

Abstract
We analyzed 27 578 CpG sites that map to 14 495 genes in omental arteries of normal pregnant and preeclamptic women for DNA methylation status using the Illumina platform. We found 1685 genes with a significant difference in DNA methylation at a false discovery rate of <10% with many inflammatory genes having reduced methylation. Unsupervised hierarchical clustering revealed natural clustering by diagnosis and methylation status. Of the genes with significant methylation differences, 236 were significant at a false discovery rate of <5%. When data were analyzed more stringently to a false discovery rate of <5% and difference in methylation of >0.10, 65 genes were identified, all of which showed reduced methylation in preeclampsia. When these genes were mapped to gene ontology for molecular functions and biological processes, 75 molecular functions and 149 biological processes were overrepresented in the preeclamptic vessels. These included smooth muscle contraction, thrombosis, inflammation, redox homeostasis, sugar metabolism, and amino acid metabolism. We speculate that reduced methylation may contribute to the pathogenesis of preeclampsia and that alterations in DNA methylation resulting from preeclampsia may increase maternal risk of cardiovascular disease later in life.
Keywords: preeclampsia, DNA methylation, epigenetics, omental blood vessels, inflammation.
Introduction
Preeclampsia is a pregnancy disorder that remains a worldwide health problem and a leading cause of preterm delivery and of maternal and infant mortality and morbidity.1 It is a complex, multisystem disease which is diagnosed clinically by new onset of hypertension and proteinuria that occur after 20 weeks of gestation in pregnant women who are otherwise healthy.1 Preeclampsia is often associated with other end organ problems, such as cerebral, pulmonary, and systemic edema, activation of the coagulation system, reduced uteroplacental blood flow, and intrauterine growth restriction. The pathophysiology of preeclampsia is not well understood. It has been considered as a 2-stage disease where the first stage is poor placentation and the second stage is development of exaggerated maternal systemic inflammatory response.2
Many studies have reported possible genetic contribution to preeclampsia based on inheritance patterns or mutations in candidate genes,3 ,4 however, few studies have examined the possible contribution of epigenetic mechanisms. Epigenetics is the study of changes in gene expression, which are mediated through mechanisms that do not involve changes in the DNA sequence. DNA methylation is the best characterized epigenetic mechanism.5 It refers to the reversible addition of a methyl group (CH3) at the 5′ carbon of a cytosine in a CpG dinucleotide context. DNA methylation patterns can change in response to age, environmental stimuli, diet, and oxidative stress.6 –8 The pattern of DNA methylation is maintained by DNA methyltransferase enzymes during future cell divisions.7 In general, DNA hypomethylation is associated with increased gene expression, whereas DNA hypermethylation is associated with decreased gene expression.9 DNA methylation is involved in important biological processes such as tissue-specific gene regulation during development, X-chromosome inactivation, and genomic imprinting.10
Changes in DNA methylation patterns have been detected in a growing number of complex diseases including cancer,11 schizophrenia,12 neural tube defect,13 and cardiovascular diseases such as atherosclerosis,14 ischemic heart disease and stroke.15 Little information is available regarding DNA methylation in preeclampsia, but it might be an important pathologic mechanism because preeclampsia is associated with oxidative stress16 ,17 and obesity,18 both of which can influence DNA methylation. It has been suggested the increased risk of preeclampsia in assisted reproductive technology pregnancies may be due to early epigenetic changes in gametes and/or embryos that contributes to abnormal placentation and development of preeclampsia.19 Epigenetic changes associated with imprinted and nonimprinted genes have been demonstrated in preeclamptic placentas,20–22 and deletion of imprinted gene p57-Kip2 in mice is associated with hypertension and proteinuria,23 which are manifestations of preeclampsia.1 In a study that investigated global DNA methylation profiles of placentas using Illumina GoldenGate Methylation Cancer panel I array, investigators reported promoter hypomethylation of 34 CpG sites in early-onset preeclampsia.24
To date, there have been no reports dealing with DNA methylation in systemic blood vessels of the mother with preeclampsia. In this study, we used the Illumina HumanMethylation27 platform to simultaneously profile more than 27 000 CpG sites, representing more than 14 000 genes in samples of omental fat arteries isolated from normal pregnant and preeclamptic women. Omental arteries were used because these arteries are a component of maternal systemic vasculature, and they play a role in blood pressure regulation by contributing to total peripheral vascular resistance.
Materials and Methods
Study Participants
Omental fat biopsies of approximately 2 cm × 2 cm × 0.5 cm in size were collected from normal pregnant (n = 5) and preeclamptic (n = 7) women (26-40 weeks of gestation) during medically indicated cesarean sections performed at MCV Hospital, Virginia Commonwealth University Medical Center, Richmond, Virginia. Once obtained, the biopsy was placed in a sterile container on ice and transferred to the laboratory for processing as soon as possible. Preeclampsia was diagnosed in gravid females by new onset of hypertension (systolic blood pressure of ≥140 mm Hg and/or diastolic blood pressure ≥90 mm Hg) and proteinuria (300 mg or more of protein in the urine per 24-hour collection) that occurred in women who were otherwise normal.1 Women with chorioamnionitis, maternal infections, active sexually transmitted diseases, lupus, or diabetes, and women who were smokers or in labor were excluded because these conditions are associated with inflammatory changes. Patient clinical data are shown in Table 1. All participants gave informed consent. The Office of Research Subjects Protection at Virginia Commonwealth University approved this study.
Table 1.
Clinical Characteristics of Patient Groupsa
| Variable | Normal Pregnant (n = 5) | Preeclamptic (n = 7) |
|---|---|---|
| Maternal age (years) | 23.0 ± 1.7 | 24.9 ± 1.4 |
| Pre-pregnancy BMI (kg/m2) | 24.4 ± 1.5 | 29.9 ± 2.9 |
| Systolic blood pressure (mm Hg) | 115.0 ± 3.2 | 168.0 ± 2.8b |
| Diastolic blood pressure (mm Hg) | 69.6 ± 1.3 | 91.1 ± 3.9c |
| Proteinuria (mg/24 h) | ND | 307.8 ± 26.7 (n = 4) |
| Dipstick | ND | 2.3 ± 0.9 (n = 3) |
| Parity | ||
| Primiparous | 2 | 2 |
| Multiparous | 3 | 5 |
| Gestational age (week) | 39.2 ± 0.2 | 34.1 ± 1.7d |
| Infant birth weight (g) | 3424 ± 110 | 2304 ± 393 |
Abbreviations: ND, not detectable; SE, standard error.
a Values are presented as mean ± SE.
b P < .001.
c P < .01.
d P < .05 by t test.
Sample Processing, DNA Extraction, and Bisulfite Treatment
Fat biopsies from normal pregnant and preeclamptic women were dissected for omental arteries under a dissection microscope. Arteries and veins were distinguished from each other based on their morphology. In general, arteries were thicker, narrower, and their branching points were more acute as compared to veins. Omental arteries were dissected and cleared carefully of adhering fat. DNA was extracted from the arteries (∼10 mg in weight) using QuickGene DNA tissue kit S kit and QuickGene-Mini80 system (AutoGen, Holliston, Massachusetts) as recommended by the user manual. RNase treatment was performed with RNase A (Qiagen, Valencia, California). DNA concentration was measured and its quality was assessed using NanoDrop 2000 spectrophotometer (Thermo Scientific, Wilmington, Delaware). DNA samples with A260/280 ratio of 1.8 were considered to be pure and used in the study. DNA (1 µg) was bisulfite treated using MethylEasy Exceed kit (Human Genetic Signatures, Randwick, Australia). DNA was eluted in 20 µL elution buffer for a final concentration of 50 ng/µL.
Methylation Assay
The high throughput Illumina Infinium HumanMethylation27 BeadChip assay (Illumina, San Diego, California) was used for DNA methylation analysis. This assay allows the examination of 27 578 CpG dinucleotides located within the proximal promoters of 14 495 genes. The BeadChip was run by the Nucleic Acids Research Facilities at VCU using the standard protocol provided by Illumina. In brief, 10 µL of bisulfite-treated DNA (50 ng/µl) was isothermally amplified at 37°C for 24 hours. The amplification product was enzymatically fragmented and the fragmented DNA was purified and applied to the BeadChip for hybridization. During hybridization, DNA anneals to specific single-stranded oligonucleotides, which are covalently linked to different types of beads. Each CpG site is presented by 2 types of beads, which corresponds to the nucleotide identity, and thus the methylation status, of the bisulfite-treated cytosine at that site. Hybridization was followed by single-base extension of the oligonucleotides where the hybridized DNA was used as a template to incorporate hapten-labeled dideoxynucleotides. After that, the BeadChip was fluorescently stained and then scanned for the intensities of the methylated and the unmethylated bead types using the Illumina BeadArray Reader. The scanned arrays were processed using Illumina’s GenomeStudio Methylation Analysis Module to obtain the β values for each CpG site, where βij represents proportion methylated for the ith CpG site and the jth array, and where β = 1 would indicate complete methylation and β = 0 would indicate no methylation. Validation of the BeadChip assay was performed by evaluating the quality of the hybridizations for 6 X-linked genes, and by using COBRA25–27 for the −1538 CpG site upstream from the transcription start site (TSS) for the matrix metalloproteinase 1 (MMP1) gene, as we previously described.28
Statistical Analysis
Following the seminal article of Bibikova et al,29 for each CpG site a 2-sample t test was performed comparing the 7 severe preeclampsia samples to the 5 normal samples with respect to proportion methylated. Specifically, for CpG sites i = 1, …, 27 578, the hypotheses tested were:
Hypothesis 0:
Hypothesis A:
The P values from the 2-sample t tests were obtained for all CpG sites. The P value is the probability of obtaining a test statistic as or more extreme as the one observed under the conditions of the null hypothesis. In most scientific endeavors, an α = .05 threshold is customarily applied so that a P value <.05 is considered as evidence for the alternative hypothesis, that is, P <.05 typically indicates a significant finding. However, application of α = .05 threshold to univariable tests of significance in high-throughput genomic settings will yield a large number of Type I errors simply by chance. When analyzing high-throughput genomic data, the multiple comparison problem is most often addressed through estimation of the false discovery rate (FDR). To control for multiple hypothesis testing, the P values were subsequently used in estimating the FDR using the q value method.30 More specifically, when testing m null hypotheses, we may want to control the number of rejected null hypotheses that are truly null (F) among all those null hypotheses that are rejected (S). The q value is such a method that for a particular feature is defined to be the expected proportion of false positives among all features as or more extreme than the one observed.30 For a given P value threshold t where 0 ≤ t ≤ 1, we want to estimate FDR(t) = E (F(t)/S(t)) ≈ E(F(t))/E(S(t)). F(t) represents the number of false discoveries at threshold t while S(t) represents the number of null hypotheses considered significant at threshold t. Formally, FDR(t) is estimated as
where π0 represents the proportion of hypotheses for which the null is true and I (P g ≤ t) is 1 if the P value for the gth CpG site is less than or equal to the threshold t. For application data sets, π0 is typically unknown and so is estimated using the distribution of raw P values using either a smoothing or A bootstrap method. In this study, CpG sites with a FDR<10% were considered significant.
A sample size calculation based on a preliminary analysis indicated that an n = 6 would have a power of 80% to detect a difference in average β values between normal pregnant and preeclamptic patients of at least 0.10, assuming a common standard deviation (sd) of 0.03 (the median SD for CpG sites) with a 2-sided α level of .001.
For CpG sites identified as significant, the CpG sites were mapped to Gene Ontology terms (molecular function [MF], biological process [BP]) and a hypergeometric test was performed to determine whether any term was overrepresented among those CpG sites identified as significant compared to the number of CpG sites mapping to that term on the entire array. For MF and BP terms, a P value <.05 was considered significant.
Canonical Pathway and Network Identification
Genes with statistically differentially methylated CpG sites were imported into Ingenuity Pathways Analysis software (IPA, v9; Ingenuity systems, Redwood City, California) to identify canonical pathways and gene-to-gene interaction networks within our data set. Canonical pathways and gene networks were algorithmically constructed based on published literature in the Ingenuity Knowledge Base, where each relation was supported by at least one reference.
Results
Due to the role of methylation in X-chromosome inactivation, differential methylation patterns between males and females have been observed for CpG sites of genes located on the X chromosome and are used as quality control checks.31 Therefore, the quality of the hybridizations was examined by plotting the proportion methylated for the 6 X-linked genes (EFNB1, ELK1, FMR1, G6PD, GPC3, and GLA), which are expected to show hemi-methylation for females as an indication of good quality.29 Based upon this examination, there were no quality concerns for the 12 samples included in this study (Figure 1). There were also no quality concerns for the methylated and unmethylated Jurkat cell DNA run as controls. As an additional validation, COBRA quantification of methylation was used for the −1538 CpG site in the MMP1 promoter, which we previously showed to have reduced methylation associated with increased expression of MMP-1 in amnions of patients with preterm premature rupture of the membranes.28 COBRA quantification results were directionally similar to the BeadChip assay findings. Methylation by COBRA of an omental artery from a normal pregnant woman was 63%, and methylation of an omental artery from a preeclamptic woman was 52% for a Δβ of −0.11. This compared favorably with the BeadChip analysis of the −1298 CpG site for MMP1 of 81.6% for normal pregnancy versus 73.6% for preeclamptic pregnancy for a Δβ of −0.08 (P = .016, FDR = 0.1069). Although the methylation percentage of the −1538 CpG site varied from that of the −1298 CpG site, this is not unexpected because methylation percentage varies for CpG sites in the MMP1 promoter.28
Figure 1.

Box plots of proportion methylated (β-values) in omental arteries for the 6 X-linked genes examined for quality assessment of hybridization. A β-value of 1 indicates complete methylation and a β-value of 0 indicates no methylation. There were no quality concerns.
Unsupervised hierarchical clustering using Euclidean distance and Ward’s method was applied to determine whether the samples naturally clustered by diagnosis. The methylation matrix was first filtered to include CpG sites having a mean proportion methylated across all samples of β >0.15 and a standard deviation across all samples among the top 90% of standard deviations, leaving 1233 CpG sites. As such, the filtering procedure did not take any phenotypic data (eg, class labels such as normal and preeclampsia) into account so that unsupervised hierarchical clustering could be applied. Unsupervised hierarchical clustering (Figure 2) revealed natural clustering of the samples by diagnosis, normal pregnant versus preeclamptic and natural clustering of the CpG sites by methylation status, reduced methylated versus relatively more methylated.
Figure 2.
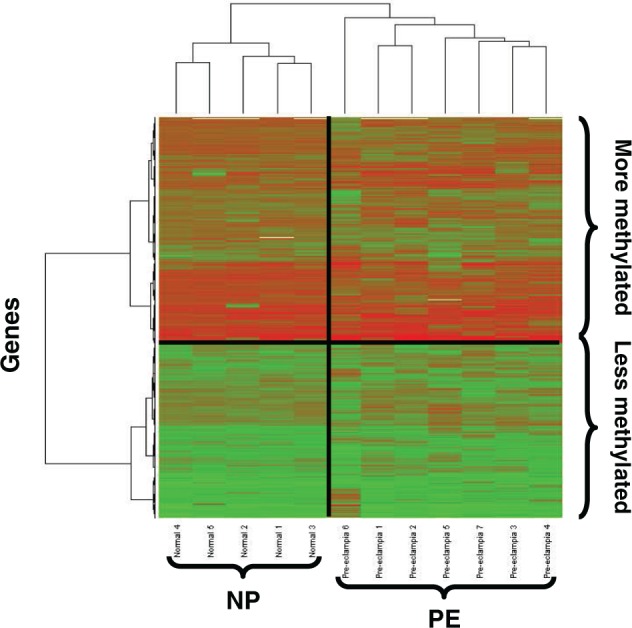
Heatmap of unsupervised hierarchical clustering. Unsupervised hierarchical clustering revealed natural grouping by diagnosis and methylation status. NP indicates normal pregnant, PE, preeclampsia, red indicates hypermethylated CpG sites, green indicates hypomethylated CpG sites. Please see online version for color reference.
Statistical analysis revealed 4184 CpG sites, corresponding to 3736 genes, with significant differential methylation when comparing between normal pregnant and preeclamptic omental arteries at P value of less than .05. This is considerably greater than what is expected by chance alone. Of these CpG sites, 1771 CpG sites, mapped to 1685 genes, had an FDR of less than 10% (Supplementary Table 1) and 237 CpG sites, representing 236 genes, had an FDR of less than 5%. Interestingly, many of these genes were related to inflammation. Of the 237 CpG sites, 155 CpG sites (∼65%) had reduced methylation in preeclamptic omental arteries. For each CpG detected in the BeadChip, the distance from the TSS, and whether it was located within a CpG island, was provided by Illumina. Transcription factor binding sites are often located within the nucleosome free region of the gene promoter.32 The position of the CpG sites relative to nucleosomes was not provided in this assay. However, one could infer that the nucleosome-free region is most likely 200 base pairs upstream of the TSS.33 The median distance from TSS for the 237 CpG sites was −299 (mean = −335, SD = −333, minimum = −3, maximum = −1487), which was significantly different from the median distance from the TSS for the remaining CpG sites on the BeadChip, which had a median distance from the TSS of 275 (P = .03, Wilcoxon rank sum test). Also, of these CpG sites, 112 CpG sites (∼47%) were located within CpG islands. Interestingly, 67% of CpG sites that had significantly reduced methylation were not located in CpG islands, whereas 91% of CpG sites that were significantly more methylated were located in CpG islands. When using an FDR of 10% and identifying 1771 CpG sites as significant, we would expect 177 to potentially be false discoveries. Likewise, when using an FDR of 5% and identifying 237 CpG sites as significant, we would only expect 12 to potentially be false discoveries. When data were analyzed more stringently to an FDR of less than 5% and difference in methylation of more than 0.10, 65 genes were identified, all of which were less methylated in preeclamptic women.
To visualize the distribution of the CpG sites based on their biological and statistical significance, a volcano plot was created by plotting the difference between average proportion methylated for normal pregnant and preeclamptic women for all 27 578 CpG sites analyzed by the BeadChip on the X axis, and the negative log10 transformed P values from the 2 class comparison analysis performed for each CpG site on the Y axis. The plot was divided into 4 quadrants by the line X = 0, which divided the CpG sites into CpG sites that are less methylated in preeclamptic omental arteries compared to normal pregnant counterparts on the left and CpG sites that were more methylated in preeclamptic omental arteries on the right, and the line Y= −log10 (P value equivalent to FDR of 10%) which divided the CpG sites that were significantly differentially methylated above that line and CpG site with no significant difference in methylation below the line (Figure 3). The larger population of CpG sites that was observed in the upper left quadrant of the plot indicated preeclamptic samples had a significant decrease in their DNA methylation compared to normal pregnant.
Figure 3.
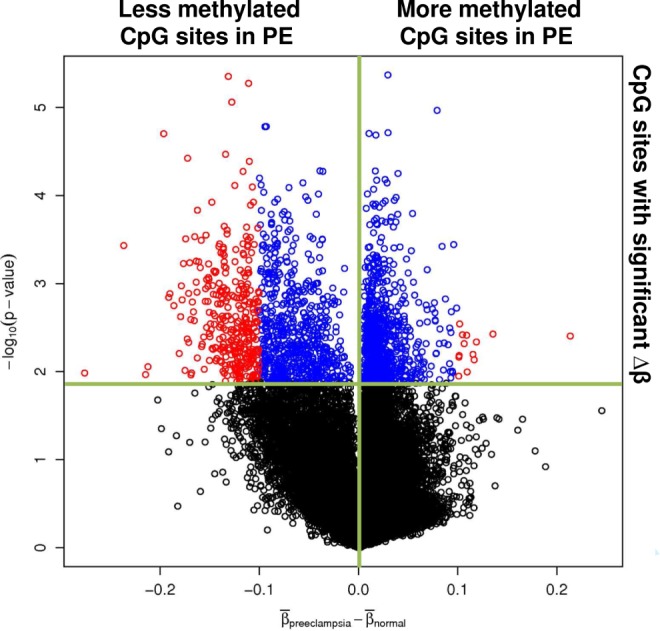
Volcano plot comparing severe preeclampsia versus normal pregnancy. The difference in average proportion methylated between severe preeclampsia and normal pregnancy is plotted on the x-axis and –log10 (P value) is plotted on the y-axis. Blue points represent CpG sites significant using an FDR of 10%; red points represent CpG sites significant using an FDR of 10% and having a difference between the 2 groups exceeding |0.10|. FDR indicates false discovery rate. Please see online version for color reference.
When genes with differentially methylated CpG sites at FDR cutoff of 0.05 were mapped to gene ontology for MFs and BPs, 75 MFs and 149 BPs were overrepresented in the preeclamptic vessels (P < .05), many of which were pertinent to the pathophysiology of preeclampsia (Table 2).
Table 2.
List of Molecular Functions and Biological Processes That Were Overrepresented in Preeclamptic Blood Vessels and Are Pertinent to the Pathophysiology of Preeclampsia
| Biological Processes | Molecular Functions |
|---|---|
| Activation of Rac GTPase activity | Amino acid transmembrane transporter activity |
| Cellular glucan metabolic process | Beta-galactosidase activity |
| GDP-l-fucose metabolic process | CCR chemokine receptor binding |
| GDP-mannose metabolic process | CCR2 chemokine receptor binding |
| Glucan metabolic process | CXCR chemokine receptor binding |
| Glycogen metabolic process | Cyclase inhibitor activity |
| Hemostasis | Galactose binding |
| Histidine catabolic process | Galactosidase activity |
| Inactivation of MAPK activity | GDP-mannose 4,6-dehydratase activity |
| l-amino acid transport | Glycogen (starch) synthase activity |
| Leukocyte migration during inflammatory response | Glycogen phosphorylase activity |
| Negative regulation of cGMP production | Glycosylceramidase activity |
| Negative regulation of MAP kinase activity | GTPase regulator activity |
| Negative regulation of nitric oxide production | Guanylate cyclase inhibitor activity |
| Negative regulation of phosphoinositide 3-kinase cascade | Heparanase activity |
| Negative regulation of vasodilation | Interferon-γ receptor activity |
| Positive regulation of calcium ion transport | Interleukin 8 receptor binding |
| Positive regulation of calcium ion transport into cytosol | Lactase activity |
| Positive regulation of GTPase activity | Phosphoprotein phosphatase inhibitor activity |
| Positive regulation of interferon-α production | vitamin E binding |
| Positive regulation of interferon-β production | |
| Positive regulation of Rac GTPase activity | |
| Positive regulation of vasoconstriction | |
| vitamin E metabolic process |
Abbreviations: GTPase, guanosine triphosphate hydrolase; GDP, guanosine diphosphate; CCR, C-C chemokine receptor; CXCR, C-X-C chemokine receptor; MAPK, mitogen-activated protein kinase; cGMP, cyclic guanosine monophosphate.
Genes with differentially methylated CpG sites at an FDR cutoff of 0.1 were analyzed with IPA software to assess the interconnectivity between these genes and other related genes within a canonical pathway or a gene-to-gene interaction network which may indicate functional importance of these genes in the biology of the disease. Twenty-five significant canonical pathways (P < .05) were identified (Table 3). Some of these pathways were associated with inflammation, such as retinoid X receptor (RXR) inhibition, peroxisome proliferator–activated receptor (PPAR) signaling, and high-mobility group box chromosomal protein 1 (HMGB1) signaling. Using IPA network analysis, 50 gene-to-gene interaction networks were identified. Four of them included inflammatory response, hematological disease, inflammatory disease, and/or cardiovascular disease among their top 3 associated biological functions (P < .005). A network is shown in Figure 4 for inflammatory genes.
Table 3.
Canonical Pathways Identified by Ingenuity Pathway Analysis Software at a P Value <.05
| Ingenuity Canonical Pathways | Molecules |
|---|---|
| LPS/IL-1-mediated inhibition of RXR function | IL18RAP, ABCG5, IL1RL1, APOC4, GSTA5, ABCG1, FMO5, NR0B2, ALDH1A3, NR1I2, CYP3A7, ACSL5, GSTM4, FABP1, CHST11, FABP4, ACSL4, CHST10, FMO1, ALDH3A1, IL1RAP, FMO3, MYD88, MGMT, CHST12, IL1R1, ACSBG2, ALDH9A1, TLR4, NR1H2, MGST2, SULT1A1, CPT2, SULT1B1, MGST3 |
| Role of cytokines in mediating communication between immune cells | IL1A, IL5, IFNA8, IL10, IL27, IL1F10, IFNA16, IL24, IL25, IL17A, IL1F9, IFNA21, IL4 |
| IL-10 signaling | IL18RAP, MAP3K14, IL1A, IL1RL1, IL10, FCGR2A, NFKBIE, IL1F10, MAPK13, IL1R1, NFKB2, FOS, IL1F9, SP1, IL1RAP |
| LXR/RXR activation | IL18RAP, ABCG5, IL1A, MSR1, IL1RL1, APOC4, ABCG1, NFKB2, IL1F10, IL1R1, TLR4, IL1F9, NR1H2, PTGS2, NCOR2, IL1RAP |
| Mismatch Repair in Eukaryotes | PCNA, MSH6, FEN1, RFC1, MLH1, EXO1 |
| Aryl hydrocarbon receptor signaling | SRC, IL1A, TP73, NQO2, GSTA5, NFKB2, CCND1, ALDH9A1, AHRR, FOS, RB1, HSP90B1, CCND2, SP1, MGST2, NR0B2, ALDH1A3, GSTM4, NCOR2, ALDH3A1, CDK2, MGST3 |
| PPAR signaling | IL18RAP, MAP3K14, IL1A, IL1RL1, RRAS, NFKBIE, CREBBP, IL1F10, IL1R1, NFKB2, FOS, HSP90B1, IL1F9, NR0B2, PTGS2, NCOR2, IL1RAP |
| Altered T cell and B cell signaling in rheumatoid arthritis | MAP3K14, IL1A, IL10, LTB, IL1F10, NFKB2, TNFRSF17, IL17A, TLR4, IL1F9, LTA, TLR6, CCL21, FCER1G, IL4 |
| Xenobiotic metabolism signaling | IL1A, MAP3K11, NQO2, GSTA5, MAPK13, FMO5, CEL, HSP90B1, CAMK2A, UGT2B4, NR1I2, CYP3A7, ALDH1A3, GSTM4, CHST11, CHST10, FMO1, PRKD3, ALDH3A1, MAP3K14, FMO3, RRAS, MGMT, CREBBP, CHST12, NFKB2, UGT1A1, ALDH9A1, AHRR, MGST2, SULT1A1, NCOR2, MAP2K5, SULT1B1, MGST3 |
| Glycosphingolipid biosynthesis—neolactoseries | ST8SIA4, FUT5, ABO, UGT2A3, GCNT2, B3GNT1, ST3GAL5 |
| IL-1 signaling | MAP3K14, IL1A, MYD88, NFKBIE, MAPK13, IL1R1, NFKB2, GNG7, FOS, GNAT2, PRKACG, GNAO1, GNB1 L, IL1RAP, GNG4 |
| HMGB1 signaling | IL1A, RRAS, DIRAS3, RBBP7, MAPK13, IL1R1, NFKB2, FOS, TLR4, KAT2B, RND3, SP1, RHOD, AKT3, MAP2K5 |
| Hepatic cholestasis | IL18RAP, MAP3K14, CYP7B1, ABCG5, IL1A, IL1RL1, MYD88, NFKBIE, NFKB2, IL1F10, IL1R1, TLR4, IL1F9, ABCB4, NR0B2, NR1I2, PRKACG, HNF4A, IL1RAP, PRKD3, FGF19 |
| Starch and sucrose metabolism | PLA2R1, ENPP5, MTAP, UGT1A1, LTK, GYS2, ENPP3, HK1, DDX6, PYGM, UGT2B4, GCK, SLC3A1, AMY2B |
| CCR5 signaling in macrophages | CD3G, FOS, PLCG2, FCER1G, MAPK13, CCL5, GNB1 L, PRKD3, CD3D, GNG4, GNG7 |
| Parkinson's signaling | CASP3, PARK7, CYCS, MAPK13, SNCA |
| FXR/RXR activation | IL1A, ABCG5, CREBBP, IL1F10, IL1F9, UGT2B4, ABCB4, NR0B2, CYP19A1, NR1I2, AKT3, FBP1, HNF4A, FGF19 |
| Biosynthesis of steroids | FDPS, CYP7B1, NQO2, FNTB, SC5DL, GGPS1 |
| DNA methylation and transcriptional repression signaling | MECP2, RBBP7, SIN3A, SAP18, RBBP4 |
| Role of hypercytokinemia/hyperchemokinemia in the Pathogenesis of Influenza | IL1F9, IL1A, IFNA8, IFNA21, IL1F10, CCL5, IFNA16, IL17A |
| Nicotinate and nicotinamide metabolism | DAPK1, CDK7, ENPP5, QPRT, TTK, VNN3, BRAF, ENPP3, NAPRT1, VNN2, PRKAA2, NMNAT2, CD38, MAK, CDK2 |
| Role of macrophages, fibroblasts, and endothelial cells in rheumatoid arthritis | IL18RAP, IL1A, SFRP2, IL1RL1, NFKBIE, MMP13, LTB, IL1F10, CCL5, CCND1, KLK11, IL1F9, CAMK2A, TMPRSS3, SFRP5, AKT3, IL1RAP, PRKD3, MMP1, KLK10, SRC, MAP3K14, IL10, RRAS, MYD88, C1 S, IL1R1, NFATC4, IL7, APC, IL17A, PLCZ1, TLR4, FOS, FZD4, LTA, PLCG2, GNAO1, TLR6 |
| Assembly of RNA polymerase I complex | POLR1C, UBTF, TAF1B |
| Fructose and mannose metabolism | HK1, ALDOB, UGT2A3, GCK, FBP1, PFKFB2, LTK, PMM2, MPI |
| Airway inflammation in asthma | IL5, IL4 |
Abbreviations: PPAR, peroxisome proliferator–activated receptor; RXR, retinoid X receptor inhibition.
Figure 4.

A gene-to-gene network identified by IPA analysis in our data set. The network includes multiple inflammatory genes, which were hypomethylated in preeclampsia (green) and show the direct (indicated by solid line) and indirect (indicated by broken line) interaction between these genes and other genes in the network. The figure also shows the interaction between genes in the network with canonical pathways related to inflammation. IPA indicates Ingenuity Pathways Analysis. Please see online version for color reference.
Discussion
In this study, we used a high-throughput assay to investigate the global DNA methylation profiles in omental arteries from normal pregnant women and women with severe preeclampsia. We identified 3736 genes with significantly differentially methylated CpG sites between the 2 groups, which is significantly greater than expected by chance alone. Unsupervised hierarchical clustering revealed natural clustering by diagnosis demonstrating that DNA methylation profiles distinguished each group. Volcano plot demonstrated that the most significant differences in DNA methylation of CpG sites were “hypomethylated” genes in preeclamptic women. Of genes with significant methylation differences, 236 were significant at an FDR of <5%. When data were analyzed more stringently to FDR of 5% and difference in methylation of >0.10, 65 genes were identified, all of which had lower levels of methylation in preeclampsia.
The thromboxane synthase gene (TBXAS1) was the most hypomethylated gene in preeclamptic women. TBXAS1 encodes an enzyme that catalyzes the isomerization of prostaglandin H2 into thromboxane.34 Preeclampsia is associated with an imbalance of prostanoids: increased thromboxane and decreased prostacyclin, which was first described in the placenta,35 and later confirmed for maternal blood,36 and maternal urine.37 This imbalance provides a plausible explanation for hypertension, reduced uteroplacental blood flow, and hypercoagulopathy observed in women with preeclampsia because thromboxane is a potent vasoconstrictor and platelet activator, whereas prostacyclin is a potent vasodilator and inhibitor of platelet activation.38 Our findings suggest epigenetic regulation of the TBXAS1 gene because we found that reduced promoter methylation is associated with increased expression in omental arteries of preeclamptic women.39
Another gene that was significantly less methylated in our analysis was MMP1 (P = .016, FDR = 0.1069, Δβ = −0.08). This gene also appears to be epigenetically regulated because its reduced methylation is associated with its increased expression in omental arteries of preeclamptic women,40 and experimentally induced hypomethylation results in its increased expression in human vascular smooth muscle cells in culture.41 Interestingly, increased expression of MMP-1 is also associated with reduced DNA methylation in amnion of patients with preterm premature rupture of the membranes.28
The thromboxane synthase gene was the most significantly hypomethylated gene in preeclamptic women with a Δβ = −0.24. However, a more significant difference in methylation does not necessarily mean it is more important. The Δβ for MMP-1 was −0.08, but the increase in gene and protein expression for MMP-1 in omental arteries of preeclamptic women40 was just as great as for thromboxane synthase.39
Normal pregnancy is associated with mild systemic inflammatory changes which increase as pregnancy progresses.42 These changes include increased plasma levels of fibrinogen,43 plasminogen activator inhibitor type 1,44 ceruloplasmin,45 interleukin 6,46 and tumor necrosis factor-α.47 ,48 They also include leukocytosis,49 leukocyte activation,50–52 increased circulating markers of oxidative stress such as lipid hydroperoxides and malondialdehyde,53 and hypertriglyceridemia.54 All these inflammatory changes are exacerbated in preeclampsia.55 This suggests that preeclampsia may arise due to an inability of maternal homeostasis to adapt to a pregnancy-induced systemic inflammation. In our study, many inflammation-related genes were significantly hypomethylated in preeclamptic women as compared to normal pregnant women. DNA hypomethylation is generally associated with increased gene expression, so reduced methylation may place a woman in a more responsive state to inflammatory stimuli.
Ingenuity pathway analysis demonstrated that genes with significant changes in methylation in preeclamptic women were associated with multiple canonical pathways related to inflammation and immune response. These pathways included inhibition of RXR function and PPAR signaling. Both PPARs and RXRs help regulate inflammatory responses in endothelial and vascular smooth muscle cells. Activation of these receptors results in the formation of PPAR/RXR heterodimers which exert anti-inflammatory effects in these cells.56 Preeclampsia is associated with inflammation and dysfunction of maternal systemic endothelial and vascular smooth muscle cells,57 so the inhibition of these pathways may contribute to the pathophysiology of preeclampsia. High-mobility group box chromosomal protein 1 signaling is another canonical pathway identified by IPA software in our data set. High-mobility group box chromosomal protein 1 is a nuclear transcription factor that possesses potent cytokine activity when released from innate immune cells, mediating the late response to inflammation58 through toll-like receptors 4 and 2.59 High-mobility group box chromosomal protein 1 is also released from cultured human vascular smooth muscle cells in response to C-reactive protein,60 which is elevated in preeclampsia.61 ,62 High-mobility group box chromosomal protein 1 causes increased expression of intercellular adhesion molecule 1 (ICAM-1) and interleukin 8 (IL-8) in cultured human endothelial cells63 and increases expression of C-reactive protein, MMP-2 and MMP-9, in cultured human vascular smooth muscles.60 These changes are similar to what is found in preeclampsia where the expression of ICAM-1 and IL-8 are increased in endothelial cells of maternal systemic blood vessels,64 and levels of C-reactive protein,61 ,62 MMP-265 and MMP-966 are elevated in the plasma. This suggests that hypomethylation related to HMGB1 signaling may be involved in the pathophysiology of preeclampsia. Analysis of IPA also indicated that genes in our data set were involved in gene-to-gene interaction networks, which had inflammatory response, inflammatory disease, and cardiovascular disease among their top associated biological functions. These relationships suggest that DNA methylation may contribute to preeclampsia by influencing multiple pathways and gene-to-gene networks related to inflammation in a woman’s systemic blood vessels.
When genes with an FDR of <5% were mapped for MFs and BFs, 75 MFs and 149 BFs were overrepresented in preeclampsia representing functions and pathways pertinent to pathophysiology. These included smooth muscle contraction, thrombosis, inflammation, redox homeostasis, sugar metabolism, and amino acid metabolism. These are particularly relevant to preeclampsia because it is associated with increased vascular resistance, increased arterial pressure,67 reduced uteroplacental blood flow,68 hypercoagulopathy,69 inflammation,55 oxidative stress,16 ,17 and altered carbohydrate and amino acid metabolism.70
Preexisting maternal genetic variation in key genes may also have an impact on DNA methylation. For example, allelic variations in the methylenetetrahydrofolate reductase gene have been associated with preeclampsia.71 Methylenetetrahydrofolate reductase is an important enzyme regulating the synthesis of S-adenosylmethionine that provides methyl groups for DNA methylation.72 S-adenosylmethionine synthase is also inhibited by oxidative stress, which reduces the availability of glutathione necessary for its activity.72
The changes in methylation observed may have been preexisting or the result of preeclampsia. If they were preexisting, they could have contributed to an increased risk of preeclampsia. The changes could also be the result of the oxidative stress of preeclampsia because oxidative stress and DNA oxidation can cause a loss in DNA methylation.8 ,72,73 Once the DNA pattern is altered, it is maintained by DNA methyltransferase 1 during future cell divisions, so alterations in DNA methylation occurring as a result of preeclampsia may contribute to the increased risk of cardiovascular disease later in life experienced by women who have had preeclampsia. Additional alterations in the DNA methylation pattern may occur after preeclampsia because many factors affect it, such as age, diet, environmental toxins, and oxidative stress.6 –8
Our study had some methodological limitations. The number of samples analyzed was limited due to the difficulty in obtaining omental arterial samples. There was also a difference in gestational ages between groups. This was inevitable because we evaluated patients with severe preeclampsia, which is a leading cause of preterm delivery. There are no studies that address the effect of gestational age on DNA methylation to suggest that a difference of 5 weeks could account for the differences in methylation patterns we observed. In addition, although the Illumina Human Methylation27 platform is a reliable way to assess the methylation status of large numbers of CpG sites in the promoter region of genes,29 it does not cover all CpG sites in the human genome. Methylation of CpG sites outside of the gene promoter regions are also likely important. Consequently, some important epigenetic alterations may have been overlooked in our analysis. Another limitation is the cellular heterogeneity of omental arteries, which did not allow us to specify the cell type where methylation changes were occurring. Because of the extensive infiltration of neutrophils into the mother’s blood vessels in preeclampsia,57 ,64,74 neutrophils are just as likely to be the source of the DNA methylation changes as are endothelial or vascular smooth muscle cells.
In summary, our study is the first to compare DNA methylation profiles in systemic blood vessels of normal pregnant and preeclamptic women. Reduced methylation of inflammatory genes was prominent in omental arteries of preeclamptic women. Genes with significant differential methylation were involved in multiple pathways and gene-to-gene networks pertinent to the clinical features of preeclampsia. It is possible that epigenetic changes could be both a cause and an effect of preeclampsia because DNA methylation is affected by factors, such as diet, obesity, and oxidative stress.8 ,72,73 It is possible that these factors reduce DNA methylation prior to pregnancy and thereby increase the risk of getting preeclampsia. On the other hand, vascular infiltration of neutrophils and oxidative stress during preeclampsia may reduce DNA methylation in the blood vessels of the mother, leading to increased risk of cardiovascular disease later in life.75 Prospective and follow-up studies will be necessary to more fully evaluate the role of DNA methylation as a cause of preeclampsia and its follow-up sequelae.
Our study may have clinical implications for the treatment of preeclampsia. Folate is an essential vitamin for the synthesis of methyl donors utilized by methyltransferases to methylate DNA. Therefore, dietary supplementation with folate might restore reduced methylation in inflammatory genes and protect against the development of preeclampsia. In this regard, a large study of almost 3000 pregnant women found supplementation with multivitamins containing folic acid was associated with reduced risk of preeclampsia.76
Acknowledgments
We greatly appreciate the help of Sonya Washington and the faculty, residents, and staff of labor and delivery in obtaining omental fat samples.
Footnotes
Author’s Note: The online data supplements are available at http://rsx.sagepub.com/supplemental.
Declaration of Conflicting Interests: The authors declare no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding: The author(s) disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: This research was supported in part by grants from the NHLBI, RO1 HL069851 (SWW) and the NCMHD P60 MD002256 (JFS). Methylation analysis was performed at the VCU Nucleic Acids Research Facility core laboratory, which is supported, in part, by funding from NIH-NCI Cancer Center Support Grant (P30 CA016059). Dr. Estrada-Gutierrez was supported by Fogarty grant 1D43 TW007692.
References
- 1. Cunningham FG, Leveno KJ, Bloom SL, Gilstrap III LC, Hauth JC, Wenstrom KD. Williams Obstetrics, 22nd ed New York, NY: McGraw-Hill; 2005 [Google Scholar]
- 2. Roberts JM, Gammill HS. Preeclampsia: recent insights. Hypertension. 2005;46(6):1243–1249 [DOI] [PubMed] [Google Scholar]
- 3. Pridjian G, Puschett JB. Preeclampsia. Part 2: experimental and genetic considerations. Obstet Gynecol Surv. 2002;57(9):619–640 [DOI] [PubMed] [Google Scholar]
- 4. Williams PJ, Pipkin FB. The genetics of pre-eclampsia and other hypertensive disorders of pregnancy. Best practice & research. Clin obstet Gynaecol. 2011;25(4):405–417 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Wilson CB, Makar KW, Shnyreva M, Fitzpatrick DR. DNA methylation and the expanding epigenetics of T cell lineage commitment. Semin Immunol. 2005;17(2):105–119 [DOI] [PubMed] [Google Scholar]
- 6. Fraga MF, Esteller M. Epigenetics and aging: the targets and the marks. Trends Genet. 2007;23(8):413–418 [DOI] [PubMed] [Google Scholar]
- 7. Tost J. DNA methylation: an introduction to the biology and the disease-associated changes of a promising biomarker. Mol Biotechnol. 2010;44(1):71–81 [DOI] [PubMed] [Google Scholar]
- 8. Franco R, Schoneveld O, Georgakilas AG, Panayiotidis MI. Oxidative stress, DNA methylation and carcinogenesis. Cancer Lett. 2008;266(1):6–11 [DOI] [PubMed] [Google Scholar]
- 9. Zaina S, Lindholm MW, Lund G. Nutrition and aberrant DNA methylation patterns in atherosclerosis: more than just hyperhomocysteinemia? J Nutr. 2005;135(1):5–8 [DOI] [PubMed] [Google Scholar]
- 10. Paulsen M, Ferguson-Smith AC. DNA methylation in genomic imprinting, development, and disease. J Pathol. 2001;195(1):97–110 [DOI] [PubMed] [Google Scholar]
- 11. Agrawal A, Murphy RF, Agrawal DK. DNA methylation in breast and colorectal cancers. Mod Pathol. 2007;20(7):711–721 [DOI] [PubMed] [Google Scholar]
- 12. Connor CM, Akbarian S. DNA methylation changes in schizophrenia and bipolar disorder. Epigenetics. 2008;3:55–58 [DOI] [PubMed] [Google Scholar]
- 13. van der Linden IJ, Heil SG, van Egmont Petersen M, van Straaten HW, den Heijer M, Blom HJ. Inhibition of methylation and changes in gene expression in relation to neural tube defects. Birth Defects Res A Clin Mol Teratol. 2008;82(10):676–683 [DOI] [PubMed] [Google Scholar]
- 14. Dong C, Yoon W, Goldschmidt-Clermont PJ. DNA methylation and atherosclerosis. J Nutr. 2002;132:2406S–2409 S. [DOI] [PubMed] [Google Scholar]
- 15. Baccarelli A, Wright R, Bollati V, Litonjua A, Zanobetti A, Tarantini L, Sparrow D, Vokonas P, Schwartz J. Ischemic heart disease and stroke in relation to blood DNA methylation. Epidemiology. 2010;21(6):819–828 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Hubel CA. Oxidative stress in the pathogenesis of preeclampsia. Proc Soc Exp Biol Med. 1999;222(3):222–235 [DOI] [PubMed] [Google Scholar]
- 17. Walsh SW. Maternal-placental interactions of oxidative stress and antioxidants in preeclampsia. Semin Reprod Endocrinol. 1998;16(1):93–104 [DOI] [PubMed] [Google Scholar]
- 18. Walsh SW. Obesity: a risk factor for preeclampsia. Trends in endocrinology and metabolism: TEM. 2007;18(10):365–370 [DOI] [PubMed] [Google Scholar]
- 19. Wang JX, Knottnerus AM, Schuit G, Norman RJ, Chan A, Dekker GA. Surgically obtained sperm, and risk of gestational hypertension and pre-eclampsia. Lancet. 2002;359(9307):673–674 [DOI] [PubMed] [Google Scholar]
- 20. Yu L, Chen M, Zhao D, et al. The H19 gene imprinting in normal pregnancy and pre-eclampsia. Placenta. 2009;30(5):443–447 [DOI] [PubMed] [Google Scholar]
- 21. Chelbi ST, Vaiman D. Genetic and epigenetic factors contribute to the onset of preeclampsia. Mol Cell Endocrinol. 2008;282(1-2):120–129 [DOI] [PubMed] [Google Scholar]
- 22. Chelbi ST, Mondon F, Jammes H, et al. Expressional and epigenetic alterations of placental serine protease inhibitors: SERPINA3 is a potential marker of preeclampsia. Hypertension. 2007;49(1):76–83 [DOI] [PubMed] [Google Scholar]
- 23. Kanayama N, Takahashi K, Matsuura T, et al. Deficiency in p57Kip2 expression induces preeclampsia-like symptoms in mice. Mol Hum Reprod. 2002;8(12):1129–1135 [DOI] [PubMed] [Google Scholar]
- 24. Yuen RK, Penaherrera MS, von Dadelszen P, McFadden DE, Robinson WP. DNA methylation profiling of human placentas reveals promoter hypomethylation of multiple genes in early-onset preeclampsia. Eur J Hum Genet. 2010;18(9):1006–1012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Clark SJ, Statham A, Stirzaker C, Molloy PL, Frommer M. DNA methylation: bisulphite modification and analysis. Nat Protoc. 2006;1:2353–2364 [DOI] [PubMed] [Google Scholar]
- 26. Grunau C, Clark SJ, Rosenthal A. Bisulfite genomic sequencing: systematic investigation of critical experimental parameters. Nucleic Acids Res. 2001;29(13):E65–5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Xiong Z, Laird PW. COBRA: a sensitive and quantitative DNA methylation assay. Nucleic Acids Res. 1997;25(12):2532–2534 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Wang H, Ogawa M, Wood JR, et al. Genetic and epigenetic mechanisms combine to control MMP1 expression and its association with preterm premature rupture of membranes. Hum Mol Genet. 2008;17(8):1087–1096 [DOI] [PubMed] [Google Scholar]
- 29. Bibikova M, Lin Z, Zhou L, et al. High-throughput DNA methylation profiling using universal bead arrays. Genome Res. 2006;16(3):383–393 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Storey JD, Tibshirani R. Statistical significance for genomewide studies. Proc Natl Acad Sci U S A. 2003;100(16):9440–9445 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Weisenberger DJ, Berg DVD., Pan F, Berman BP, Laird PW. Comprehensive DNA Methylation Analysis on the Illumina Infinium Assay Platform. San Diego, CA: Illumina and Inc; 2008 [Google Scholar]
- 32. Li B, Carey M, Workman JL. The role of chromatin during transcription. Cell. 2007;128(4):707–719 [DOI] [PubMed] [Google Scholar]
- 33. Archer KJ, Mas VR, Maluf DG, Fisher RA. High-throughput assessment of CpG site methylation for distinguishing between HCV-cirrhosis and HCV-associated hepatocellular carcinoma. Mol Genet Genomics. 2010;283(4):341–349 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Tanabe T, Ullrich V. Prostacyclin and thromboxane synthases. J Lipid Mediat Cell Signal. 1995;12:243–255 [DOI] [PubMed] [Google Scholar]
- 35. Walsh SW. Preeclampsia: an imbalance in placental prostacyclin and thromboxane production. Am J Obstet Gynecol. 1985;152(3):335–340 [DOI] [PubMed] [Google Scholar]
- 36. Chavarria ME, Lara-Gonzalez L, Gonzalez-Gleason A, Garcia-Paleta Y, Vital-Reyes VS, Reyes A. Prostacyclin/thromboxane early changes in pregnancies that are complicated by preeclampsia. Am J Obstet Gynecol. 2003;188(4):986–992 [DOI] [PubMed] [Google Scholar]
- 37. Mills JL, DerSimonian R, Raymond E, et al. Prostacyclin and thromboxane changes predating clinical onset of preeclampsia: a multicenter prospective study. JAMA. 1999;282(4):356–362 [DOI] [PubMed] [Google Scholar]
- 38. Walsh SW. Eicosanoids in preeclampsia. Prostaglandins Leukot Essent Fatty Acids. 2004;70:223–232 [DOI] [PubMed] [Google Scholar]
- 39. Mousa AA, Strauss III JF, Walsh SW. Reduced methylation of thromboxane synthase gene is correlated with its increased vascular expression in preeclampsia. Hypertension. 2012;59(6):1249–1255 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Estrada-Gutierrez G, Cappello RE, Mishra N, Romero R, Strauss JF, 3rd, Walsh SW. Increased expression of matrix metalloproteinase-1 in systemic vessels of preeclamptic women: a critical mediator of vascular dysfunction. Am J Pathol. 2011;178(1):451–460 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Cappello R, Estrada-Gutierrez G, Gerk PM, Strauss III JF, Walsh SW. Epigenetic control of collagen regulating genes in vascular smooth muscle. Reprod Sci. 2008;15(supplement):73A [Google Scholar]
- 42. Redman CW, Sacks GP, Sargent IL. Preeclampsia: an excessive maternal inflammatory response to pregnancy. Am J Obstet Gynecol. 1999;180(2 pt 1):499–506 [DOI] [PubMed] [Google Scholar]
- 43. Gatti L, Tenconi PM, Guarneri D, et al. Hemostatic parameters and platelet activation by flow-cytometry in normal pregnancy: a longitudinal study. Int J Clin Lab Res. 1994;24(4):217–219 [DOI] [PubMed] [Google Scholar]
- 44. Halligan A, Bonnar J, Sheppard B, Darling M, Walshe J. Haemostatic, fibrinolytic and endothelial variables in normal pregnancies and pre-eclampsia. Br J Obstet Gynaecol. 1994;101(6):488–492 [DOI] [PubMed] [Google Scholar]
- 45. Haram K, Augensen K, Elsayed S. Serum protein pattern in normal pregnancy with special reference to acute-phase reactants. Br J Obstet Gynaecol. 1983;90(2):139–145 [DOI] [PubMed] [Google Scholar]
- 46. Austgulen R, Lien E, Liabakk NB, Jacobsen G, Arntzen KJ. Increased levels of cytokines and cytokine activity modifiers in normal pregnancy. Eur J Obstet Gynecol Reprod Biol. 1994;57(3):149–155 [DOI] [PubMed] [Google Scholar]
- 47. Melczer Z, Banhidy F, Csomor S, et al. Influence of leptin and the TNF system on insulin resistance in pregnancy and their effect on anthropometric parameters of newborns. Acta Obstet Gynecol Scand. 2003;82(5):432–438 [PubMed] [Google Scholar]
- 48. Arntzen KJ, Liabakk NB, Jacobsen G, Espevik T, Austgulen R. Soluble tumor necrosis factor receptor in serum and urine throughout normal pregnancy and at delivery. Am J Reprod Immunol. 1995;34(3):163–169 [DOI] [PubMed] [Google Scholar]
- 49. Lurie S, Rahamim E, Piper I, Golan A, Sadan O. Total and differential leukocyte counts percentiles in normal pregnancy. Eur J Obstet Gynecol Reprod Biol. 2008;136(1):16–19 [DOI] [PubMed] [Google Scholar]
- 50. Barriga C, Rodriguez AB, Ortega E. Increased phagocytic activity of polymorphonuclear leukocytes during pregnancy. Eur J Obstet Gynecol Reprod Biol. 1994;57(1):43–46 [DOI] [PubMed] [Google Scholar]
- 51. Smarason AK, Gunnarsson A, Alfredsson JH, Valdimarsson H. Monocytosis and monocytic infiltration of decidua in early pregnancy. J Clin Lab Immunol. 1986;21(1):1–5 [PubMed] [Google Scholar]
- 52. Sacks GP, Studena K, Sargent K, Redman CW. Normal pregnancy and preeclampsia both produce inflammatory changes in peripheral blood leukocytes akin to those of sepsis. Am J Obstet Gynecol. 1998;179(1):80–86 [DOI] [PubMed] [Google Scholar]
- 53. Barden A. Circulating markers of oxidative stress are raised in normal pregnancy and pre-eclampsia. Br J Obstet Gynaecol. 1999;106(11):1232. [DOI] [PubMed] [Google Scholar]
- 54. Martin U, Davies C, Hayavi S, Hartland A, Dunne F. Is normal pregnancy atherogenic? Clin Sci (Lond). 1999;96(4):421–425 [DOI] [PubMed] [Google Scholar]
- 55. Borzychowski AM, Sargent IL, Redman CW. Inflammation and pre-eclampsia. Semin Fetal Neonatal Med. 2006;11(5):309–316 [DOI] [PubMed] [Google Scholar]
- 56. Plutzky J. The PPAR-RXR transcriptional complex in the vasculature: energy in the balance. Circ Res. 2011;108(8):1002–1016 [DOI] [PubMed] [Google Scholar]
- 57. Shah TJ, Walsh SW. Activation of NF-kappaB and expression of COX-2 in association with neutrophil infiltration in systemic vascular tissue of women with preeclampsia. Am J Obstet Gynecol. 2007;196(1):48 e1–8 [DOI] [PubMed] [Google Scholar]
- 58. Lotze MT, Tracey KJ. High-mobility group box 1 protein (HMGB1): nuclear weapon in the immune arsenal. Nat Rev Immunol. 2005;5(4):331–342 [DOI] [PubMed] [Google Scholar]
- 59. Yu M, Wang H, Ding A, et al. HMGB1 signals through toll-like receptor (TLR) 4 and TLR2. Shock. 2006;26(2):174–179 [DOI] [PubMed] [Google Scholar]
- 60. Inoue K, Kawahara K, Biswas KK, et al. HMGB1 expression by activated vascular smooth muscle cells in advanced human atherosclerosis plaques. Cardiovasc Pathol. 2007;16(3):136–143 [DOI] [PubMed] [Google Scholar]
- 61. Mihu D, Costin N, Mihu CM, Blaga LD, Pop RB. C-reactive protein, marker for evaluation of systemic inflammatory response in preeclampsia. Rev Med Chir Soc Med Nat Iasi. 2008;112(4):1019–1025 [PubMed] [Google Scholar]
- 62. Qiu C, Luthy DA, Zhang C, Walsh SW, Leisenring WM, Williams MA. A prospective study of maternal serum C-reactive protein concentrations and risk of preeclampsia. Am J Hypertens. 2004;17(2):154–160 [DOI] [PubMed] [Google Scholar]
- 63. Fiuza C, Bustin M, Talwar S, Tropea M, Gerstenberger E, Shelhamer JH, Suffredini AF. Inflammation-promoting activity of HMGB1 on human microvascular endothelial cells. Blood. 2003;101(7):2652–2660 [DOI] [PubMed] [Google Scholar]
- 64. Leik CE, Walsh SW. Neutrophils infiltrate resistance-sized vessels of subcutaneous fat in women with preeclampsia. Hypertension. 2004;44:72–77 [DOI] [PubMed] [Google Scholar]
- 65. Narumiya H, Zhang Y, Fernandez-Patron C, Guilbert LJ, Davidge ST. Matrix metalloproteinase-2 is elevated in the plasma of women with preeclampsia. Hypertens Pregnancy. 2001;20(2):185–194 [DOI] [PubMed] [Google Scholar]
- 66. Poon LC, Nekrasova E, Anastassopoulos P, Livanos P, Nicolaides KH. First-trimester maternal serum matrix metalloproteinase-9 (MMP-9) and adverse pregnancy outcome. Prenat Diagn. 2009;29(6):553–559 [DOI] [PubMed] [Google Scholar]
- 67. Khalil RA, Granger JP. Vascular mechanisms of increased arterial pressure in preeclampsia: lessons from animal models. Am J Physiol Regul Integr Comp Physiol. 2002;283(1):R29–45 [DOI] [PubMed] [Google Scholar]
- 68. Lunell NO, Nylund LE, Lewander R, Sarby B. Uteroplacental blood flow in pre-eclampsia measurements with indium-113m and a computer-linked gamma camera. Clin Exp Hypertens B. 1982;1(1):105–117 [DOI] [PubMed] [Google Scholar]
- 69. Perry KG., Jr., , Martin JN,, Jr Abnormal hemostasis and coagulopathy in preeclampsia and eclampsia. Clin Obstet Gynecol. 1992;35(2):338–350 [PubMed] [Google Scholar]
- 70. von Versen-Hoeynck FM, Powers RW. Maternal-fetal metabolism in normal pregnancy and preeclampsia. Front Biosci. 2007;12:2457–2470 [DOI] [PubMed] [Google Scholar]
- 71. Hill LD, York TP, Kusanovic JP, et al. Epistasis between COMT and MTHFR in maternal-fetal dyads increases risk for preeclampsia. PloS one. 2011;6(1):e16681. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Hitchler MJ, Domann FE. An epigenetic perspective on the free radical theory of development. Free Radic Biol Med. 2007;43(7):1023–1036 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Weitzman SA, Turk PW, Milkowski DH, Kozlowski K. Free radical adducts induce alterations in DNA cytosine methylation. Proc Natl Acad Sci U S A. 1994;91(4):1261–1264 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Mishra N, Nugent WH, Mahavadi S, Walsh SW. Mechanisms of enhanced vascular reactivity in preeclampsia. Hypertension. 2011;58(5):867–873 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Bellamy L, Casas JP, Hingorani AD, Williams DJ. Pre-eclampsia and risk of cardiovascular disease and cancer in later life: systematic review and meta-analysis. BMJ. 2007;335(7627):974. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. Wen SW, Chen XK, Rodger M, et al. Folic acid supplementation in early second trimester and the risk of preeclampsia. Am J Obstet Gynecol. 2008;198(1):45 e1–7 [DOI] [PubMed] [Google Scholar]