOG Press; 2003:189–207 [Google Scholar]
- 3. Samadi A, Mayberry R, Zaidi A, et al. Maternal hypertension and associated pregnancy complications among African-American and other women in the United States. Obstet Gynecol. 1996;87(4):557–563 [DOI] [PubMed] [Google Scholar]
- 4. Caughey AB, Stotland NE, Washington AE, Escobar GJ. Maternal ethnicity, paternal ethnicity, and parental ethnic discordance: predictors of preeclampsia. Obstet Gynecol. 2005;106(1):156–161 [DOI] [PubMed] [Google Scholar]
- 5. Tanaka M, Jaamaa G, Kaiser M, et al. Racial disparity in hypertensive disorders of pregnancy in New York State: a 10-year longitudinal population-based study. Am J Public Health. 2006;97(1):163–170 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Roberts JM, Redman CW. Pre-eclampsia: more than pregnancy-induced hypertension. Lancet. 1993;341(8858):1447–1451 [DOI] [PubMed] [Google Scholar]
- 7. Redman CW. Current topic: pre-eclampsia and the placenta. Placenta. 1991;12(4):301–308 [DOI] [PubMed] [Google Scholar]
- 8. Wang Z, Zhang G, Lin M. Mitochondrial tRNA(leu)(UUR) gene mutation and the decreased activity of cytochrome c oxidase in preeclampsia. J Tongji Med Univ. 1999;19(3):209–211 [DOI] [PubMed] [Google Scholar]
- 9. Wang Y. Placental mitochondria as a source of oxidative stress in pre-eclampsia. Placenta. 1998;19(8):581–586 [DOI] [PubMed] [Google Scholar]
- 10. Wang Y, Walsh SW, Kay HH. Placental lipid peroxides and thromboxane are increased and prostacyclin is decreased in women with preeclampsia. Am J Obstet Gynecol. 1992;167(4 pt 1):946–949 [DOI] [PubMed] [Google Scholar]
- 11. Gupta S, Agarwal A, Sharma RK. The role of placental oxidative stress and lipid peroxidation in preeclampsia. Obstet Gynecol Surv. 2005;60(12):807–816 [DOI] [PubMed] [Google Scholar]
- 12. Balaban RS, Nemoto S, Finkel T. Mitochondria, oxidants, and aging. Cell. 2005;120(4):483–495 [DOI] [PubMed] [Google Scholar]
- 13. Torbergsen T, Oian P, Mathiesen E, Borud O. Pre-eclampsia—a mitochondrial disease? Acta Obstet Gynecol Scand. 1989;68(2):145–148 [DOI] [PubMed] [Google Scholar]
- 14. Berkowitz K, Monteagudo A, Marks F, Jackson U, Baxi L. Mitochondrial myopathy and preeclampsia associated with pregnancy. Am J Obstet Gynecol. 1990;162(1):146–147 [DOI] [PubMed] [Google Scholar]
- 15. Folgerø T, Storbakk N, Torbergsen T, Oian P. Mutations in mitochondrial transfer ribonucleic acid genes in preeclampsia. Am J Obstet Gynecol. 1996;174(5):1626–1630 [DOI] [PubMed] [Google Scholar]
- 16. Jauniaux E, Poston L, Burton GJ. Placental-related diseases of pregnancy: involvement of oxidative stress and implications in human evolution. Hum Reprod Update. 2006;12(6):747–755 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Shanklin DR, Sibai BM. Ultrastructural aspects of preeclampsia. II. Mitochondrial changes. Am J Obstet Gynecol. 1990;163(3):943–953 [DOI] [PubMed] [Google Scholar]
- 18. Widschwendter M, Schrocksnadel H, Mortl M. Pre-eclampsia: a disorder of placental mitochondria? Mol Med Today. 1998;4(7):286–291 [DOI] [PubMed] [Google Scholar]
- 19. DiMauro S, Schon EA. Mitochondrial respiratory-chain diseases. N Engl J Med. 2003;348(26):2656–2668 [DOI] [PubMed] [Google Scholar]
- 20. Vuorinen K, Remes A, Sormunen R, Tapanainen J, Hassinen IE. Placental mitochondrial DNA and respiratory chain enzymes in the etiology of preeclampsia. Obstet Gynecol. 1998;91(6):950–955 [DOI] [PubMed] [Google Scholar]
- 21. Metzker ML. Sequencing technologies—the next generation. Nat Rev Genet. 2010;11(1):31–46 [DOI] [PubMed] [Google Scholar]
- 22. Lindheimer MD, Taler SJ, Cunningham FG. Hypertension in pregnancy. J Am Soc Hypertens. 2010;4(2):68–78 [DOI] [PubMed] [Google Scholar]
- 23. Anon. Report of the National High Blood Pressure Education Program Working Group on High Blood Pressure in Pregnancy. Am J Obstet Gynecol. 2000;183(1):S1–S22 [PubMed] [Google Scholar]
- 24. Langmead B, Trapnell C, Pop M, Salzberg SL. Ultrafast and memory-efficient alignment of short DNA sequences to the human genome. Genome Biol. 2009;10(3):R25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Cann RL, Stoneking M, Wilson AC. Mitochondrial DNA and human evolution. Nature. 1987;325(6099):31–36 [DOI] [PubMed] [Google Scholar]
- 26. van Oven M, Kayser M. Updated comprehensive phylogenetic tree of global human mitochondrial DNA variation. Hum Mutat. 2009;30(2):E386–394 [DOI] [PubMed] [Google Scholar]
- 27. Behar DM, Villems R, Soodyall H, et al. The Dawn of Human Matrilineal Diversity. Am J Hum Genet. 2008;82(5):1130–1140 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Salas A, Richards M, Lareu M-V, et al. The African diaspora: mitochondrial DNA and the Atlantic slave trade. Am J Hum Genet. 2004;74(3):454–465 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Gonder MK, Mortensen HM, Reed FA, de Sousa A, Tishkoff SA. Whole-mtDNA genome sequence analysis of ancient African lineages. Mol Biol Evol. 2006;24(3):757–768 [DOI] [PubMed] [Google Scholar]
- 30. Eltsov N, Volodko N, Starikovskaya E, Sukernik R. New tool (mtPHYL) proposed for phylogenetic analysis of human complete mitochondrial genomes. http://eltsov.org/mtphyl.aspx Accessed February 2, 2011
- 31. Ingman M, Gyllensten U. Analysis of the complete human mtDNA genome: methodology and inferences for human evolution. J Hered. 2001;92(6):454–461 [DOI] [PubMed] [Google Scholar]
- 32. Alexander GR, Himes JH, Kaufman RB, Mor J, Kogan M. A United States national reference for fetal growth. Obstet Gynecol. 1996;87(2):163–168 [DOI] [PubMed] [Google Scholar]
- 33. Pichler I, Fuchsberger C, Platzer C, et al. Drawing the history of the Hutterite population on a genetic landscape: inference from Y-chromosome and mtDNA genotypes. Eur J Hum Genet. 2009;18(4):463–470 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Chaiworapongsa T, Romero R, Espinoza J, et al. Evidence supporting a role for blockade of the vascular endothelial growth factor system in the pathophysiology of preeclampsia. Young Investigator Award. Am J Obstet Gynecol. 2004;190(6):1541–1547; discussion 1547-1550 [DOI] [PubMed] [Google Scholar]
- 35. Kusanovic JP, Romero R, Jodicke C, et al. Amniotic fluid soluble human leukocyte antigen-G in term and preterm parturition, and intra-amniotic infection/inflammation. J Matern Fetal Neonatal Med. 2009;22(12):1151–1166 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Levine RJ, Lam C, Qian C, et al. Soluble endoglin and other circulating antiangiogenic factors in preeclampsia. N Engl J Med. 2006;355(10):992–1005 [DOI] [PubMed] [Google Scholar]
- 37. Romero R, Chaiworapongsa T, Erez O, et al. An imbalance between angiogenic and anti-angiogenic factors precedes fetal death in a subset of patients: results of a longitudinal study. J Matern Fetal Neonatal Med 2010;23(12):1384–1399 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Romero R, Nien JK, Espinoza J, et al. A longitudinal study of angiogenic (placental growth factor) and anti-angiogenic (soluble endoglin and soluble vascular endothelial growth factor receptor-1) factors in normal pregnancy and patients destined to develop preeclampsia and deliver a small for gestational age neonate. J Matern Fetal Neonatal Med. 2008;21(1):9–23 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Liu X-L, Zhou X, Zhou J, et al. Leber’s hereditary optic neuropathy is associated with the T12338C mutation in mitochondrial ND5 gene in six Han Chinese families. Ophthalmology. 2010. http://www.ncbi.nlm.nih.gov/pubmed/21131053 [DOI] [PubMed]
- 40. Zou Y, Jia X, Zhang A-M, et al. The MT-ND1 and MT-ND5 genes are mutational hotspots for Chinese families with clinical features of LHON but lacking the three primary mutations. Biochem Biophys Res Commun. 2010;399(2):179–185 [DOI] [PubMed] [Google Scholar]
- 41. Liolitsa D, Rahman S, Benton S, Carr LJ, Hanna MG. Is the mitochondrial complex I ND5 gene a hot-spot for MELAS causing mutations? Ann Neurol. 2003;53(1):128–132 [DOI] [PubMed] [Google Scholar]
- 42. Shanske S, Coku J, Lu J, et al. The G13513A mutation in the ND5 gene of mitochondrial DNA as a common cause of MELAS or Leigh syndrome: evidence from 12 cases. Arch Neurol. 2008;65(3):368–372 [DOI] [PubMed] [Google Scholar]
- 43. Bai Y, Shakeley RM, Attardi G. Tight control of respiration by NADH dehydrogenase ND5 subunit gene expression in mouse mitochondria. Mol Cell Biol. 2000;20(3):805–815 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Brown WM, George M, Jr, , Wilson AC. Rapid evolution of animal mitochondrial DNA. Proc Natl Acad Sci U S A. 1979;76(4):1967–1971 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Fay JC, Wu CI. A human population bottleneck can account for the discordance between patterns of mitochondrial versus nuclear DNA variation. Mol Biol Evol. 1999;16(7):1003–1005 [DOI] [PubMed] [Google Scholar]
- 46. Sigurõardóttir S, Helgason A, Gulcher JR, Stefansson K, Donnelly P. The mutation rate in the human mtDNA control region. Am J Hum Genet. 2000;66(5):1599–1609 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Chen YS, Torroni A, Excoffier L, Santachiara-Benerecetti AS, Wallace DC. Analysis of mtDNA variation in African populations reveals the most ancient of all human continent-specific haplogroups. Am J Hum Genet. 1995;57(1):133–149 [PMC free article] [PubMed] [Google Scholar]
- 48. Ingman M, Kaessmann H, Pääbo S, Gyllensten U. Mitochondrial genome variation and the origin of modern humans. Nature. 2000;408(6813):708–713 [DOI] [PubMed] [Google Scholar]
- 49. Parra EJ, Marcini A, Akey J, et al. Estimating African American admixture proportions by use of population-specific alleles. Am J Hum Genet. 1998;63(6):1839–1851 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Zakharia F, Basu A, Absher D, et al. Characterizing the admixed African ancestry of African Americans. Genome Biol. 2009;10(12):R141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Chevrollier A, Guillet V, Loiseau D, et al. Hereditary optic neuropathies share a common mitochondrial coupling defect. Ann Neurol. 2008;63(6):794–798 [DOI] [PubMed] [Google Scholar]
- 52. Santorelli FM, Shanske S, Macaya A, DeVivo DC, DiMauro S. The mutation at nt 8993 of mitochondrial DNA is a common cause of Leigh’s syndrome. Ann Neurol. 1993;34(6):827–834 [DOI] [PubMed] [Google Scholar]
- 53. Yu-Wai-Man P, Griffiths PG, Chinnery PF. Mitochondrial optic neuropathies—disease mechanisms and therapeutic strategies. Prog Retin Eye Res. 2011;30(2):81–114 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Bu XD, Rotter JI. X chromosome-linked and mitochondrial gene control of Leber hereditary optic neuropathy: evidence from segregation analysis for dependence on X chromosome inactivation. Proc Natl Acad Sci U S A. 1991;88(18):8198–8202 [DOI] [PMC free article] [PubMed] [Google Scholar]